

Abstract
Endogenous adenosine in the brain is thought to prevent the development and spread of seizures via a tonic anticonvulsant effect. Brain levels of adenosine are primarily regulated by the activity of adenosine kinase. To establish a link between adenosine kinase expression and seizure activity, we analyzed the expression of adenosine kinase in the brain of control mice and in a kainic acid-induced mouse model of mesial temporal lobe epilepsy. Immunohistochemical analysis of brain sections of control mice revealed intense staining for adenosine kinase, mainly in astrocytes, which were more or less evenly distributed throughout the brain, as well as in some neurons, particularly in olfactory bulb, striatum, and brainstem. In contrast, hippocampi lesioned by a unilateral kainic acid injection displayed profound astrogliosis and therefore a significant increase in adenosine kinase immunoreactivity accompanied by a corresponding increase of enzyme activity, which paralleled chronic recurrent seizure activity in this brain region. Accordingly, seizures and interictal spikes were suppressed by the injection of a low dose of the adenosine kinase inhibitor 5-iodotubercidin. We conclude that overexpression of adenosine kinase in discrete parts of the epileptic hippocampus may contribute to the development and progression of seizure activity.
Keywords: adenosine kinase, adenosine, epilepsy, seizure susceptibility, kainic acid, iodotubercidin
Introduction
Seizure susceptibility may be caused by abnormalities in central neurotransmitter systems, such as impaired inhibitory transmission, excessive excitatory transmission, or a combination of both. Previous studies, as well as pharmacological intervention, have thus primarily focused on the GABAergic and glutamatergic systems. During the last decade, adenosine has emerged as an important neuromodulator with mostly inhibitory effects on neuronal activity (Dunwiddie and Masino, 2001). Brain concentrations of adenosine in the range of 25-250 nm (Dunwiddie and Diao, 1994) generally lead to an inhibitory tone by activation of both adenosine A1 and A2A receptors (Fredholm et al., 2001). This tonic inhibition may be relieved by receptor antagonists such as caffeine, which therefore have a stimulatory action on CNS functions (Fredholm et al., 1999). In contrast, activation of A1 and A2A receptors by selective agonists (Huber et al., 2002; Gouder et al., 2003) or by adenosine delivered by implanted cells (Huber et al., 2001; Boison et al., 2002a) provides for seizure suppression. Taken together, these findings suggest that epilepsy may be associated with a dysfunction of the adenosine-mediated inhibitory tone.
Endogenous adenosine levels are mainly dependent on the activity of adenosine kinase (ADK; EC 2.7.1.20), the key enzyme of adenosine metabolism. This notion is based on several lines of evidence: (1) Pharmacological inhibition of ADK in hippocampal slices increases A1 receptor-mediated presynaptic inhibition (Pak et al., 1994). (2) Pharmacological inhibition of ADK provides seizure suppression in various models of epilepsy (Kowaluk et al., 1998; Wiesner et al., 1999; Ugarkar et al., 2000a,b) (3) Inhibition of adenosine deaminase has little or no influence on the concentration of extracellular adenosine (Pak et al., 1994; Zhu and Krnjevic, 1994; Huber et al., 2001). (4) In certain types of cells there is a high flux rate in a futile cycle involving ADK and 5′-nucleotidase (Bontemps et al., 1983). As a consequence, inhibition of ADK may lead to rapid and large increases in adenosine, whereas overexpression of ADK may aggravate seizure activity.
To investigate whether potential changes of endogenous ADK expression may contribute to epileptogenesis, a mouse model based on a unilateral injection of a low dose of kainate into the dorsal hippocampus of adult mice (Bouilleret et al., 1999, 2000) was used in this study. In this model, the histopathological changes occurring in human mesial temporal lobe epilepsy are closely replicated and accompanied by chronic recurrent seizures that are resistant to classic antiepileptic drugs (Riban et al., 2002), but are suppressed by activation of A1 receptors (Gouder et al., 2003). This model is therefore well suited to investigate the role of the regulation of endogenous adenosine levels in the process of epileptogenesis.
In the present study the expression levels of ADK were therefore determined immunohistochemically in brain sections of kainate-treated epileptic mice and untreated mice as controls. Hippocampi of treated mice were characterized by an overexpression of ADK and increased enzyme activity, which may contribute to seizure activity, because seizures could be suppressed by the ADK inhibitor 5-iodotubercidin.
Materials and Methods
Production of recombinant adenosine kinase. For the production of recombinant adenosine kinase, an 1865 bp full-length Adk cDNA was generated from mouse liver using a Marathon cDNA Amplification Kit (Clontech, Palo Alto, CA) and the internal Adk PCR primers 5′-CCAATGCATCCGAAGAACGTTGCTGC-3′ (5′ RACE) and 5′-GTGAACTCGAAGAG GCAGAGGACCG-3′ (3′-RACE). The cDNA was inserted into an EcoRV site of the pBluescriptIIKS+ vector (Stratagene, La Jolla, CA). The insert was sequenced and identified as a full-length mouse Adk cDNA being homologous to the short isoform (calculated size of 38.7 kDa) of Adk described in humans (McNally et al., 1997). After subcloning into the bacterial expression vector pQe60 (Qiagen, Hilden, Germany) the mouse Adk cDNA was expressed in Escherichia coli SM15 cells (Qiagen) and found to display ADK activity (Boison et al., 1999).
Immunization. Immunizations were performed in rabbits (Institute of Laboratory Animal Sciences, University of Zurich, Switzerland) using recombinant ADK. Polyclonal serum was collected and found to be highly specific for both isoforms of ADK, as determined in a Western blot analysis performed on liver extracts from Adk+/+, Adk+/-, and Adk-/- mice (Boison et al., 2002a).
Preparation of organ extracts. Aqueous extracts from whole brains, livers, lungs, kidneys, and hearts derived from Adk+/+, Adk+/-, and Adk-/- pups at postnatal day 4 were prepared by homogenizing and solubilizing the tissue in two volumes of a 5 mm EDTA solution and by removing unsolubilized material by centrifugation (100,000 × g, 15 min, 4°C). The protein content in the supernatants was determined using a commercial Bradford assay (Bio-Rad, Hercules, CA).
Western blot. Twenty-five micrograms of each supernatant was separated on an SDS-10% PAGE gel and blotted onto a nitrocellulose membrane according to standard procedures. The blots were probed for 1 hr with a 1:6000 dilution of the polyclonal rabbit antiserum in 5% blocking reagent in TBST (10 mm Tris, 150 mm NaCl, 0.05% Tween 20 in H2O). After washing (5 × 5 min in TBST), blots were then probed with a peroxidase-linked anti-rabbit IgG, and bands were visualized with a commercial enhanced bioluminescence detection method (ECL) kit (PerkinElmer Life Sciences, Boston, MA).
Mouse model of partial epilepsy. Experiments were conducted on male Naval Medical Research Institute wild-type (Adk+/+) mice (35-40 gm) (Harlan Netherland, Horst, The Netherlands) that were housed in individual cages in a 12 hr light/dark cycle (lights on from 7:00 A.M. to 7:00 P.M.) with food and water provided ad libitum. All animal procedures were conducted in accordance with the regulations of the local animal welfare authority. All efforts were made to minimize animal suffering and to reduce the number of animals used.
Under general anesthesia using equithesin (4 ml/kg, i.p.), the experimental mice (n = 47) were stereotactically injected with 50 nl of a 20 mm solution of kainic acid (KA) in 0.9% NaCl (i.e., 1 nmol KA) (n = 41 or vehicle n = 6) into the right dorsal hippocampus [coordinates with bregma as reference: anteroposterior (AP) = -1.5, mediolateral (ML) = -1.8, dorsoventral (DV) = -1.9 mm] using a stainless steel cannula (outer diameter, 0.28 mm) connected to a 0.5 μl microsyringe (Hamilton, Bonaduz, Switzerland). Each injection was performed over a period of 1 min. At the end of the injection, the cannula was left in place for an additional period of 1 min to limit reflux along the cannula track.
To observe the effect of the adenosine kinase inhibitor 5-iodotubercidin on seizure activity, 26 mice were then implanted with a bipolar electrode inserted into the injected hippocampus, and a monopolar surface electrode was placed over the cerebellum (reference electrode). The bipolar electrode was formed of two twisted enamel insulated stainless steel wires (diameter, 170 μm, distance between the tips, 0.4 mm) connected to a male connector. It was aimed at the right hippocampus with the same coordinates as for the injection site. The monopolar electrode was made of the same enamel insulated stainless steel wire (diameter, 250 μm) soldered on a male connector (Wirepro, Farnell, France). It was inserted in the skull so that only the tip (0.5 mm) protruded onto cerebellar tissue. The electrodes were fixed to the skull with cyanoacrylate and dental acrylic cement. The mice were then allowed to recover from anesthesia before being placed in the EEG recording chamber.
Histology. Mice were transcardially perfused with 4% paraformaldehyde and 15% saturated picric acid solution in phosphate buffer (0.15 m, pH 7.4). The brains from pups or adults were then postfixed in the same fixative at 4°C for 48 or 6 hr, respectively. The brains were then cryoprotected in 10% DMSO in PBS (v/v) before being cut into 40 μm sagittal or coronal sections using a sliding microtome. Histological analysis was performed after cresyl violet staining to verify (1) the location of the KA injection, and (2) the pattern of neuronal loss and the dispersion of dentate gyrus granule cells, as reported previously (Bouilleret et al., 1999; Knuesel et al., 2001).
Immunoperoxidase staining. For the immunohistochemical detection of ADK, brain sections were incubated overnight at 4°C with primary anti-ADK antiserum diluted 1:5000 in Tris-Triton, pH 7.4, containing 2% normal goat serum and 0.2% Triton X-100. The sections were then washed 3 × 10 min in Tris-buffered saline (TBS) at pH 7.4, incubated for 30 min with biotinylated goat anti-rabbit antibody diluted 1:300 in Tris-Triton, pH 7.4, containing 2% normal goat serum, washed again three times in TBS, then incubated for 20-30 min with avidin-biotin enzyme complex (Vectastain Elite Kit; Vector Laboratories, Burlingame, CA). After washing again three times with TBS, the tissue antigen was localized by incubation with diaminobenzidine hydrochloride (Sigma, St. Louis, MO), which acts as a chromogen. The sections were then mounted on gelatin-coated slides, air-dried, dehydrated, and coverslipped.
Immunofluorescence staining. To assess in which brain cells adenosine kinase is expressed, a double immunofluorescence staining was performed on coronal brain sections from saline-treated control mice and mice killed 4 weeks after the injection of KA. The anti-adenosine kinase serum was used together with either a monoclonal mouse antibody against the astrocytic marker glial fibrillary acidic protein (GFAP) (MAB360; Chemicon International, Temecula, CA) or a monoclonal mouse antibody against the neuron-specific nuclear protein NeuN (MAB377; Chemicon International).
Brain sections, stored in anti-freeze at -20°C, were washed under agitation in PBS for 30 min, changing the solution three times, and were then incubated overnight at 4°C in a solution containing the primary antibodies diluted 1:5000 in Tris-Triton, pH 7.4, which contained 2% normal goat serum and 0.2% Triton X-100. Sections were then washed three times for 10 min in Tris-Triton, pH 7.4, followed by a 30 min incubation at room temperature in a solution containing the secondary antibodies goat anti-mouse conjugated to Cy2 (Jackson Immuno Research, West Grove, PA) to detect the GFAP and NeuN antibodies, and goat anti-rabbit conjugated to Cy3 (Jackson ImmunoResearch) to detect the ADK antibody. The secondary antibodies were diluted 1:200 and 1:300, respectively, in Tris-Triton containing 2% NGS. Sections were washed a further three times for 10 min, mounted on gelatinized slides, and air-dried for 1 hr before being coverslipped with a few drops of Dako (Carpinteria, CA) fluorescent mounting medium. To verify the specificity of the monoclonal antibodies, control stainings were performed with secondary antibody only. These control stainings were devoid of localized immunofluorescence (data not shown).
Immunochemical data analysis. Immunoperoxidase-stained sections were analyzed with a Zeiss (Jena, Germany) AxioCam microscope equipped with bright-field microscopy, using a high-resolution digital camera. High-resolution images were analyzed by laser-scanning confocal microscopy (LSM510-Meta; Zeiss), using simultaneous dual-channel recording of double-labeled sections. Stacks of 12-20 images spaced by 0.4-0.6 μm were color-coded and superimposed (maximal intensity pixel mode). The digitized images were processed with the software Imaris (Bitplane, Zurich, Switzerland).
Electroencephalographic recordings. EEG activities of freely moving animals placed in a Faraday cage were recorded using a digital acquisition computer-based system (MP100WSW System; Biopac Systems, Inc., Santa Barbara, CA; six channels, sampling rate 200 Hz). Before starting the EEG recordings, a period of 1 hr was allowed for the habituation of the animals to the test cage.
Drug administration. The effects of the ADK inhibitor 5-iodotubercidin on the frequency of recurrent seizures were assessed in mice exhibiting typical hippocampal paroxysmal discharges. 5-iodotubercidin (Research Biochemicals, Natick, MA) was dissolved in 20% DMSO at a final concentration of 1.24 mg/ml. An intraperitoneal dose of 3.1 mg/kg was injected into each test animal (n = 4). Control injections of vehicle [20% DMSO in 0.9% saline (v/v)] alone were performed (n = 4). To exclude potential effects of the activation of peripheral adenosine receptors by ADK inhibition-induced adenosine release, 8-sulfophenyltheophylline, a nonselective non-brain-permeable adenosine receptor antagonist, dissolved in 10% polyethylene glycol (PEG) in saline (v/v), was used. In control experiments 8-SPT (20 mg/kg, i.p.) was injected 15 min before 5-iodotubercidin or vehicle administration (n = 4, each). To demonstrate the selectivity of the 5-iodotubercidin response, coinjections were performed with 3.1 mg/kg of 5-iodotubercidin and the selective A1-receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) (1 mg/kg, i.p.) in five additional animals. All injections were performed during the fourth week after KA treatment, when all animals were in the chronic phase of seizure activity. After a reference EEG recording period of 2 hr, the animals were treated accordingly and were recorded another 6 hr.
Determination of adenosine kinase activity in isolated hippocampi. KA-lesioned and contralateral control hippocampi (n = 5 each) were carefully dissected out from freshly killed animals 8 weeks after KA injection. Ten hippocampi dissected out from five naive mice were used as controls. Each hippocampus was processed individually at 4°C by 1 burst of 5 sec of ultrasound homogenization in 200 μl of 50 mm NaCl, 20 mm Na2HPO4, and 5 mm EDTA, pH 6.5. The homogenates were then centrifuged at 100,000 × g for 10 min. The protein contents of the supernatants were quantified with a commercial Bradford assay kit (Bio-Rad, Glattbrugg, Switzerland). To remove nucleotides and nucleosides from the supernatants, 75 μl of each sample was applied to a Micro Bio-Spin 6 column (catalog #732-6221; Bio-Rad), which was equilibrated with 500 μl of the adenosine 5′-triphosphate assay mix dilution buffer (FL-AAB) (Sigma, Buchs, Switzerland). After centrifugation at 1000 × g for 4 min, the flow-through was diluted with FL-AAB to a final protein concentration of 1.0 mg/ml. We used 5 μl of each diluted sample for adenosine kinase activity determinations with an enzyme-linked bioluminescent assay according to the following principle: an excess of adenosine, added to the sample is phosphorylated by adenosine kinase to AMP, which is then phosphorylated further to ATP by an excess of recombinant GTP:AMP phosphotransferase (own production) and pyruvate kinase (Roche, Rotkreuz, Switzerland). The resulting ATP increase is then quantified with a luciferase assay (ATP Bioluminescent Assay kit; Sigma). To run the reactions in a 96 well microtiter plate, 5 μl of each diluted sample containing an unknown activity of ADK was added to 20 μl of a solution containing 5 mU/ml GTP:AMP phosphotransferase, 2 U/ml pyruvate kinase, 400 μm GTP, 400 μm phosphoenolpyruvate, 10 μm erythro-9-[3-(2-hydroxynonyl)]adenine, and 100 μm adenosine in 30% adenosine 5′-phosphate Assay Mix (v/v) in FL-AAB. The reactions were followed for 15 min at room temperature in a luminometer (Microlumat plus LB96V; EG&G Berthold). Adenosine kinase activity was determined as an increase of relative light units per time. ADK activity values were normalized to endogenous lactate dehydrogenase (LDH) activity, which was taken as an internal standard. LDH activity was determined with a commercial assay kit according to the manufacturer's protocol (Roche). The assay was validated by performing a standard curve of different ratios of samples from Adk+/+ mice mixed with those from adenosine kinase knock-out mice.
Statistical analysis. ADK activity values were first analyzed in the Kruskal-Wallis test for the comparison of three groups. Results were found to be highly significant (p < 0.001). Statistical analysis of paired groups was then performed by Mann-Whitney U tests.
Results
Characterization of antibodies to adenosine kinase
Antibodies were raised against recombinant full-length adenosine kinase expressed in Escherichia coli by using the expression vector pQe60. The antibody has previously been tested in Western blots from liver extracts of ADK knock-out mice and found to be specific for ADK (Boison et al., 2002b).
Adenosine kinase-deficient mice (Adk-/-) die shortly after birth because of severe microvesicular hepatic steatosis (Boison et al., 2002b) and represent a valuable tool to test the specificity of our anti-ADK antibody. In the present study we further verified the specificity of the antibody by performing Western blot analyses of tissue extracts from heart, kidney, lung, brain, and liver derived from Adk+/+, Adk+/-, and Adk-/- pups at postnatal day 4 (P4) (Fig. 1A). In wild-type mice (Adk+/+) and heterozygous mutants (Adk+/-), double bands were detected in the size range of 44-46 kDa, being consistent with two alternatively spliced products, whose functional differences are yet unknown (McNally et al., 1997). As expected, organs from heterozygous Adk+/- pups contained ∼50% of the ADK protein levels of Adk+/+ pups, whereas no ADK-specific protein was detected in liver extracts from homozygous Adk-/- mutants (Fig. 1A). These findings confirm the specificity of the ADK antiserum. A comparison of ADK expression levels in different organs in P4 Adk+/+ and Adk+/- pups revealed the strongest expression levels in liver being consistent with the fact that ADK is essential for maintaining transmethylation reactions, of which 85% take place within the liver (Boison et al., 2002b). Apart from the liver, a strong expression of ADK became evident in heart and lung, whereas moderate levels of ADK were found in brain and kidney. It is noteworthy to recognize that the relative contributions of the long and short isoforms of ADK differ among the organs investigated. Whereas in lung the long isoform of ADK appears to predominate, in brain the short isoform of ADK is exclusively expressed.
Figure 1.
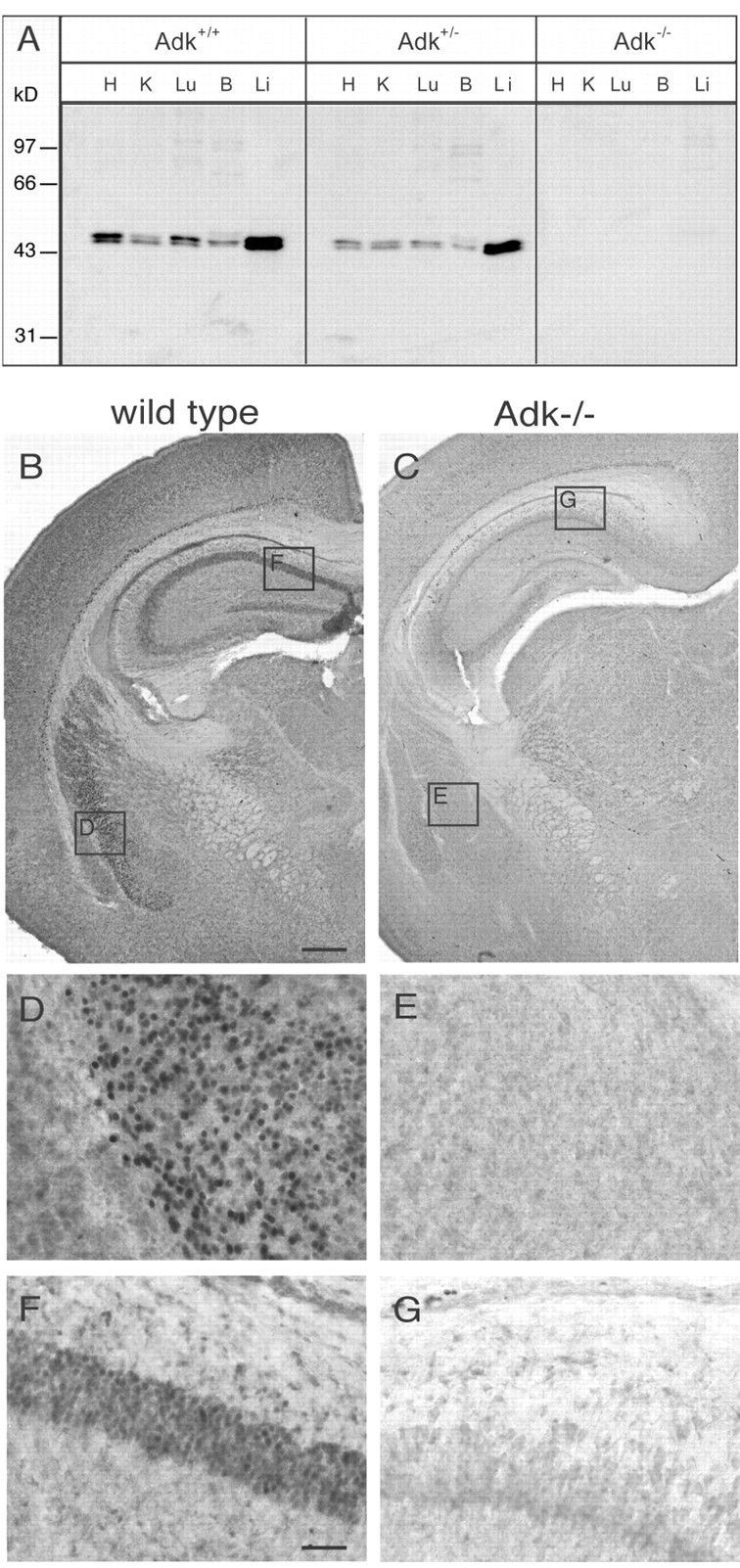
Specificity of the anti-ADK antiserum. A, Western blot analysis of aqueous protein extracts derived from various organs from P4 pups from an Adk+/- x Adk+/- intercross. Extracts corresponding to 25 μg of total protein each from wild-type (Adk+/+), heterozygous (Adk+/-), and homozygous (Adk-/-) mutant pups were probed with a polyclonal rabbit antiserum raised against recombinant mouse ADK. H, Heart; K, kidney; Lu, lung; B, brain; Li, liver. B, Part of coronal brain section of an Adk+/+ pup taken at P4 and stained with diaminobenzidine hydrochloride (DAB) for ADK immunoreactivity. C, Part of a transverse brain section of a mutant Adk-/- pup taken at P4 and stained with DAB for ADK immunoreactivity. Note the lack of ADK staining in the brain taken from the knock-out animal. D-G, Insets are magnified in the corresponding panels. Scale bars: (in B) B, C, 350 μm; (in F) D-G, 50 μm.
To verify the specificity of the antibody for immunohistochemical applications, brain sections taken from Adk+/+, Adk+/-, and Adk-/- pups at postnatal day 4 were analyzed. A distinct expression pattern of ADK became apparent in brains taken from Adk+/+ (Fig. 1B) and Adk+/- (data not shown) pups, whereas ADK was not detected in brains taken from Adk-/- pups (Fig. 1C). These findings demonstrate the specificity of the immunoreactivity produced by the ADK antiserum. It is therefore an ideal tool to investigate the distribution of ADK in brain.
In sections taken from Adk+/+ pups, a diffuse staining for ADK became apparent in most brain regions (Fig. 1B). However, at least four brain areas were characterized by the presence of strongly labeled cells: (1) the striatum where labeled cells showed a nuclear and perinuclear localization as seen in a close up presentation (Fig. 1D), whereas the corresponding area in Adk-/- pups was devoid of staining (Fig. 1E); (2) the pyramidal cells in the CA1 region of the hippocampus of Adk+/+ pups were strongly labeled with ADK immunoreactivity (Fig. 1F), whereas corresponding cells in the hippocampus taken from Adk-/- pups were devoid of ADK (Fig. 1G); (3) in Adk+/+ pups, ADK-positive cells were present in the ependyme of the lateral ventricle (Fig. 1B), and (4) layer VIb of the neocortex (Fig. 1B). It is important to note that these ADK-positive cells of the developing brain (P4) displayed nuclear or perinuclear staining, indicating a requirement of ADK as a constituent of purine salvage pathways for nucleotide synthesis.
Expression of adenosine kinase in adult brain
Adenosine kinase in adult brain is expected to be crucial in the regulation of endogenous adenosine levels. To determine the distribution pattern of ADK in adult brain, coronal and sagittal sections taken from 8-week-old Adk+/+ mice were analyzed immunohistochemically. Weak ADK immunoreactivity was apparent by a diffuse staining of the neuropil throughout the brain, whereas strong ADK immunoreactivity appeared to predominate in ubiquitously present, discrete selected cell bodies. With few exceptions, ADK immunoreactivity could not be detected in cellular processes (Fig. 2).
Figure 2.
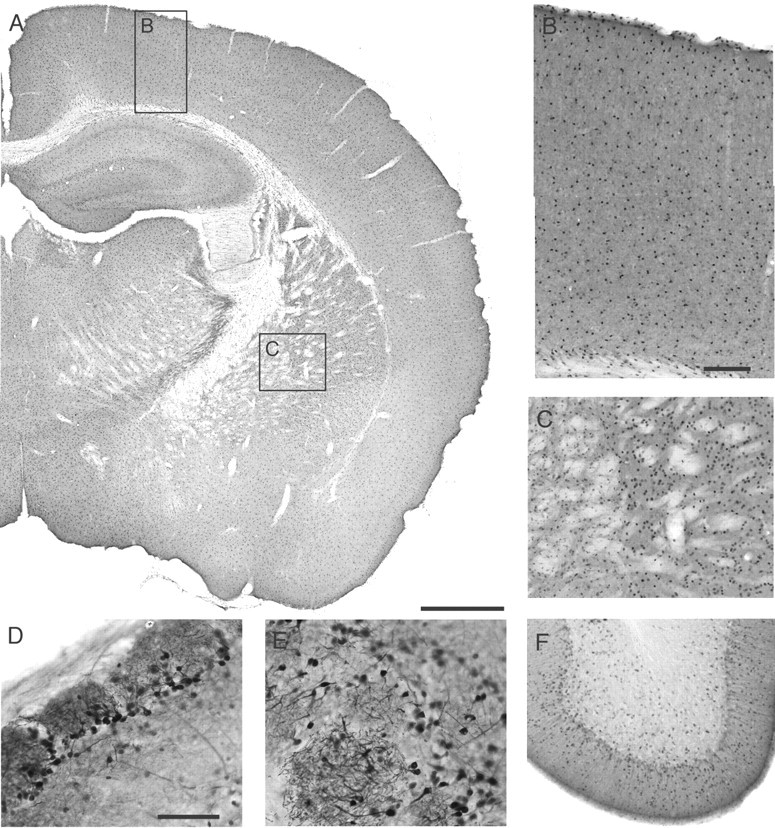
Photographs of ADK immunoreactivity in transverse and parasagittal brain sections of an adult Adk+/+ mouse, processed for immunoperoxidase staining. A, Transverse section of a brain hemisphere showing a rather homogenous distribution of individual cells expressing ADK. B, Neocortex at higher magnification (see inset in A) showing an even distribution of ADK-positive cells in all cortical layers. C, Striatum and globus pallidus at higher magnification (see inset in A) showing individual ADK-positive cells. D, E, Olfactory bulb at higher magnification taken from a parasagittal section. Note the strong staining of ADK-positive neurons (D) and their processes in glomeruli (E). F, Cerebellum at higher magnification taken from a parasagittal section. Note the concentration of ADK-positive cells in the molecular layer. Scale bars: A, 800 μm; (in B), B, C, F, 135 μm; (in D), D, E, 90 μm.
Large numbers of ADK-positive cells were observed in the olfactory bulb (Fig. 2D,E) and in the striatum (Fig. 2A,C), whereas lower numbers of ADK-positive cells were present in white matter tracts (Fig. 2A). It is striking to note that the overall immunoreactivity for ADK in each brain region appears to be determined by the number and density of individual cells expressing high levels of adenosine kinase. Therefore, it is tempting to speculate that cells specialized for adenosine removal are distributed throughout the brain. Interestingly, in brains taken from P4 Adk+/+ pups, the exclusive localization of ADK-positive cells in the four brain regions described above (Fig. 1B,D,F), has given way to a dispersion of ADK-positive cells into all regions of the adult brain (Fig. 2).
The cerebral cortex of adult Adk+/+ mice displays a rather homogenous distribution of nearly evenly spaced ADK-positive cells (Fig. 2B), which can be found in all cortical layers. In contrast, ADK-positive cells appear at high density within the striatum (Fig. 2C), an area that also displayed a high density of ADK-positive cells in P4 Adk+/+ pups (Fig. 1D). ADK immunoreactivity changed dramatically within the hippocampal formation during the course of postnatal development. Whereas immunoreactivity in P4 Adk+/+ pups was confined exclusively to the pyramidal and granular cell layers (Fig. 1B,F), ADK-positive cells have dispersed throughout the hippocampus in adult mice, with a near complete lack of ADK-positive cells within the granular cell layer of the dentate gyrus. Instead, a higher density of ADK-positive cells was observed at the border between the granule cells and the hilus of the dentate gyrus (Figs. 2A, 3F,K). The expression of ADK within the hippocampal formation is of interest with regards to the vulnerability of this brain structure for the development of epileptic activity. Colocalization studies with GFAP demonstrated immunoreactivity in hippocampal astrocytes (Fig. 4) as well as in distinct astrocytes in all areas of the brain (data not shown). In these cells ADK immunoreactivity failed to colocalize with the neuronal marker NeuN. However, inspection of close-up views taken from sagittal sections of adult Adk+/+ brains revealed a strong staining for ADK in periglomerular neurons of the olfactory bulb (Fig. 2D,E), which was colocalized with NeuN (data not shown) as well as in dispersed single cells within the cerebellum (Fig. 2F). Taken together our findings, the rather homogenous distribution of ADK-expressing astrocytes and neurons throughout the whole brain suggests that brain adenosine levels are under the control of these ADK-expressing cells.
Figure 3.
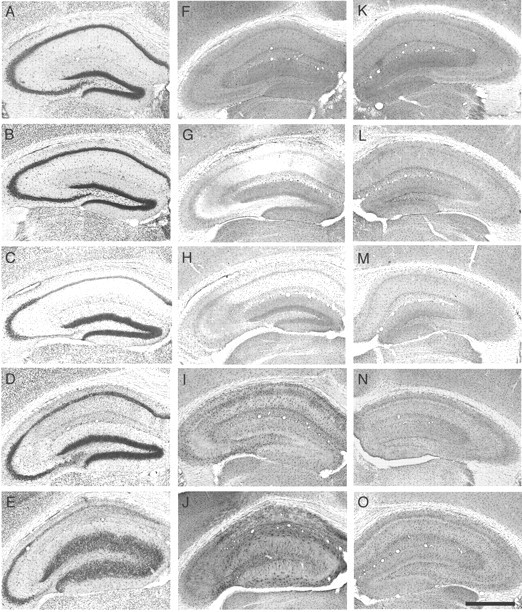
Changes in hippocampal cytoarchitecture ADK immunoreactivity during the course of epileptogenesis. Brains from KA-treated mice were taken at different time points after either intrahippocampal KA or saline injection (n = 4 animals for each time point). Transverse brain sections of the KA-injected brain hemisphere were stained with either cresyl violet (A-E) or for ADK immunoreactivity (F-O). F-J, Ipsilateral side; K-O, Contralateral side. A, F, K, Two hours after saline injection. B, G, L, Two hours after KA injection. C, H, M, One day after KA injection. D, I, N, One week after KA injection. E, J, O, Four weeks after KA injection. Note the biphasic regulation of ADK immunoreactivity after intrahippocampal KA injection (F-J). An initial loss of ADK immunoreactivity within 2 hr after KA injection (G) is gradually followed by a significant overexpression (J), which becomes also evident, although to a lesser degree, within the contralateral hippocampus (O). Scale bar: (in O), A-O, 1000 μm.
Figure 4.
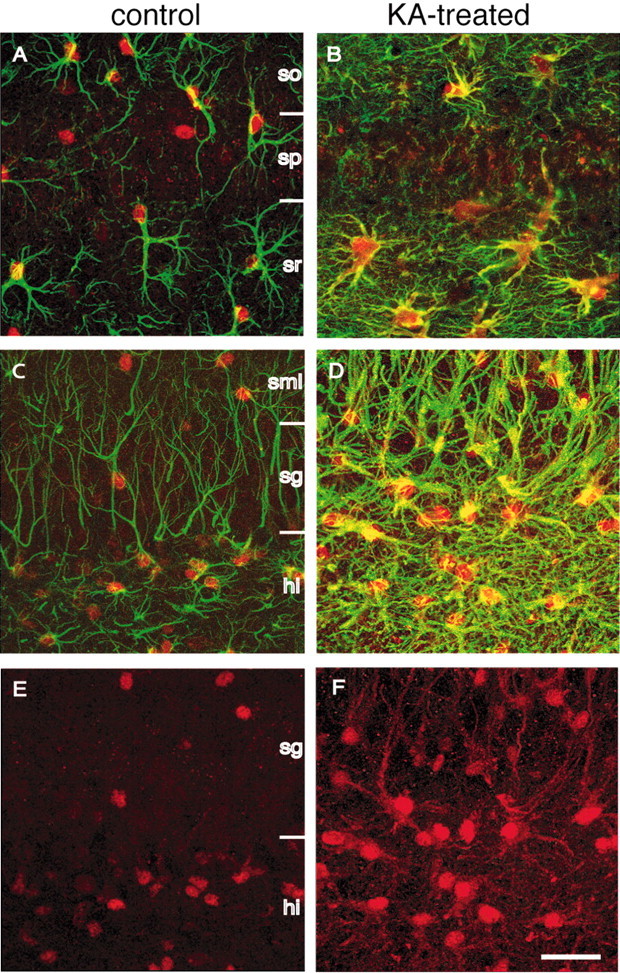
Colocalization of ADK and GFAP immunofluorescence, as seen by confocal laser-scanning microscopy. Transverse brain sections of a KA-injected animal taken 4 weeks after the injection and those from a naive control animal were double-stained for ADK (red) and the astrocyte marker GFAP (green). Optical sections were digitized at high magnification and superimposed for display. A, CA1 of a control animal. Note the red nuclei stained for ADK and astrocytic processes stained for GFAP (green). B, CA1 of a KA-injected animal. Note the characteristic pattern of gliosis characterized by an increase of astrocytic processes and by a spread of ADK immunoreactivity into the processes (colocalization of ADK and GFAP, yellow). C, Dentate gyrus of a control animal. Note that the cell bodies of individual astrocytes (green processes) are stained for ADK. D, Dentate gyrus of a KA-injected animal. Note the massive gliosis characterized by the swelling of cell bodies, the enlargement of astrocytic processes, and the expansion of ADK immunoreactivity into the processes (colocalization of ADK and GFAP, yellow). E, Same as in C showing ADK immunofluorescence only, which is mainly confined to the nucleus. F, Same as in D showing ADK immunofluorescence only, which becomes evident in astrocytic processes. so, Stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; sml, stratum moleculare; sg, stratum granulosum; hi, hilus. Scale bar: (in F), A-F, 30 μm.
Overexpression of adenosine kinase in epileptic hippocampus
To determine whether changes in brain expression of ADK contribute to epileptogenesis, we chose the mouse kainate model of hippocampal (focal) chronic epilepsy. In this model after a latent period of 2 weeks, spontaneous seizures begin to occur accompanied by reactive gliosis and progressive cell loss in the CA1 and CA3 regions of the injected hippocampus over 4-6 weeks (Suzuki et al., 1995; Bouilleret et al., 2000). Adult Adk+/+ mice were injected with a single unilateral intrahippocampal dose of 50 nl of 20 mm kainic acid (KA; n = 15) or saline (control; n = 6) and killed at different time points thereafter (2 hr, n = 4;1d, n = 4; 1 week, n = 4; and 4 weeks n = 3 after KA injection). Transverse brain sections were stained with cresyl violet to visualize the KA-induced hippocampal lesion or with the anti-ADK antibody to monitor ADK expression during epileptogenesis.
During 4 weeks after the KA injection, progressive histopathological changes within the injected hippocampus became apparent (Fig. 3, first column). Two hours after KA injection, a time point during which the experimental animals experienced a status epilepticus, the injected brain hemisphere did not show any cellular rearrangements (Fig. 3B) compared with saline-injected controls (Fig. 3A) or to control sections from untreated mice (data not shown). However, as soon as 1 d after KA injection, a profound loss of cells within the hilus of the dentate gyrus became apparent (Fig. 3C). In addition, cell loss was observed in the CA1 region of the hippocampus. One week after KA injection, during the latent phase of epileptogenesis, histopathological changes within the hippocampus continued to increase (Fig. 3D), culminating in severe cell loss in CA1 and CA3 regions of the hippocampus and dispersion of granule cells in the dentate gyrus, as observed 4 weeks after KA injection during the chronic phase of seizure activity (Fig. 3E).
In contrast to these cellular rearrangements, the pattern of immunoreactivity and the distribution of ADK-positive cells changed more rapidly after the KA injection. It is important to note that ADK immunoreactivity only changed in the injected hippocampus and, to a lesser degree, in the contralateral hippocampus. Other brain areas were not affected by the KA lesion. As soon as 2 hr after the injection, surrounding areas of the hippocampus (hilus and stratum lucidum, oriens, and radiatum) appeared transiently devoid of any ADK immunoreactivity (Fig. 3G). In contrast, the hippocampus contralateral to the injected side appeared unaffected at this time point (Fig. 3L). One day after KA injection, strong labeling of ADK was observed in the granule cells of the dentate gyrus (Fig. 3H), whereas ADK-expressing cells were distributed more or less homogeneously within the affected hippocampus. In contrast, the contralateral hippocampus (Fig. 3M) was still similar to control (Figs. 2, 3A). One week after KA injection, the distribution of ADK-positive cells again was observed to be completely different: immunoreactivity within the granular cells of the dentate gyrus had disappeared, whereas the remaining hippocampal formation appeared to be infiltrated with large numbers of ADK-expressing cells, a process paralleling ongoing astrogliosis (Bouilleret et al., 1999). These ADK-expressing cells were particularly prominent within the hilus of the dentate gyrus and the stratum lacunosum-moleculare (Fig. 3I). A slight increase of ADK-positive cells became apparent also in the contralateral hippocampus (Fig. 3N). During the progression of astrogliosis and epileptogenesis, the ADK expression was further increased bilaterally. This finding is reflected by a high density of ADK-positive cells within the dentate gyrus, the stratum lacunosum, and the stratum oriens (Fig. 3J). The contralateral hippocampus was also affected by an increase of ADK immunoreactivity (Fig. 3O). It is important to note that 1 and 4 weeks after KA injection, ADK immunoreactivity became also evident at a perinuclear localization and in the processes of the ADK-expressing cells (Fig. 3I,K,J,O), a finding that became most prominent 4 weeks after KA injection in the injected hippocampus (Fig. 3J). Taken together, 4 weeks after KA injection the injected hippocampus displays a strong increase of ADK immunoreactivity in certain subregions, whereas in other regions such as the CA3 region ADK immunoreactivity remains unchanged.
Expression of ADK in hippocampal astrocytes
To determine the nature of ADK-positive cells, we performed double immunofluorescence studies combining ADK staining (red) with staining for GFAP (green), a marker for astrocytes. Analysis of hippocampal sections from control animals revealed ADK immunoreactivity largely confined to astrocytes, being strongest in the nucleus of GFAP-positive cells (Fig. 4A,C). Four weeks after KA injection, the dentate gyrus and the molecular layer of the dentate gyrus were characterized by massive astrogliosis, becoming evident by the expansion of a GFAP-positive network of astrocyte processes (Fig. 4B,D), which is a characteristic feature of hippocampal sclerosis. It is striking to note that in epileptic hippocampus an increase of ADK immunoreactivity was observed becoming evident by the detection of ADK immunoreactivity in large-diameter processes, as revealed by a colocalization of GFAP and ADK (yellow) (Fig. 4B,D). The increased detection of ADK immunoreactivity in astrocyte processes in KA-treated hippocampus became also evident in single stainings for ADK (red) (Fig. 4F) and is in marked contrast to the predominantly nuclear localization of ADK in control hippocampus (Fig. 4E).
Kainic acid-lesioned hippocampi display enhanced adenosine kinase activity
To establish a causal relationship between increased ADK immunoreactivity in KA-lesioned hippocampi and enhanced adenosine metabolism, we compared the enzymatic activity of ADK in homogenates derived from KA-lesioned hippocampi with those from contralateral and control hippocampi.
Lesioned and contralateral hippocampi from five KA-treated mice were carefully excised 8 weeks after the lesion. Hippocampi from age-matched naive mice (n = 5) were isolated as controls. Enzyme activity of ADK was determined by performing an enzyme-coupled bioluminescent assay. Whereas ADK extracted from naive hippocampi (n = 10) metabolized 10.1 ± 1.5 ng of adenosine per minute per milligram of protein, extracts from KA-lesioned hippocampi (n = 5) and to a lesser degree those from contralateral hippocampi (n = 5) displayed a significant enhancement of ADK activity (p < 0.01 for both groups) that was reflected by the metabolism of 18.0 ± 1.7 ng of adenosine per minute per milligram of protein and of 14.4 ± 2.2 ng of adenosine per minute per milligram of protein, respectively. These results were normalized to the ADK activity found in naive hippocampi (Fig. 5). It is important to note that ADK activity in the KA-lesioned hippocampi was found to be 1.24-fold higher (p < 0.05) than ADK activity in the noninjected contralateral hippocampi.
Figure 5.

Increase of ADK enzyme activity in epileptic hippocampus. KA-injected and contralateral hippocampi were dissected out, homogenized, and analyzed in an enzyme coupled bioluminescent assay 8 weeks after the lesion (n = 5 mice). Hippocampi (n = 10) from age-matched naive mice (n = 5) were dissected as controls. Adenosine kinase enzyme activity, measured as phosphorylation of nanograms of adenosine per minute per milligram of protein, was determined by the increase of relative light units per time. The results were normalized to the ADK activity found in naive control hippocampi (100%). Samples from both KA-injected and contralateral hippocampi displayed a significant increase in ADK activity of 177.2 ± 16% and 142.4 ± 21.9%, respectively. Data were analyzed by a Kruskal-Wallis test (p < 0.001), and paired comparisons were done with Mann-Whitney U tests (**p < 0.01; *p < 0.05). Means are given with ± SDs.
These results demonstrate a significant hippocampal increase of ADK activity as a consequence of the KA lesion, which is in line with the enhanced ADK immunoreactivity. It is important to note that the contralateral hippocampus is also affected by an increase in ADK enzyme activity. Given the fact that we measured global hippocampal ADK activity and found increased ADK immunoreactivity in subregions of the hippocampal formation, we conclude that in these subregions even higher increases of ADK enzyme activity can be expected, which may lead to a profound local reduction of adenosine.
Inhibition of ADK provides seizure suppression
To investigate whether overexpression of ADK in the affected hippocampus contributes to ongoing chronic seizure activity in KA-treated mice during the chronic phase of epileptogenesis, 4 weeks after KA injection the selective ADK inhibitor 5-iodotubercidin was injected at a dose of 3.1 mg/kg intraperitoneally (n = 4), which is half of the ED50 (Wiesner et al., 1999) for the compound. Before drug injection, the animals displayed 25 ± 2 seizures per hour, each lasting 31.2 ± 17.7 sec, as determined by intrahippocampal EEG recordings. However, after the injection of 5-iodotubercidin in all animals, seizures were completely suppressed for at least 6 hr before individual spikes started reappearing on the EEG, whereas animals receiving vehicle injections (n = 4) continued to display unaltered seizure activity. It is important to note that a low dose of 5-iodotuberdidin (50% of ED50) was sufficient to suppress seizure activity, while causing little sedation. This side effect became very evident at higher doses (100% of ED50). To exclude a contribution to seizure suppression by peripheral activation of adenosine receptors after systemic inhibition of ADK, coinjections of 5-iodotubercidin (3.1 mg/kg, i.p.) and 8-sulfophenyltheophylline (20 mg/kg, i.p.), a nonselective non-brain-permeable adenosine receptor antagonist, were performed in KA-lesioned mice (n = 4). After coinjection of both drugs, seizure suppression was comparable to 5-iodotubercidin alone, indicating that inhibition of central ADK is responsible for this effect. To verify the selectivity of the 5-iodotubercidin-response, the selective A1-receptor antagonist DPCPX (1 mg/kg, i.p.) was injected in five additional mice 60 min after the 5-iodotubercidin (3.1 mg/kg, i.p.) injection. Before the 5-iodotubercidin injection, the animals averaged 32 ± 4 seizures per hour, each lasting 48.5 ± 9.7 sec (Fig. 6A). Again, after the injection of 5-iodotubercidin in all animals, seizures were completely suppressed (Fig. 6B). However, as soon as 15 min after the injection of DPCPX, seizure activity was restored with 12 ± 4 seizures per hour, each lasting 58.5 ± 15.6 sec (Fig. 6C). Likewise seizure activity became evident, when DPCPX was injected 20 min before 5-iodotubercidin or together with 5-iodotubercidin (data not shown). The abrogation of seizure suppression by DPCPX indicates the dependence of A1-receptor activation for seizure suppression.
Figure 6.
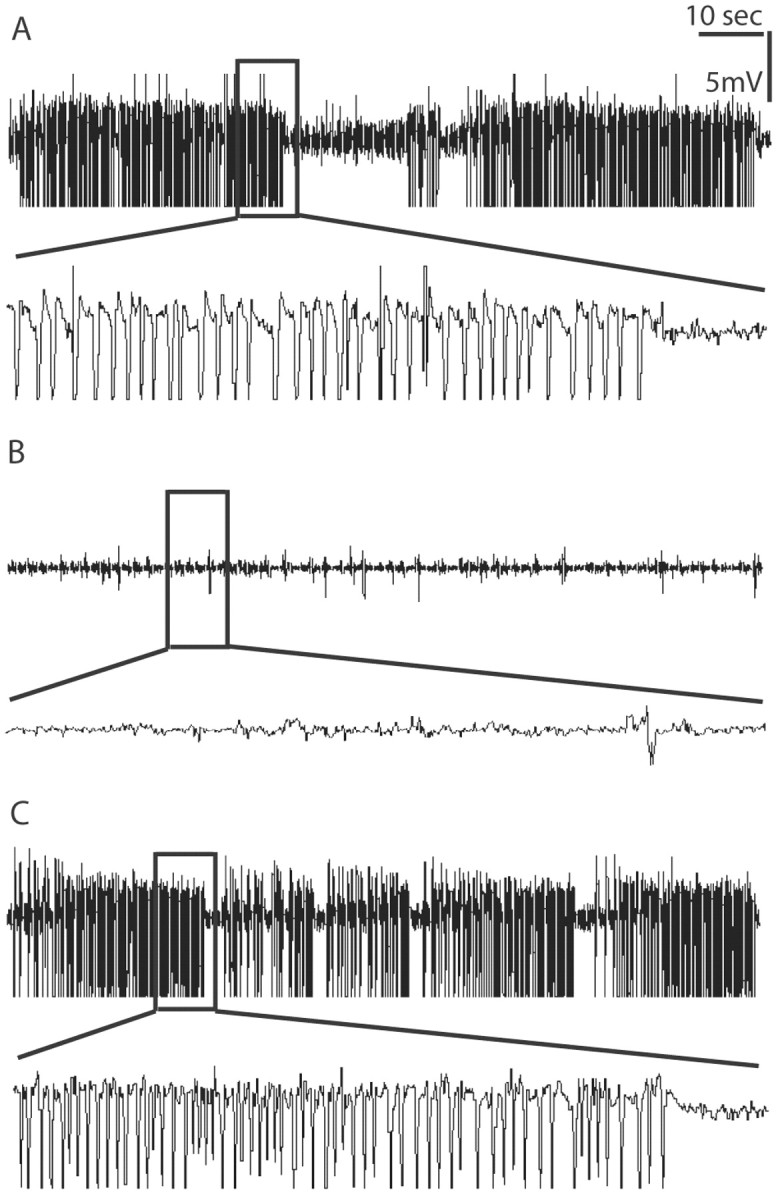
Seizure suppression by 5-iodotubercidin. Representative intrahippocampal EEG recordings taken from one animal before and after intraperitoneal injection of 5-iodotubercidin and DPCPX. Four weeks after kainic acid treatment 5-iodotubercidin was administered, followed after 60 min by DPCPX (n = 5 animals). The animals were recorded for a total of 2 hr before and 6 hr after 5-iodotubercidin injection. Representative sections of 120 sec duration (top traces) are displayed. Close-up views of 10 sec each are displayed in the bottom traces. The recordings displayed were performed on the same animal 1 hr before 5-iodotubercidin injection (A) (note the occurrence of high-amplitude and high-frequency discharges) 30 min after 5-iodotubercidin injection (B) (3.1 mg/kg, i.p.) (note the suppression of seizure activity), and 30 min after DPCPX (C) (1 mg/kg i.p.) (note the recurrence of full seizure activity).
Discussion
In the present study we provide for the first time an analysis of ADK expression in brain. So far, ADK was considered to be a ubiquitous protein responsible for the regulation of adenosine levels, which are of importance in several pathological states, including epilepsy (Dunwiddie and Masino, 2001). Because ADK is not essential for de novo purine biosynthesis, cells are able to survive in the absence of this enzyme (Huber et al., 2001), and physiological regulation of ADK expression is not likely to affect cell survival. In adult healthy mouse brain our findings demonstrate highest levels of ADK expression in the nuclear or perinuclear region of specific cells, which are distributed rather homogeneously throughout the whole brain (Fig. 2).
Biphasic regulation of ADK expression in epileptic hippocampus
In brain taken from KA-injected epileptic mice, ADK immunoreactivity followed a biphasic regulation in the injected hippocampus: (1) during the initial KA-induced status epilepticus we observed a rapid and profound reduction of ADK immunoreactivity within large parts of the injected hippocampus (Fig. 3G). Regulatory mechanisms based on reduced transcription or translation appear unlikely in such a short time span. However, inactivation of the enzyme by aggregation (Chakraborty et al., 2002) or rapid turnover could account for rapid changes in ADK activity. It is therefore tempting to speculate that the decrease of ADK would lead initially to increased adenosine levels within the hippocampus, which may contribute to the termination of the status epilepticus. This notion is in line with data supporting the role of adenosine in mediating seizure arrest and postictal refractoriness (During and Spencer, 1992). (2) After a silent phase of seizure activity, a significant overexpression of ADK immunoreactivity occurred in certain parts of the hippocampus (Fig. 3), which was in parallel with ongoing astrogliosis (Bouilleret et al., 1999). In addition, this expression pattern paralleled changes in EEG activity, in which initial seizure suppression during the latent phase of epileptogenesis is followed by gradual development of chronic epilepsy. The increase of ADK expression in epileptic hippocampus resulted in an apparent increase of ADK-positive astrocytes and increased staining of their processes, thus providing a three-dimensional network of ADK immunoreactivity (Fig. 4). The increased ADK expression in specific subregions of the lesioned hippocampi was paralleled by a significant increase of global hippocampal ADK enzyme activity (Fig. 5), suggesting a dysregulation of the adenosinergic inhibitory tone on a subregional or cellular level. Interestingly, the hippocampus contralateral to the KA lesion also displayed mild gliosis and increased ADK enzyme activity. It is important to note that the spread of ADK expression (Fig. 3I) appears before gross morphological alterations of the hippocampus (Fig. 3D). It is likely to conceive that overexpression of ADK in the hippocampus may contribute to chronic seizure activity by reduction of protective endogenous adenosine levels.
Regulation of hippocampal adenosine levels by ADK
Apart from ADK, adenosine may be metabolized by adenosine deaminase (ADA) and S-adenosylhomocysteine (SAH) hydrolase. However, three lines of evidence suggest that adenosine levels in the hippocampus are exclusively regulated by ADK.
Although ADK is ubiquitously expressed throughout the whole body, ADA activity varies between organs with brain activity being only 1-10% of ADA activity found in thymus (Geiger and Nagy, 1986). In contrast to the mostly astrocyte-specific brain-wide expression of ADK, several reports describe a selective expression of ADA only in discrete neuronal populations in rat and mouse brain, with the strongest immunoreactivity found in neurons of the basal hypothalamus (Patel and Tudball, 1986; Nagy et al., 1996). It is important to note that among all brain regions investigated, hippocampus displayed the lowest activity of ADA (Geiger and Nagy, 1986).
Another possible route of adenosine inactivation is the reversible reaction catalyzed by SAH hydrolase, forming SAH from adenosine and homocysteine. This pathway is limited by the availability of homocysteine, and because homocysteine levels are very low in brain, SAH synthesis represents only a minor pathway of adenosine metabolism in brain (Reddington and Pusch, 1983).
The specific inhibitor of ADK, 5-iodotubercidin, has been demonstrated to be more effective than ADA inhibitors at increasing extracellular adenosine levels in the brain (Pazzagli et al., 1995). In addition, inhibition of ADK reduced synaptic transmission in the hippocampus, indicating a significant role of ADK in the regulation of basal extracellular adenosine (Pak et al., 1994).
Adenosine levels in brain
Because of difficulties to perform correct local adenosine measurements in brain tissue and because of large variations according to the methods used (Delaney and Geiger, 1996), little is known on the regional and subregional concentrations of adenosine. However, in one investigation using immunohistochemistry with an antiserum directed against the adenosine derivative laevulinic acid, immunoreactivity was detected exclusively in neurons with high levels found in the pyramidal cells of the hippocampus and the granule cells of the dentate gyrus (Braas et al., 1986). It is interesting to note that these areas lack ADK expression (Fig. 2), suggesting a reciprocal distribution of adenosine and ADK, which would be a further support of the notion that adenosine levels in brain are regulated by ADK.
Based on our findings and literature data, it is likely that the upregulation of ADK described here leads to reduced levels of adenosine, which in turn may cause chronic seizure activity. This hypothesis is further supported by our recent findings that during the chronic seizure phase in KA-induced epilepsy the activation of A1 receptors leads to prolonged suppression of seizures (Gouder et al., 2003), a finding indicating that binding sites on A1 receptors are not saturated in KA-induced epilepsy, which may be attributable to reduced adenosine levels. To support these findings, application of a low dose of the specific ADK inhibitor 5-iodotubercidin during the chronic phase of epilepsy also resulted in complete lack of seizures for a period of at least 6 hr (Fig. 6).
Astrogliosis may contribute to epileptogenesis by increased ADK expression
Astrogliosis, characterized by hypertrophy and proliferation of astrocytes, is a hallmark of epileptic foci (Khurgel and Ivy, 1996). However, whether it plays a causal role in the development of seizures and the persistence of seizure disorders, or whether gliosis is merely an adaptive response to seizures is still an unresolved issue. Here we demonstrate that ADK is coexpressed in GFAP-positive reactive hypertrophied astrocytes, which are characterized by an increase in the size of their soma as well as an increase in the length and width of astrocytic stellae (Fig. 4). These findings demonstrate that astrogliosis is accompanied by an overexpression of ADK and thus may contribute to the persistence of recurrent seizure activity.
Our results are of importance for the following reasons: (1) In healthy brain, highest ADK expression occurs in astrocytes, suggesting that these cells regulate adenosine levels. (2) During chronic seizure activity, ADK expression and enzyme activity parallels gliosis. ADK expression was found to be increased in reactive astrocytes (Fig. 4). Therefore, gliosis may contribute to epileptogenesis by reducing endogenous adenosine levels. (3) During the chronic phase of epilepsy, increased ADK immunoreactivity and enzyme activity became also obvious in the contralateral hippocampus, although to a much lesser extent (Figs. 3N,O, 5). This could be indicative for an involvement of the adenosine system, even in the side contralateral to the primary lesion. (4) Our observation of rapid reduction of ADK immunoreactivity as an immediate consequence of intrahippocampal KA injection is in line with a marked downregulation of ADK activity during oxygen-glucose deprivation in an in vitro model of ischemia (Lynch et al., 1998). This downregulation of ADK was proposed to provide an endogenous mechanism contributing to the accumulation of extracellular adenosine, which provides an A1 receptor-mediated neuroprotective effect (Lynch et al., 1998). Likewise, the rapid downregulation of ADK in the KA-injected hippocampus described here, may lead to transiently increased intrahippocampal adenosine concentrations, which may provide seizure suppression during the latent phase of epileptogenesis. (5) A pathological increase of ADK enzyme activity in epileptic hippocampus would be an explanation for the efficacy of antiepileptic drugs that inhibit ADK. ADK inhibition was demonstrated to potentiate local extracellular adenosine levels at cell and tissue sites, which are undergoing accelerated adenosine release (Kowaluk et al., 1998; Kowaluk and Jarvis, 2000), and cultured cells with a complete lack of ADK released substantial amounts of adenosine (Huber et al., 2001; Zumsteg and Boison, 2002). (6) Finally, the contribution of ADK expression to chronic seizure activity would favor the development of local adenosine-based therapeutic interventions for chronic epilepsy, e.g., by grafting of adenosine-releasing cells and/or by pharmacological inhibition of ADK. Local adenosine-based treatment restricted to an epileptic focus would thus be sufficient and an option to circumvent pronounced side effects.
Footnotes
This work was supported by Grant 3100A0-100841 from the Swiss National Science Foundation and by the National Center for Competence in Research on Neural Plasticity and Repair. We thank Denise Fedele for assistance in surgery and Cornelia Schwerdel and Corinne Sidler for technical assistance.
Correspondence should be addressed to Dr. D. Boison, Institute of Pharmacology and Toxicology, University of Zurich, Winterthurerstrasse 190, CH-8057 Zürich, Switzerland. E-mail: boison@pharma.unizh.ch.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240692-10$15.00/0
References
- Boison D, Scheurer L, Tseng JL, Aebischer P, Mohler H (1999) Seizure suppression in kindled rats by intraventricular grafting of an adenosine releasing synthetic polymer. Exp Neurol 160: 164-174. [DOI] [PubMed] [Google Scholar]
- Boison D, Huber A, Padrun V, Déglon N, Aebischer P, Möhler H (2002a) Seizure suppression by adenosine releasing cells is independent of seizure frequency. Epilepsia 43: 788-796. [DOI] [PubMed] [Google Scholar]
- Boison D, Scheurer L, Zumsteg V, Rülicke T, Litynski P, Fowler B, Brandner S, Mohler H (2002b) Neonatal hepatic steatosis by disruption of the adenosine kinase gene. Proc Natl Acad Sci USA 99: 6985-6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bontemps F, Van den Berghe G, Hers HG (1983) Evidence for a substrate cycle between AMP and adenosine in isolated hepatocytes. Proc Natl Acad Sci USA 80: 2829-2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouilleret V, Ridoux V, Depaulis A, Marescaux C, Nehlig A, Le Gal La Salle G (1999) Recurrent seizures and hippocampal sclerosis following intrahippocampal kainate injection in adult mice: electroencephalography, histopathology and synaptic reorganization similar to mesial temporal lobe epilepsy. Neuroscience 89: 717-729. [DOI] [PubMed] [Google Scholar]
- Bouilleret V, Loup F, Kiener T, Marescaux C, Fritschy JM (2000) Early loss of interneurons and delayed subunit-specific changes in GABA(A)-receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus 10: 305-324. [DOI] [PubMed] [Google Scholar]
- Braas KM, Newby AC, Wilson VS, Snyder SH (1986) Adenosine-containing neurons in the brain localized by immunocytochemistry. J Neurosci 6: 1952-1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Das I, Datta R, Sen B, Bhattacharyya D, Mandal C, Datta AK (2002) A single-domain cyclophilin from Leishmania donovani reactivates soluble aggregates of adenosine kinase by isomerase-independent chaperone function. J Biol Chem 277: 47451-47460. [DOI] [PubMed] [Google Scholar]
- Delaney SM, Geiger JD (1996) Brain regional levels of adenosine and adenosine nucleotides in rats killed by high-energy focused microwave irradiation. J Neurosci Methods 64: 151-156. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Diao L (1994) Extracellular adenosine concentrations in hippocampal brain slices and the tonic inhibitory modulation of evoked excitatory responses. J Pharmacol Exp Ther 268: 537-545. [PubMed] [Google Scholar]
- Dunwiddie TV, Masino SA (2001) The role and regulation of adenosine in the central nervous system. Annu Rev Neurosci 24: 31-55. [DOI] [PubMed] [Google Scholar]
- During MJ, Spencer DD (1992) Adenosine: a potential mediator of seizure arrest and postictal refractoriness. Ann Neurol 32: 618-624. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Battig K, Holmen J, Nehlig A, Zvartau EE (1999) Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacol Rev 51: 83-133. [PubMed] [Google Scholar]
- Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J (2001) International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev 53: 527-552. [PMC free article] [PubMed] [Google Scholar]
- Geiger JD, Nagy JI (1986) Distribution of adenosine deaminase activity in rat brain and spinal cord. J Neurosci 6: 2707-2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouder N, Fritschy JM, Boison D (2003) Seizure suppression by adenosine A1 receptor activation in a mouse model of pharmacoresistant epilepsy. Epilepsia 44: 877-885. [DOI] [PubMed] [Google Scholar]
- Huber A, Padrun V, Déglon N, Aebischer P, Möhler H, Boison D (2001) Grafts of adenosine releasing cells suppress seizures in kindling epilepsy. Proc Natl Acad Sci USA 98: 7611-7616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A, Güttinger M, Möhler H, Boison D (2002) Seizure suppression by adenosine A2A receptor activation in a rat model of audiogenic brainstem epilepsy. Neurosci Lett 329: 289-292. [DOI] [PubMed] [Google Scholar]
- Khurgel M, Ivy GO (1996) Astrocytes in kindling: relevance to epileptogenesis. Epilepsy Res 26: 163-175. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Zuellig RA, Schaub MC, Fritschy JM (2001) Alterations in dystrophin and utrophin expression parallel the reorganization of GABAergic synapses in a mouse model of temporal lobe epilepsy. Eur J Neurosci 13: 1113-1124. [DOI] [PubMed] [Google Scholar]
- Kowaluk EA, Jarvis MF (2000) Therapeutic potential of adenosine kinase inhibitors. Expert Opin Investig Drugs 9: 551-564. [DOI] [PubMed] [Google Scholar]
- Kowaluk EA, Bhagwat SS, Jarvis MF (1998) Adenosine kinase inhibitors. Curr Pharm Des 4: 403-416. [PubMed] [Google Scholar]
- Lynch III JJ, Alexander KM, Jarvis MF, Kowaluk EA (1998) Inhibition of adenosine kinase during oxygen-glucose deprivation in rat cortical neuronal cultures. Neurosci Lett 252: 207-210. [DOI] [PubMed] [Google Scholar]
- McNally T, Helfrich RJ, Cowart M, Dorwin SA, Meuth JL, Idler KB, Klute KA, Simmer RL, Kowaluk EA, Halbert DN (1997) Cloning and expression of the adenosine kinase gene from rat and human tissues. Biochem Biophys Res Commun 231: 645-650. [DOI] [PubMed] [Google Scholar]
- Nagy JI, Yamamoto T, Uemura H, Schrader WP (1996) Adenosine deaminase in rodent median eminence: detection by antibody to the mouse enzyme and co-localization with adenosine deaminase-complexing protein (CD26). Neuroscience 73: 459-471. [DOI] [PubMed] [Google Scholar]
- Pak MA, Haas HL, Decking UKM, Schrader J (1994) Inhibition of adenosine kinase increases endogenous adenosine and depresses neuronal activity in hippocampal slices. Neuropharmacology 33: 1049-1053. [DOI] [PubMed] [Google Scholar]
- Patel BT, Tudball N (1986) Localization of S-adenosylhomocysteine hydrolase and adenosine deaminase immunoreactivities in rat brain. Brain Res 370: 250-264. [DOI] [PubMed] [Google Scholar]
- Pazzagli M, Corsi C, Fratti S, Pedata F, Pepeu G (1995) Regulation of extracellular adenosine levels in the striatum of aging rats. Brain Res 684: 103-106. [DOI] [PubMed] [Google Scholar]
- Reddington M, Pusch R (1983) Adenosine metabolism in a rat hippocampal slice preparation: incorporation into S-adenosylhomocysteine. J Neurochem 40: 285-290. [DOI] [PubMed] [Google Scholar]
- Riban V, Bouilleret V, Pham-Le BT, Fritschy JM, Marescaux C, Depaulis A (2002) Evolution of hippocampal epileptic activity during the development of hippocampal sclerosis in a mouse model of temporal lobe epilepsy. Neuroscience 112: 101-111. [DOI] [PubMed] [Google Scholar]
- Suzuki F, Junier MP, Guilhem D, Sorensen JC, Onteniente B (1995) Morphogenetic effect of kainate on adult hippocampal neurons associated with a prolonged expression of brain-derived neurotrophic factor. Neuroscience 64: 665-674. [DOI] [PubMed] [Google Scholar]
- Ugarkar BG, Castellino AJ, DaRe JM, Kopcho JJ, Wiesner JB, Schanzer JM, Erion MD (2000a) Adenosine kinase inhibitors. 2. Synthesis, enzyme inhibition, and antiseizure activity of diaryltubercidin analogues. J Med Chem 43: 2894-2905. [DOI] [PubMed] [Google Scholar]
- Ugarkar BG, DaRe JM, Kopcho JJ, Browne CE, 3rd, Schanzer JM, Wiesner JB, Erion MD (2000b) Adenosine kinase inhibitors. 1. Synthesis, enzyme inhibition, and antiseizure activity of 5-iodotubercidin analogues. J Med Chem 43: 2883-2893. [DOI] [PubMed] [Google Scholar]
- Wiesner JB, Ugarkar BG, Castellino AJ, Barankiewicz J, Dumas DP, Gruber HE, Foster AC, Erion MD (1999) Adenosine kinase inhibitors as a novel approach to anticonvulsant therapy. J Pharmacol Exp Ther 289: 1669-1677. [PubMed] [Google Scholar]
- Zhu PJ, Krnjevic K (1994) Endogenous adenosine deaminase does not modulate synaptic transmission in rat hippocampal slices under normoxic or hypoxic conditions. Neuroscience 63: 489-497. [DOI] [PubMed] [Google Scholar]
- Zumsteg V, Boison D (2002) The use of real-time PCR with fluorogenic probes for the rapid selection of mutant neuroectodermal grafts. J Neurosci Methods 120: 85-94. [DOI] [PubMed] [Google Scholar]