

Abstract
KCNQ subunits encode for the M current (IKM), a neuron-specific voltage-dependent K+ current with a well established role in the control of neuronal excitability. In this study, by means of a combined biochemical, pharmacological, and electrophysiological approach, the role of presynaptic IKM in the release of previously taken up tritiated norepineprine (NE), GABA, and d-aspartate (d-ASP) from hippocampal nerve terminals (synaptosomes) has been evaluated. Retigabine (RT) (0.01-30 μm), a specific activator of IKM, inhibited [3H]NE, [3H]d-ASP, and [3H]GABA release evoked by 9 mm extracellular K+ ([K+]e). RT-induced inhibition of [3H]NE release was prevented by synaptosomal entrapment of polyclonal antibodies directed against KCNQ2 subunits, an effect that was abolished by antibody preabsorption with the KCNQ2 immunizing peptide; antibodies against KCNQ3 subunits were ineffective. Flupirtine (FP), a structural analog of RT, also inhibited 9 mm [K+]e-induced [3H]NE release, although its maximal inhibition was lower than that of RT. Electrophysiological studies in KCNQ2-transfected Chinese hamster ovary cells revealed that RT and FP (10 μm) caused a -19 and -9 mV hyperpolarizing shift, respectively, in the voltage dependence of activation of KCNQ2 K+ channels. In the same cells, the cognition enhancer 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone (XE-991) (10 μm) blocked KCNQ2 channels and prevented their activation by RT (1-10 μm). Finally, both XE-991 (10-100 μm) and tetraethylammonium ions (100 μm) abolished the inhibitory effect of RT (1 μm) on [3H]NE release. These findings provide novel evidence for a major regulatory role of KCNQ2 K+ channel subunits in neurotransmitter release from rat hippocampal nerve endings.
Keywords: epilepsy, potassium channels, KCNQ2 subunits, retigabine, hippocampus, norepinephrine release
Introduction
K+ channels control neuronal excitability by regulating interspike membrane potential, action potential waveform, and firing frequency. The M current (IKM) is a slowly activating and noninactivating voltage-dependent K+ current that repolarizes the neuronal membrane back toward the K+ equilibrium potential (EK) during long-lasting depolarizing inputs, thus limiting repetitive firing and causing spike-frequency adaptation (Brown and Adams, 1980). Receptor-dependent regulation of the M current is a primary mechanism by which neurotransmitters control neuronal excitability (Marrion, 1997). Heteromeric assembly of subunits encoded by two members of the KCNQ gene family, namely KCNQ2 and KCNQ3, recapitulate the functional properties of IKM (Wang et al., 1998), although KCNQ5 and possibly KCNQ4 subunits might also participate in IKM heterogeneity (Rogawski, 2000).
IKM plays a crucial role in epileptogenesis; mutations in KCNQ2 or KCNQ3 genes cause benign neonatal familial convulsions, a rare dominantly inherited form of human epilepsy (Jentsch, 2000). Furthermore, drug-induced enhancement of IKM function may oppose epileptic activity; in fact, the novel anticonvulsant N-[2-amino-4-(4-fluorobenzylamino)-phenyl]-carbamic acid ethyl ester [retigabine (RT)], which is structurally unrelated to currently available antiepileptics, specifically activates voltage-gated K+ currents carried by some members of the KCNQ family (Main et al., 2000; Rundfeldt and Netzer, 2000a; Wickenden et al., 2000). In keeping with a K+ channel-opening mechanism, RT hyperpolarized and reduced repetitive firing of entorhinal cortical neurons in cortical slices (Hetka et al., 1999), inhibited firing of mouse primary cortical neurons (Otto et al., 2002), and activated native IKM in rat sympathetic neurons (Tatulian et al., 2001). Together, these studies highlight the role of IKM at the postsynaptic level. However, IKM may also play a relevant presynaptic role; in fact, a drug class known as “cognition enhancers” increases neurotransmitter release by suppressing IKM (for review, see Brown and Yu, 2000), and immunohistochemical studies have demonstrated a widespread presynaptic distribution of some KCNQ subunits within rodents and the human brain (Cooper et al., 2000, 2001).
Because multiple KCNQ-type subunits underlie IKM, the aims of the present study have been: (1) to define the specific influence exerted by IKM-encoding KCNQ subunits in the release of previously taken up 1-[7,8-3H]norepinephrine ([3H]NE), d-[2,3-3H]aspartic acid ([3H]d-ASP), and 4-amino-N-[2,3-3H]butyric acid ([3H]GABA) from rat hippocampal presynaptic terminals and (2) to correlate the effects of IKM modulators on KCNQ-type subunits expressed in Chinese hamster ovary (CHO) cells with their ability to modulate neurotransmitter release. The results obtained, which showed that activation of M channels inhibited depolarization-induced neurotransmitter release and that blockade of KCNQ2 subunits prevented this inhibition, suggest that M channels containing KCNQ2 subunits are crucial regulators of neurotransmitter release from rat hippocampal nerve endings.
Materials and Methods
Synaptosomal preparation and release experiments. Synaptosomes were prepared from the hippocampi of adult male rats (Martire et al., 2000). In experiments with antibodies, these were entrapped into the synaptosomes during hippocampal tissue homogenization (1:100 dilution) (Raiteri et al., 2000). Synaptosomal pellets were resuspended in a medium containing the following (in mm): 125 NaCl, 3 KCl, 1.2 MgSO4, 1.2 CaCl2, 1 NaH2PO4, 22 NaHCO3, and 10 glucose, pH 7.2-7.4, aerated with 95% O2 and 5% CO2 (standard medium). Synaptosomes were incubated for 15 min at 37°C with 0.04 μm [3H]NE, 0.04 μm [3H]d-ASP, or 0.02 μm [3H]GABA. Either 500 μm dihydrokainic acid or 100 μm β-alanine was added with [3H]d-ASP or [3H]GABA, respectively, to prevent glial uptake of the neurotransmitters (O'Shea, 2002; Raiteri et al., 2002). Identical aliquots of the synaptosomal suspension were placed at the bottom of parallel superfusion chambers maintained at 37°C and superfused (0.6 ml/min) with standard medium. In [3H]GABA release experiments, 50 μm amino-oxyacetic acid was added to prevent the catabolism of the neurotransmitter. After a 46 min equilibration period, perfusate fractions were collected every 2 min; after collection of the first fraction (to measure basal release), synaptosomes were exposed for 3 min to a medium containing elevated KCl concentrations (9, 15, 30, or 50 mm) plus different test drugs. Neurotransmitter efflux into the samples collected was calculated as a fractional rate. Drug effects were evaluated by determining the integrated area under the time course release curve (AUC) after subtraction of the area corresponding to basal release. Data, expressed as means ± SEM, were analyzed by one-way ANOVA followed by Dunnett's multiple comparison test (p < 0.05).
Heterologous expression of KCNQ subunits. KCNQ2 or KCNQ3 subunits were expressed in CHO cells (Wickenden et al., 2000) by transient transfection with KCNQ2 or KCNQ3 cDNA, respectively, using Lipofectamine 2000 (Invitrogen, Milan, Italy). Enhanced green fluorescent protein (Clontech, Palo Alto, CA) was used as a transfection marker. For electrophysiological and biochemical experiments, the cells were seeded on glass coverslips, transfected after 24 hr, and analyzed 1-2 d after transfection.
Electrophysiology. Currents were recorded at room temperature (20-22°C) using an Axopatch 200A amplifier and pClamp 6.0.4 software (Axon Instruments, Foster City, CA). The “perforated-patch” configuration of the patch-clamp technique was adopted using glass micropipettes of 2-3 MΩ resistance, because KCNQ2 currents showed a fast “run-down” in the conventional whole-cell mode (Shah et al., 2002). The extracellular solution contained the following (in mm): 150 NaCl, 2 CaCl2, 2 KCl, 1 MgCl2, 11 glucose, and 10 HEPES, pH 7.4, with NaOH. Increases in [KCl]e were compensated with equimolar decreases in [NaCl]e. The intracellular (pipette) solution contained the following (in mm): 120 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, 1 Mg-ATP, and 0.25 mm cAMP, pH 7.3-7.4, with KOH. Nystatin (120-240 μg/ml) was added to this solution immediately before each recording session. KCNQ2 channels were activated by means of voltage ramps from -100 mV to 0 or +20 mV at a 0.5-1 sec/20 mV speed and a frequency of 0.05 Hz. Given the small volume of the recording chamber (≈200 μl) and the fast perfusion speed (1 ml/min), solution exchanges were complete after two to three pulses. To quantify drug effects on KCNQ2 channels, the integrated AUCs of ramp-evoked K+ currents recorded from KCNQ2-transfected cells in the presence of drug(s) were expressed as a percentage of the AUCs of ramp-evoked K+ currents recorded in control conditions.
Indirect immunofluorescence. Cells were fixed with 4% formaldehyde for 20 min at room temperature and permeabilized with 0.1% Triton X-100 in PBS. Cells were labeled with the appropriate primary antibodies (1:200 dilution) followed by rhodamine-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) and analyzed on an Eclipse 400 photomicroscope (Nikon, Tokyo, Japan) with a 60× objective.
Materials. [3H]NE (specific activity, 36.0 Ci/mmol), [3H]GABA (specific activity, 100 Ci/mmol), and [3H]d-ASP (specific activity, 13.0 Ci/mmol) were from Amersham Biosciences (Buckinghamshire, UK). Retigabine and ethyl-2-amino-6-[4-(4-fluorbenzyl)amino]-pyridine-3-carbamic acid maleate [flupirtine (FP)] were from ASTA Medica (Radebeul, Germany). 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone (XE-991) was from DuPont NEN (Wilmington, DE). KCNQ2- and KCNQ3-specific antibodies (sc-7793 and sc-7795, respectively) and neutralizing peptides were from Santa Cruz Biotechnology (Santa Cruz, CA).
Results
RT inhibits previously taken up [3H]neurotransmitter release from isolated nerve endings
Synaptosomal release of [3H]neurotransmitters is exquisitely sensitive to mild depolarizing stimuli such as those produced by small changes in extracellular K+ ([K+]e). In fact, a 3 min exposure of hippocampal synaptosomes to 9 mm [K+]e enhanced the release of previously taken up [3H]NE (Fig. 1A). RT (0.01-30 μm) dose-dependently inhibited 9 mm [K+]e-elicited [3H]NE release, with a maximal inhibition (EMax) of 42.2 ± 2.1% and an EC50 of 1.00 ± 0.06 μm (Fig. 1A,B). No effect was exerted by RT on basal [3H]NE release (data not shown). RT also inhibited [3H]NE release from cortical synaptosomes with a potency and efficacy similar to that seen in hippocampal nerve terminals (EMax, 41.3 ± 2.3%; EC50, 1.00 ± 0.06; data not shown). Furthermore, in hippocampal synaptosomes, [3H]NE release evoked by exposure to 100 μm 4-aminopyridine (4-AP) (AUC, 16.58 ± 1.16%) was also inhibited by RT (EMax, 43.5 ± 2.1%; EC50, 1.13 ± 0.05 μm; data not shown).
Figure 1.

Effect of RT and FP on [3H]neurotransmitter release from hippocampal synaptosomes. A, Time course and dose dependence of RT-induced inhibition of [3H]NE release. Synaptosomes were exposed to 9 mm [K+]e with or without the indicated concentrations of RT for 3 min (boxed region). B, RT (filled symbols)-induced and FP (open symbols)-induced inhibition of [3H]NE (squares), [3H]GABA (circles), and [3H]d-ASP (diamonds) release evoked by 9 mm [K+]e. Results are expressed as the percentage of inhibition produced by each drug concentration of the integrated AUCs of [3H]neurotransmitter release evoked by 9 mm [K+]e ([3H]NE, 6.85 ± 0.48%; [3H]d-ASP, 2.15 ± 0.15%; [3H]GABA, 3.90 ± 0.27%). Values are means ± SEM of 5-10 separate experiments run in triplicate. *p < 0.05 versus respective control.
To verify whether RT also inhibited the release of other hippocampal neurotransmitters, we evaluated its effects on the release of glutamate and GABA. To label glutamatergic terminals, [3H]d-ASP was used because it is more resistant to catabolism (Davies and Johnston, 1976); furthermore, synaptosomal [3H]d-ASP uptake (Gundersen et al., 1995) and release (Martire et al., 2000) resemble that of endogenous glutamate. RT (0.01-30 μm) dose-dependently inhibited both [3H]d-ASP and [3H]GABA release evoked by 9 mm [K+]e from hippocampal synaptosomes (Fig. 1B), with an EMax and an EC50 of 45.5 ± 3.3% and 1.10 ± 0.36 μm for [3H]d-ASP and 47.8 ± 2.9% and 1.08 ± 0.06 μm for [3H]GABA, respectively.
K+ channel-containing KCNQ2 subunits underlie RT-induced inhibition of hippocampal [3H]NE release
To confirm that M channels were involved in RT-induced inhibition of synaptosomal neurotransmitter release, and to identify the contribution of specific KCNQ subunits, we used commercially available polyclonal antibodies specifically directed against KCNQ2 or KCNQ3 subunits. Immunofluorescence experiments revealed that anti-KCNQ2 antibodies labeled CHO cells transfected with KCNQ2 but not with KCNQ3 cDNAs, whereas the opposite was observed with anti-KCNQ3 antibodies (Fig. 2A). In both cases, labeling was abolished by antibody preabsorption with the respective immunizing peptide, arguing in favor of a specific interaction between these antibodies and KCNQ subunits. Notably, these antibodies were also used to detect KCNQ2 and KCNQ3 subunits in immunocytochemistry experiments in native rat hippocampal neurons (Shah et al., 2002). These results suggest that both antibodies bind to KCNQ subunit epitopes in their native, nondenatured conformation, making them useful tools to dissect the role of KCNQ subunits in synaptosomal [3H]NE release. Because both antibodies recognized intracellular epitopes, we used the synaptosomal entrapping technique, which allows membrane-impermeant proteins to gain access to the synaptosomal cytoplasm (Raiteri et al., 2000). Synaptosomal entrapment of anti-KCNQ2 antibodies completely prevented 1 μm RT-induced inhibition of 9 mm [K+]e-evoked hippocampal [3H]NE release (Fig. 2B), an effect that was abolished after antibody preabsorption, before entrapment, with the KCNQ2 immunizing peptide; in contrast, anti-KCNQ3 antibodies were ineffective. Entrapment of both anti-KCNQ2 and anti-KCNQ3 antibodies also abolished RT-induced inhibition of 9 mm [K+]e-evoked [3H]NE release.
Figure 2.

Antibody (Ab) characterization by immunocytochemistry in transfected CHO cells and effects on RT-induced inhibition of hippocampal [3H]NE release. A, Immunocytochemistry in CHO cells. Anti-KCNQ antibodies (1:200 dilution) were probed with (Q2Ab+Q2P; Q3Ab+Q3P) or without (Q2Ab; Q3Ab) preabsorption with the respective immunizing peptides in cells transfected with KCNQ2 (pcQ2) or KCNQ3 (pcQ3) cDNAs, as indicated in each respective panel. B, Effects of antibodies on [3H]NE release. Hippocampi were homogenized in the absence (NoAb) or in the presence of anti-KCNQ2 (Q2Ab), anti-KCNQ3 (Q3Ab), or anti-KCNQ2 plus anti-KCNQ3 (Q2Ab+Q3Ab) antibodies; when indicated, the antibodies were preabsorbed with the respective immunizing peptides (Q2P and Q3P; antibody:peptide ratio, 1:2). Data are the mean ± SEM of 3-12 experiments run in triplicate. Asterisks denote values of RT-treated synaptosomes that are significantly different (p < 0.05) from their respective controls (no RT treatment).
RT-induced inhibition of hippocampal [3H]NE release is dependent on [K+]e
Depolarization of hippocampal synaptosomes with increasing [K+]e progressively enhanced [3H]NE outflow; the ability of 10 μm RT to inhibit the release of the catecholamine decreased with increasing [K+]e (Fig. 3A). To investigate whether RT interference with KCNQ2 channels depended on [K+]e, electrophysiological studies in KCNQ2-transfected CHO cells were performed to assess the effects of RT on KCNQ2 K+ channel activity with 2 and 50 mm [K+]e. In the presence of 2 mm [K+]e, 10 μm RT hyperpolarized the voltage dependence of KCNQ2 K+ channel activation by -19.6 ± 0.9 mV (n = 13) (Fig. 3B). In fact, KCNQ2 currents reached 10% of the peak at -40 ± 1.9 mV in controls and at -60.3 ± 2.2 mV after exposure to 10 μm RT (p < 0.05) (Fig. 3B). A similar negative shift of -16.7 ± 1.6 mV in the voltage dependence of activation of KCNQ2 currents was induced by 10 μm RT when 50 mm [K+]e was used (controls, -63.8 ± 3.1 mV; 10 μm RT, -80.5 ± 3.6 mV; n = 4; p < 0.05). Thus, RT hyperpolarized the activation threshold of KCNQ2 K+ channels regardless of [K+]e, arguing against a possible displacement of RT binding to KCNQ2 subunits with increasing [K+]e. Interestingly, the RT-induced KCNQ2 K+ current enhancement was maximal at approximately -65 mV, a value close to EK in the presence of 9 mm [K+]e, although it declined progressively with increasing depolarizations, becoming negligible at approximately -20 mV, a value close to EK in the presence of 50 mm [K+]e (Fig. 3C).
Figure 3.
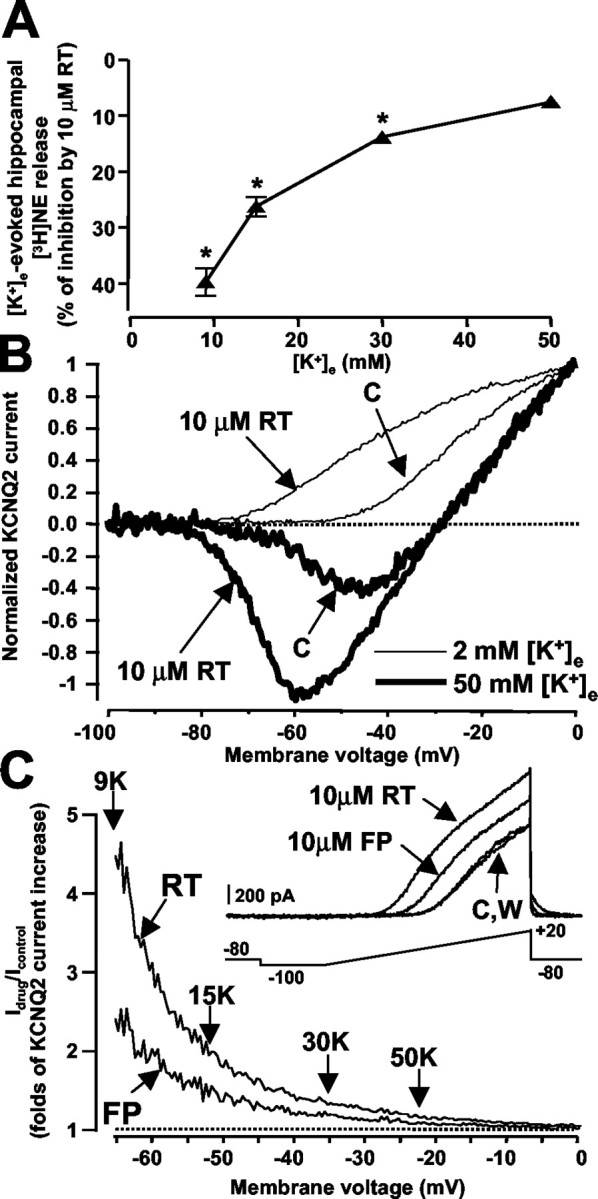
Effects of RT and FP on K+- evoked release of hippocampal [3H]NE release and on KCNQ2 channels expressed in CHO cells. A, Effects of 10 μm RT on [3H]NE release evoked by increasing [K+]e. The integrated AUCs of hippocampal [3H]NE release were 6.08 ± 0.42, 12.95 ± 0.78, 18.20 ± 1.26, and 23.60 ± 1.65% of total synaptosomal radioactivity with 9, 15, 30, and 50 mm [K+]e, respectively. Filled triangles are the percentage of RT (10 μm)-induced inhibition of depolarization-induced [3H]NE release observed at the indicated [K+]e. Data represent the mean ± SEM of three experiments run in triplicate. *p < 0.05 versus respective controls. B, Effect of 10 μm RT on KCNQ2 channels with 2 and 50 mm [K+]e. Representative voltage ramp-evoked current traces from a single KCNQ2-transfected cell are recorded in 2 mm (thin lines) or 50 mm (thick lines) [K+]e. C, Enhancement of KCNQ2 K+ currents by RT and FP is maximal at negative membrane potentials. KCNQ2 K+ currents (2 mm [K+]e) were activated by the voltage-ramp protocol described in Materials and Methods under control conditions (C), after exposure to 10 μm RT or to 10 μm FP and after washout (W), as indicated in the inset. After normalization to the maximal value, the currents recorded in the presence of the drug were divided by the control currents, and the results were expressed as a function of membrane potential. The vertical arrows indicate the theoretical EK values predicted by the Nernst equation (EK = RT/F × ln [K+]e/[K+]i) in the presence of the indicated [K+]e. In, Logarithm. Similar results were obtained in four cells with 10 μm RT and in three cells with 10 μm FP.
Hippocampal [3H]NE release is influenced by IKM modulators
The nonopioid analgesic FP (0.01-30 μm), a structural analog of RT (Friedel and Fitton, 1993), inhibited 9 mm [K+]e-induced [3H]NE release from hippocampal synaptosomes; when compared with RT, FP appeared to be equipotent (EC50, 1.27 ± 0.08 μm) but less effective (EMax, 24.7 ± 1.5%) (Fig. 1B). Electrophysiological studies in KCNQ2-transfected cells showed that 10 μm FP caused a voltage-dependent hyperpolarizing shift in KCNQ2 activation threshold of -9 ± 1 mV (n = 4), approximately half that caused by RT (Fig. 3C); furthermore, just as with RT, maximal FP-induced current enhancement was observed at potentials of approximately -65 mV (Fig. 3C).
Low micromolar concentrations of the cognition enhancer XE-991 (Zaczek et al., 1998) specifically inhibit K+ channels formed by KCNQ-type subunits (Wang et al., 1998). XE-991 (10-100 μm) completely prevented the inhibitory effects on 9 mm [K+]e-induced [3H]NE release exerted by 1 μm RT (Fig. 4A). These findings are consistent with those obtained in CHO cells, in which XE-991 blocked KCNQ2 K+ channels and essentially prevented their activation by RT (Fig. 4B). In fact, in the absence of XE-991, the integrated AUCs of ramp-evoked KCNQ2 K+ currents were 163.5 ± 9.6 and 211.6 ± 15.9% with 1 and 10 μm RT, respectively, compared with control KCNQ2 currents (n = 4; p < 0.05 vs controls); in the presence of 10 μm XE-991, these values were 6.1 ± 2.7, 10.9 ± 2.1, and 12.8 ± 3.6% of control KCNQ2 currents with 0, 1, and 10 μm RT, respectively (n = 4; p < 0.05 vs controls).
Figure 4.

XE-991 and TEA prevent RT-induced inhibition of 9 mm K+-evoked hippocampal [3H]NE. A, Effect of RT on 9 mm K+-evoked [3H]NE release in the presence of XE-991 or TEA. [3H]NE release was evaluated with (filled bars) or without (open bars) 1 μm RT in synaptosomes that had been preincubated for 8 min with XE-991 (10 or 100 μm) or TEA (100 μm), as indicated. Each bar is the mean ± SEM of three to four experiments performed in triplicate. *p < 0.05 versus respective control; **p < 0.05 versus controls without drug (XE-991 or TEA) preincubation but not different from each other. B, RT does not counteract XE-991 (XE)-induced K+ current blockade in KCNQ2-transfected CHO cells. Representative ramp-evoked K+ currents from a single KCNQ2-transfected cell were recorded under control conditions and after subsequent perfusion with 1 μm RT, 10 μm XE-991, and 10 μm XE-991 plus 1 μm RT (3-4 min each; see Results).
M channels containing KCNQ2 subunits are exquisitely sensitive to blockade by submillimolar concentrations of tetraethylammonium ions (TEA), whereas KCNQ3-containing channels are highly resistant (Wang et al., 1998). The fact that 100 μm TEA completely prevented RT-induced inhibition (Fig. 4A) provided strong support for the hypothesis that KCNQ2-type subunits were involved in RT-induced inhibition of NE release. Furthermore, 100 μm TEA, similar to 100 μm XE-991, potentiated 9 mm [K+]e-evoked [3H]NE release (Fig. 4A), possibly because at this concentration, both drugs inhibited other voltage-gated K+ channels controlling NE release (Wang et al., 1998).
Discussion
In the present study, we examined the possible role of presynaptic M channels in [3H]neurotransmitter release from hippocampal synaptosomes evoked by exposure to 9 mm [K+]e; neurotransmitter release induced by this small depolaring stimulus is entirely (as in the case of [3H]NE) or for the most part (as with [3H]d-ASP and [3H]GABA) calcium dependent and exocytotic in nature (Raiteri et al., 2002). Furthermore, dihydrokainate or β-alanine was used during synaptosomal labeling with [3H]d-ASP and [3H]GABA, respectively, to prevent glial incorporation and subsequent release of labeled transmitters.
The IKM activator RT produced a remarkable (>40%) inhibition of depolarization-induced [3H]NE, [3H]d-ASP, and [3H]GABA release evoked by 9 mm [K+]e. A similar degree of inhibition of [3H]NE release was also observed when synaptosomes were isolated from the cerebral cortex or when hippocampal [3H]NE release was triggered by 4-AP, a nonselective blocker of various K+ channel subtypes that does not significantly affect IKM (Robbins et al., 1992). The concentrations of RT required to inhibit [3H]NE release (EC50 ≈ 1 μm) were comparable with those required to activate homomeric and heteromeric channels formed by KCNQ2, KCNQ3, KCNQ4, and KCNQ5 but not KCNQ1 subunits (Rundfeldt and Netzer, 2000a; Tatulian et al., 2001) and ∼30 times lower than those needed to exert other neurochemical actions such as an enhancement of GABAA receptor-mediated postsynaptic currents (Rundfeldt and Netzer, 2000b; Otto et al., 2002).
The correspondence between the concentrations of RT inhibiting [3H]NE release and those activating KCNQ-type channels strongly suggested that KCNQ K+ channels were involved in RT-induced inhibition of hippocampal neurotransmitter release. In agreement with this hypothesis, other IKM modulators profoundly influenced hippocampal [3H]NE release. In fact, the centrally acting nonopioid analgesic FP, a structural analog of RT, also inhibited 9 mm [K+]e-induced [3H]NE release from hippocampal synaptosomes, although its efficacy was lower than that of RT. Our electrophysiological analysis revealed that in KCNQ2-transfected cells, FP hyperpolarized the voltage dependence of activation of KCNQ2 K+ channels by -9 mV, half that produced by RT (-19 mV), thus providing a plausible explanation for the lower efficacy of FP in modulating [3H]NE release when compared with RT.
XE-991 is a novel cognition-enhancing compound with an anthracenone structural backbone (Zaczek et al., 1998) that selectively blocks KCNQ subunits (Wang et al., 1998). XE-991 counteracted both the RT-induced KCNQ2 channel activation and the inhibition of 9 mm [K+]e-enhanced hippocampal [3H]NE release prompted by the antiepileptic compound. The lack of effect of 10 μm XE-991 as well as of anti-KCNQ2 antibodies in enhancing [3H]NE release evoked by 9 mm [K+]e is in line with the results obtained by Vickroy (1993), who showed that the potentiation of [3H]ACh release from hippocampal synaptosomes by the cognition enhancer linopirdine required higher (15-30 mm) [K+]e. Thus, the small synaptosomal depolarization prompted by exposure to 9 mm [K+]e seems insufficient to activate presynaptic IKM. As a matter of fact, the inhibitory effect of RT on hippocampal [3H]NE release was inversely correlated with the [K+]e triggering the release of the catecholamine. The negative shift in KCNQ2 voltage dependence of activation produced by RT regardless of [K+]e (Tatulian et al., 2001) predicted that the influence of RT on hippocampal [3H]NE release would be maximal when relatively low (9 mm) [K+]e is used to trigger neurotransmitter release. In fact, at relatively negative values of EM (less than -60 mV), RT would dramatically increase the activity of KCNQ2 channels, enhancing their participation in synaptosomal membrane potential control. In contrast, the effects of RT would be negligible when [3H]NE release is elicited by higher [K+]e, because stronger depolarizations (an EM greater than -30 mV) would already cause a maximal degree of KCNQ2 K+ channel activation. Although other factors (swelling and Ca2+ release from internal sources) may also influence neurotransmitter release evoked by high (>9 mm) [K+]e (Raiteri et al., 2002), these results suggest that IKM controls depolarization-induced neurotransmitter release.
Synaptosomal entrapment experiments with KCNQ-specific antibodies revealed that KCNQ2 subunits play a major role in RT-induced modulation of depolarization-induced [3H]NE release. The negative results of the release experiments with anti-KCNQ3 antibodies cannot rule out the involvement of KCNQ3 subunits in neurotransmitter release. In fact, we cannot exclude the possibility that in our experiments, the intraterminal concentration of these antibodies might not have been sufficient for binding to KCNQ3, or that despite effectively binding to KCNQ3, they might not have blocked the activity of this K+ channel subunit. However, the fact that TEA, when used at a concentration (100 μm) that specifically inhibited M channels containing KCNQ2 subunits (Wang et al., 1998), completely counteracted RT-induced inhibition of [3H]NE release further underlies the contribution of KCNQ2 subunits in hippocampal neurotransmitter release. Interestingly, KCNQ2 and KCNQ3 subunit expression has been found postsynaptically on somata and dendrites of hippocampal and neocortical pyramidal and polymorphic neurons; in contrast, in the hippocampus, KCNQ2 but not KCNQ3 immunoreactivity is located presynaptically on axons and nerve terminals of the mossy fiber pathway in the dentate hilus and the stratum lucidum of the CA3 region in both mouse and human brain (Cooper et al., 2000, 2001). The expression of KCNQ2 subunits in axon terminals within CA1 and CA3 hippocampal subfields has been confirmed by immunoelectron microscopy in the macaque monkey brain (Fieles et al., 2002).
In conclusion, the present findings reveal a novel role for KCNQ2 subunit-containing M-type channels in the modulation of hippocampal [3H]NE, [3H]d-ASP, and [3H]GABA release. The release of all three transmitters evoked by 9 mm [K+]e is primarily calcium dependent (Raiteri et al., 2002); therefore, activation of presynaptic IKM may hyperpolarize hippocampal nerve endings, thus reducing Ca2+ influx through multiple voltage-gated Ca2+ channels (Alvarez Maubecin et al., 1995). Because of its slow activation kinetics, IKM would participate in presynaptic membrane potential control mainly during repetitive activity or prolonged depolarization such as that induced by increased [K+]e. Given the multiplicity of receptors regulating IKM, the modulation of presynaptic M channels containing KCNQ2 subunits may be a powerful means by which multiple neurotransmitters affect synaptic function in the hippocampus. Finally, additional work is needed to clarify whether the activation of presynaptic M channels controlling neurotransmitter release contributes to the antiepileptic action of RT.
Footnotes
The present study was supported by grants from the Italian Ministry of the University and Research (Cofinanziamento 2001 and 2003, and Fondo per gli Investimenti della Ricerca di Base RBNE01XMP4 and RBNE01E7YX), the Italian Ministry of Health (Special Project “Alzheimer 2001/2004” and “Epilepsy 2003/2006”), Telethon (GP030209), European Commission (STREP n. 503038), and Regione Campania (Legge Regionale n. 5/2002 and Programma Operativo Regionale 2000/2006). We are deeply indebted to Prof. Thomas J. Jentsch (Zentrum fur Molekulare Neurobiologie, Hamburg, Germany) for KCNQ2 and KCNQ3 cDNAs.
Correspondence should be addressed to Dr. Maurizio Taglialatela, Division of Pharmacology, Department of Neuroscience, School of Medicine, University of Naples Federico II, Building 19, Via Pansini 5, 80131 Naples, Italy. E-mail mtaglial@unina.it.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240592-06$15.00/0
References
- Alvarez Maubecin V, Sanchez VN, Rosato Siri MD, Cherksey BD, Sugimori M, Llinas R, Uchitel OD (1995) Pharmacological characterization of the voltage-dependent Ca2+ channels present in synaptosomes from rat and chicken central nervous system. J Neurochem 64: 2544-2551. [DOI] [PubMed] [Google Scholar]
- Brown BS, Yu SP (2000) Modulation and genetic identification of the M channel. Prog Biophys Mol Biol 73: 135-166. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR (1980) Muscarinic suppression of a novel voltage sensitive K+ current in a vertebrate neurone. Nature 283: 673-676. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY (2000) Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA 97: 4914-4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY (2001) M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci 21: 9529-9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LP, Johnston GAR (1976) Uptake and release of d- and l-aspartate by rat brain slices. J Neurochem 26: 1007-1014. [DOI] [PubMed] [Google Scholar]
- Fieles WE, Smith J, Christian EP, Cross A, Mrzljak L (2002) Subcellular localization of KCNQ2 potassium channel subunit in the macaque monkey cerebral cortex and hippocampus. Soc Neurosci Abstr 28: 438.8. [Google Scholar]
- Friedel HA, Fitton A (1993) Flupirtine. A review of its pharmacological properties, and therapeutic efficacy in pain states. Drugs 45: 548-569. [DOI] [PubMed] [Google Scholar]
- Gundersen V, Shupliakov O, Brodin L, Ottersen OP, Storm-Mathisen J (1995) Quantification of excitatory amino acid uptake at intact glutamatergic synapses by immunocytochemistry of exogenous d-aspartate. J Neurosci 15: 4417-4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetka R, Rundfeldt C, Heinemann U, Schmitz D (1999) Retigabine strongly reduces repetitive firing in rat entorhinal cortex. Eur J Pharmacol 386: 165-171. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ (2000) Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci 1: 21-30. [DOI] [PubMed] [Google Scholar]
- Main MJ, Cryan JE, Dupere JR, Cox B, Clare JJ, Burbidge SA (2000) Modulation of KCNQ2/3 potassium channels by the novel anticonvulsant retigabine. Mol Pharmacol 58: 253-262. [DOI] [PubMed] [Google Scholar]
- Marrion NV (1997) Control of M-current. Annu Rev Physiol 59: 483-504. [DOI] [PubMed] [Google Scholar]
- Martire M, Altobelli D, Maurizi S, Preziosi P, Fuxe K (2000) K+-evoked [3H]d-aspartate release in rat spinal cord synaptosomes: modulation by neuropeptide Y and calcium channel antagonists. J Neurosci Res 62: 722-729. [DOI] [PubMed] [Google Scholar]
- O'Shea RD (2002) Roles and regulation of glutamate transporters in the central nervous system. Clin Exp Pharmacol Physiol 29: 1018-1029. [DOI] [PubMed] [Google Scholar]
- Otto JF, Kimball MM, Wilcox KS (2002) Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol 61: 921-927. [DOI] [PubMed] [Google Scholar]
- Raiteri L, Stigliani S, Zedda L, Raiteri M, Bonanno G (2002) Multiple mechanisms of transmitter release evoked by “pathologically” elevated extracellular [K+]: involvement of transporter reversal and mitochondrial calcium. J Neurochem 80: 706-714. [DOI] [PubMed] [Google Scholar]
- Raiteri M, Sala R, Fassio A, Rossetto O, Bonanno G (2000) Entrapping of impermeant probes of different size into nonpermeabilized synaptosomes as a method to study presynaptic mechanisms. J Neurochem 74: 423-431. [DOI] [PubMed] [Google Scholar]
- Robbins J, Trouslard J, Marsh SJ, Brown DA (1992) Kinetic and pharmacological properties of the M-current in rodent neuroblastoma × glioma hybrid cells. J Physiol (Lond) 451: 159-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA (2000) KCNQ2/KCNQ3 K+ channels and the molecular pathogenesis of epilepsy: implications for therapy. Trends Neurosci 23: 393-398. [DOI] [PubMed] [Google Scholar]
- Rundfeldt C, Netzer R (2000a) The novel anticonvulsant retigabine activates M-currents in Chinese hamster ovary-cells transfected with human KCNQ2/3 subunits. Neurosci Lett 282: 73-76. [DOI] [PubMed] [Google Scholar]
- Rundfeldt C, Netzer R (2000b) Investigations into the mechanism of action of the new anticonvulsant retigabine. Interaction with GABAergic and glutamatergic neurotransmission and with voltage gated ion channels. Arzneimittelforschung 50: 1063-1070. [DOI] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P (2002) Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol (Lond) 544: 29-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Delmas P, Abogadie FC, Brown DA (2001) Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci 21: 5535-5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vickroy TW (1993) Presynaptic cholinergic actions by the putative cognitive enhancing agent DuP 996. J Pharmacol Exp Ther 264: 910-917. [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D (1998) KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science 282: 1890-1893. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK (2000) Retigabine, a novel anticonvulsant, enhances activation of KCNQ2/3 potassium channels. Mol Pharmacol 58: 591-600. [DOI] [PubMed] [Google Scholar]
- Zaczek R, Chorvat RJ, Saye JA, Pierdomenico ME, Maciag CM, Logue AR, Fisher BN, Rominger DH, Earl RA (1998) Two new potent neurotransmitter release enhancers, 10,10-bis(4-pyridinylmethyl)-9(10H)-anthracenone and 10,10-bis(2-fluoro-4-pyridinylmethyl)-9(10H)-anthracenone: comparison to linopirdine. J Pharmacol Exp Ther 285: 724-730. [PubMed] [Google Scholar]