

Abstract
In the present study a possible role of glycosphingolipids (GSLs) in inducible nitric oxide synthase (iNOS) gene expression and nitric oxide (NO) production after spinal cord injury (SCI) in rats has been established. In primary rat astrocytes lipopolysaccharide (LPS) and interferon-γ (IFN-γ) treatment increased the intracellular levels of lactosylceramide (LacCer) and induced iNOS gene expression. d-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol·HCI (PDMP), a glucosylceramide synthase and LacCer synthase (galactosyltransferase, GalT-2) inhibitor, inhibited LPS/IFN-γ induced iNOS expression, which was reversed by exogenously supplied LacCer, but not by other glycosphingolipids. LPS/IFN-γ caused a rapid increase in the activity of GalT-2 and synthesis of LacCer. Silencing of GalT-2 gene with the use of antisense oligonucleotides resulted in decreased LPS/IFN-γ-induced iNOS, TNF-α, and IL-1β gene expression. The PDMP-mediated reduction in LacCer production and inhibition of iNOS expression correlated with decreased Ras and ERK1/2 activation along with decreased IκB phosphorylation, NF-κB DNA binding activity, and NF-κB-luciferase reporter activity. LacCer-mediated Ras activation was redox-mediated and was attenuated by antioxidants N-acetyl cysteine (NAC) and pyrrolidine dithiocarbamate (PDTC). In vivo administration of PDMP after SCI resulted in improved functional outcome (Basso, Beattie, Bresnahan score); inhibition of iNOS, TNF-α, and IL-1β expression; decreased neuronal apoptosis; and decreased tissue necrosis and demyelination. The in vivo studies supported the conclusions drawn from cell culture studies and provided evidence for the possible role of GalT-2 and LacCer in SCI-induced inflammation and pathology. To our knowledge this is the first report of a role of LacCer in iNOS expression and the advantage of GSL depletion in attenuating post-SCI inflammation to improve the outcome of SCI.
Keywords: iNOS, astrocytes, lactosylceramide, lipopolysaccharide, interferon-γ, spinal cord injury
Introduction
Excessive production of nitric oxide (NO) has been implicated in neuronal cell death and demyelination in a number of CNS diseases such as multiple sclerosis, Parkinson's, Alzheimer's, and Krabbe's disease; bacterial/viral infections; cerebral ischemia; spinal cord injury (SCI); and in an inherited metabolic disorder of peroxisomes, X-adrenoleukodystrophy (Dawson et al., 1993; Koprowski et al., 1993; Bo et al., 1994; Vodovotz et al., 1996; Wada et al., 1998a,b; Akiyama et al., 2000; Gilg et al., 2000; Satake et al., 2000; Giri et al., 2002).
Of the three isoforms of nitric oxide synthase (NOS), two isoforms are calcium-dependent and expressed constitutively (neuronal, nNOS, and endothelial, eNOS). The third is a calcium-independent and inducible isoform (iNOS). iNOS, once induced in response to a number of stress-inducing factors such as proinflammatory cytokines, bacterial/viral components, et cetera, produces high amounts of NO (Simmons and Murphy, 1992; Zielasek et al., 1992). The pathologically high levels of NO produced by iNOS in the CNS are associated with inhibition of mitochondrial functions, rapid glutamate release from both astrocytes and neurons, and excitotoxic death of neurons (Leist et al., 1997; Sequeira et al., 1997; Bal-Price and Brown, 2001). iNOS expression in reactive astrocytes has been implicated in the development of post-traumatic spinal cord cavitation and neurological impairment (Matsuyama et al., 1998; Suzuki et al., 2001). Strategies for iNOS inhibition to improve neurological outcome are an active area of investigation in neuroinflammatory diseases.
Studies from our laboratory and others have shown the involvement of sphingolipids such as ceramide and psychosine in the regulation of cytokine-mediated iNOS expression (Pahan et al., 1998b; Fern, 2001; Giri et al., 2002). In the present study the involvement of glycosphingolipids (GSLs) in the regulation of cytokine-mediated iNOS gene expression was investigated. GSL biosynthesis is initiated by transfer of UDP-glucose onto ceramide by the action of glucosylceramide synthase to form glucosylceramide (GluCer). Lactosylceramide (LacCer) is generated from GluCer by the action of LacCer synthase (galactosyltransferase, GalT-2). LacCer is a precursor for complex GSLs, including gangliosides.
In this report we have identified a novel role of LacCer that mediates lipopolysaccharide-induced (LPS) and interferon-γ-induced (IFN-γ) iNOS gene expression through the Ras/ERK1/2 and IκB/NF-κB pathways. The possible role of GSLs and the advantage of inhibition of their synthesis in suppressing inflammation after CNS trauma were demonstrated by observing an inhibition of iNOS, tumor necrosis factor-α (TNF-α), and interleukin-1β (IL-1β) gene expression and reactive astrogliosis by a GSL biosynthesis inhibitor, d-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol· HCI(PDMP) in a rat model of SCI. Furthermore, PDMP treatment improved the neurological outcome after SCI and also attenuated SCI-induced neuronal apoptosis. Histological examination of the spinal cord tissue showed a marked decrease in SCI-induced white matter vacuolization as well as loss of myelin with PDMP treatment. This study establishes for the first time the role of LacCer as a key signaling modulator in the regulation of iNOS gene expression via regulation of Ras/ERK1/2 and nuclear factor-κB (NF-κB) pathways. It further demonstrates the effectiveness of PDMP in the attenuation of inflammation-mediated secondary damage for amelioration of CNS pathology, as in SCI.
Materials and Methods
Reagents. Recombinant rat IFN-γ was obtained from Calbiochem (La Jolla, CA). N-acetyl cysteine (NAC), pyrrolidine dithiocarbamate (PDTC), and lipopolysaccharide (from Escherichia coli serotype 0111:B4) were obtained from Sigma (St. Louis, MO). DMEM and FBS were from Invitrogen (San Diego, CA). GluCer, LacCer, galactosylceramide (Gal-Cer), gangliosides, and PDMP were from Matreya (Pleasant Gap, PA). [14C]galactose and [3H]UDP-galactose were obtained from American Radiolabeled Chemicals (St. Louis, MO).
Cell culture. Primary astrocyte-enriched cultures were prepared from the whole cortex of 1-d-old Sprague Dawley rats as described earlier (Pahan et al., 1998b). Briefly, the cortex was dissected rapidly in ice-cold calcium/magnesium-free HBSS (Invitrogen), pH 7.4, as described previously (Won et al., 2001). Then the tissue was minced, incubated in HBSS containing trypsin (2 mg/ml) for 20 min, and washed twice in plating medium containing 10% FBS and 10 μg/ml gentamicin; next it was disrupted by triturating through a Pasteur pipette, after which the cells were seeded in 75 cm2 culture flasks (Falcon, Franklin, NJ). After incubation at 37°C in 5% CO2 for 1 d, the medium was changed completely to the culture medium (DMEM containing 5% FBS and 10 μg/ml gentamicin). The cultures received half-exchanges with fresh medium twice a week. After 14-15 d the cells were shaken for at least 24 hr on an orbital shaker to remove the microglia and then seeded on multi-well tissue culture dishes. The cells were incubated with serum-free DMEM for 24 hr before the incubation with drugs.
Assay for NO production. Cells were cultured in 12-well plastic tissue culture plates. After appropriate treatment the production of NO was determined by an assay of the culture supernatant for nitrite (Green et al., 1982). Briefly, 100 μl of culture supernatant was allowed to react with 100 μl of Griess reagent. The optical density of the assay samples was measured spectrophotometrically at 570 nm. Nitrite concentrations were calculated from a standard curve derived from the reaction of NaNO2 in fresh media.
Western blot analysis. For iNOS protein the cells were washed with cold Tris-buffered saline (TBS; 20 mm Trizma base and 137 mm NaCl, pH 7.5) and lysed in 1× SDS sample loading buffer (62.5 mm Trizma base, 2% w/v SDS, 10% glycerol); after sonication and centrifugation at 15,000 × g for 5 min, the supernatant was used for the iNOS Western immunoblot assay. The protein concentration of samples was determined with the detergent-compatible protein assay reagent (Bio-Rad, Hercules, CA), using bovine serum albumin (BSA) as the standard. Sample was boiled for 3 min with 0.1 volume of 10% β-mercaptoethanol and 0.5% bromophenol blue mix. Total cellular protein (50 μg) was resolved by electrophoresis on 8 or 12% polyacrylamide gels, electro-transferred to a polyvinylidene difluoride (PVDF) filter, and blocked with Tween 20 containing Tris-buffered saline (TBST; 10 mm Trizma base, pH 7.4, 1% Tween 20, and 150 mm NaCl) with 5% skim milk. After incubation with antiserum against iNOS (BD Biosciences PharMingen, San Diego, CA) or H-Ras (Upstate Biotechnology, Lake Placid, NY) or phospho-specific ERK1/2 or pIκB (Signal Transduction) in PVDF buffer for 2 hr at room temperature, the filters were washed three times with TBST buffer and then incubated with horseradish peroxidase-conjugated anti-rabbit or mouse IgG for 1 hr. The membranes were detected by autoradiography, using ECL-plus (Amersham Biosciences, Arlington Heights, IL) after being washed with TBST buffer.
Nuclear extraction and electrophoretic mobility shift assay. Nuclear extracts from cells (1 × 107 cells) were prepared via a previously published method (Dignam et al., 1983) with slight modification. Cells were harvested, washed twice with ice-cold TBS, and lysed in 400 μl of buffer A containing (in mm): 10 KCl, 2 MgCl2, 0.5 dithiothreitol plus protease inhibitor mixture (Sigma) and 0.1% Nonidet P-40 in 10 mm HEPES, pH 7.9, for 10 min on ice. After centrifugation at 5000 × g for 10 min the pelleted nuclei were washed with buffer A without Nonidet P-40 and resuspended in 40 μl of buffer B containing 25% (v/v) glycerol, 0.42 m NaCl, and (in mm) 1.5 MgCl2, 0.2 EDTA, 0.5 dithiothreitol, and Complete protease inhibitor mixture (Roche Molecular Biochemicals, Indianapolis, IN) in 20 mm HEPES, pH 7.9, for 30 min on ice. The lysates were centrifuged at 15,000 × g for 15 min, and the supernatants containing the nuclear proteins were stored at -70°C until use. Nuclear proteins (10 μg) were used for the electrophoretic mobility shift assay for the detection of NF-κB DNA binding activities. DNA-protein binding reactions were performed at room temperature for 20 min in (in mm) 10 Trizma base, pH 7.9, 50 NaCl, 5 MgCl2, 1 EDTA, 1 dithiothreitol, plus 1 μg of poly (dI-dC), 5% (v/v) glycerol, and ∼0.3 pmol of NF-κB probe (Santa Cruz Biotechnology, Santa Cruz, CA) labeled with DIG-ddUTP, using terminal deoxynucleotidyl transferase (Roche Molecular Biochemicals). Protein-DNA complexes were resolved from protein-free DNA in 5% polyacrylamide gels at room temperature in 50 mm Tris, pH 8.3, 0.38 m glycine, and 2 mm EDTA and were electroblotted onto positively charged nylon membranes. The chemiluminescent autoradiography detection was performed as suggested by the manufacturer (Roche Molecular Biochemicals), using an alkaline phosphatase-conjugated anti-DIG Fab fragment and disodium-3-[4-methoxyspiro{1,2-dioxetane-3,2′-(5′-chloro)tricycle[3.3.1.137]decan}-4-yl]phenyl phosphate (CSPD) (both from Roche Molecular Biochemicals).
Plamids and transient transfections and reporter gene assay. Dominant-negative and constitutively active Ras expression vector (pCMVrasN17 and pCMVrasv12) and κB repeat luciferase reporter construct (pNF-κBLuc) were purchased from BD Biosciences (San Jose, CA). Then 3 × 105 cells/well were cultured in six-well plates for 1 d before the transfection. Transfection was performed with plasmid concentration constant (2.5 μg/transfection) and 8 μl of Fugene transfection reagent (Roche Molecular Biochemicals). At 24 hr after transfection the cells were placed in serum-free media overnight. After appropriate treatment the cells were washed with PBS, scraped, and resuspended with 100 μl of lysis buffer [containing (in mm): 40 tricine, pH 7.8, 50 NaCl, 2 EDTA, 1 MgSO4, 5 dithiothreitol plus 1% of Triton X-100]. After incubation at room temperature for 15 min with occasional vortexing, the samples were centrifuged. The luciferase and β-galactosidase activities were measured by using a luciferase assay kit (Stratagene, La Jolla, CA) and β-gal assay kit (Invitrogen), respectively. The emitted light and optical absorbance was measured with SpectraMax/Gemini XG and SpectraMax 190 (both from Molecular Devices, Sunnyvale, CA).
Quantification of Ras activation. After stimulation the primary astrocytes in six-well plates were washed with ice-cold PBS and lysed in membrane lysis buffer (MLB; 0.5 ml of (in mm) 25 HEPES, pH 7.5, 150 NaCl, 10 MgCl2, 1 EDTA, 25 NaF, 1 sodium orthovanadate plus 1% Igepal CA-630, 0.25% sodium deoxycholate, 10% glycerol, and EDTA-free Complete protease inhibitor mixture). After centrifugation (5000 × g)at 4°C for 5 min the supernatant was used for Ras activation assay. Supernatant (100 μg) was used for binding with agarose-conjugated Rasbinding domain (RBD) of Raf-1, which was expressed in BL21 (Invitrogen), Escherichia coli strain, transformed by pGEX-2T-GST-RBD in the presence of 0.1 mm of isopropyl β-d-thiogalactoside (IPTG) as described previously (Herrmann et al., 1995). The binding reaction was performed at 4°C for 30 min in MLB. After being washed with MLB three times, the Ras-RBD complex was denatured by the addition of 2× SDS sample buffer. Ras protein was identified by Western blot analysis with Ras antibodies from Upstate Biotechnology.
Measurement of LacCer synthesis. Cultured cells were incubated in growth medium containing [14C]galactose (5 μCi/ml) for 24 hr as described previously. The medium was removed, and the cell monolayer was washed with sterile PBS. After the stimulation with LPS/IFN-γ (1 μg/ml; 10 U/ml) for various durations, the cells were harvested, washed with ice-cold PBS, and lysed by sonication. Then 200 μg of protein was used for extraction of lipids, using chloroform/methanol/HCl (100:100:1). The organic phase was dried under nitrogen. Glycosphingolipids were resolved by high-performance thin-layer chromatography (HPTLC), using chloroform/methanol/0.25% KCl (70:30:4, v/v/v) as the developing solvent. The gel area corresponding to LacCer was scraped, and radioactivity was measured by using Liquiscint (NEN Life Science Products, Boston, MA) as a scintillating fluid.
Identification and analysis of purified LacCer. LacCer from LPS-treated cells was resolved by a silica gel 60 HPTLC plate. Fatty acid methyl ester (FAME) was prepared as described earlier (Khan et al., 1998; Pahan et al., 1998b). FAME was analyzed by gas chromatography (Shimadzu GC 17A gas chromatograph, Kyoto, Japan) on a silica capillary column and quantified as a percentage of total fatty acids that were identified. Mass spectrometry data were recorded as Finnegan LCQ classic (ion trap quadrupole) mass spectrometer.
GalT-2 activity assay. The activity of GalT-2 was measured by using [3H]UDP-galactose as the galactose donor and GluCer as the acceptor as described previously (Yeh et al., 2001). Briefly, the cells were harvested in PBS, and the cell pellets were suspended in Triton X-100 lysis buffer. Cell lysates were sonicated; after protein quantification 100 μg of cell lysate was added to reaction mixture containing 20 μm cacodylate buffer, pH 6.8, 1 mm Mn/Mg, 0.2 mg/ml Triton X-100 (1:2 v/v), 30 nmol GluCer, and 0.1 mmol UDP-[3H]galactose in a total volume of 100 μl. The reaction was terminated by adding 10 μl of 0.25 m EDTA, 10 μl of 0.5 m KCl, and 500 μl of chloroform/methanol (2:1 v/v); the products were separated by centrifugation. The lower phase was collected and dried under nitrogen. After resolution on HPTLC plates the gel was cut out, and radioactivity was measured in a scintillation counter. Assay without exogenous GluCer served as blank, and their radioactivity counts were subtracted from all respective data points.
GalT-2 antisense oligonucleotides. A 20-mer antisense oligonucleotide of the following sequence, 5′-CGC TTG AGC GCA GAC ATC TT-3′, targeted against rat lactosylceramide synthase (GalT-2) was synthesized by Integrated DNA Technology (Coralville, IA). A scrambled oligonucleotide (5′-CTG ATA TCG TCG ATA TCG AT-3′) also was synthesized and used as control. Cells were counted and plated 1 d before transfection and the next day were treated with Oligofectamine-oligonucleotide complexes (200 nm oligo; Invitrogen) under serum-free conditions. At 48 hr after transfection the protein levels of GalT-2 were analyzed by using polyclonal antibodies raised against rat GalT-2 (Abjent). The transfected cells were stimulated with LPS/IFN-γ (1 μg/ml), and the levels of nitric oxide were checked 24 hr after stimulation. iNOS mRNA and protein levels were checked at 6 and 24 hr, respectively, after stimulation of the transfected cells.
Reverse transcriptase-PCR amplification. After total RNA extraction with the use of Trizol (Invitrogen) as per the manufacturer's protocol, single-stranded cDNA was synthesized from total RNA. Total RNA (5 μg) was treated with 2 U DNase I (bovine pancreas; Sigma) for 15 min at room temperature in 18 μl of volume containing 1× PCR buffer and 2 mm MgCl2. Then it was inactivated by incubation with 2 μl of 25 mm EDTA at 65°C for 15 min. Random primers (2 μl) were added and annealed to the RNA according to the manufacturer's protocol. cDNA was synthesized in a 50 μl reaction containing 5 μg of total RNA and 50-100 U reverse transcriptase by incubating the tubes at 42°C for 60 min. PCR amplification was conducted in 25 μl of reaction mixture with 1.0 μl of cDNA and 0.5 mm of each primer under the manufacturer's Taq polymerase conditions (Takara, Takara Shuzo, Kyoto, Japan). The sequences of primers used for PCR amplification are as follows: iNOS (forward, 5′-ctccttcaaagaggcaaaaata-3′, and reverse, 5′-cacttcctccaggatgttgt-3′); GalT-2 (forward, 5′-tggtacaagctagaggc-3′, and reverse, 5′-gcatggcacattgaa C-3′); glyceraldehyde phosphate dehydrogenase (GAPDH; forward, 5′-cgggatcgtggaagggctaatga-3′, and reverse, 5′-cttcacgaagttgtcattgagggca-3′); TNF-α (forward, 5′-ccgagatgtggaactggcaga g-3′, and reverse, 5′-cggagaggaggctgactttctc-3′); and IL-1β (forward, 5′-ccacctcaatggacagaacat-3′, and reverse, 5′-ccatctttaggaagacacgggt-3′). The PCR program included preincubation at 95°C for 4 min, amplification for 30 cycles at 94°C for 1 min plus 50°C annealing for 1 min plus 74°C extension for 1 min, and a final 74°C for a 10 min extension. Then 10 μl of the PCR products was separated on 1.2% agarose gel and visualized under UV.
Real-time PCR. Total RNA isolation from rat spinal cord sections was performed by using Trizol (Invitrogen) according to the manufacturer's protocol. Real-time PCR was conducted with the Bio-Rad iCycler (iCycler iQ Multi-Color Real-Time PCR Detection System). Single-stranded cDNA was synthesized from total RNA as described. The primer sets for use were designed (Oligoperfect Designer, Invitrogen) and synthesized from Integrated DNA Technologies. The primer sequences for iNOS were as follows: (forward, 5′-gaaagaggaacaactactgct ggt-3′, and reverse, 5′-gaactgagggtacatgctggagc); GAPDH (forward, 5′-cctacccccaatgtatccgttgtg-3′, and reverse, 5′-ggaggaatgggagttgctgttgaa-3′); TNF-α (forward, 5′cttctgtctactgaacttcggggt-3′, and reverse, 5′-tgg aac tga tga gag gga gcc-3′); and IL-1β (forward, 5′-gagagacaagca acgacaaaatcc-3′, and reverse, 5′-ttcccatcttcttctttgggt att-3′). IQTM SYBR Green Supermix was purchased from Bio-Rad. Thermal cycling conditions were as follows: activation of DNA polymerase at 95°C for 10 min, followed by 40 cycles of amplification at 95°C for 30 sec and at 58.3°C for 30 sec. The normalized expression of target gene with respect to GAPDH was computed for all samples by using Microsoft Excel data spreadsheet.
Induction of SCI in rats. Sprague Dawley female rats (225-250 gm weight) were purchased (Harlan Laboratories, Durham, NC) for the induction of SCI. All rats were given water and food pellets ad libitum and maintained in accordance with the Guide for the Care and Use of Laboratory Animals of the US Department of Health and Human Services (National Institutes of Health, Bethesda, MD). We used a clinically relevant weight-drop device for the induction of SCI in rats as described earlier (Gruner, 1992). Briefly, the rats were anesthetized by intraperitoneal administration of ketamine (80 mg/kg) plus xylazine (10 mg/kg), followed by laminectomy at T12. While the spine was immobilized with a stereotactic device, injury (30 gm/cm force) was induced by dropping a weight of 5 gm from a height of 6 cm onto an impounder gently placed on the spinal cord. Sham-operated animals underwent laminectomy only. After recovery from anesthesia the animals were evaluated neurologically and monitored for food and water intake. However, no prophylactic antibiotics or analgesics were used to avoid their possible interactions with the experimental therapy of SCI.
Treatment of SCI. Rats received the glycosphingolipid inhibitor, PDMP (Matreya), at various time points after SCI. PDMP was dissolved in 5% Tween 80 in saline and diluted with sterile saline (0.85% NaCl) at the time of intraperitoneal administration to sham and SCI rats. Animals (six per group) were selected randomly to form four different groups: vehicle-treated (5% Tween 80 in saline) sham (laminectomy only) and SCI, and PDMP-treated (20 mg/kg in 5% Tween 80) sham and SCI. The first dose of PDMP was administered (10 and 30 min and 1, 2, and 12 hr) after SCI, followed by the second dose at 24 hr (day 1), third dose at 48 hr (day 2), and fourth dose at 72 hr (day 3) after SCI. Animals were killed under anesthesia at 1, 4, 12, 24, 48, and 72 hr after treatment.
Assessment of neurological (functional) recovery was performed by an open-field test via the 21 point Basso, Beattie, Bresnahan (BBB) locomotor rating scale (Basso et al., 1996) until day 15 after SCI. The animals were observed by a blinded observer before assignment of grade.
Preparation of spinal cord sections. Rats were anesthetized and decapitated. Spinal cord sections with the site of injury as the epicenter (lesion epicenter) were extracted carefully from VHC-treated sham and SCI as well as PDMP-treated sham and SCI animals. Tissue targeted to be used for RNA and protein extraction was homogenized immediately in Trizol (Invitrogen), snap-frozen in liquid nitrogen, and stored at -80°C until further use. Total RNA was extracted as per the manufacturer's protocol and used for cDNA synthesis as described earlier. Sections of spinal cord to be used for histological examination as well as immunohistochemistry were fixed in 10% neutral-buffered formalin (Stephens Scientific, Riverdale, NJ) The tissues were embedded in paraffin and sectioned at 4 μm thickness.
Immunohistochemical analysis. Spinal cord sections were deparaffinized and rehydrated sequentially in graded alcohol. Slides were boiled in antigen unmasking fluids (Vector Laboratories, Burlingame, CA) for 10 min, cooled in the same solution for another 20 min, and then washed three times for 5 min each in Tris-sodium buffer (0.1 m Tris-HCl, pH 7.4, 0.15 m NaCl) with 0.05% Tween 20 (TNT). Sections were treated with trypsin (0.1% for 10 min) and immersed for 10 min in 3% hydrogen peroxide to eliminate endogenous peroxidase activity. Sections were blocked in Tris sodium buffer with 0.5% blocking reagent (TNB; supplied with TSA-Direct kit, NEN Life Science Products) for 30 min to reduce nonspecific staining. For immunofluorescent labeling the sections were incubated overnight with anti-iNOS, TNF-α, or IL-1β antibody (1:100; mouse monoclonal, Santa Cruz), followed by antibodies against the astrocyte marker glial fibrillary acidic protein (GFAP; 1:100; rabbit polyclonal, Dako, Kyoto, Japan) for 1 hr (in case of double staining). Anti-iNOS was visualized by fluorescein isothiocyanate-conjugated (FITC) anti-mouse IgG (1:100, Sigma) and GFAP, using tetramethylrhodamine isothiocyanate-conjugated (TRITC) anti-rabbit IgG (1:100; Sigma). The sections were mounted in mounting media (EMS, Fort Washington, PA) and visualized by immunofluorescence microscopy (Olympus, Melville, NY) with Adobe Photoshop software. Rabbit polyclonal IgG was used as control primary antibody. Sections also were incubated with conjugated FITC anti-rabbit IgG (1:100; Sigma) or TRITC-conjugated IgG (1:100) without the primary antibody as negative control. Hematoxylin and eosin (H&E) staining was performed as described by Kiernan (1990). Luxol fast blue (LFB)-PAS was performed according to Lassmann and Wisniewski (1979).
Fluorescent terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) assay was performed by using Apoptag Fluorescein In Situ Apoptosis detection kit (Serological Corporation, Norcross, GA) according to the manufacturer's protocol. For double labeling, the sections were incubated with mouse anti-neuronal nuclei 1:100 (NeuN, Chemicon, Temecula, CA). Sections were incubated with TRITC-conjugated mouse IgG 1:100 (Sigma), mounted in mounting media, and visualized by fluorescence microscopy.
Statistical analysis. All values shown in the figures are expressed as the mean ± SD obtained from at least three independent experiments. The results were examined by one- and two-way ANOVA; then individual group means were compared with the Bonferroni test. A p value <0.05 was considered significant.
Results
LPS/IFN-γ-induced NO production and iNOS gene expression are mediated by GSL
LPS/IFN-γ stimulation of primary astrocytes resulting in iNOS gene expression is a complex multi-step process. In the present study we tested whether GSLs somehow were involved in iNOS induction. Primary astrocytes pretreated for 0.5 hr with several concentrations of the glycosphingolipid inhibitor PDMP (0, 10, 25, and 50 μm), followed by stimulation with LPS/IFN-γ (1 μg/ml; 10 U/ml), showed a dose-dependent decrease in the production of NO as well as mRNA and protein levels of iNOS (Fig. 1A). However, in the presence of increasing doses of LacCer, PDMP-mediated inhibition of NO production and iNOS gene expression (Fig. 1B) was blunted. To prove that this was a LacCer-specific effect, we also exogenously supplemented other glycosphingolipid derivatives. However, the presence of GluCer (Fig. 2A), GalCer (Fig. 2B), or gangliosides GM1 (Fig. 2C), GM3 (Fig. 2D), and GD3 (Fig. 2E) did not reverse PDMP-mediated inhibition of LPS/IFN-γ-induced NO production as LacCer, provided exogenously, did. These studies indicate that a metabolite of the glycosphingolipid pathway, LacCer, may play a role in the regulation of LPS/IFN-γ-mediated induction of iNOS gene expression and NO production.
Figure 1.

LacCer regulates LPS/IFN-γ-induced NO production and iNOS gene expression in rat primary astrocytes. A, Effect of PDMP (10, 25, and 50 μm) on NO production and the induction of iNOS mRNA and protein expression after 6 hr (for iNOS mRNA level) or 24 hr (for iNOS protein and NO levels) after LPS/IFN-γ (1 μg/ml; 10 U/ml) stimulation. The cells were pretreated with PDMP for 0.5 hr before LPS/IFN-γ stimulation. B, The effect of LacCer on PDMP-mediated inhibition of iNOS gene expression in astrocytes was examined also. The cells were pretreated with PDMP (50 μm) and/or LacCer (5 and 10 μm) for 0.5 hr before LPS/IFN-γ stimulation. NO production and iNOS mRNA and protein levels were quantified at 6 and 24 hr after LPS/IFN-γ stimulation, respectively. Levels of GAPDH were used as an internal standard for mRNA levels. The procedures for measurement of mRNA and of protein and NO are described in Materials and Methods. Data are represented as the mean ± SD from three independent experiments. ***p < 0.001 in A and B compared with unstimulated control; **p < 0.01 in A compared with LPS/IFN-γ-stimulated cells. #p < 0.001 in A and B compared with LPS/IFN-γ-stimulated cells; *p < 0.01 and **p < 0.001 in B compared with PDMP-treated cells.
Figure 2.

Effect of various metabolites of the glycosphingolipid pathway on PDMP-mediated inhibition of LPS-induced NO production. Primary astrocytes were pretreated with PDMP and GluCer (A), GalCer (B), GM1 (C), GM3 (D), or GD3 (E) all at individual concentrations of 5 and 10 μm for 0.5 hr before stimulation with LPS/IFN-γ. NO production was assayed at 24 hr after LPS/IFN-γ stimulation, as described in Figure 1.
LPS/IFN-γ stimulation results in increased synthesis and altered fatty acid composition of LacCer
To understand the mechanism of LPS/IFN-γ-induced iNOS gene expression by LacCer, we quantified the in situ levels of lactosylceramide. [14C]LacCer was resolved and characterized by Rf value, using commercially available standard LacCer by HPTLC as described in Materials and Methods. As shown in Figure 3A, a sharp increase in [14C]LacCer levels was observed within 2-5 min after stimulation with LPS/IFN-γ. With LPS/IFN-γ stimulation the LacCer levels increased ∼1.5-fold of those observed in unstimulated cells. Inhibition of LacCer synthase (GalT-2, the enzyme responsible for LacCer biosynthesis) by PDMP inhibited this increase in [14C]LacCer biosynthesis after LPS/IFN-γ stimulation. Additionally, when GalT-2 activity was assayed after LPS/IFN-γ stimulation, a rapid increase in enzyme activity with a peak at 5 min after LPS/IFN-γ stimulation was observed (Fig. 3B). The role of GalT-2 and its product LacCer in iNOS gene regulation was confirmed further by silencing GalT-2 gene with the use of antisense (AS) DNA oligomers against rat GalT-2 mRNA and a sequence-scrambled (Scr) oligomer as a control. As shown in Figure 3C, diminished protein levels of GalT-2 by AS GalT-2 oligonucleotides correlated with diminished synthesis of [14C]LacCer with LPS/IFN-γ stimulation. Silencing of GalT-2 with AS oligomers decreased the LPS/IFN-γ-mediated NO production (Fig. 3D) and iNOS mRNA and protein levels (Fig. 3E), whereas supplementing LacCer exogenously reversed the inhibition, presumably because the signaling events downstream of LacCer could be triggered with the addition of LacCer. In addition to iNOS, inhibition of LacCer synthesis with LPS/IFN-γ stimulation by using AS oligonucleotides also suppressed the mRNA expression of two other potent proinflammatory cytokines, TNF-α and IL-1β (Fig. 3E), both of which are known to be critical players in causing secondary damage by inducing inflammation in neurological disorders (Andersson et al., 1992; Renno et al., 1995; Saklatvala et al., 1996; Perry et al., 2001). The AS-mediated inhibition of the expression of these inflammatory cytokines also was reversed by exogenous supplementation of LacCer, suggestive of the fact that, along with iNOS expression, LacCer may exacerbate inflammation in general by mediating expression of potent inflammatory mediators.
Figure 3.

The effect of LPS/IFN-γ stimulation on the biosynthesis of LacCer. A, Primary astrocytes were treated with [14C]galactose overnight. After pretreatment with PDMP 0.5 hr before LPS/IFN-γ stimulation, the cells were harvested at the time points indicated, and LacCer was analyzed by HPTLC as described in Materials and Methods. B, The enzyme activity of LacCer synthase (GalT-2) was assayed as described in Materials and Methods, using cell lysates derived from cells stimulated with LPS/IFN-γ for various durations as shown. C, For the silencing of GalT-2 gene, the cells were transfected with either GalT-2 antisense DNA oligomer or its sequence-scrambled DNA oligomer (Scr) as described in Materials and Methods. At 48 hr after transfection GalT-2 protein levels were analyzed by immunoblot analysis, and [14C]LacCer synthesis was examined in transfected and nontransfected cells. At 48 hr after transfection with AS oligonucleotides the cells were stimulated with LPS/IFN-γ and NO production (D), and the mRNA levels of iNOS, TNF-α, and IL-1β (E) were measured as described previously. Data are represented as the mean ± SD of three independent experiments. ***p < 0.001 in A and B compared with unstimulated control. ***p < 0.001 in D compared with stimulated, untransfected cells; #p < 0.001 in D compared with transfected cells without LacCer.
Furthermore, to investigate the possible role of LacCer and GalT-2 in iNOS gene regulation, we also investigated LacCer, isolated and purified from these experiments, for its compositional and structural study. The structure of LacCer obtained from LPS/IFN-γ-stimulated cells or from unstimulated cells was studied by its mass spectrometric (MS) analysis. LacCer consisting of 18:0 had the diagnostic peak at m/z 889 (M, 1.1%), m/z 890 (M+H, 1.4%), and m/z 740 (M-[5 × OH + 2 × CH3OH], 41.6%). Similarly, 16:0 species of LacCer had the significant peaks present at m/z 861 (M+, 0.8%), 862 (M+H, 1.2%), m/z 860 (M-H, 1.1%), and m/z 711 (860-[5 × OH + 2 × CH3OH], 51.9%). The species of LacCer consisting of oleic acid (18:1) had a significant peak present at m/z 888 (M+H, 1.8%) and m/z 739 (888-[5 × OH + 2 × CH3OH], 100%). Two more important peaks present were at m/z 342 (M-sphingolipid backbone, 4.4%) and m/z 529 (M-LacCer backbone-H2O, 1.5%). In addition, LPS/IFN-γ-stimulated cells had the altered fatty acid profile measured as a percentage of total fatty acids and compared with the levels of the same fatty acid in unstimulated cells. Gas chromatograph (GC) analysis identified three major fatty acids (18:0, 56.2%; 18:1, 26.4%; 16:0, 12.9%) in LPS/IFN-γ-stimulated cells. Furthermore, LPS/IFN-γ-stimulated cells had increased levels (measured as a percentage of total fatty acids) of saturated fatty acids including 14:0 (167%), 16:0 (65.8%), 18:0 (7.3%), and 20:0 (5.7%) when compared with unstimulated cells. Taken together, the data from the GC and MS confirmed that LacCer from LPS/IFN-γ-stimulated cells has three major species consisting of stearic, oleic, and palmitic acids.
LacCer-mediated regulation of LPS/IFN-γ-induced iNOS gene expression is reactive oxygen species-dependent
To elucidate further the mechanism of LacCer-mediated regulation of LPS/IFN-γ-induced cellular signaling for induction of iNOS expression, we investigated whether these events are reactive oxygen species-mediated (ROS-mediated). An earlier report by Pahan et al. (1998a) has shown LPS/cytokine-induced iNOS gene expression and NO production to be ROS-mediated (e.g., H2O2,O2- and OH.); however, the source of ROS production has not been defined clearly so far. Furthermore, a number of reports have shown that LacCer can stimulate superoxide production and generate oxidative stress in endothelial cells and neutrophils (Bhunia et al., 1997; Iwabuchi and Nagaoka, 2002). In primary astrocytes, pretreatment with increasing concentrations of the membrane-permeant antioxidant NAC, a ROS scavenger and precursor for glutathione (Pahan et al., 1998a), and PDTC, another antioxidant, blocked LPS/IFN-γ-induced NO production and iNOS protein and mRNA expression (Fig. 4A). As shown in Figure 4B, despite the supplementation of LacCer exogenously, the NAC- and PDTC-mediated inhibition of LPS/IFN-γ-stimulated iNOS gene expression was not reversed. Furthermore, although LacCer could reverse PDMP-mediated inhibition of iNOS gene expression and NO production effectively, in the presence of NAC and PDTC LacCer was not able to reverse PDMP-mediated inhibition of iNOS expression (Fig. 4C). These results clearly indicate that LacCer regulates LPS/IFN-γ-induced iNOS gene expression via a ROS-dependent mechanism because, in the presence of antioxidants, the signaling cascade is blocked, as is iNOS expression.
Figure 4.

LacCer-mediated regulation of LPS/IFN-γ-induced iNOS gene expression is ROS mediated. A, Effect of NAC (5, 10 mm) and PDTC (50, 100 μm) pretreatment 1 hr before LPS/IFN stimulation was analyzed; NO production and the induction of iNOS mRNA and protein expression were examined after 6 hr (for iNOS mRNA) or 24 hr (for iNOS protein and NO levels) after LPS/IFN-γ (1 μg/ml; 10 U/ml) stimulation. B, The effect of LacCer on NAC- and PDTC-mediated inhibition of iNOS gene expression was analyzed also. The cells were pretreated with NAC (10 mm) or PDTC (100 μm) for 1 hr before LPS/IFN-γ and LacCer stimulation. NO production and iNOS protein and mRNA levels were quantified at 24 and 6 hr after LPS/IFN-γ stimulation, respectively. C, NAC and PDTC were pretreated at 1 hr and PDMP/LacCer at 0.5 hr before LPS/IFN-γ stimulation, after which NO production and iNOS protein and mRNA levels were analyzed. Data are represented as the mean ± SD of three independent experiments. ***p < 0.001 in A compared with LPS/IFN-stimulated cells without NAC. #p < 0.001 in A compared with LPS/IFN-stimulated cells without PDTC.
Activation of small GTPase Ras and ERK1/2 is involved in LacCer-mediated regulation of LPS/IFN-γ-induced iNOS gene expression and is ROS-dependent
Because a recent study indicated that small GTPase Ras is critical for LPS/IFN-γ-induced iNOS gene expression (Pahan et al., 2000), compounded with the fact that this protein is redox-sensitive (Lander et al., 1995), the role of Ras in LacCer-mediated regulation of iNOS expression was investigated. Transient transfection with dominant-negative Ras, DN-Ras (hras N17 mutant), inhibited LPS/IFN-γ-mediated iNOS gene expression that could not be reversed by supplementation of exogenous LacCer. The inability of exogenous LacCer to bypass the inhibition by DN-Ras demonstrated that Ras is necessary for LacCer-mediated iNOS gene expression and suggests that Ras is downstream of LacCer in the signaling cascade that induces iNOS expression and NO production (Fig. 5A). Moreover, transient transfection with constitutively active Ras, CA-Ras (hras G12V mutant), completely bypassed PDMP-mediated inhibition of iNOS gene expression and NO production (Fig. 5B), which substantiated further the conclusion that functional Ras downstream of LacCer is critical for mediating the induction of iNOS expression. Because neither DN-Ras nor CA-Ras had any effect on LPS/IFN-stimulated [14C]LacCer synthesis, the possible role of Ras in regulating LacCer synthesis was ruled out (Fig. 5C).
Figure 5.

The involvement of small GTPase Ras and ERK1/2 in LacCer-mediated regulation of LPS-induced iNOS gene expression in primary astrocytes. A, Dominant-negative Ras (DN-Ras) was transfected transiently in primary astrocytes, followed by stimulation with LPS/IFN-γ and/or LacCer. NO production and iNOS protein and mRNA levels were analyzed as described previously. B, Constitutively active Ras (CA-Ras) was transfected transiently, followed by PDMP pretreatment 0.5 hr before LPS/IFN-γ stimulation. NO production and iNOS protein and mRNA expression are shown. C, After transient transfection with DN-Ras and CA-Ras, synthesis of [14C]LacCer with LPS/IFN-γ stimulation of primary astrocytes was analyzed as described in Materials and Methods. Ras activation was examined by using GST-tagged Raf-1 Ras binding domain (GST-RBD) as described in Materials and Methods. D, Ras activation was checked after LPS/IFN-γ stimulation for different durations of time. After pretreatment with LacCer and/or PDMP (50 μm) for 0.5 hr followed by LPS/IFN-γ stimulation for 5 min, cell lysates were used to assay levels of activated Ras, which also is represented as a graph after densitometric analysis of the autoradiograph. E, After pretreatment with NAC (10 mm) or PDTC (100 μm) for 1 hr followed by LacCer stimulation for 5 min, cell lysates were used to assay levels of activated Ras, which also is represented as a graph after densitometric analysis of the autoradiograph. F, ERK1/2 activation was assayed with pretreatment of cells with LacCer and/or PDMP for 0.5 hr, followed by stimulation with LPS/IFN-γ for 20 min; immunoblot analysis used anti-phosphorylated ERK1/2 antibodies as described in Materials and Methods. G, For the examination of MEK/ERK pathway involvement, after pretreatment for 0.5 hr with PD98059 (a MEK1/2 inhibitor) followed by stimulation with LPS/IFN-γ for 24 hr, NO production and iNOS protein levels were assayed.
As expected, the stimulation of cells with LPS/IFN-γ enhanced the activation of Ras (maximal activation was detected within 5 min after LPS/IFN treatment; Fig. 5D). This LPS/IFN-γ-mediated activation of Ras was reduced by pretreatment with the GSL inhibitor PDMP, which was reversed fully by the addition of LacCer, indicating that LacCer mediates iNOS gene expression by activation of Ras. Furthermore, LacCer-mediated Ras activation was inhibited with pretreatment with NAC and PDTC, thus showing that LacCer-mediated Ras activation is ROS-mediated (Fig. 5E). In addition to Ras, activation of ERK1/2 (which are downstream targets of Ras) also was observed with LPS/IFN-γ stimulation. Pretreatment with PDMP inhibited the LPS/IFN-γ-induced phosphorylation of ERK1/2, which was reversed in the presence of exogenous LacCer (Fig. 5F). Additionally, inhibition of a kinase responsible for ERK phosphorylation and activation, MEK1/2, by PD98059 resulted in inhibition of NO production and iNOS expression, further proving the involvement of ERK pathway in iNOS gene expression (Fig. 5G). Supplementation of exogenous LacCer had no effect on PD98059-mediated inhibition of iNOS gene expression, thus placing LacCer upstream of the MEK/ERK cascade as a second messenger molecule mediating the regulation of LPS/IFN-γ-induced iNOS gene expression through this pathway. These findings suggest that LacCer regulates iNOS gene expression via ROS-mediated activation of the small GTPase Ras/MEK/ERK pathway.
The role of NF-κB in LacCer-mediated regulation of iNOS gene expression
Because the activation of NF-κB is necessary for the induction of iNOS (Xie et al., 1994) and Ras is involved in NF-κB activation resulting in iNOS expression (Pahan et al., 2000), the observed inhibition of LPS/IFN-γ-mediated iNOS gene expression by PDMP in rat primary astrocytes may be attributable to the inhibition of NF-κB. To demonstrate this possibility, we observed the effect of PDMP on luciferase activity in κB-repeat luciferase-transfected cells. LPS/IFN-γ-induced luciferase activity was abolished with PDMP pretreatment and was bypassed effectively by exogenously supplemented LacCer (Fig. 6A). As shown in Figure 6B, NF-κB DNA binding activity tested by electrophoresis mobility shift assay was inhibited by increasing doses of PDMP but was reversed in the presence of exogenous LacCer. Specificity of NF-κB probe binding was proven by using 50× cold probe, which out-competed labeled NF-κB binding activity. Because IκB phosphorylation and degradation are required for NF-κB activation and translocation to the nucleus, phosphorylated IκB levels were examined also. Decreased phosphorylation of IκB was observed in the presence of PDMP. However, when LacCer was added, the levels of phosphorylated IκB were increased, which correlated with increased NF-κB nuclear translocation and DNA-binding activity (Fig. 6C). These results show that LacCer may mediate transcriptional regulation of LPS/IFN-γ-induced iNOS gene expression via the IκB/NF-κB pathway.
Figure 6.

Involvement of LacCer in LPS/IFN-γ-mediated NF-κB activation and iNOS gene expression. A, At 24 hr after transient transfection of cells with κB-luciferase gene construct, the cells were pretreated with PDMP for 0.5 hr before stimulation with LPS/IFN-γ. The cellular luciferase activity was measured as described in Materials and Methods. B, The NF-κB DNA binding activity was detected by gel shift assay, using 10 μg of nuclear extract from cells pretreated for 0.5 hr with LacCer and/or increasing doses of PDMP, followed by stimulation with LPS/IFN-γ for 45 min. C, The cytoplasmic extract was used to detect the levels of phosphorylated IκB and total IκB levels by immunoblot, using antibodies against phosphorylated IκB and total IκB. Data are represented as the mean ± SD of three independent experiments.
Attenuation of tissue destruction and demyelination by treatment with PDMP after SCI
To test the physiological relevance of our observations and investigate further the role of LacCer in the induction of iNOS in neuroinflammatory diseases, we examined the effect of PDMP in the rat SCI model. PDMP (20 mg/kg) was administered at various time points (10, 15, and 30 min and 1, 2, and 12 hr) after SCI, and the spinal cord tissue was fixed and analyzed 24 hr after SCI. SCI induced white matter vacuolization, and tissue necrosis (Fig. 7B) observed by histological examination of injured rat spinal cord sections was decreased markedly in tissue sections of PDMP-treated SCI rats. Treatment with PDMP for 10 min (Fig. 7D), 30 min (Fig. 7E), 1 hr (Fig. 7F), and 2 hr (Fig. 7F) was efficacious in protecting against tissue damage compared with VHC-treated SCI (Fig. 7B). Treatment with PDMP 12 hr after SCI showed some damage but still was able to provide a substantial amount of protection against tissue destruction compared with VHC-treated SCI (Fig. 7H). The weight-drop injury is known also to result in a loss of myelin, resulting in locomotor dysfunction of the hindlimbs (Suzuki et al., 2001). LFB staining of spinal cord sections for myelin from VHC-treated SCI rats showed profound demyelination (Fig. 7J), which also was attenuated by PDMP treatment until 12 hr after SCI (Fig. 7P). Taken together, these results document that treatment with PDMP protects against white matter vacuolization, tissue destruction, and demyelination after SCI and is effective when administered within minutes or until 12 hr after SCI.
Figure 7.
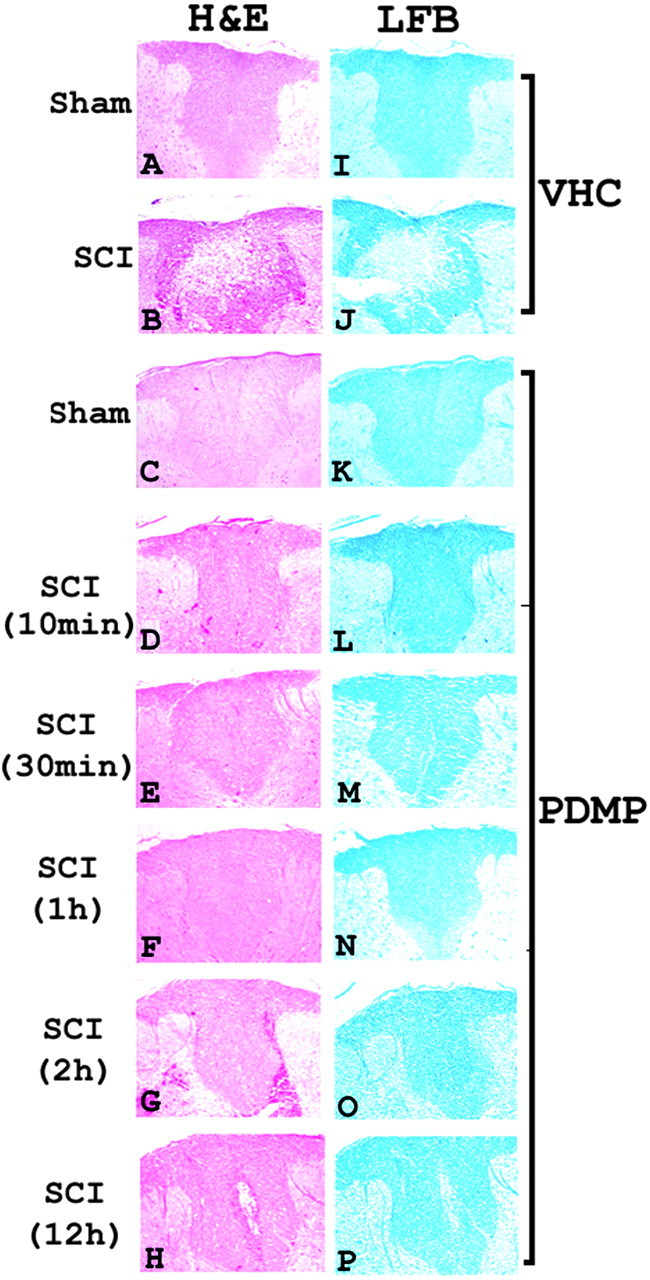
Histology and myelin content examination of spinal cord sections from the lesion epicenter of sham and SCI rats. Shown is H&E examination of spinal cord sections from VHC-treated sham (A), VHC-treated SCI (B), PDMP-treated sham (C), and PDMP-treated SCI (D-H); also shown is LFB-PAS staining for myelin in VHC-treated sham (I), VHC-treated SCI (J), PDMP-treated sham (K), and PDMP-treated SCI (L-P) at 24 hr after SCI. PDMP was administered intraperitoneally at the indicated times (10 and 30 min and 1, 2, and 12 hr) after SCI; tissue sections were extracted and analyzed at day 1 (24 hr) after SCI.
PDMP treatment after SCI shows improved locomotor function
Necrosis and apoptosis, which develops in a delayed manner, are reported to play an important role in secondary injury after SCI, especially because neurological deficit to a large extent is determined by the lesion size in the white matter (Wrathall, 1992; Wrathall et al., 1996). The locomotor function of rats after SCI was assessed on the basis of the 21 point BBB score that evaluates various criteria of hindlimb mobility after SCI (Basso et al., 1996). The first dose of PDMP was administered 10 min after SCI, the second dose at 24 hr (day 1) after SCI, the third dose at 48 hr (day 2) after SCI, and the last dose at 72 hr (day 3) after SCI. From day 4 until day 15 after SCI the rats were cared for without treatment and monitored for locomotor functions. As shown in Figure 8A, all animals started with a normal score of 21 pre-spinal cord injury (pre-SCI). The score plummeted to 0 at day 1, with bilateral hindlimb paralysis in all animals after SCI. However, PDMP-treated animals regained hindlimb function much sooner than the VHC-treated animals. PDMP-treated rats showed a score of 6.9 ± 0.2 at day 3 after SCI, which reflects extensive movement of hip, knee, and ankle; however, the VHC-treated rats showed profound hindlimb paralysis, with a score of 0.9 ± 0.2 with no observable hindlimb movement. Even when PDMP treatment was stopped at day 3 after SCI, the PDMP-treated SCI rats steadily gained hindlimb function. At day 15 after SCI the PDMP-treated rats had a BBB score of 13.9 ± 0.1, demonstrating consistent weight-supported plantar steps and forelimb-hindlimb (FL-HL) coordination. The improved locomotor functions at days 2 and 3 with PDMP treatment also correlated with reduced tissue necrosis (Fig. 8F,G, respectively) and demyelination (Fig. 8L,M, respectively) at the lesion epicenter. Spinal cord sections from VHC-treated rats at days 2 and 3 after SCI showed a large necrotic core at the lesion epicenter (Fig. 8C,D, respectively) and profound demyelination (Fig. 8I,J, respectively). The VHC-treated SCI rats showed very slow recovery of hindlimb motor function as well, with a BBB score of 6.7 ± 0.4 at day 15 after SCI. These results clearly demonstrate the efficacy of PDMP in reducing SCI-induced pathology possibly via attenuation of post-SCI inflammation, resulting in improved functional outcome.
Figure 8.

Locomotor function of PDMP- and VHC-treated rats after SCI. A, BBB locomotor scores of PDMP- and VHC-treated SCI animals at various days after contusion injury. Normal locomotion is represented by 21; 0 represents no observable movement. An increase in BBB score reflects a gain in hindlimb function and recovery. Histology and myelin content examination of spinal cord sections from the lesion epicenter of sham and SCI rats at days 2 and 3 after SCI. Shown is H&E examination of spinal cord sections from VHC-treated sham (B), VHC-treated SCI at day 2 (C), and VHC-treated SCI at day 3 after SCI (D); PDMP-treated sham (E), PDMP-treated SCI at day 2 (F), and PDMP-treated SCI at day 3 after SCI (G). Also shown is LFB-PAS staining for myelin in VHC-treated sham (H), VHC-treated SCI at day 2 (I), and VHC-treated SCI at day 3 after SCI (J); PDMP-treated sham (K), PDMP-treated SCI at day 2 (L), and PDMP-treated SCI at day 3 after SCI (M). Dose 1 of PDMP was administered 10 min after SCI, dose 2 at day 1 (24 hr), dose 3 at day 2 (48 hr), and dose 4 at day 3 (72 hr) after SCI. Tissue sections were extracted and analyzed at day 2 (48 hr) and day 3 (72 hr) after SCI. Data are represented as the mean ± SD; ***p < 0.001 in A compared with VHC-treated SCI at day 3 and #p < 0.001 in A compared with VHC-treated SCI at day 15 after SCI.
Efficacy of PDMP in controlling inflammation and iNOS induction in SCI
Secondary damage as a result of inflammation in response to primary injury is widely believed to exacerbate the impact of the primary injury and impede neuronal recovery. Inflammation composed of proinflammatory cytokine expression and iNOS, TNF-α, and IL-1β gene expression resulting in NO production by reactive astrocytes and macrophages significantly contributes to apoptosis, axonal destruction, and functional deficit in SCI (Wada et al., 1998a,b). To demonstrate the possibility that protection against white matter destruction and demyelination by PDMP might be via attenuation of iNOS expression, we analyzed iNOS expression after SCI. As shown in Figure 9, a robust induction of iNOS mRNA as measured by real-time PCR (Fig. 9A) and protein expression (Fig. 9B) is observed 12 hr after SCI in the VHC-treated SCI group compared with the naive or sham-operated animals. PDMP treatment after SCI markedly suppressed this increase in iNOS gene expression. Double-immunofluorescence analysis of spinal cord sections from the lesion epicenter of VHC-treated SCI rats showed a significant increase in GFAP, a marker for reactive astrogliosis (Fig. 9F), and iNOS (Fig. 9G) levels and their colocalization (Fig. 9H), at 24 hr after SCI, whereas PDMP-treated SCI rats showed significantly reduced GFAP (Fig. 9L) as well as iNOS (Fig. 9M) expression and their colocalization (Fig. 9N), thus demonstrating the efficacy of PDMP in vivo in attenuating iNOS gene expression as well as reactive astrogliosis. In addition to iNOS induction, treatment with PDMP was equally effective in suppressing the expression of proinflammatory cytokines such as TNF-α and IL-1β. A robust increase in mRNA levels of TNF-α at 1 hr (Fig. 10A) and IL-1β at 4 hr (Fig. 10B) after SCI was observed that was inhibited markedly with PDMP treatment. Immunofluorescence detection showed increased protein levels of TNF-α (Fig. 10D) and IL-1β (Fig. 10H) after SCI in the VHC-treated SCI group, which was suppressed significantly with PDMP treatment (Fig. 10F,J, respectively). These studies demonstrate the efficacy of PDMP in attenuating iNOS expression by reactive astrocytes at the site of lesion in an in vivo model of SCI. In addition to iNOS, PDMP also attenuated the production of proinflammatory cytokines such as TNF-α and IL-1β, both of which initiate deadly cascades, causing neuronal apoptosis and massive secondary injury in SCI. The observed anti-inflammatory potential of PDMP finds critical relevance in a number of other neuroinflammatory diseases as well, because iNOS, TNF-α, and IL-1β expression and their related pathology are common to a number of CNS diseases.
Figure 9.

iNOS mRNA and protein expression at the lesion epicenter after SCI. iNOS mRNA levels were quantified by real-time PCR analysis (A), and protein levels were quantified by immunoblot analysis (B) from RNA and protein samples derived from spinal cords sections of VHC- or PDMP-treated sham-operated or SCI rats. Data are represented as the mean ± SD; ***p < 0.001 in A compared with VHC-treated sham and #p < 0.001 compared with VHC-treated for 12 hr. Included is double-immunofluorescence staining of spinal cord sections from the lesion epicenter for iNOS/GFAP colocalization; also included are immunofluorescent microscopy images of spinal cord sections from sham and SCI rats stained with antibodies to iNOS (green) and GFAP (red) as described in Materials and Methods. Shown are GFAP (C), iNOS (D), and their colocalization (E) in VHC-treated sham; GFAP (F), iNOS (G), and their colocalization (H) in VHC-treated SCI; GFAP (I), iNOS (J), and their colocalization (K) in PDMP-treated sham; GFAP (L), iNOS (M), and their colocalization (N) in PDMP-treated SCI rats.
Figure 10.

TNF-α and IL-1β mRNA and protein expression at the lesion epicenter after SCI. TNF-α (A) and IL-1β (B) mRNA levels were quantified by real-time PCR analysis at various durations after SCI. Immunofluorescent microscopy images of spinal cord sections from sham and SCI rats stained with antibodies to TNF-α show VHC-treated sham (C), VHC-treated SCI (D), PDMP-treated sham (E), and PDMP-treated SCI (F) extracted 1 hr after SCI. Immunofluorescent microscopy images of spinal cord sections from sham and SCI rats stained with antibodies to IL-1β as described in Materials and Methods show VHC-treated sham (G), VHC-treated SCI (H), PDMP-treated sham (I), and PDMP-treated SCI (J) extracted 4 hr after SCI. Data are represented as the mean ± SD; ***p < 0.001 in A and B compared with VHC-treated sham; #p < 0.001 in A compared with VHC-treated at 1 hr; #p < 0.001 in B compared with VHC-treated at 4 hr.
Attenuation of apoptosis and demyelination by attenuation of iNOS gene expression after SCI by PDMP
With respect to spinal cord impairment after trauma at the molecular level, NO has been reported to be involved closely in the development of post-traumatic cavitation, neuronal death, axonal degeneration and myelin disruption. Significantly numerous TUNEL-positive cells were scattered in the lesion epicenter after SCI (Fig. 11E), which were identified to be neurons by double-immunofluorescence staining via the use of anti-neuronal nuclei (NeuN) antibodies (Fig. 11D,F). PDMP had a dual beneficial effect in the rat model of SCI. It could attenuate iNOS and proinflammatory cytokine expression after SCI; furthermore, as shown in Figure 11J-L, it also provided protection against apoptosis of neurons. This is of significant importance because no adverse effect of PDMP was observed on neuronal survival in sham-operated animals (Fig. 11G-I), showing that the dose that was administered effectively attenuated inflammation without any obvious adverse effects, which also translates in reduced SCI-related pathology in terms of neuronal loss. Taken together, these studies document the anti-inflammatory potential of PDMP in SCI and possibly other neuroinflammatory disorders, because it effectively can block inflammatory events such as iNOS and cytokine expression, thus providing protection against white matter vacuolization, neuronal apoptosis, and demyelination.
Figure 11.

Double-immunofluorescence staining of spinal cord sections from the lesion epicenter for TUNEL-positive nuclei and neuron-specific marker (NeuN). Immunofluorescent microscopy images of spinal cord sections were taken 24 hr after SCI from sham and SCI rats stained for TUNEL-positive cells (green), using the Apoptag detection kit and antibodies to NeuN (red), as described in Materials and Methods. Shown are NeuN (A), TUNEL (B), and their colocalization (C) in VHC-treated sham; NeuN (D), TUNEL (E), and their colocalization (F) in VHC-treated SCI; NeuN (G), TUNEL (H), and their colocalization (I) in PDMP-treated sham; NeuN (J), TUNEL (K), and their colocalization (L) in PDMP-treated SCI rats.
Discussion
Nitric oxide-mediated pathophysiology is common to a number of neuroinflammatory diseases, including stroke and SCI. Because the factors that induce and regulate iNOS gene expression in inflammatory diseases are not known completely, in this study we investigated the involvement of GSL and demonstrated a novel pathway of iNOS gene regulation by LacCer-mediated events involving the Ras/ERK1/2 and the IκB/NF-κB pathways in primary astrocytes. These conclusions are based on the following findings. (1) LPS/IFN-γ stimulation induced the activity of GalT-2 and increased the production of LacCer. (2) The inhibition of GSL synthesis by PDMP or antisense oligonucleotides to GalT-2 inhibited iNOS gene expression that was reversed by LacCer, but not by other GSLs (GluCer, GalCer, GM1, GM3, and GD3). (3) Inhibition of LacCer synthesis also inhibited the activation of the Ras, ERK, and NF-κB pathways. (4) LacCer-stimulated activation of the Ras/ERK signaling cascade was found to be necessary and ROS-dependent, because the presence of antioxidants NAC and PDTC abolished LacCer-mediated Ras activation as well as LPS/IFN-γ-stimulated iNOS expression. Figure 12 shows a schematic representation of the possible regulation of the Ras/ERK/NF-κB pathways by LacCer. Activation of the small GTPase Ras could be via the direct activation of Src kinases associated with the LacCer-enriched glycosphingolipid-signaling domains (GSDs) present on the cell surface. A number of studies have shown that several transducer molecules such as Src kinase associate with these GSDs and form functional units that mediate signal transduction and cellular functions (Brown and London, 1998). In particular, an Src kinase, Lyn, has been found to associate directly with LacCer, resulting in superoxide generation via NADPH oxidase activation in neutrophils (Iwabuchi and Nagaoka, 2002). Src kinase activation possibly leading to ROS generation may be followed by Grb/SOS-mediated Ras activation that triggers the downstream MEK1/2-ERK1/2 pathway. Activation of the small G-protein Ras and the downstream ERK1/2 has been demonstrated earlier to mediate cytokine-induced iNOS gene expression and NF-κB activation (Pahan et al., 1998b; Marcus et al., 2003). Because Ras-mediated NF-κB regulation has been demonstrated earlier (Won et al., 2004), LacCer-mediated activation of the IκB-NFκB pathway well could be mediated by Ras activation. The critical role for NF-κB in the transcriptional regulation of iNOS gene expression via phosphorylation and degradation of Iκ-B also has been demonstrated earlier (Pahan et al., 1998b). Furthermore, the potentiation of cytokine-mediated expression of iNOS by sphingolipids has been well documented (Pahan et al., 1998b; Giri et al., 2002). The data presented in this study identify a glycosphingolipid, LacCer, as a signaling molecule regulating iNOS gene expression. In addition, the blockade of SCI-mediated iNOS and of proinflammatory gene expression of cytokines in the rat SCI model by PDMP further establishes LacCer, generated via GalT-2 stimulation, to be a potent signaling lipid molecule that triggers inflammation and mediates NO-mediated pathophysiology in various neuroinflammatory diseases.
Figure 12.

Schematic representation of the model for LacCer-mediated regulation of LPS/IFN-γ-induced iNOS gene expression in rat primary astrocytes.
Since the discovery of the sphingomyelin (SM) cycle, which involves sphingomyelin hydrolysis by sphingomyelinases (SMases) resulting in ceramide generation, several inducers (1a,25 dihydroxyvitamin D3, radiation, antibody cross-linking, TNF-α, IFN-γ, IL-1β, nerve growth factor, and brefeldin A) have been shown to be coupled to sphingomyelin-ceramide signaling events (Hannun, 1994; Kolesnick et al., 1994; Kanety et al., 1995; Linardic et al., 1996). Ceramide thus generated plays a role in growth suppression and apoptosis in various cell types, including glial and neuronal cells (Brugg et al., 1996; Wiesner and Dawson, 1996). Impairment of mitochondrial function results in enhanced production of ROS and a decrease in mitochondrial glutathione levels. Depletion of glutathione has been established as one of the major causes of ceramide-induced cytotoxicity/apoptosis in CNS (Singh et al., 1998). Ceramide generated as result of neutral sphingomyelinase activation has been shown to potentiate LPS- and cytokine-mediated induction of iNOS in astrocytes and C6 glioma cells (Pahan et al., 1998b). Furthermore, ceramide generation and its mediated iNOS gene expression are known to be through the Ras/ERK/NF-κB pathway, which is shown to be a redox-sensitive process (Pahan et al., 1998b; Singh et al., 1998). Instead of enzymes of sphingolipid metabolism being viewed as isolated signaling modules, these pathways now are accepted to be highly interconnected, with the product of one enzyme serving as a substrate for the other. This is also true of ceramide generated via the SM cycle or de novo, because ceramide can be converted into other bioactive molecules such as sphingosine, sphingosine-1-phosphate (S-1-P), or glycosphingolipids. The complexity of these bioactive sphingolipids is accentuated by growing evidence of the presence of ceramide and other derivatives such as LacCer and gangliosides in lipid-enriched microdomains within membranes. These microdomains, called “lipid rafts,” have a number of receptors and signaling molecules clustered within or associated with them, thus making them hot spots for signaling events (Hakomori and Handa, 2003). The metabolic interconnections of ceramide and other lipids mediators such as sphingosine, S-1-P, and glycosphingolipids make predicting the specific actions of these intermediates and the enzymes regulating their levels rather complex. For example, although sphingosine has proapoptotic effects like ceramide, depending on cell type (Spiegel and Merrill, 1996), its rapid conversion to S-1-P has proliferative properties antagonistic to those of sphingosine and ceramide (Spiegel and Milstien, 2000). Of the GSLs, GluCer and LacCer have been shown to promote the drug resistance state (Liu et al., 1999) and to mediate oxidized LDL and TNF-α effects on superoxide formation, the activation of MAP kinase, and the induction of proliferation in aortic smooth muscle cells, respectively (Bhunia et al., 1996, 1998; Chatterjee et al., 1997; Chatterjee, 1998).
Traumatic SCI results in pathophysiological changes that can be characterized as acute, secondary, and chronic, which extend from minutes to years after the injury. Numerous detrimental events occur in the acute phase that begins at the moment of injury and extends over the first few days. Mechanical lesions induce immediate damage to the neuronal tracts; blood flow is reduced, creating substantial ischemia along with production of potent proinflammatory cytokines such as TNF-α and IL-1β. In the secondary phase of tissue damage, which occurs over a time course of minutes to weeks after injury, increased production of ROS and RNS (reactive nitrogen species), excessive release of excitatory neurotransmitters, and inflammatory reactions occur. In addition to massive ischemic necrosis, apoptotic cell death also is observed. The size and GFAP content of astrocytes increase in a process of reactive astrogliosis (Bareyre and Schwab, 2003). Traumatic injury also leads to a strong inflammatory response with the recruitment of peripheral-derived immune cells. As with most neurodegenerative conditions including SCI, therapies that use NOS inhibitors and antioxidants aimed at preserving the spared tissue after injury, along with blocking the ensuing inflammation and apoptosis, have shown profound beneficial effects on the behavioral outcome and recovery after injury, because these are able to suppress the acute inflammatory reactions and minimize secondary damage (Blight, 1983; Young, 1993; Liu et al., 1997). However, therapies so far have aimed at inhibiting individual events. In this report we found that PDMP treatment after SCI resulted in profoundly improved hindlimb functional outcome (Fig. 8A). PDMP treatment was found to be effective in (1) blocking trauma-mediated iNOS gene expression in the spinal cord in the rat model of SCI, (2) attenuating proinflammatory cytokine production, (3) attenuating reactive astrogliosis evident by reduced GFAP immunoreactivity, and (4) markedly decreasing neuronal apoptosis and demyelination. Protection of neuronal apoptosis well could be attributable to the inhibition of iNOS expression and NO production. In addition to that, protection against apoptosis possibly may be via depletion of GD3, a LacCer-derived ganglioside, as well. GD3 is a minor ganglioside in normal adult brains; however, its levels are elevated in activated microglia and reactive astrocytes (Kawai et al., 1994). Increased GD3 has been found in multiple sclerosis plaques (Yu et al., 1974) and in brain tissue from patients with various neurodegenerative disorders, such as Creutzfeldt-Jakob disease and subacute sclerosis encephalitis (Ando et al., 1984; Ohtani et al., 1996). It is known now that GD3 causes apoptosis of murine cortex neurons (Simon et al., 2002) and murine primary oligodendrocytes (Castro-Palomino et al., 2001). However, in contrast to the toxicity related to GD3,GM1, another LacCer-derived ganglioside, is known to be essential for neuronal survival (Inokuchi et al., 1998). Because we did not observe a reversal of PDMP-mediated iNOS gene expression by GM1, we expect that PDMP therapy along with GM1 administration will be effective in bypassing the inflammatory reaction while preserving the GM1-mediated prosurvival signals.
In conclusion, this report documents a tight link of LacCer with the regulation of iNOS gene expression in inflammatory disease processes and unravels a novel, potential therapeutic target of glycosphingolipid modulation for the amelioration of pathophysiology in neuroinflammatory disorders.
Footnotes
This work was supported by National Institutes of Health Grants NS-22576, NS-34741, NS-37766, NS-40144, and NS-40810. We acknowledge the help of Jason Wallace from the Medical University of South Carolina Mass Spectrometry Facility and Joyce Bryan for laboratory assistance.
Correspondence should be addressed to Dr. Inderjit Singh, Division of Developmental Neurogenetics, Department of Pediatrics, Medical University of South Carolina, 316 CSB, Charleston, SC 29425. E-mail: singhi@musc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245942-14$15.00/0
References
- Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K (2000) Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord 14[Suppl 1]: S47-S53. [DOI] [PubMed] [Google Scholar]
- Andersson PB, Perry VH, Gordon S (1992) Intracerebral injection of pro-inflammatory cytokines or leukocyte chemotaxins induces minimal myelomonocytic cell recruitment to the parenchyma of the central nervous system. J Exp Med 176: 255-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ando S, Toyoda Y, Nagai Y, Ikuta F (1984) Alterations in brain gangliosides and other lipids of patients with Creutzfeldt-Jakob disease and subacute sclerosing panencephalitis (SSPE). Jpn J Exp Med 54: 229-234. [PubMed] [Google Scholar]
- Bal-Price A, Brown GC (2001) Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci 21: 6480-6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre FM, Schwab ME (2003) Inflammation, degeneration, and regeneration in the injured spinal cord: insights from DNA microarrays. Trends Neurosci 26: 555-563. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC (1996) Graded histological and locomotor outcomes after spinal cord contusion using the NYU weight-drop device versus transection. Exp Neurol 139: 244-256. [DOI] [PubMed] [Google Scholar]
- Bhunia AK, Han H, Snowden A, Chatterjee S (1996) Lactosylceramide stimulates Ras-GTP loading, kinases (MEK, Raf), p44 mitogen-activated protein kinase, and c-fos expression in human aortic smooth muscle cells. J Biol Chem 271: 10660-10666. [DOI] [PubMed] [Google Scholar]
- Bhunia AK, Han H, Snowden A, Chatterjee S (1997) Redox-regulated signaling by lactosylceramide in the proliferation of human aortic smooth muscle cells. J Biol Chem 272: 15642-15649. [DOI] [PubMed] [Google Scholar]
- Bhunia AK, Arai T, Bulkley G, Chatterjee S (1998) Lactosylceramide mediates tumor necrosis factor-α-induced intercellular adhesion molecule-1 (ICAM-1) expression and the adhesion of neutrophil in human umbilical vein endothelial cells. J Biol Chem 273: 34349-34357. [DOI] [PubMed] [Google Scholar]
- Blight AR (1983) Axonal physiology of chronic spinal cord injury in the cat: intracellular recording in vitro Neuroscience 10: 1471-1486. [DOI] [PubMed] [Google Scholar]
- Bo L, Dawson TM, Wesselingh S, Mork S, Choi S, Kong PA, Hanley D, Trapp BD (1994) Induction of nitric oxide synthase in demyelinating regions of multiple sclerosis brains. Ann Neurol 36: 778-786. [DOI] [PubMed] [Google Scholar]
- Brown DA, London E (1998) Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 14: 111-136. [DOI] [PubMed] [Google Scholar]
- Brugg B, Michel PP, Agid Y, Ruberg M (1996) Ceramide induces apoptosis in cultured mesencephalic neurons. J Neurochem 66: 733-739. [DOI] [PubMed] [Google Scholar]
- Castro-Palomino JC, Simon B, Speer O, Leist M, Schmidt RR (2001) Synthesis of ganglioside GD3 and its comparison with bovine GD3 with regard to oligodendrocyte apoptosis mitochondrial damage. Chemistry 7: 2178-2184. [DOI] [PubMed] [Google Scholar]
- Chatterjee S (1998) Sphingolipids in atherosclerosis and vascular biology. Arterioscler Thromb Vasc Biol 18: 1523-1533. [DOI] [PubMed] [Google Scholar]
- Chatterjee S, Bhunia AK, Snowden A, Han H (1997) Oxidized low density lipoproteins stimulate galactosyltransferase activity, Ras activation, p44 mitogen-activated protein kinase and c-fos expression in aortic smooth muscle cells. Glycobiology 7: 703-710. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL, Snyder SH (1993) Nitric oxide as a mediator of neurotoxicity. NIDA Res Monogr 136: 258-273. [PubMed] [Google Scholar]
- Dignam JD, Martin PL, Shastry BS, Roeder RG (1983) Eukaryotic gene transcription with purified components. Methods Enzymol 101: 582-598. [DOI] [PubMed] [Google Scholar]
- Fern R (2001) Ischemia: astrocytes show their sensitive side. Prog Brain Res 132: 405-411. [DOI] [PubMed] [Google Scholar]
- Gilg AG, Singh AK, Singh I (2000) Inducible nitric oxide synthase in the central nervous system of patients with X-adrenoleukodystrophy. J Neuropathol Exp Neurol 59: 1063-1069. [DOI] [PubMed] [Google Scholar]
- Giri S, Jatana M, Rattan R, Won JS, Singh I, Singh AK (2002) Galactosylsphingosine (psychosine)-induced expression of cytokine-mediated inducible nitric oxide synthases via AP-1 and C/EBP: implications for Krabbe disease. FASEB J 16: 661-672. [DOI] [PubMed] [Google Scholar]
- Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fluids. Anal Biochem 126: 131-138. [DOI] [PubMed] [Google Scholar]
- Gruner JA (1992) A monitored contusion model of spinal cord injury in the rat. J Neurotrauma 9: 123-128. [DOI] [PubMed] [Google Scholar]
- Hakomori S, Handa K (2003) Interaction of glycosphingolipids with signal transducers and membrane proteins in glycosphingolipid-enriched microdomains. Methods Enzymol 363: 191-207. [DOI] [PubMed] [Google Scholar]
- Hannun YA (1994) The sphingomyelin cycle and the second messenger function of ceramide. J Biol Chem 269: 3125-3128. [PubMed] [Google Scholar]
- Herrmann C, Martin GA, Wittinghofer A (1995) Quantitative analysis of the complex between p21ras and the Ras-binding domain of the human Raf-1 protein kinase. J Biol Chem 270: 2901-2905. [DOI] [PubMed] [Google Scholar]
- Inokuchi J, Kuroda Y, Kosaka S, Fujiwara M (1998) l-Threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol stimulates ganglioside biosynthesis, neurite outgrowth and synapse formation in cultured cortical neurons, and ameliorates memory deficits in ischemic rats. Acta Biochim Pol 45: 479-492. [PubMed] [Google Scholar]
- Iwabuchi K, Nagaoka I (2002) Lactosylceramide-enriched glycosphingo-lipid signaling domain mediates superoxide generation from human neutrophils. Blood 100: 1454-1464. [PubMed] [Google Scholar]
- Kanety H, Feinstein R, Papa MZ, Hemi R, Karasik A (1995) Tumor necrosis factor α-induced phosphorylation of insulin receptor substrate-1 (IRS-1). Possible mechanism for suppression of insulin-stimulated tyrosine phosphorylation of IRS-1. J Biol Chem 270: 23780-23784. [DOI] [PubMed] [Google Scholar]
- Kawai K, Kuroda S, Watarai S, Takahashi H, Ikuta F (1994) Occurrence of GD3 ganglioside in reactive astrocytes—an immunocytochemical study in the rat brain. Neurosci Lett 174: 225-227. [DOI] [PubMed] [Google Scholar]
- Khan M, Pahan K, Singh AK, Singh I (1998) Cytokine-induced accumulation of very long-chain fatty acids in rat C6 glial cells: implication for X-adrenoleukodystrophy. J Neurochem 71: 78-87. [DOI] [PubMed] [Google Scholar]
- Kiernan J (1990) Interactions between mast cells and nerves. Neurogenic inflammation. Trends Pharmacol Sci 11: 316. [DOI] [PubMed] [Google Scholar]
- Kolesnick RN, Haimovitz-Friedman A, Fuks Z (1994) The sphingomyelin signal transduction pathway mediates apoptosis for tumor necrosis factor, Fas, and ionizing radiation. Biochem Cell Biol 72: 471-474. [DOI] [PubMed] [Google Scholar]
- Koprowski H, Zheng YM, Heber-Katz E, Fraser N, Rorke L, Fu ZF, Hanlon C, Dietzschold B (1993) In vivo expression of inducible nitric oxide synthase in experimentally induced neurologic diseases. Proc Natl Acad Sci USA 90: 3024-3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander HM, Ogiste JS, Teng KK, Novogrodsky A (1995) p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem 270: 21195-21198. [DOI] [PubMed] [Google Scholar]
- Lassmann H, Wisniewski HM (1979) Chronic relapsing experimental allergic encephalomyelitis: clinicopathological comparison with multiple sclerosis. Arch Neurol 36: 490-497. [DOI] [PubMed] [Google Scholar]
- Leist M, Fava E, Montecucco C, Nicotera P (1997) Peroxynitrite and nitric oxide donors induce neuronal apoptosis by eliciting autocrine excitotoxicity. Eur J Neurosci 9: 1488-1498. [DOI] [PubMed] [Google Scholar]
- Linardic CM, Jayadev S, Hannun YA (1996) Activation of the sphingomyelin cycle by brefeldin A: effects of brefeldin A on differentiation and implications for a role for ceramide in regulation of protein trafficking. Cell Growth Differ 7: 765-774. [PubMed] [Google Scholar]
- Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW (1997) Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci 17: 5395-5406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YY, Han TY, Giuliano AE, Cabot MC (1999) Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J Biol Chem 274: 1140-1146. [DOI] [PubMed] [Google Scholar]
- Marcus JS, Karackattu SL, Fleegal MA, Sumners C (2003) Cytokine-stimulated inducible nitric oxide synthase expression in astroglia: role of ERK mitogen-activated protein kinase and NF-κB. Glia 41: 152-160. [DOI] [PubMed] [Google Scholar]
- Matsuyama Y, Sato K, Kamiya M, Yano J, Iwata H, Isobe K (1998) Nitric oxide: a possible etiologic factor in spinal cord cavitation. J Spinal Disord 11: 248-252. [PubMed] [Google Scholar]
- Ohtani Y, Tamai Y, Ohnuki Y, Miura S (1996) Ganglioside alterations in the central and peripheral nervous systems of patients with Creutzfeldt-Jakob disease. Neurodegeneration 5: 331-338. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Namboodiri AM, Singh I (1998a) N-acetyl cysteine inhibits induction of NO production by endotoxin or cytokine-stimulated rat peritoneal macrophages, C6 glial cells, and astrocytes. Free Radic Biol Med 24: 39-48. [DOI] [PubMed] [Google Scholar]
- Pahan K, Sheikh FG, Khan M, Namboodiri AM, Singh I (1998b) Sphingomyelinase and ceramide stimulate the expression of inducible nitric oxide synthase in rat primary astrocytes. J Biol Chem 273: 2591-2600. [DOI] [PubMed] [Google Scholar]
- Pahan K, Liu X, McKinney MJ, Wood C, Sheikh FG, Raymond JR (2000) Expression of a dominant-negative mutant of p21ras inhibits induction of nitric oxide synthase and activation of nuclear factor-κB in primary astrocytes. J Neurochem 74: 2288-2295. [DOI] [PubMed] [Google Scholar]
- Perry RT, Collins JS, Wiener H, Acton R, Go RC (2001) The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging 22: 873-883. [DOI] [PubMed] [Google Scholar]
- Renno T, Krakowski M, Piccirillo C, Lin JY, Owens T (1995) TNF-α expression by resident microglia and infiltrating leukocytes in the central nervous system of mice with experimental allergic encephalomyelitis. Regulation by Th1 cytokines. J Immunol 154: 944-953. [PubMed] [Google Scholar]
- Saklatvala J, Davis W, Guesdon F (1996) Interleukin 1 (IL1) and tumour necrosis factor (TNF) signal transduction. Philos Trans R Soc Lond B Biol Sci 351: 151-157. [DOI] [PubMed] [Google Scholar]
- Satake K, Matsuyama Y, Kamiya M, Kawakami H, Iwata H, Adachi K, Kiuchi K (2000) Nitric oxide via macrophage iNOS induces apoptosis following traumatic spinal cord injury. Brain Res Mol Brain Res 85: 114-122. [DOI] [PubMed] [Google Scholar]
- Sequeira SM, Ambrosio AF, Malva JO, Carvalho AP, Carvalho CM (1997) Modulation of glutamate release from rat hippocampal synaptosomes by nitric oxide. Nitric Oxide 1: 315-329. [DOI] [PubMed] [Google Scholar]
- Simmons ML, Murphy S (1992) Induction of nitric oxide synthase in glial cells. J Neurochem 59: 897-905. [DOI] [PubMed] [Google Scholar]
- Simon BM, Malisan F, Testi R, Nicotera P, Leist M (2002) Disialoganglioside GD3 is released by microglia and induces oligodendrocyte apoptosis. Cell Death Differ 9: 758-767. [DOI] [PubMed] [Google Scholar]
- Singh I, Pahan K, Khan M, Singh AK (1998) Cytokine-mediated induction of ceramide production is redox-sensitive. Implications to proinflammatory cytokine-mediated apoptosis in demyelinating diseases. J Biol Chem 273: 20354-20362. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Merrill Jr AH (1996) Sphingolipid metabolism and cell growth regulation. FASEB J 10: 1388-1397. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S (2000) Sphingosine-1-phosphate: signaling inside and out. FEBS Lett 476: 55-57. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Tatsuoka H, Chiba T, Sekikawa T, Nemoto T, Moriya H, Sakuraba S, Nakaya H (2001) Beneficial effects of nitric oxide synthase inhibition on the recovery of neurological function after spinal cord injury in rats. Naunyn Schmiedebergs Arch Pharmacol 363: 94-100. [DOI] [PubMed] [Google Scholar]
- Vodovotz Y, Lucia MS, Flanders KC, Chesler L, Xie QW, Smith TW, Weidner J, Mumford R, Webber R, Nathan C, Roberts AB, Lippa CF, Sporn MB (1996) Inducible nitric oxide synthase in tangle-bearing neurons of patients with Alzheimer's disease. J Exp Med 184: 1425-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Busto R, Dietrich WD (1998a) Role of nitric oxide in traumatic brain injury in the rat. J Neurosurg 89: 807-818. [DOI] [PubMed] [Google Scholar]
- Wada K, Chatzipanteli K, Kraydieh S, Busto R, Dietrich WD (1998b) Inducible nitric oxide synthase expression after traumatic brain injury and neuroprotection with aminoguanidine treatment in rats. Neurosurgery 43: 1427-1436. [DOI] [PubMed] [Google Scholar]
- Wiesner DA, Dawson G (1996) Staurosporine induces programmed cell death in embryonic neurons and activation of the ceramide pathway. J Neurochem 66: 1418-1425. [DOI] [PubMed] [Google Scholar]
- Won JS, Choi MR, Suh HW (2001) Stimulation of astrocyte-enriched culture with C2 ceramide increases proenkephalin mRNA: involvement of cAMP-response element binding protein and mitogen-activated protein kinases. Brain Res 903: 207-215. [DOI] [PubMed] [Google Scholar]
- Won JS, Im YB, Khan M, Singh AK, Singh I (2004) The role of neutral sphingomyelinase produced ceramide in lipopolysaccharide-mediated expression of inducible nitric oxide synthase. J Neurochem 88: 583-593. [DOI] [PubMed] [Google Scholar]
- Wrathall JR (1992) Spinal cord injury models. J Neurotrauma 9[Suppl 1]: S129-S134. [PubMed] [Google Scholar]
- Wrathall JR, Teng YD, Choiniere D (1996) Amelioration of functional deficits from spinal cord trauma with systemically administered NBQX, an antagonist of non-N-methyl-d-aspartate receptors. Exp Neurol 137: 119-126. [DOI] [PubMed] [Google Scholar]
- Xie QW, Kashiwabara Y, Nathan C (1994) Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J Biol Chem 269: 4705-4708. [PubMed] [Google Scholar]
- Yeh LH, Kinsey AM, Chatterjee S, Alevriadou BR (2001) Lactosylceramide mediates shear-induced endothelial superoxide production and intercellular adhesion molecule-1 expression. J Vasc Res 38: 551-559. [DOI] [PubMed] [Google Scholar]
- Young W (1993) Secondary injury mechanisms in acute spinal cord injury. J Emerg Med 11[Suppl 1]: 13-22. [PubMed] [Google Scholar]
- Yu RK, Ledeen RW, Eng LF (1974) Ganglioside abnormalities in multiple sclerosis. J Neurochem 23: 169-174. [DOI] [PubMed] [Google Scholar]
- Zielasek J, Tausch M, Toyka KV, Hartung HP (1992) Production of nitrite by neonatal rat microglial cells/brain macrophages. Cell Immunol 141: 111-120. [DOI] [PubMed] [Google Scholar]