

Abstract
We investigated the mechanisms by which activation of group I metabotropic glutamate receptors (mGluRs) and CB1 cannabinoid receptors (CB1Rs) leads to inhibition of synaptic currents at the calyx of Held synapse in the medial nucleus of the trapezoid body (MNTB) of the rat auditory brainstem. In ∼50% of the MNTB neurons tested, activation of group I mGluRs by the specific agonist (s)-3,5-dihydroxyphenylglycine (DHPG) reversibly inhibited AMPA receptor- and NMDA receptor-mediated EPSCs to a similar extent and reduced paired-pulse depression, suggestive of an inhibition of glutamate release. Presynaptic voltage-clamp experiments revealed a reversible reduction of Ca2+ currents by DHPG, with no significant modification of the presynaptic action potential waveform. Likewise, in ∼50% of the tested cells, the CB1 receptor agonist (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone (WIN) reversibly inhibited EPSCs, presynaptic Ca2+ currents, and exocytosis. For a given cell, the amount of inhibition by DHPG correlated with that by WIN. Moreover, the inhibitory action of DHPG was blocked by the CB1R antagonist N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251) and occluded by WIN, indicating that DHPG and WIN operate via a common pathway. The inhibition of EPSCs by DHPG, but not by WIN, was abolished after dialyzing 40 mm BAPTA into the postsynaptic cell, suggesting that DHPG activated postsynaptic mGluRs. Light and electron microscopy immunolabeling indicated a presynaptic expression of CB1Rs and postsynaptic localization of mGluR1a. Our data suggest that activation of postsynaptic mGluRs triggers the Ca2+-dependent release of endocannabinoids that activate CB1 receptors on the calyx terminal, which leads to a reduction of presynaptic Ca2+ current and glutamate release.
Keywords: presynaptic Ca channels, CB1 receptor, cannabinoids, MNTB, calyx of Held, retrograde signaling, group I mGluRs, exocytosis
Introduction
Metabotropic glutamate receptors (mGluRs) are expressed widely in the CNS where they regulate neurotransmission (Conn and Pin, 1997; Anwyl, 1999). Although anatomical and physiological evidence point to a mostly presynaptic expression of group II and III mGluRs, the localization of group I mGluRs is less clear. Physiological and neurochemical evidence exist for presynaptic modulation of neurotransmission by group I mGluRs (Morishita et al., 1998; Rodriguez-Moreno et al., 1998; Faas et al., 2002), yet most anatomical evidence suggests that these receptors are expressed exclusively postsynaptically (Cartmell and Schoepp, 2000; Elezgarai et al., 2003). This paradox may be resolved by interactions between group I mGluRs and presynaptic CB1 receptors (CB1Rs), the main cannabinoid receptor in the CNS (Doherty and Dingledine, 2003). In the hippocampus and cerebellum, activation of group I mGluRs can lead to release of endocannabinoids (via Ca2+-dependent and -independent pathways) and CB1R-dependent inhibition of neurotransmitter release (Maejima et al., 2001; Varma et al., 2001; Ohno-Shosaku et al., 2002; Chevaleyre and Castillo, 2003).
Activation of CB1R inhibits neurotransmitter release in many brain regions but the mechanism of action is debated (Alger, 2002). In expression systems and cell bodies, CB1R couples to activation of K+ channels or inhibition of neuronal Ca2+ channels, or both (Howlett et al., 2002). Either of these mechanisms can reduce Ca2+ influx at nerve terminals and thereby inhibit transmitter release. Activation of K+ channels may change the presynaptic action potential (AP) and thus indirectly modulate Ca2+ channel activity (Diana and Marty, 2003). Alternatively, CB1R activation may directly inhibit presynaptic Ca2+ channels coupled to exocytosis. Investigations of CB1R action in nerve terminals have relied on measurements of cytosolic Ca2+ concentration with indicator dyes (Kreitzer and Regehr, 2001) and occlusion experiments using specific Ca2+ channel blockers (Sullivan, 1999; Robbe et al., 2001; Wilson and Nicoll, 2001); however, direct recordings of CB1R-dependent modulation of presynaptic Ca2+ currents or APs are still missing.
The calyx of Held, a glutamatergic nerve terminal located in the medial nucleus of the trapezoid body (MNTB), permits direct recording of presynaptic APs and Ca2+ currents (Forsythe, 1994; Borst and Sakmann, 1998). This synapse expresses presynaptic group II and III mGluRs that are negatively coupled to neurotransmitter release (Barnes-Davies and Forsythe, 1995; von Gersdorff et al., 1997; Elezgarai et al., 1999, 2001). In contrast, the expression and function of group I mGluRs and CB1Rs at the calyx of Held have not been explored. Recent attention has focused on CB1R-dependent inhibition of neurotransmitter release in synapses in which presynaptic Ca2+ influx is mediated largely by Q- or N-type Ca2+ channels (Sullivan, 1999; Wilson et al., 2001). In contrast, a role for P-type Ca2+ channels in presynaptic cannabinoid signaling has not been demonstrated. The calyx of Held, after postnatal day 10 (P10), uses exclusively P-type Ca2+ channels for glutamate release (Iwasaki and Takahashi, 1998) and thus provides an opportunity to study the role of these Ca2+ channels in CB1R-mediated inhibition. Here we investigate how CB1Rs and group I mGluRs interact to regulate glutamate release from the calyx of Held.
Materials and Methods
Slice preparation
Brainstem slices were obtained from P5-P14 Sprague Dawley rats. After decapitation the brainstem was quickly immersed in ice-cold low-calcium artificial CSF (aCSF) containing (in mm): 125 NaCl, 2.5 KCl, 3 MgCl2, 0.1 CaCl2, 25 glucose, 25 NaHCO3, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo-inositol, 2 Na-pyruvate, pH 7.3, when bubbled with carbogen (95% O2, 5% CO2). The brainstem was then glued onto the stage of a Vibratome slicer (Leica, Nussloch, Germany), and 180- to 200-μm-thick slices were cut proceeding from caudal to rostral. Slices were transferred to an incubation chamber containing normal aCSF bubbled with carbogen and maintained at 35°C for 60 min and thereafter at room temperature (RT) (22-24°C). The normal aCSF was the same as the low-calcium aCSF except that 1.0 mm MgCl2 and 2.0 mm CaCl2 were used.
Electrophysiology
Standard whole-cell voltage and current-clamp recordings were made from MNTB principal neurons and calyx of Held nerve terminals with an EPC-9 amplifier (HEKA, Lambrecht/Pfalz, Germany). Sampling intervals and filter settings were 10-25 μsec and 4 kHz, respectively. Slices were visualized by infrared-differential interference contrast microscopy through a 40× water-immersion objective (Axioskop, Zeiss, Oberkochen, Germany) coupled to a 2× premagnification (Optovart, Zeiss) and a CCD camera (C79; Hamamatsu, Bridgewater, NJ). Most experiments were performed at RT. Where indicated, the bath temperature was raised to 35°C using a Warner Instruments heater system.
Patch pipettes were pulled from standard wall borosilicate or Kimax glass (WPI, Sarasota, FL) on a Narishige puller (PP-830; Tokyo, Japan) or a Sutter puller (P-97; Novato, CA). Open tip resistance was 1-3 MΩ for recording synaptic currents and 2-4 MΩ for recording postsynaptic holding current and presynaptic currents and APs. Pipettes were coated with dental wax to reduce noise and pipette capacitance. Access resistance (RS) was <20 MΩ for calyx recordings and <6.0 MΩ for postsynaptic recordings. RS was compensated ∼60% for presynaptic and >70% for postsynaptic recordings. No corrections were made for liquid junction potentials.
EPSCs were recorded under voltage clamp (-70 or -80 mV) of the MNTB neuron during stimulation of the calyx axon via a bipolar tungsten electrode (FHC, Bowdoinham, ME) placed at the brainstem mid-line. Stimulation pulses (100 μsec) were applied through a Master-8 stimulator (AMPI, Jerusalem, Israel), and stimulation voltage was set to 20-30% above threshold (<30 V). In some experiments, paired stimuli were delivered using an interstimulus interval of 50-100 msec. The paired-pulse ratio (PPR) in these experiments was calculated as the ratio between the mean values of the second and first EPSC [as recommended by Kim and Alger (2001)]. Usually, mean EPSC amplitudes were obtained by averaging 10 consecutive EPSCs. For experiments with DHPG, mean EPSCs were obtained by averaging at least three EPSCs at the peak of inhibition in cells that showed desensitization of the DHPG response. Pipette solution for measuring EPSCs consisted of (in mm): 75 K-gluconate, 75 KCl, 2 Na2-phosphocreatine, 10 HEPES, 0.5-5 EGTA, 4 ATP-Mg, 0.3 GTP, pH 7.3 with KOH. To record NMDA receptor (NMDAR)-mediated EPSCs, CsCl was substituted for K-gluconate and KCl, and 10 mm TEA was added. For experiments using high intracellular BAPTA, the pipette solution contained (in mm): 40 K4-BAPTA, 10-20 HEPES, 4 MgATP, 2 Na2-phosphocreatine, 0.3 NaGTP, 90-100 sucrose (for osmotic balance), pH 7.3 with KOH. In some experiments, the postsynaptic holding current was recorded at depolarized membrane potentials to measure the slope input conductance about the zero-current potential (see Fig. 5A). In these experiments, the pipette solution was as described above for synaptic currents, and 1 μm TTX was added to the aCSF to block APs. The osmolarity of all external and internal solutions was 305-310 mOsm.
Figure 5.

Signaling through the postsynaptic cell is required for EPSC inhibition by DHPG. A1, DHPG (100 μm) changes the holding current and decreases input conductance of the MNTB neuron. This shift in holding current does not require CB1 receptors and is accompanied by a decrease in conductance. Steady-state membrane current was measured every 5 sec at -60 mV (□) and -70 mV (○) during application of DHPG (indicated by the thick line) in the presence of AM251 (included throughout the experiment). The DC slope conductance was determined as the difference between these two currents divided by the voltage difference (10 mV) and is shown as the open triangles. A2, Example of an EPSC that was insensitive to DHPG. A3, Time course of EPSC amplitude (□) and postsynaptic holding current (○; Vh = -70 mV) recorded simultaneously in control aCSF and during application of DHPG (same cell as B1). B, Buffering Ca2+ in the MNTB neuron with 40 mm BAPTA blocks the action of DHPG, but not that of WIN, on postsynaptic currents. B1, EPSCs recorded with a patch-clamp pipette containing 40 mm BAPTA are insensitive to DHPG but reversibly inhibited by WIN. B2, Average inhibition of EPSC amplitudes with 40 mm BAPTA included in the postsynaptic pipette solution. Strong Ca2+ buffer in the postsynaptic cell selectively inhibits DHPG actions in WIN-sensitive cells (n = 7). Another seven cells tested under these conditions were insensitive to both DHPG and WIN.
Presynaptic Ca2+ currents were recorded in aCSF containing 1 μm TTX and 10 mm TEA. Pipette solution contained (in mm): 135 Cs-gluconate, 20 TEA-Cl, 10 HEPES, 0.5 EGTA, 4 Mg-ATP, 0.3 GTP, 5 Na2-phosphocreatine, pH 7.3 with CsOH. In some experiments, a Cs-methanesulfonate-based internal solution was used, as described by Leão and von Gersdorff (2002). Results obtained with these two internal solutions were comparable and therefore pooled. The calyces were held at -80 mV, and step depolarizations were applied as described in Results. All Ca2+ currents have been leak subtracted using a P/5 protocol. Membrane capacitance was monitored using a software lock-in amplifier (Pulse, HEKA), which implements the “sin + DC” algorithm. The sinusoidal excitation had a peak-to-peak amplitude of 60 mV and a frequency of 1 kHz.
Presynaptic APs were recorded in normal aCSF, using the fast current-clamp mode of the EPC-9 after adjusting the fast-capacitance cancellation while in cell-attached mode (Taschenberger and von Gersdorff, 2000). Pipette solution for measuring presynaptic APs consisted of (in mm): 110 K-methylsulfate, 30 KCl, 1 MgCl2, 10 HEPES, 0.2 EGTA, 2 Mg-ATP, 0.5 GTP, 10 Na2-phosphocreatine, pH 7.3 with KOH. Presynaptic APs were elicited by afferent fiber stimulation as described above for synaptic currents. After the whole-cell mode of recording was established, holding current was applied to set the membrane potential to -80 mV. The holding current was then left constant throughout the experiment.
Drug application
Where indicated in the text, the following drugs were bath applied: picrotoxin (50 μm), strychnine (2 μm), d,l-2-amino-5-phosphonovaleric acid (APV; 100 μm), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f] quinoxaline-7-sulfonamide (NBQX; 10 μm), (RS)-α-cyclopropyl-4-phosphonophenylglycine (CPPG; 300 μm), DHPG (100 μm), scopolamine (1 μm), CGP 56999a (1 μm), N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251; 5 μm), (R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo [1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone [WIN 55212-2 (WIN); 2-5 μm], and 2S-2-amino-2-(1S,2S-2-carboxycycloprop-1-yl)-3-(xanth-9-yl) propanoic acid (LY341495; 200 μm). APV, CPPG, NBQX, AM251, WIN 55212-2, LY341495, 2-methyl-6-(phenylethynyl)pyridine hydrochloride (MPEP), and DHPG were from Tocris Cookson (Bristol, UK). CGP 56999a was a gift from Novartis Pharmaceuticals (Basel, Switzerland). All other salts and chemicals were from Sigma (St. Louis, MO). Off-line analysis was done with “IgorPro” software (Wavemetrics, Lake Oswego, OR). Average data are reported as mean ± SEM.
Confocal immunofluorescence
For confocal immunofluorescence staining experiments, 200-μm-thick transverse slices containing MNTB were made from P10-P14 rat brainstem using a Leica VT1000S Vibratome and postfixed in 0.1 m PBS containing 4% paraformaldehyde or 4% paraformaldehyde, 0.025% glutaraldehyde, and 0.1% picric acid for up to 2 hr at room temperature. The slices were washed and blocked in 0.1 m PBS containing 3% calf serum, 0.5% Triton X-100, and 0.025% azide for ∼60 min and incubated with primary antibody for 2 d at 4°C. Monoclonal mouse antibody against Rab3a (Transduction Laboratories, Lexington, KY) was used at 1:1000 in PBS blocking solution. Polyclonal rabbit antibodies against mGluR1a (Chemicon, Temecula, CA) were used at 1:500, and affinity-purified goat antibodies against the C terminus of rat CB1 (gift from Dr. Ken Mackie, University of Washington, Seattle, WA) were used at 1:300. Slices were then washed in 0.1 m PBS and incubated with Alexa dye-conjugated secondary antibodies (Molecular Probes, Eugene, OR) for 24 hr at 4°Cat 1:500-800 dilution. Slices were washed again in 0.1 m PBS and then mounted in Prolong mounting gel (Molecular Probes), coverslipped, and imaged on a scanning laser confocal microscope (Zeiss). Photomicrographs (1- to 2-μm-thick optical slices) were taken with a Zeiss LSM510 META confocal microscope and presented using Adobe Photoshop 7 software.
Light microscopic immunocytochemistry
Brains were removed from the skull, and brainstem blocks containing MNTB were transferred first to a 15% (w/v) aqueous sucrose solution and later to a 30% sucrose solution after sinking of the tissue. Cryostat sections (30 μm thick) were quickly thawed in PBS at room temperature. Sections were preincubated with 1.5% normal goat serum (NGS) prepared in PBS for 1 hr at room temperature and incubated with an immunopurified antisera to CB1 or mGluR1a (1 mg/ml diluted in 1.5% normal goat serum/PBS; Chemicon) for 2 d at 4°C. Finally, tissue sections were processed using a conventional avidin-biotin horseradish peroxidase complex method (ABC; Elite, Vector Laboratories, Burlingame, CA). Briefly, sections were incubated with a biotinylated secondary antibody for 1 hr and with the avidin-biotin complex for 1 hr, both at room temperature. To visualize the immunoreaction in the light microscope, sections were preincubated with 0.05% 3,3′-diaminobenzidine (DAB) for 5 min and subsequently incubated by adding 0.01% hydrogen peroxide to the same solution for 5 min. Stained sections were mounted, dried, dehydrated, and coverslipped with DPX (Fluka Chemie, Buchs, Switzerland).
To assess the specificity of the commercial CB1 antiserum, we performed several positive controls on cerebellar and brainstem tissues expressing high levels of mRNA coding for CB1 (Matsuda et al., 1993). Both tissues were processed exactly as described above. As a negative control, MNTB sections were incubated with nonimmune serum instead of the primary CB1 antiserum. Previous work has demonstrated a high specificity of the immunostaining when using the mGluR1a antiserum used in this study (Mateos et al., 2000).
Electron microscopy
Five Sprague Dawley rats (OF1; Iffa-Credo, L'Arbresle, France) from P14 were used in this study. After they were anesthetized with sodium pentobarbital (60 mg/kg body weight; Sanofi, Libourne, France), the animals were transcardially perfused at room temperature (20-25°C) with PBS, pH 7.4, for 20 sec, followed by 500 ml of ice-cold fixative containing 4% formaldehyde, 0.025% glutaraldehyde, and 0.2% picric acid in 0.1 m phosphate buffer (PB), pH 7.4, for 10-15 min. After perfusion, brainstem blocks were washed thoroughly in 0.1 m PB. Parasagittal vibratome sections (50 μm) containing MNTB were cut and collected in 0.1 m PB at room temperature. Floating sections were preincubated in 20% NGS/PBS for 1 hr at room temperature and incubated with affinity-purified antisera (1 mg/ml in 1.5% NGS/PBS) recognizing mGluR1a or C terminus of the CB1 receptor (Chemicon) for 3 d at 4°C. After several washes in PBS, tissue sections were incubated in 1.4 nm gold-labeled goat anti-rabbit IgG (Fab′ fragment, 1:100 in 1.5% NGS/PBS; Nanoprobes Inc., Stony Brook, NY) for 3 hr on a shaker at room temperature. Thereafter, the MNTB sections were washed in PBS overnight and postfixed in 1% glutaraldehyde for 10 min. After washes in double-distilled water, gold particles were silver intensified with an HQ Silver kit (Nanoprobes Inc.) for ∼12 min. The gold-labeled, silver-intensified tissue sections were osmicated in 1% PB (0.1 m), pH 7.4, osmium tetroxide for 20 min on a shaker at room temperature, dehydrated in graded alcohols, transferred to propylene oxide, and embedded flat in Epon 812. Ultrathin sections were collected on mesh nickel grids stained with lead citrate and examined in a JEOL X-100 electron microscope.
Statistical analysis of immunohistochemistry
Light microscopy. Four P14 rats were used for analysis of the percentage of MNTB principal cells associated with immunoreactivity for CB1 or mGluR1a. The counting was done by three different observers on 30-μm-thick cryostat MNTB sections processed by the ABC method, as described above. The sections selected showed good and reproducible immunostaining. Principal neurons with immunoreactive rings, typical calyx-like structures, or finger-like tendrils characteristics of calycine terminals were counted as positive for mGluR1a or CB1.
Electron microscopy. Five P14 rats processed as described were used for this purpose. Positive labeling was considered if immunoparticles were in close proximity to the plasmalemma of principal cell bodies or to membranes of calyces of Held in apposition to principal neurons. The total area studied was ∼2298 μm2 for mGluR1a and ∼1016 μm2 for CB1. Statistical analysis of the data was performed using the SPSS program (version 7.5, 1995). For preliminary analysis of the data, we applied the test of normality of Kolmogorov-Smirnov and the Levenes test of equality of error variances. Then, the Wilcoxon test was used to detect differences in gold-silver particle densities between the presynaptic and postsynaptic membrane compartments under study (p < 0.0001).
Results
Group I mGluRs inhibit EPSCs and presynaptic Ca2+ currents
To study the effect of group I mGluR activation on glutamatergic transmission at the calyx of Held synapse, we applied the specific agonist DHPG (100 μm) (Ito et al., 1992; Schoepp et al., 1994) while recording AMPA receptor (AMPAR)- or NMDAR-mediated EPSCs elicited by low-frequency afferent fiber stimulation (0.05-0.2 Hz) in MNTB neurons. In 65% of synapses tested (52 of 80), bath application of DHPG caused a reversible inhibition of EPSCs (Fig. 1). In responding cells (defined as those with >10% inhibition), DHPG inhibited the AMPAR- and NMDAR-mediated EPSCs to a similar extent (41 ± 3%, n = 52 vs 65 ± 11%, n = 3; p > 0.5; for AMPAR-EPSC vs NMDAR-EPSC inhibition, respectively). Inhibition of both AMPAR- and NMDAR-EPSCs was accompanied by an increase in the PPR by 33 ± 8% (from 0.62 ± 0.22 for control conditions to 0.78 ± 0.25 in the presence of DHPG; p < 0.05; paired t test; n = 28), consistent with a reduction in release probability. In many recordings, we observed partial recovery of AMPAR-EPSC amplitudes while in the continued presence of DHPG (Fig. 1A2). Similar desensitization has been described for other group I mGluR-dependent processes (Guatteo et al., 1999; Francesconi and Duvoisin, 2000; Maejima et al., 2001; Dale et al., 2002). Interestingly, neurons in the lateral superior olive (LSO) (an auditory brainstem nucleus) display a DHPG-induced increase in free cytoplasmic calcium ([Ca2+]i) consisting of a peak followed by a plateau reminiscent of the behavior seen in Figure 1A2 (Ene et al., 2003). On washout of DHPG, both EPSC amplitude and PPR returned to control levels. Together with the similar AMPAR- and NMDAR-EPSC inhibition (Fig. 1B), the observed reduction in PPR points to a presynaptic origin of the inhibition by DHPG.
Figure 1.

The group I metabotropic glutamate receptor agonist DHPG (100 μm) inhibits EPSCs in the MNTB. A, DHPG inhibits amplitudes of AMPAR-mediated EPSCs and decreases paired-pulse depression. A1, Paired EPSCs recorded during afferent fiber stimulation of the calyx of Held (interstimulus interval, 100 msec; stimulation frequency, 0.05 Hz; Vh = -70 mV). Shown are example traces obtained in response to the paired stimuli (axis break represents 90 msec) in control (ctrl), during peak inhibition by DHPG, and after washout of DHPG. A2, Time course of inhibition of the first EPSC by DHPG, partial recovery in the continued presence of DHPG, and full recovery on washout. B, DHPG inhibits NMDAR-mediated EPSC amplitudes and decreases paired-pulse depression. Composite EPSCs (mediated by both AMPAR and NMDAR) were evoked with paired stimuli (interstimulus interval and stimulation frequency as in A1; Vh = +30 mV) and recorded from MNTB principal neurons. The peak outward current is a good estimate of the NMDAR-mediated EPSC component because the AMPAR-mediated EPSC component has decayed to a negligible amplitude at that point. C, The mGluR antagonist LY341495 (200 μm) blocks the inhibition of synaptic currents by DHPG. The antagonist effect of LY341495 is reversible, because a third application of DHPG after washout of LY341495 resulted in an inhibition of the EPSC comparable with that obtained during the first application of DHPG.
To clarify whether the inhibitory effect of DHPG on EPSCs is mediated by group I mGluRs, we used the broad spectrum antagonist LY341495 (200 μm), which blocks group I mGluRs (IC50 = 7-8 μm) (Kingston et al., 1998). As illustrated in Figure 1C, LY341495 reversibly blocked inhibition of the EPSC by DHPG (Fig 1C)(n = 5). In contrast, CPPG (300 μm), which completely antagonizes group II-III mGluRs at this synapse (von Gersdorff et al., 1997), did not antagonize the inhibitory action of DHPG (n = 4; data not shown). These data indicate that DHPG is indeed activating group I mGluRs. Furthermore, MPEP (0.5 or 10 μm), a high-affinity noncompetitive antagonist of mGluR5, did not block the effect of DHPG (n = 2 cells), suggesting that mGluR1 receptors mediate the DHPG effect.
We observed inhibition of EPSCs by DHPG in calyces from P5-P14 animals. During this period, a number of developmental changes occur, including a switch in presynaptic Ca2+ channel types coupled to glutamate release from a mixture of N-, P-, and R-type to predominantly P-type (Iwasaki and Takahashi, 1998). To examine possible differences in sensitivity to DHPG during postnatal development, we compared responses in cells from P5-P7 versus P12-P14. The fraction of DHPG-sensitive synapses was not different at P5-P7 and P12-P14 (29 of 45 vs 8 of 15; p = 0.5; Fisher's exact test). The average inhibition obtained in DHPG-sensitive synapses also did not change during development (40 ± 3.3 vs 50 ± 5.8% inhibition at P5-P7 and P12-P14, respectively; p = 0.15; t test). Because rat pups begin to hear at P12, this observation also indicates that group I mGluR-mediated modulation of glutamate release is present in a large subset of calyx synapses in hearing animals.
To examine possible presynaptic mechanisms for the inhibitory actions of DHPG, we recorded presynaptic Ca2+ currents (ICa(pre)) and APs in the calyx of Held. As shown in Figure 2, application of DHPG reduced ICa(pre). The Ca2+ current inhibition partially desensitized in the continued presence of DHPG, and the DHPG-induced inhibition of ICa(pre) was fully reversible on washout. In an attempt to quantify the inhibition of ICa(pre) by DHPG under more physiological conditions, some experiments were also performed at 35°C. The degree of inhibition of ICa(pre) was similar, however, at room temperature and 35°C, and therefore the data were pooled. DHPG reduced ICa(pre) in 13 of 20 cells tested. The average peak inhibition of ICa(pre) in DHPG-sensitive terminals amounted to 13.4 ± 1.8%. In most DHPG-sensitive terminals (9 of 13), an apparent desensitization of the ICa(pre) inhibition was observed in the continued presence of DHPG, similar to that seen for inhibition of EPSCs described above (compare Figs. 1A2, 2A2). As shown in Figure 2B, a voltage-dependent relief of inhibition of ICa(pre) using a strong depolarization (100 msec to +100 mV) between two test depolarizations was obtained, suggesting that inhibition by DHPG is mediated by G-protein βγ subunits, which are sensitive to membrane voltage depolarization (Ikeda, 1996; Leão and von Gersdorff, 2002).
Figure 2.
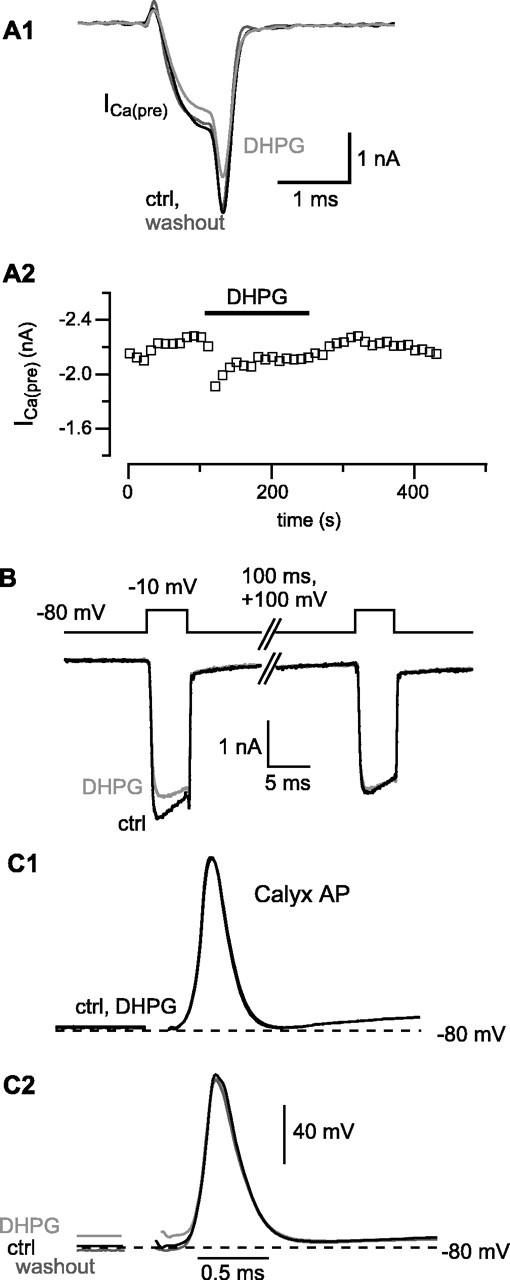
DHPG inhibits the presynaptic Ca2+ current in calyx of Held terminals. A1, Presynaptic Ca2+ current elicited every 10 sec by 1 msec depolarizations to 0 mV from a holding potential of -80 mV, in control, at the peak of inhibition by DHPG (100 μm) and during washout of the drug. A2, Time course of inhibition of ICa(pre) by DHPG, its partial recovery in the continued presence of DHPG, and full recovery on washout of the drug. B, Relief of block by a strong depolarization. A double depolarization protocol was used in which two 5 msec depolarizations to -10 mV were separated by a 100 msec depolarization to +100 mV, in control, and during the peak inhibition of the first Ca2+ current by DHPG. This experiment was done at 35°C. C, Lack of significant effect of DHPG on presynaptic AP waveform. APs were recorded under whole-cell current clamp of the calyx of Held terminal during afferent fiber stimulation. In most recordings, no effect of DHPG was observed, as illustrated in C1. In a minority of cells (2 of 7), DHPG caused a small depolarization of the calyx, which reversed on washout of the drug, as shown in C2.
We next considered the possibility that DHPG could also inhibit EPSCs by modulating the presynaptic AP waveform. To test this, we measured presynaptic APs evoked by afferent fiber stimulation. Application of DHPG did not change the AP waveform (Fig. 2C) (n = 7). In two calyces studied, application of DHPG caused a small (<10 mV) depolarization, which was reversible on washout. Thus we were able to demonstrate a small effect of DHPG on some calyces; however, this small depolarization was apparently insufficient to modify the ionic currents that determine the AP waveform, because half-width, rise time, maximal overshoot, and fall time of the APs remained unchanged. As positive controls, bath application of TEA reversibly broadened the AP (n = 2), whereas raising bath temperature to 35°C reversibly shortened the AP half-width (n = 3). Thus our current-clamp measurements were sufficiently sensitive to detect small changes in AP kinetics.
Inhibitory actions of the CB1 receptor in the MNTB
In the hippocampus and cerebellum, group I mGluRs can trigger release of endogenous CB1 receptor agonists (endocannabinoids) that inhibit EPSCs via presynaptic CB1Rs (Alger, 2002). To investigate whether such a signaling pathway is also present in the auditory CNS, and particularly in the MNTB, and to elucidate mechanisms by which CB1 receptors modulate neurotransmitter release, we studied the effects of the CB1 receptor agonist WIN (5 μm) (D'Ambra et al., 1992) on EPSCs and ICa(pre). Similar to the results described above for DHPG, application of WIN decreased EPSC amplitudes and increased the PPR in 50% (18 of 36) of the tested synapses (Fig. 3A1). Unlike the inhibition observed after application of DHPG, however, we never observed desensitization of the WIN responses (Fig. 3A2). On average, WIN inhibited EPSCs by 46 ± 4.0% (n = 18) and increased the paired-pulse ratio by 47 ± 11% (n = 5). Both the inhibition of the EPSC and the increase in PPR evoked by WIN were reversed by the CB1 receptor antagonist AM251 (5 μm) (Fig. 3A2), which had no effect on EPSCs when applied alone (see below).
Figure 3.
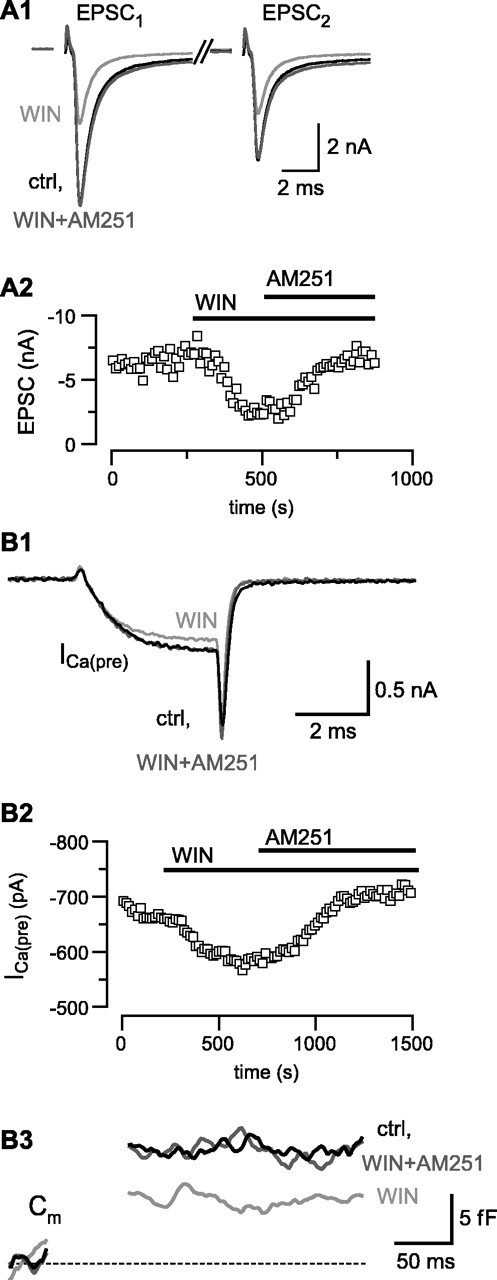
Activation of CB1R inhibits Ca2+ channels coupled to exocytosis in the calyx of Held nerve terminal. A, The CB1 agonist WIN (5 μm) inhibits amplitudes of EPSCs recorded in MNTB neurons and decreases paired-pulse depression. A1, AMPAR-mediated EPSCs recorded during afferent fiber stimulation of the calyx of Held (interstimulus interval, 50 msec; stimulation frequency, 0.2 Hz; Vh = -70 mV). Shown are the currents obtained in response to the paired stimuli (axis break represents 40 msec) in control (ctrl), after application of WIN, and in the combined presence of WIN and the CB1 antagonist AM251 (5 μm). A2, Time course of inhibition of the first EPSC by WIN and reversal of inhibition by AM251 (same cell as shown in A1). B, Activation of CB1 inhibits the presynaptic Ca2+ current, ICa(pre), and Ca2+-dependent exocytosis. B1, Presynaptic Ca2+ currents obtained under whole-cell patch clamp of the calyx of Held. The terminal was held at -80 mV and stepped to 0 mV for 3 msec at 15 sec intervals. Currents have been leak-subtracted using a P/5 protocol. B2, Time course of inhibition of the presynaptic Ca current by WIN (5 μm) and reversal of the inhibition by AM251 (5 μm). B3, Average membrane capacitance jumps elicited by a 3 msec depolarization in control (ctrl), during inhibition of the Ca2+ current with WIN, and after recovery with AM251.
We then examined the effect of CB1R activation on ICa(pre). Application of WIN inhibited ICa(pre), and this inhibition reversed on subsequent application of AM251 (Fig. 3B1,B2). In WIN-sensitive calyces (those with >5% inhibition), ICa(pre) was inhibited by 13.4 ± 1.6% (n = 18). The magnitude of ICa(pre) inhibition by WIN was thus similar to that seen with DHPG. As illustrated in Figure 3B3, application of WIN also inhibited Ca2+-dependent exocytosis, measured as membrane capacitance (Cm) jumps elicited by depolarization under voltage clamp (Sun et al., 2002; Taschenberger et al., 2002).
Inhibition of EPSCs by group I mGluRs requires CB1 receptor activation
We next tested whether CB1R-specific agonists and antagonists interfere with the inhibition of EPSCs by DHPG. In DHPG-sensitive cells, repeated application of DHPG after washout of the previous application resulted in robust inhibition of EPSCs (EPSC inhibition during second application of DHPG = 80 ± 4.8% of the inhibition during first application; n = 14) (Fig. 4A). To test the involvement of CB1 receptors in the response to DHPG, AM251 (5 μm) was applied to DHPG-sensitive synapses, followed by a second application of DHPG in the presence of AM251 (Fig. 4B). AM251 alone had no effect on EPSCs (102 ± 2% of control; n = 8), suggesting the absence of tonic inhibition by CB1Rs (Losonczy et al., 2004). When applied in the presence of AM251, inhibition by DHPG was abolished (EPSC amplitudes in DHPG plus AM251 were 107 ± 3% of control; n = 6). Thus, antagonizing CB1Rs blocks group I mGluR-dependent inhibition of glutamate release from the calyx of Held.
Figure 4.

Inhibition of glutamate release by DHPG requires CB1 receptor activation. A, B, The CB1R antagonist AM251 (5 μm) blocks the action of the group I mGluR agonist DHPG (100μm). A, Repeated applications of DHPG in control aCSF cause reproducible inhibition of the AMPAR-EPSC. B2, Coapplication of AM251 prevents a second inhibition by DHPG. C, The CB1R agonist WIN (5 μm) occludes inhibition of synaptic currents by DHPG. After application of WIN, no further inhibition by DHPG is observed. D, Sensitivity to WIN and DHPG are correlated. Inhibition of the EPSC by DHPG was measured, and then after washout of DHPG, inhibition by WIN was measured. Another five cells studied were insensitive to both drugs. The best-fit slope of a regression line through the data was 0.93. The dashed curves represent the 95% confidence band for the regression.
If inhibition of EPSCs by DHPG is mediated by presynaptic CB1Rs, then CB1 agonists should occlude the response to DHPG. This is indeed the case, as illustrated in Figure 4C. When applied in the presence of WIN, DHPG caused no further reduction in EPSC amplitude (n = 5). In contrast, the average EPSC inhibition by DHPG was not different from control when picrotoxin (50 μm; n = 8), strychnine (2 μm; n = 8), CPPG (300 μm; n = 4), CGP 56999a (1 μm; n = 2), or scopolamine (1 μm; n = 2) was added to the bath solution (data not shown), eliminating any role for ionotropic GABAA, GABAC, and glycine receptors, group II and III mGluRs, GABAB receptors, or muscarinic receptors in this signaling pathway. These results indicate that the group I mGluR-mediated inhibition is caused solely by activation of presynaptic CB1 receptors.
Additional evidence for an involvement of presynaptic CB1 receptors in the DHPG-mediated inhibition of EPSCs was obtained from 10 cells in which sensitivity to both DHPG and WIN (Fig. 4D) was tested. In this set of cells, EPSCs that were sensitive DHPG were also sensitive to WIN (5 of 10), whereas EPSCs that were insensitive to DHPG were also insensitive to WIN. This correlation is consistent with the pharmacological experiments described above and reinforces the hypothesis that inhibition of EPSCs by DHPG requires CB1 receptors.
DHPG acts via postsynaptic mGluRs
As shown in Figure 5A, DHPG increased the postsynaptic voltage-clamp holding current by -36 ± 4 pA(n = 15; Vh=-70 mV), which fully recovered on washout of DHPG. The observed shift in holding current was accompanied by a decrease in the membrane conductance from 5.6 ± 0.48 to 4.8 ± 0.48 nS (n = 11; p < 0.005; paired t test), suggesting that it was caused by inhibition of an outward current. With K+ channels blocked (Cs/TEA in the pipette solution, TEA in the bath solution), this DHPG-induced current was reduced by 65% (p < 0.05; n = 5 cells) and lacked the strong voltage dependence observed under control conditions (data not shown). The change in voltage dependence on K channel blockade suggests that this current consists of multiple components with different voltage dependencies and pharmacology. The shift in membrane current during DHPG application was observed in the presence of picrotoxin (50 μm) and strychnine (2 μm; n = 11), APV (100 μm; n = 6), and NBQX (10 μm; n = 5), and thus was not caused by activation of ionotropic GABA, glycine, or glutamate receptors. Similar to the effect of DHPG on EPSCs, the shift in the holding current was blocked by the mGluR antagonist LY341495 (200 μm; n = 5) but insensitive to the group II-III mGluR antagonist CPPG (300 μm; n = 5), suggesting activation of group I mGluRs (data not shown). Unlike DHPGs action on EPSCs, however, this effect was insensitive to AM251 (5 μm; n = 7), indicating that it does not depend on cannabinoid receptors and that AM251 at the concentration used did not interfere with group I mGluR activation. Application of DHPG decreased the membrane conductance in all MNTB neurons tested, regardless of the sensitivity of their EPSCs to DHPG (Fig. 5A1,A3), suggesting that most if not all MNTB neurons express group I mGluRs. This observation may thus serve as a positive control that the drug reached all of the cells studied and moreover indicates that DHPG does not affect postsynaptic AMPA receptors.
We next asked whether DHPG activated postsynaptic group I mGluRs when inhibiting EPSCs. In other synapses, endocannabinoids can be released from postsynaptic cells via Ca2+-dependent mechanisms and mediate a phenomenon known as depolarization-induced suppression of inhibition/excitation (DSI/DSE) (Kreitzer and Regehr, 2001, 2002; Alger, 2002; Diana et al., 2002; Wilson and Nicoll, 2002). In the cerebellum, blocking of DSI required intracellular perfusion with the Ca2+ chelator, BAPTA (Glitsch et al., 2000). We therefore used a pipette solution containing 40 mm BAPTA and examined the ability of DHPG and WIN to inhibit EPSCs under these conditions. With 40 mm BAPTA in the postsynaptic recording pipette, application of WIN still inhibited EPSCs in 50% of the cells tested (7 of 14) (Fig. 5B2), whereas EPSCs were insensitive to DHPG in all tested cells (n = 14) (Fig. 5B2). Including 40 mm BAPTA in the postsynaptic recording pipette also blocked the negative shift in holding current, suggesting that this current is Ca2+ dependent. This may suggest that a transient receptor potential (TRP)-like conductance may mediate part of the shift in membrane current during DHPG application because some TRP channels are activated by increases in [Ca2+]i levels (Gee et al., 2003). A Ca2+-permeable TRP channel activated by DHPG may be present in LSO neurons of mice auditory brainstem (Ene et al., 2003).
In summary, a strong increase in postsynaptic free Ca2+, which overcomes 5 mm intracellular EGTA Ca2+ buffers (the standard concentration of EGTA used in our pipette solution), triggers a Ca2+-dependent pathway in postsynaptic cells that is necessary for the group I mGluR-mediated inhibition of EPSCs. This suggests that the release of endocannabinoids in the MNTB requires high levels of free Ca2+ as described for the cerebellum (Brenowitz and Regehr, 2003). Furthermore, the inhibition of glutamate release by endocannabinoids released from a given principal cell in the MNTB seems to be restricted spatially to its corresponding calyx terminal, so that endocannabinoid release from nearby MNTB neurons or glia does not mediate the effect of DHPG. Finally, these BAPTA experiments (and the AM251 data of Fig. 4B) also indicate that putative presynaptic group I mGluRs (Elezgarai et al., 2003) do not mediate a reduction of glutamate release from the calyx of Held by DHPG.
DSE in the MNTB
To investigate whether endocannabinoid release can be elicited from the MNTB neuron independently of mGluR activation, we examined the effect of DSE protocols (Kreitzer and Regehr, 2001) applied to this cell on glutamate release from its associated calyx. A 5 sec depolarization of the postsynaptic MNTB neuron to 0 mV caused a transient inhibition of EPSC amplitudes by 54 ± 8% in 8 of 26 cells tested (data not shown). This inhibition recovered over a period of 15-20 sec, much slower than recovery of external Ca2+ depletion from the synaptic cleft (Borst and Sakmann, 1999). In cells that exhibited DSE initially, it could not always be elicited a second time, which precluded testing of its dependence on CB1 receptors with AM251. Thus prolonged depolarizations of a small minority of MNTB neurons can evoke a form of DSE, but we cannot conclude that retrograde synaptic signaling to the calyx via DSE is mediated by CB1 receptors. The rarity and unreliability of DSE in the MNTB thus precluded further study.
Localization of mGluR1a and CB1 receptors in the MNTB
To characterize the distribution of group I mGluRs and CB1 receptors in the MNTB, we used antibodies shown previously to target these receptors to determine their presynaptic or postsynaptic locus (Hajos et al., 2000; Mateos et al., 2000). Both mGluR1a (group I mGluR) and CB1 immunoreactivity were seen in cells of the MNTB at the light and ultrastructural electron microscopy (EM) level. Fluorescence confocal photomicrographs of stained slices allowed discrimination between presynaptic or postsynaptic expression loci, as shown in Figure 6. Immunoreactivity for mGluR1a appears in a majority of the principal cells in the MNTB of P10-P14 rats (Fig. 6A). This staining never colocalized with Rab3a, a presynaptic marker (Mizoguchi et al., 1990), but was distributed in the cell membrane apposing the presynaptic terminal (Fig. 6A, and inset). Staining was confined to the cell soma and was not seen in efferent principal cell axons (data not shown). CB1 receptor antibodies showed a similar pattern of staining as Rab3a at the calyx of Held presynaptic terminal but labeled only a subpopulation of calyces (Fig. 6B) and no principal cells. When present, CB1 immunoreactivity was seen in the terminal with Rab3a but was also present in the afferent axon (Fig. 6B, arrowheads). Three to five independent staining experiments were performed for each antibody, and staining patterns were consistent among experiments. In control experiments, primary antibody was omitted to confirm secondary antibody specificity. These results verify that CB1 and mGluR1a are located in complementary compartments (presynaptic and postsynaptic, respectively) at calyx of Held synapses.
Figure 6.

Confocal immunofluorescence. Postsynaptic mGluR1a immunoreactivity is localized opposite presynaptic CB1 at the calyx of Held. A, mGluR1a is restricted to the postsynaptic principal cell in the MNTB of a P11 rat. Rab3, a vesicle-associated protein, defines the presynaptic calyx of Held terminal (red). Immunoreactivity to mGluR1a (green) is concentrated at the plasma membrane of a majority of principal cells (stars), but does not colocalize with Rab3a immunoreactivity (merge, and inset). B, CB1 and mGluR1a are located on opposite sides of the calyceal synapse in the MNTB of a P14 rat. mGluR1 (green) is located opposite Rab3a (red) at the calyceal synapse (small arrow, left panels). CB1 (cyan) and Rab3a are colocalized presynaptically in the calyx of Held terminal (large arrow, middle bottom panel). The three arrowheads point to the location of the axon of the calyx terminal. CB1 stained the axon but Rab3a did not.
To further study the distribution of mGluR1 and CB1 receptors, we quantified staining of MNTB cells with an immunoperoxidase method, using mGluR1a and CB1 antibodies (Fig. 7A,B). We analyzed 1614 principal cells obtained from four different animals for mGluR1a staining. Of these, 1391 (86 ± 7%; average of four rats used in this experiment) were positive for mGluR1a staining, which was observed as very fine rings around the cell body (Fig. 7A). Staining for CB1 receptor presented a thicker immunoreactive profile, which wrapped tightly around the perikarya of principal neurons forming a structure reminiscent of calyces of Held (Fig. 7B). Such structures were observed in 45 ± 14% (687 of 1517) principal cells stained for CB1 receptor.
Figure 7.

Immunogold EM and peroxidase (DAB) immunohistochemistry. A, Using the immunoperoxidase method, P14 MNTB shows thin rings of intense mGluR1a immunoreactivity around the principal cell bodies (arrows), similar to the immunofluorescence staining of Figure 6 A. B, Use of a second independent primary antibody directed against the C terminus of CB1 shows a similar staining pattern as immunofluorescence (Fig. 6 B) (see Materials and Methods). Immunoperoxidase method of detection shows strong immunostaining for CB1 at the MNTB of P14 rats. Staining is limited to terminal-like profiles reminiscent of calyces of Held, closely apposed to MNTB principal cell bodies (arrows). Immuno-EM: Antibody staining with labeled immunogold particles shows discrete presynaptic CB1 or postsynaptic mGluR1a immunoreactivity at the ultrastructural level. C, D, Electron micrograph of the calyx of Held (P14 rat) shows presynaptic location of silver-enhanced immunoparticles directed against CB1. Silver-intensified immunogold particles directed against CB1 are found throughout the presynaptic plasma membrane of the calyx of Held. Arrows indicate the location of active zones where synaptic vesicles cluster. No CB1 staining was seen at the postsynaptic plasmalemma of the principal cell in the MNTB. Note the narrow synaptic cleft. E, F, Dense silver-enhanced staining for mGluR1a using the same immunogold method reveals exclusive staining of the postsynaptic principal cell membrane. No staining was seen for mGluR1a presynaptically. Arrows indicate puncta adherentia between presynaptic and postsynaptic membranes. The calyx terminal is filled with synaptic vesicles. Note that staining for mGluR1a was not found near active zones but in perisynaptic or extrasynaptic regions of the plasma membrane.
To study subcellular distribution of mGluR1a and CB1 receptor expression, we used electron microscopy coupled with immunogold labeling to define immunopositive sites at the calyx of Held synapse (Fig. 7C-F). Here we used the same antibodies as for the immunoperoxidase method shown above. Presynaptic terminals of calyces of Held were immunoreactive for CB1 (Fig. 7C,D). Silver-intensified CB1-immunogold particles decorated presynaptic membrane specializations at calyx of Held-principal cell synapses. Statistical analysis indicated that there was an approximately threefold higher density of silver-intensified gold particles on membranes of calyces of Held than on the plasmalemma of principal cell bodies. Silver-intensified gold particles indicating the presence of mGluR1a were distributed over membranes of principal cell bodies (Fig. 7E,F). Label for mGluR1a was markedly accumulated at perisynaptic sites of the numerous postsynaptic specializations of the calyciform synapses. Labeling for mGluR1a could be observed in perikaryal membranes up to the more distal digits of the calyx (data not shown). Furthermore, immunoparticles were also localized to plasmalemmal portions not directly associated with presynaptic membranes, suggesting the presence of extrasynaptic mGluR1a receptors (Fig. 6F). The prevalent postsynaptic localization of mGluR1a was confirmed by quantitative evaluation of immunoparticles, which revealed that membranes of principal perikarya were immunolabeled approximately three times more frequently than the presynaptic membranes of calyces of Held. Thus, both group I mGluR and CB1 are expressed in the MNTB, with CB1 located primarily in the calyx and mGluR1a in MNTB principal neurons, consistent with our results at the light microscope level.
To rule out the possibility that the CB1 antiserum was revealing only a part of the immunoreactivity in MNTB, we performed several positive controls on tissues expressing high levels of mRNA coding for CB1. Thus, granule cells and occasional Golgi cells were immunoreactive in the cerebellar cortex (supplemental Figure 1A, available at www.jneurosci.org), as reported previously (Matsuda et al., 1993). In addition, a dense mesh of very strongly stained fibers was observed in the interpeduncular nucleus (supplemental Figure 1B, available at www.jneurosci.org). This nucleus is known to receive a heavy axonal projection from the habenula, which expresses high levels of CB1 mRNA (Matsuda et al., 1993). Finally, no immunostaining was detected in sections processed without primary antiserum or when primary antiserum was substituted with goat serum (supplemental Figure 1C, available at www.jneurosci.org).
Discussion
We investigated inhibition of glutamate release and modulation of presynaptic Ca2+ channels in the calyx of Held nerve terminal in response to two stimuli: DHPG, a specific agonist of group I mGluRs, and WIN, a specific agonist of CB1Rs (Freund et al., 2003). Application of either agonist reversibly inhibited EPSCs and presynaptic Ca2+ currents. Our data indicate that DHPG activates postsynaptic mGluRs in MNTB neurons (because its actions are blocked by the mGluR antagonist LY341495 or by BAPTA in the postsynaptic recording pipette) and inhibits EPSCs via a mechanism that depends on CB1Rs (because it is blocked by AM251 and occluded by WIN). Although for a given synapse the degree of inhibition by DHPG correlated with that by WIN, the results with LY341495 or postsynaptic intracellular BAPTA indicate that DHPG does not activate CB1Rs directly. Experiments using high intracellular BAPTA also indicate that the retrograde messenger generated by DHPG originates from the recorded postsynaptic principal cell and not from nearby neurons or glia. Anatomical data show that group I mGluRs (subtype mGluR1a) are indeed located in the postsynaptic cell and that CB1Rs are restricted to the calyx. On the basis of these results, we conclude that inhibition of glutamate release by DHPG is mediated by the release of endocannabinoids from MNTB principal neurons.
DHPG inhibits the EPSC via CB1R action on ICa(pre)
The mechanism by which CB1Rs inhibit evoked transmitter may vary across synapses but is generally thought to involve modulation of ion channels and Ca2+ influx during APs. Occlusion experiments have demonstrated an important role for K+ channels in mediating the effects of CB1R agonists at some synapses (Robbe et al., 2001; Diana and Marty, 2003). During the CB1R-mediated phenomenon of DSE, the Ca2+ influx into the nerve terminal is reduced (Kreitzer and Regehr, 2001), an effect that could be secondary to changes in the presynaptic AP waveform or caused by direct effects of CB1R on presynaptic Ca2+ channels.
Using a recorded presynaptic AP waveform as a voltage template to drive a model of the calyx Ca2+ current (Borst and Sakmann, 1998), and assuming a third to fourth power law relationship between Ca2+ influx and EPSC (Schneggenburger et al., 1999), we calculate that a reduction of glutamate release by 30-50% would require a 10-15% shortening of the AP leading to a 10-15% reduction in presynaptic Ca2+ influx during APs. Such shortening of the AP, however, was not observed after application of DHPG.
In two cases we did observe a small (<10 mV) depolarization of the calyx when DHPG was applied that reversed on washout. Nonetheless, even in these two calyces, we did not observe any change in the AP that would explain the effect of DHPG on EPSCs. On the basis of the I-V relationship for ICa(pre) (Borst and Sakmann, 1998), the observed depolarization in these two calyces was too small to activate Ca2+ channels. An increased inactivation of calyx Ca2+ channels caused by this depolarization is also unlikely to occur because the steady-state inactivation versus Vm relationship for these channels is flat in this range of potentials (Forsythe et al., 1998). Thus, endocannabinoids released by DHPG do not seem to modulate EPSCs through effects on the presynaptic AP waveform.
Activation of CB1Rs causes inhibition of somatic N-type and P/Q-type Ca2+ currents (Caulfield and Brown, 1992; Mackie and Hille, 1992; Twitchell et al., 1997). In hippocampal neurons, CB1-mediated inhibition of neurotransmitter release can be occluded by Ca2+ channel toxins that block P/Q-type and N-type Ca2+ channels (Sullivan, 1999; Wilson et al., 2001). At these synapses, however, Ca2+ influx through P-type channels plays little or no role in neurotransmitter release. In contrast, the Ca2+ channel subtypes that trigger glutamate release from the calyx of Held change from a mixture of P-, N-, and R-type to exclusively P-type after P10 (Iwasaki and Takahashi, 1998; Wu et al., 1999). We observed inhibition of presynaptic Ca2+ currents by WIN at P11 and inhibition of EPSCs by DHPG out to P14. These data thus indicate that CB1R activation can inhibit presynaptic P-type Ca2+ channels, and the observed inhibition of the Ca2+ current by DHPG or WIN (10-15% inhibition) accounts quantitatively for the effects of these drugs on EPSC amplitudes (30-50% inhibition).
Endocannabinoid release is Ca2+ dependent in the MNTB
Release of endocannabinoids may occur in response to elevations in intracellular Ca2+, for example during prolonged depolarizations (Alger, 2002) or during flash photolysis of caged Ca2+ (Brenowitz and Regehr, 2003). Endocannabinoid release during mGluR1 activation by DHPG, however, may also be Ca2+ independent in the cerebellum (Maejima et al., 2001) and hippocampus (Chevaleyre and Castillo, 2003). Our standard postsynaptic recording solution contained 5 mm EGTA, which presumably reduced [Ca2+]i to very low resting levels. Because of its slow kinetics, however, EGTA cannot buffer very fast changes in Ca2+ (Naraghi and Neher, 1997). The effects of DHPG on both EPSCs and postsynaptic membrane conductance, however, were completely antagonized by 40 mm BAPTA. In the latter case, WIN was always applied to ascertain that the cells expressed CB1Rs, and in half of these cells an inhibition of the EPSC was detected with WIN and 40 mm BAPTA present in the recording pipette. Thus high levels of BAPTA were required to block endocannabinoid release from MNTB neurons triggered by DHPG. This observation may suggest a very close coupling between the site of internal Ca2+ release (or Ca2+ influx) triggered by group I mGluR activation and a Ca2+-dependent step in endocannabinoid synthesis and release. These data also suggest that endocannabinoids released from one synapse do not inhibit release from neighboring calyces. Endocannabinoid signaling in the MNTB is thus spatially restricted in its action.
Group I mGluR modulation of the MNTB output
In addition to effects on glutamate release from the calyx of Held, activation of group I mGluRs caused voltage-dependent changes in the MNTB neuron membrane conductance. The net effect is to generate an inward current and a decrease in membrane conductance near the resting potential. The current shift was reduced by Cs/TEA, making it likely to be caused, at least in part, by a K+ conductance tonically active under control conditions. Inhibition of this conductance by group I mGluRs may render the postsynaptic cell more excitable. Bath application of the mGluR antagonist LY341495 blocked the shift in postsynaptic holding current by DHPG, and it was completely abolished by 40 mm BAPTA in the recording pipette solution, suggesting that it is specifically attributable to group I mGluR activation of a Ca-dependent pathway. Future studies will try to dissect the different components of this novel current shift and its possible physiological role.
Our EM data indicate perisynaptic and extrasynaptic labeling for mGluR1a in the MNTB neuron. Consistent with this finding, the response of up to 100 calyx APs at 100 Hz can be completely blocked by AMPAR and NMDAR antagonists (n = 3; data not shown). Unlike LSO neurons (Kotak and Sanes, 1995), there is thus no evidence for a slow EPSC mediated by activation of group I mGluRs in thin MNTB slice experiments; however, during prolonged high-frequency activity of multiple units in vivo, it is possible that extrasynaptic glutamate could rise to levels sufficient to activate these high-affinity receptors, perhaps via a “spillover” type of release. In addition to the calyx, the MNTB neuron also receives glutamatergic inputs via conventional bouton-type synapses (Banks and Smith, 1992). It is possible that glutamate release from these nerve terminals could also modulate mGluR1a receptors. We note that our immuno-EM data on mGluR1a and CB1R was obtained from P14 rat pups, an age at which we also observed inhibition of EPSCs by DHPG. At this age, the calyx of Held has acquired an adult-like morphology and rat pups respond to sound stimuli. Thus, unlike some group III mGluR subtypes that disappear during early development (Elezgarai et al., 1999), the mGluR1a-CB1R signaling pathway may be relevant for hearing animals and not only during early development. Group I mGluRs may also have a neuroprotective role in setting physiological [Ca2+]i levels (Zirpel and Rubel, 1996; Zirpel et al., 2000).
The MNTB is often referred to as a relay nucleus in which output faithfully follows input (Oertel, 1999). In vivo single-unit recordings in the gerbil MNTB, however, reveal that presynaptic APs may fail to trigger postsynaptic APs under certain conditions (Kopp-Scheinpflug et al., 2003). In addition, the calyx of Held is subject to modulation by a number of presynaptic receptors (Trussell, 2002). It is still not clear under what circumstances these multiple receptors become activated. Moreover, the present results also clearly demonstrate that the nucleus is heterogeneous with respect to modulation by endocannabinoids, because not all calyces express CB1R. The presence of such an elaborate array of presynaptic modulatory mechanisms suggests that the role of the MNTB may be subtler than the classification “relay nucleus” may suggest. The MNTB also contains a medial-to-lateral tonotopic map and is thus functionally heterogeneous in terms of the processing of distinct sound frequencies. Perhaps the heterogeneity in calyx receptors fine-tunes release properties, optimizing them for the particular frequencies of transmission at different locations within the MNTB.
Footnotes
This work was supported by grants from the National Institutes of Health (NIH)-National Institute on Deafness and Other Communication Disorders and Pew Biomedical Research Scholar to H.v.G., NIH-National Eye Institute to R.M.D., and Fondo de Investigación Sanitaria, FIS00/0198, Ministerio de Ciencia y Tecnología, BFI2002-01474 (Spain) to P.G. Confocal microscopy was supported by NIH Grant RR016858. We thank Ken Mackie (University of Washington, Seattle, WA) for the gift of CB1 antibody, Linda Overstreet for suggesting the CB1 and mGluR link, and Craig E. Jahr, John T. Williams, Mary J. Palmer (Medical Research Council, Bristol, UK), and Peter Jonas (University of Freiburg, Freiburg, Germany) for valuable discussions.
Correspondence should be addressed to Dr. H. von Gersdorff, The Vollum Institute, Oregon Health and Science University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239-3098. E-mail: vongersd@ohsu.edu.
C. Kushmerick's present address: Universidade Federal de Minas Gerais, Belo Horizonte, Minas Gerais 31270-901, Brazil.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245955-11$15.00/0
C.K. and G.D.P. contributed equally to this work.
References
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Anwyl R (1999) Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Rev 29: 83-120. [DOI] [PubMed] [Google Scholar]
- Banks MI, Smith PH (1992) Intracellular recordings from neurobiotin-labeled cells in brain slices of the rat medial nucleus of the trapezoid body. J Neurosci 12: 2819-2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes-Davies M, Forsythe ID (1995) Pre- and postsynaptic glutamate receptors at a giant excitatory synapse in rat auditory brainstem slices. J Physiol (Lond) 488: 387-406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1998) Calcium current during a single action potential in a large presynaptic terminal of the rat brainstem. J Physiol (Lond) 506: 143-157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1999) Depletion of calcium in the synaptic cleft of a calyx-type synapse in the rat brainstem. J Physiol (Lond) 521: 123-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG (2003) Calcium dependence of retrograde inhibition by endocannabinoids at synapses onto Purkinje cells. J Neurosci 23: 6373-6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD (2000) Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem 75: 889-907. [DOI] [PubMed] [Google Scholar]
- Caulfield MP, Brown DA (1992) Cannabinoid receptor agonists inhibit Ca current in NG108-15 neuroblastoma cells via a pertussis toxin-sensitive mechanism. Br J Pharmacol 106: 231-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE (2003) Heterosynaptic LTD of hippocampal GABAergic synapses: a novel role of endocannabinoids in regulating excitability. Neuron 38: 461-472. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205-237. [DOI] [PubMed] [Google Scholar]
- Dale LB, Babwah AV, Ferguson SS (2002) Mechanisms of metabotropic glutamate receptor desensitization: role in the patterning of effector enzyme activation. Neurochem Int 41: 319-326. [DOI] [PubMed] [Google Scholar]
- D'Ambra TE, Estep KG, Bell MR, Eissenstat MA, Josef KA, Ward SJ, Haycock DA, Baizman ER, Casiano FM, Beglin NC, Chippari SM, Grego JD, Kullnig RK, Daley GT (1992) Conformationally restrained analogues of pravadoline: nanomolar potent, enantioselective, (aminoalkyl)indole agonists of the cannabinoid receptor. J Med Chem 35: 124-135. [DOI] [PubMed] [Google Scholar]
- Diana MA, Marty A (2003) Characterization of depolarization-induced suppression of inhibition using paired interneuron-Purkinje cell recordings. J Neurosci 23: 5906-5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A (2002) Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci 22: 200-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty J, Dingledine R (2003) Functional interactions between cannabinoid and metabotropic glutamate receptors in the central nervous system. Curr Opin Pharmacol 3: 46-53. [DOI] [PubMed] [Google Scholar]
- Elezgarai I, Benitez R, Mateos JM, Lazaro E, Osorio A, Azkue JJ, Bilbao A, Lingenhoehl K, van der Putten H, Hampson DR, Kuhn R, Knöpfel T, Grandes P (1999) Developmental expression of group III metabotropic glutamate receptor mGluR4a in the medial nucleus of the trapezoid body of the rat. J Comp Neurol 411: 431-440. [DOI] [PubMed] [Google Scholar]
- Elezgarai I, Bilbao A, Mateos JM, Azkue JJ, Benitez R, Osorio A, Diez J, Puente N, Donate-Oliver F, Grandes P (2001) Group II metabotropic glutamate receptors are differentially expressed in the medial nucleus of the trapezoid body in the developing and adult rat. Neuroscience 104: 487-498. [DOI] [PubMed] [Google Scholar]
- Elezgarai I, Puente N, Diez J, Azkue J, Hermida D, Bilbao A, Donate-Oliver F, von Gersdorff H, Grandes P (2003) Group I metabotropic glutamate receptors are localized in developing calyces of Held. Soc Neurosci Abstr 29: 800.6. [Google Scholar]
- Ene FA, Kullmann PH, Gillespie DC, Kandler K (2003) Glutamatergic calcium responses in the developing lateral superior olive: receptor types and their specific activation by synaptic activity patterns. J Neurophysiol 90: 2581-2591. [DOI] [PubMed] [Google Scholar]
- Faas GC, Adwanikar H, Gereau RW, Saggau P (2002) Modulation of presynaptic calcium transients by metabotropic glutamate receptor activation: a differential role in acute depression of synaptic transmission and long-term depression. J Neurosci 22: 6885-6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID (1994) Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J Physiol (Lond) 479: 381-387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T (1998) Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron 20: 797-807. [DOI] [PubMed] [Google Scholar]
- Francesconi A, Duvoisin RM (2000) Opposing effects of protein kinase C and protein kinase A on metabotropic glutamate receptor signaling: selective desensitization of the inositol trisphosphate/Ca2+ pathway by phosphorylation of the receptor-G protein-coupling domain. Proc Natl Acad Sci USA 97: 6185-6190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017-1066. [DOI] [PubMed] [Google Scholar]
- Gee CE, Benquet P, Gerber U (2003) Group I metabotropic glutamate receptors activate a calcium-sensitive transient receptor potential-like conductance in rat hippocampus. J Physiol (Lond) 546: 655-664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch M, Parra P, Llano I (2000) The retrograde inhibition of IPSCs in rat cerebellar Purkinje cells is highly sensitive to intracellular Ca2+ Eur J Neurosci 12: 987-993. [DOI] [PubMed] [Google Scholar]
- Guatteo E, Mercuri NB, Bernardi G, Knöpfel T (1999) Group I metabotropic glutamate receptors mediate an inward current in rat substantia nigra dopamine neurons that is independent from calcium mobilization. J Neurophysiol 82: 1974-1981. [DOI] [PubMed] [Google Scholar]
- Hajos N, Katona I, Naiem SS, MacKie K, Ledent C, Mody I, Freund TF (2000) Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. Eur J Neurosci 12: 3239-3249. [DOI] [PubMed] [Google Scholar]
- Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA, Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R, Pertwee RG (2002) International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev 54: 161-202. [DOI] [PubMed] [Google Scholar]
- Ikeda SR (1996) Voltage-dependent modulation of N-type calcium channels by G-protein beta gamma subunits. Nature 380: 255-258. [DOI] [PubMed] [Google Scholar]
- Ito I, Kohda A, Tanabe S, Hirose E, Hayashi M, Mitsunaga S, Sugiyama H (1992) 3,5-Dihydroxyphenyl-glycine: a potent agonist of metabotropic glutamate receptors. NeuroReport 11: 1013-1016. [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T (1998) Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J Physiol (Lond) 509: 419-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Alger BE (2001) Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci 21: 9608-9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S, Schoepp (1998) LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology 37: 1-12. [DOI] [PubMed] [Google Scholar]
- Kopp-Scheinpflug C, Lippe WR, Dorrscheidt GJ, Rubsamen R (2003) The medial nucleus of the trapezoid body in the gerbil is more than a relay: comparison of pre- and postsynaptic activity. J Assoc Res Otolaryngol 4: 1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Sanes DH (1995) Synaptically evoked prolonged depolarizations in the developing auditory system. J Neurophysiol 74: 1611-1620. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29: 717-727. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2002) Retrograde signaling by endocannabinoids. Curr Opin Neurobiol 12: 324-330. [DOI] [PubMed] [Google Scholar]
- Leão RM, von Gersdorff H (2002) Noradrenaline increases high-frequency firing at the calyx of Held synapse during development by inhibiting glutamate release. J Neurophysiol 87: 2297-2306. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Biro AA, Nusser Z (2004) Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci USA 101: 1362-1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackie K, Hille B (1992) Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc Natl Acad Sci USA 89: 3825-3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Hashimoto K, Yoshida T, Aiba A, Kano M (2001) Presynaptic inhibition caused by retrograde signal from metabotropic glutamate to cannabinoid receptors. Neuron 31: 463-475. [DOI] [PubMed] [Google Scholar]
- Mateos JM, Benitez R, Elezgarai I, Azkue JJ, Lazaro E, Osorio A, Bilbao A, Donate F, Sarria R, Conquet F, Ferraguti F, Kuhn R, Knopfel T, Grandes P (2000) Immunolocalization of the mGluR1b splice variant of the metabotropic glutamate receptor 1 at parallel fiber-Purkinje cell synapses in the rat cerebellar cortex. J Neurochem 74: 1301-1309. [DOI] [PubMed] [Google Scholar]
- Matsuda LA, Bonner TI, Lolait SJ (1993) Localization of cannabinoid receptor mRNA in rat brain. J Comp Neurol 327: 535-550. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Kim S, Ueda T, Kikuchi A, Yorifuji H, Hirokawa N, Takai Y (1990) Localization and subcellular distribution of smg p25A, a Ras p21-like GTP-binding protein, in rat brain. J Biol Chem 265: 11872-11879. [PubMed] [Google Scholar]
- Morishita W, Kirov SA, Alger BE (1998) Evidence for metabotropic glutamate receptor activation in the induction of depolarization-induced suppression of inhibition in hippocampal CA1. J Neurosci 18: 4870-4882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M, Neher E (1997) Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+]i at the mouth of a calcium channel. J Neurosci 17: 6961-6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oertel D (1999) The role of timing in the brain stem auditory nuclei of vertebrates. Annu Rev Physiol 61: 497-519. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M (2002) Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci 15: 953-961. [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ (2001) Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 21: 109-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Sistiaga A, Lerma J, Sanchez-Prieto J (1998) Switch from facilitation to inhibition of excitatory synaptic transmission by group I mGluR desensitization. Neuron 21: 1477-1486. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E (1999) Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron 23: 399-409. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Goldsworthy J, Johnson BG, Salhoff CR, Baker SR (1994) 3,5-Dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. J Neurochem 63: 769-772. [DOI] [PubMed] [Google Scholar]
- Sullivan JM (1999) Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol 82: 1286-1294. [DOI] [PubMed] [Google Scholar]
- Sun JY, Wu XS, Wu LG (2002) Single and multiple vesicle fusion induce different rates of endocytosis at a central synapse. Nature 417: 555-559. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, von Gersdorff H (2000) Fine-tuning an auditory synapse for speed and fidelity: developmental changes in presynaptic waveform, EPSC kinetics, and synaptic plasticity. J Neurosci 20: 9162-9173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taschenberger H, Leão RM, Rowland KC, Spirou GA, von Gersdorff H (2002) Optimizing synaptic architecture and efficiency for high-frequency transmission. Neuron 36: 1127-1143. [DOI] [PubMed] [Google Scholar]
- Trussell LO (2002) Modulation of transmitter release at giant synapses of the auditory system. Curr Opin Neurobiol 12: 400-404. [DOI] [PubMed] [Google Scholar]
- Twitchell W, Brown S, Mackie K (1997) Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J Neurophysiol 78: 43-50. [DOI] [PubMed] [Google Scholar]
- Varma N, Carlson GC, Ledent C, Alger BE (2001) Metabotropic glutamate receptors drive the endocannabinoid system in hippocampus. J Neurosci 21: RC188(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E (1997) Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J Neurosci 17: 8137-8146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296: 678-682. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA (2001) Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31: 453-462. [DOI] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA, Sakmann B (1999) Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci 19: 726-736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zirpel L, Rubel EW (1996) Eighth nerve activity regulates intracellular calcium concentration of avian cochlear nucleus neurons via a metabotropic glutamate receptor. J Neurophysiol 76: 4127-4139. [DOI] [PubMed] [Google Scholar]
- Zirpel L, Janowiak MA, Taylor DA, Parks TN (2000) Developmental changes in metabotropic glutamate receptor-mediated calcium homeostasis. J Comp Neurol 421: 95-106. [PubMed] [Google Scholar]