

Abstract
The accumulation and activation of microglial cells at sites of amyloid prion deposits or plaques have been documented extensively. Here, we investigate the in vivo recruitment of microglial cells soon after intraocular injection of scrapie-infected cell homogenate (hgtsc+) using immunohistochemistry on retinal sections. A population of CD11b/CD45-positive microglia was specifically detected within the ganglion and internal plexiform retinal cell layers by 2 d after intravitreal injection of hgtsc+. Whereas no chemotactism properties were ascribed to hgtsc+ alone, a massive migration of microglial cells was observed by incubating primary cultured neurons and astrocytes with hgtsc+ in a time- and concentration-dependent manner. hgtsc+ triggered the recruitment of microglial cells by interacting with both neurons and astrocytes by upregulation of the expression levels of a broad spectrum of neuronal and glial chemokines. We show that, in vitro and in vivo, the microglia migration is at least partly under the control of chemokine receptor-5 (CCR-5) activation, because highly specific CCR-5 antagonist TAK-779 significantly reduced the migration rate of microglia. Activated microglia recruited in the vicinity of prion may, in turn, cause neuronal cell damage by inducing apoptosis. These findings provide insight into the understanding of the cell-cell communication that takes place during the development of prion diseases.
Keywords: prion, microglial cells, recruitment, chemokines, retina, neurotoxicity
Introduction
Transmissible spongiform encephalopathies (TSEs) are fatal neurodegenerative and infectious disorders affecting humans [e.g., Creutzfeldt-Jakob disease (CJD)] and animals (e.g., sheep scrapie, bovine spongiform encephalopathy). The main hallmarks of TSEs are neuronal cell death, vacuolation of neuropil, and astrogliosis. The accumulation under amyloid aggregates of an abnormally folded isoform (PrPsc or PrPres) of the cellular prion protein (PrPc) is detected within affected brains. Both isoforms, PrPsc and PrPc, are required for infection and pathogenesis (Sailer et al., 1994). Neurons and astrocytes have been emphasized as major sites of infectious agent replication (Race et al., 1995; Raeber et al., 1997), whereas the role of other cerebral cell types in TSE pathogenesis remained unclear. Although TSEs are no inflammatory disorders, growing evidence incriminates microglial cells in both brain damage and spread of the disease. The presence of activated microglial cells adjacent to PrPsc deposits is a common feature in affected brains of humans and animals (Guiroy et al., 1994; Williams et al., 1997). Moreover, the in vivo accumulation of activated microglial cells is concomitant with the PrPsc deposition and precedes neuronal death (Giese et al., 1998) and clinical signs of disease (Betmouni et al., 1996). Using prion fragment P106-126 as an in vitro model, Brown et al. (1996) have shown that the presence of activated microglia is required for neurotoxicity. More recently, P106-126 has been shown to exhibit in vitro chemotactic properties for human monocytes through direct interactions with cell surface receptors (Le et al., 2001). However, little is known concerning the nature of molecular signals and cell types responsible for microglial cell recruitment in the vicinity of PrPsc aggregates.
Because chemokines and chemokine receptors are well known regulators of leukocyte trafficking, some of these molecules could be involved in the TSE pathogenesis. A likely candidate playing a key role in microglia migration in the context of TSEs is the chemokine receptor-5 (CCR-5). CCR-5, which belongs to the G-protein-coupled receptor family, binds multiple chemokines including macrophage inflammatory protein-1 (MIP-1α, MIP-1β) and RANTES (regulated on activation, normal T-cell expressed and secreted). CCR-5 is expressed by T-cells, macrophages, and microglial cells, and its expression has been shown to be upregulated in microglia from CJD-infected animals (Baker et al., 1999). Although microglia, neurons, and glia are well known to produce chemokines, the precise role of each cell type remains to be elucidated, particularly during the development of neurodegenerative disorders (for review, see Tran and Miller, 2003). In Alzheimer's disease, astrocytes and microglia activation might be triggered by β-amyloid protein, resulting in the initiation of an inflammatory cascade (Johnstone et al., 1999).
It was of interest to determine the temporal sequence of events and the cellular mechanisms that result in the recruitment of microglial cells during the development of TSEs. Thus, we investigate the in vivo directional migration of microglial cells soon after intraocular prion infection. In vitro, PrPsc induces the upregulation of expression of CCR-5-binding chemokines from both neurons and astrocytes. We propose that glial and neuronal chemokines could constitute the chemical signal triggering for the recruitment of microglial cells soon after infection. These findings underscore the importance of early inflammatory process long before the onset of the disease and could open up new therapeutic strategies against TSEs.
Materials and Methods
Cell cultures. Neuroblastoma cells chronically infected with the murine Chandler scrapie strain (N2asc+) were grown in Opti-MEM (Invitrogen, San Diego, CA) supplemented with 10% FBS (BioWest), 100 U/ml penicillin, 100 mg/ml streptomycin (BioWhittaker, Walkersville, MD), and 1 μg/ml G418 (Invitrogen), as described previously (Nishida et al., 2000). Because the treatment of N2asc+ cells with Congo red (1 μg/ml) totally cured the cells of PrPsc, Congo red-treated N2a cells (N2asc-) were used as a control. The mouse microglia cell line N11 was grown in RPMI 1640 (BioWhittaker) containing 10% FBS, penicillin, streptomycin, 0.5 mm β-mercaptoethanol, and 2 mm l-glutamine (BioWhittaker). As shown previously, this cloned cell line presents all the phenotype markers described for adult microglial cell populations in terms of cytokine release and nitric oxide (NO) production in response to inflammatory or immune response (Righi et al., 1989).
Cortical neurons from embryonic day 13-14 C57-black mice were prepared as described previously (Chabry et al., 1990), with minor modifications. Briefly, cells were dissociated mechanically with a Pasteur pipette in Neurobasal medium containing B27 supplement (Invitrogen) and penicillin-streptomycin. Dissociated cells were plated at a density of 3 × 106 cells in 35 mm tissue-plastic dishes or 5 × 105 cells/24-well dishes precoated with polylysine (10 μg/ml). Cultures were pure of neurons up to >98%. For the different experiments described below, neurons were used after 4-5 d of culture.
Primary microglial cells were obtained from newborn C57-black mice. Meninges-free cortices were dissociated in PBS containing 50 μg/ml trypsin (BioWhittaker) and 1 mg/ml DNase I (Promega, Madison, WI) for 10 min at 37°C, then diluted 1:1 in PBS supplemented with 10% FBS. Cells were centrifuged at 200 × g for 5 min, then resuspended in DMEM (BioWhittaker) containing N2 supplement (Invitrogen) and triturated with a Pasteur pipette. Dissociated cells were pelleted and resuspended in DMEM supplemented with N2, 10% FBS, penicillin, and streptomycin, then plated in 75 mm tissue-plastic flasks precoated with polylysine. Cells were grown for 7 d at 37°C under 5% CO2. To take off microglial cells, flasks were shaken for 30 min in an orbital shaker at room temperature. Resuspended cells were pelleted and plated in the culture medium. The proportion of microglia was estimated to 93% based on the detection with FITC-conjugated CD11b antibody and examination by fluorescence microscopy (data not shown). The adherent cell population (i.e., astrocytes) was grown in DMEM, 10% FBS, penicillin, and streptomycin, split every week, and used after three passages. Astrocyte cultures contained >99% glial fibrillary acidic protein-positive cells. All cell cultures were grown at 37°C in a humidified atmosphere of 5% CO2/95% air.
N2asc- and N2asc+ homogenates (hgtsc-, hgtsc+) preparation. To use a source of PrPsc as close as possible to the physiopathological conditions, we performed homogenates from N2asc- and N2asc+ cells using a detergent-free procedure. Confluent 100 mm culture dishes of N2asc+ and N2asc- were collected in PBS supplemented with a protease inhibitor mixture (PBSi; Boehringer, Bagnolet, France) and passed repeatedly through a 26 gauge needle. The lysate was centrifuged for 5 min at 4000 rpm; the supernatant (S1) was kept, and the pellet was resuspended in PBSi and underwent the same procedure once again. Supernatants S1 and S2 were pooled, sonicated for 10 min, and centrifuged for 5 min at 4000 rpm. The supernatant was centrifuged for 2 hr at 14,000 rpm at 4°C. Finally, the pellet was resuspended in PBSi to a final volume of 60 μl/100 mm tissue culture dish. Alternatively, lysates were treated with proteinase K (PK; 20 μg/ml) for 60 min at 37°C, followed by another centrifugation at 14,000 rpm for 2 hr. After this step, purity was >75% as estimated by SDS-PAGE, followed by silver staining (Merril et al., 1981). PK digestion results in an amino-terminally truncated version of PrPsc, lacking the first 90 residues, which has been shown to keep the same physical-chemical properties of PrPsc, including infectivity (Nishida et al., 2000). Contaminant proteins with molecular masses ∼60 kDa were detectable but nonimmunoreactive with anti-PrP monoclonal antibodies, as estimated by immunoblot analysis.
Intravitreal injections and immunohistochemistry. Adult male C57-black mice (10-15 weeks old) underwent intravitreal injections, as described previously (Chabry et al., 2003), with 1 μl of hgtsc+ or noninfected hgtsc-. At the end of the experiments, the immunohistology methods were performed as described previously (Ettaiche et al., 2000). Briefly, mice were killed with an overdose of sodium pentobarbital, the eyes were enucleated and fixed in ice-cold 4% paraformaldehyde (PAF) in PBS for 24 hr, then cryoprotected overnight in PBS containing 20% sucrose at 4°C. The eyes were then embedded in optimal cutting temperature compound (Tissue-Tek; SAKURA), and frozen sections (15 μm) cut on a cryostat (Leica, Nussloch, Germany) were transferred onto 1% gelatin-precoated slides. Endogenous peroxidase activities were blocked in methanol containing 1% H2O2 for 15 min. Slides were incubated in a blocking solution for 30 min [PBS, 5% BSA, and 5% inactivated horse serum (HSi)]. A 1:1 mixture of biotinylated antibodies anti-CD11b and anti-CD45 (PharMingen, San Diego, CA) in PBS, 2.5% BSA, and 2.5% HSi was incubated onto the slides to a final concentration of 1:100 for 3 hr at room temperature. After washes in PBS, slides were incubated with ABC reagent (Vector Laboratories, Burlingame, CA) and revealed with a DAB kit (Vector Laboratories). Retina sections were counterstained with 0.5% Cresyl violet, dehydrated with alcohol, and mounted with Entellan.
Reverse transcription-PCR. The RNA preparations were done using the protocol described by Chomczynski and Sacchi (1987). One microgram of RNA was reverse transcribed using the Moloney murine leukemia virus reverse transcriptase kit (Invitrogen) according to the manufacturer's instructions. PCR conditions were optimized for linear amplification, allowing direct comparison between samples. cDNAs (50 ng) were amplified using 1× buffer containing 2 mm MgCl2 (Appligene, Heidelberg, Germany), 2.5 mm of each dNTPs, 1 U of Taq polymerase (Appligene), and 25 pmol of each primer (Eurogentec). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers were added in each sample. The PCR cycles included general denaturation at 94°C (30 sec), annealing (30 sec) at 54°C, and elongation at 72°C (90 sec), except for the first cycle with a 3 min denaturation and last cycle with a 7 min elongation. Primer sequences, PCR conditions, and expected molecular masses of PCR products are summarized in Table 1. The PCR products were separated on 2% agarose gels using the SYBR Green I (Molecular Probes, Eugene, OR) staining. DNA bands were visualized by a UV light trans-illuminator and quantified using the NIH Image software. The level of expression of GAPDH did not change for any treatment.
Table 1.
Primer sequences and PCR conditions
|
Chemokines |
Oligonucleotides |
Number of cycle |
PCR product (bp) |
|---|---|---|---|
| I CTCACCATCATCCTCACT | |||
| RANTES (CCL5) | II ATGCTGATTTCTTGGGTT | 24 | 431 |
| I GACTCCAGCCACACTTCA | |||
| MIP-2 (CXCL1) | II TTGCCTTTGTTCAGTATC | 26 | 320 |
| I AACTCTCACTGAAGCCAG | |||
| MCP-1 (CCL2) | II TTCACTGTCACACTGGTC | 23 | 480 |
| I TTGCTGTTCTTCTCTGTA | |||
| MIP-1α (CCL3) | II TCAGTGATGTATTCTTGG | 25 | 237 |
| I ATGAAGCCTTTTCATACT | |||
| MIP-1β (CCL4) | II TATTGTTTGTAGGTCCGT | 25 | 367 |
| I AGCCTCGTCCCGTAGACA | |||
| GAPDH |
II TTCAAGAGAGTAGGGAGG |
26 |
1174 |
I, Forward primer; II, reverse primer.
Cell migration assays. Neurons or astrocytes were added to the bottom wells of the Boyden chambers in the appropriate medium. N11 cells or primary cultured microglia (5 × 104 in 200 μl of DMEM plus penicillin-streptomycin) were added to the top chamber and allowed to migrate through polyester filters (pore size, 8 μm; Falcon) for 6 hr at 37°C in humidified atmosphere of 5% CO2. Cells that did not migrate and stayed on the top surface of the filter were wiped off, whereas cells that had migrated to the bottom surface were fixed in 3% PAF for 10 min, stained with 0.5% crystal violet, and counted in random fields at 40× magnification.
NO production. Confluent N11 cells were incubated in the presence of 10 μl of hgtsc+ or hgtsc- for 14 hr at 37°C. Incubations with interferon-γ (IFNγ; 10 U/ml) and a submaximal concentration of lipopolysaccharide (LPS) (100 ng/ml) were performed in parallel to estimate the nitrite production capacity of N11 cells (Hemmer et al., 2001). Sample aliquots of culture supernatants were mixed with an equal volume of Griess reagent (Sigma, St. Louis, MO) according to the manufacturer's recommendations. Measurements were performed at 550 nm, and the nitrite concentration was determined using sodium nitrite solution as standard range.
Preconditioning microglia medium and cytotoxicity assays. Microglia cells (∼5 × 106 cells/ml in 6-well tissue plates) were incubated in the presence of 10 μl of hgtsc+ or hgtsc- for 18 hr at 37°C. The culture medium was centrifuged at 14,000 rpm for 2 hr, then the supernatant was added to 4-d-old neurons (5 × 104 cells/100 μl in 96-well tissue plates) for 18 hr at 37°C under 5% CO2. Fragmentation analysis of the neuronal DNA was performed as described previously (Chabry et al., 2003).
Results
In vivo recruitment of microglial cells induced by PrPsc
We addressed the question of the in vivo recruitment of microglial cells using direct injection of homogenates from scrapie-infected neuroblastoma cells (hgtsc+) into mouse eyes, followed by immunohistology studies of retinal sections (Fig. 1). In the control mice, CD11b/CD45-immunopositive microglial cells were rare (Fig. 1a). In noninfected neuroblastoma cell homogenate (hgtsc-)-treated animals (Fig. 1b), microglia were occasional and located exclusively in the ganglion cell layer of the retina. Two days after the intraocular injection of hgtsc+, microglia were more abundant (Fig. 1c) and mainly located in the internal plexiform and ganglion cell layers of the retina. Their shapes were typically ramified and arborized (Fig. 1d). A high density of microglial cells was observed in the vicinity of the optic nerve (Fig. 1e).
Figure 1.
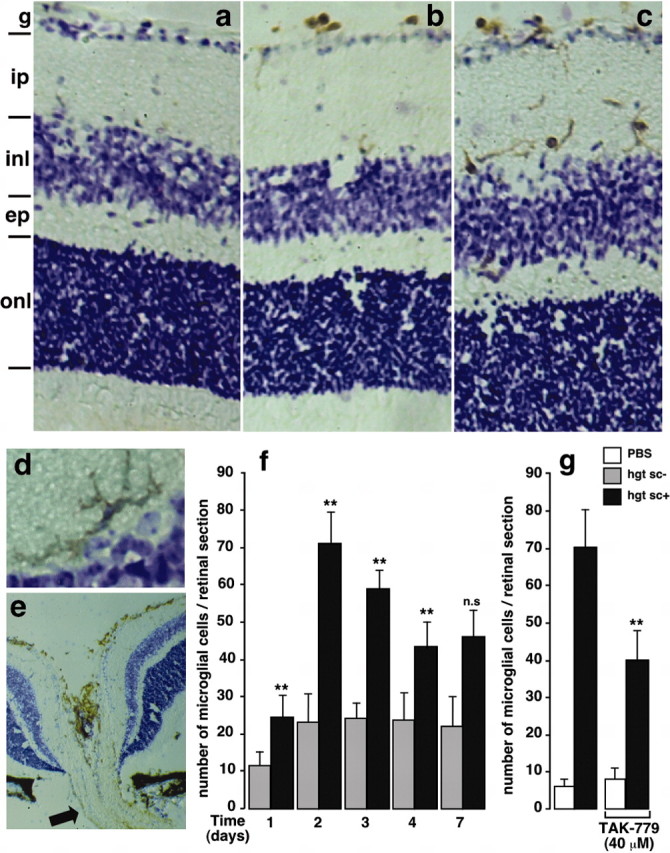
In vivo microglial cell recruitment after intraocular injection of hgtsc+. Immunohistochemical staining of retinal microglia (brown) expressing CD11b/CD45 epitopes 2 d after injection of PBS (a), hgtsc- (b), and hgtsc+ (c-e) into mouse eyes. A typically ramified microglia cell 2 d after hgtsc+ injection (d) and a representative retinal section through the optic nerve (e, arrow) are shown. Cell layers: g, ganglion cell; ip, internal plexiform; inl, inner nuclear; ep, external plexiform; onl, outer nuclear. Immunopositive microglial cells are brown; the counterstain is cresyl violet. Magnification: a-c, e, 10×; d, f, 40×. Quantification of the number of microglial cells per retinal section as a function of time after intravitreal inoculation of hgtsc- (gray bars) or hgtsc+ (black bars). The number of immunopositive cells was obtained by counting 90 independent retina sections corresponding to the whole eye from three treated mice in two separate experiments. Statistical significance as calculated by Student's t test: **p < 0.01 and not significant (n.s.) between day and d 1 in hgtsc+- treated animals, except for d 1, for which calculations were performed between hgtsc+- and hgtsc-- treated mice. g, Inhibitory effect of TAK-779 on the in vivo microglia migration 2 d after hgtsc+ injection. One microliter of hgtsc+ (black bars) or vehicle (white bars) was injected intravitreally in the presence or absence of TAK-779 (40 μm). Retinal sections were processed through the immunohistology methods described in Materials and Methods. Statistical analysis (**p < 0.01) between hgtsc+ treatment conditions with and without TAK-779 is shown. Histograms show the mean ± SD of two independent experiments (n = 2) in which each condition represents three mice injected unilaterally.
Interestingly, 7 d after hgtsc+ inoculation, microglia became more rounded and presented an amoeboid shape (data not shown). This cell morphology is thought to be associated with an activated and phagocytic stage.
We followed the time course and extent of microglia in both hgtsc- and hgtsc+ inoculated eyes by immunohistochemistry of retinal sections. During the time course of these experiments, injections with hgtsc+ always resulted in a higher number of microglial cells than injections with hgtsc- (Fig. 1f). In the hgtsc+-injected animals, the number of retinal microglia maximally increased at day 2, then declined slowly, but significantly, from day 2 to day 4 and reached a stable plateau value until day 7. Our data clearly showed that prion infection induced microglial recruitment in vivo within a few days.
TAK-779 is a highly potent and selective nonpeptide antagonist of the chemokine receptor CCR-5 (Baba et al., 1999). As shown in Figure 1g, 2 d after injection, TAK-779 provoked a ≈40% decrease in the number of retinal microglia when coinjected with the hgtsc+ inoculum. This result suggests that the activation of CCR-5 could account, at least in part, for the in vivo microglial cell migration.
Neurons and astrocytes trigger microglia migration in response to PrPsc
To investigate the precise role of both neurons and astrocytes in microglia recruitment in the context of TSEs, we used the Boyden chamber assay (Wilkinson, 1998). The N11 microglial cell line was added to the top chamber, and their migration through a filter toward the bottom chamber containing primary cultures of mouse neurons or astrocytes was monitored (Fig. 2). Cell-cell contacts being prevented, this paradigm allows cell contact-independent communication only via diffusible soluble factors. We found that the incubation of neurons or astrocytes with hgtsc+ induced the migration of N11 microglial cells in a time-dependent (Fig. 2a,c) and concentration-dependent manner (Fig. 2b,d). No significant effect on basal N11 cell migration was observed in the presence of increasing concentrations of noninfected hgtsc-. Interestingly, an additive effect of the migration was observed when neurons and astrocytes were cocultured in the bottom chamber (data not shown). To avoid possible artifact attributable to the use of the immortalized microglial cell line, primary cultures of mouse microglia cells were assessed in the Boyden chamber assay. The addition of 5 μl of hgtsc+ induced a 2.5- or 3.5-fold increase of the rate of mouse microglial cell migration from neurons or astrocytes, respectively, compared with the hgtsc- incubation (Fig. 2e,f). Because microglia from both primary cultures and the immortalized cell line exhibited similar migration behavior, the N11 microglial cell line was used in the following experiments as an accurate cell model. In the absence of cells in the bottom chamber, hgtsc+ alone did not exhibit chemotactic properties (Fig. 2g). Interestingly, incubation with hgtsc+ alone led to a basal migration rate significantly weaker than incubations with hgtsc- or vehicle; this was probably caused by direct effects of hgtsc+ on the microglial cell activation stage.
Figure 2.

In vitro characterization of the directional microglial cell migration on hgtsc+ stimulation of primary cultures of neurons and astrocytes. Incubations of 5 μl of vehicle (white bar), hgtsc- (gray bars), and hgtsc+ (black bars) on neurons (a) and astrocytes (c) were performed at 37°C for 18, 24, or 48 hr in the presence of the N11 microglial cell line in the top chamber. Microglia were challenged for migration through an 8 μm pore size insert for 6 hr at 37°C, then migrated cells were fixed, stained, and counted on an inverted microscope (5 fields counted/filter). Dose-responses were performed in the presence of a 24 hr stimulation time with the indicated volume of hgtsc- (gray bars) or hgtsc+ (black bars) on neurons (b) and astrocytes (d). The microglial migration assay and cell counts were done as described above. Migration assays performed with mouse primary cultures of microglia in the presence of 4-d-old neurons (e) or astrocytes (f) in the bottom chamber were stimulated for 24 hr with 5 μl of vehicle (white bar), hgtsc- (gray bar), and hgtsc+ (black bar). g, Migration assays performed with 5 μl of vehicle (white bar), hgtsc- (gray bar), and hgtsc+ (black bar) in the absence of plated cells in the bottom chamber. Histograms represent the mean of five independent experiments ± SD with two determinations each. Statistical analyses calculated with Student's t test [**p < 0.01; * p < 0.05 and not significant (n.s.)] compared hgtsc+ with hgtsc- incubation. For kinetics and dose-response experiments, statistical calculations were performed compared with the preceding concentration or incubation time.
PK resistance is a common feature of scrapie-affected brain-derived PrPsc (Prusiner et al., 1984). As expected, the PK treatment of both hgtsc- and hgtsc+ cleared off a large number of proteins, except PrPsc, as shown by silver staining of electrophoresis gels (Fig. 3a). To test the effect of a semipurified PrPsc preparation, hgtsc+ was PK digested and further assayed in the Boyden chamber assay in the presence of neurons in the bottom chamber. PK-treated hgtsc+ showed no significant difference in the microglia migration rate compared with PK-untreated hgtsc+ (Fig. 3b).
Figure 3.

Effect of PK treatment of hgtsc- and hgtsc+ on microglial cell migration. a, Untreated or PK-digested hgtsc- and hgtsc+ (5 μl), as described in Materials and Methods, were analyzed by SDS-PAGE and silver stain. Molecular mass markers are indicated on the left (kDa). The bracket indicates PK-resistant PrP (PrPres). b, Untreated or PK-digested hgtsc- (gray bars) and hgtsc+ (black bars) (5 μl) were incubated for 24 hr at 37°C on mouse primary cultured neurons seeded in the bottom chamber. Then, N11 cells were plated on the top chamber and challenged for migration for 6 hr. At least five fields of migrated cells/condition were counted. Histograms are means of three independent experiments ± SD. Statistical analyses calculated with Student's t test (**p < 0.01) compared hgtsc+ with hgtsc- incubation with or without PK digestion.
In summary, our results demonstrate that PrPsc triggered microglial cell migration via interactions with neurons and astrocytes by releasing diffusible factors but exhibited no chemotactism properties by itself.
Chemokines involved in microglia recruitment in response to PrPsc
To identify neuronal and glial chemokines involved in microglia attraction, reverse transcription-PCR experiments were performed on cDNA prepared from hgtsc+- and hgtsc--stimulated neurons and astrocytes. Here, we focused on secreted chemokines with inducible expression related to inflammatory responses and CNS injuries (Zlotnik and Yoshie, 2000). In both primary cultures of neurons and astrocytes, mRNA expression levels of all tested chemokines were upregulated in hgtsc+ stimulation compared with hgtsc- stimulation conditions. However, the highest activation factor was found for RANTES (CCL5) and MIP-1β (CCL4) mRNAs in hgtsc+-stimulated neurons and astrocytes, respectively (6.62 ± 1.57 and 5.59 ± 2.21) (Fig. 4b). Thus, the interaction of PrPsc with neurons and astrocytes upregulated the expression of messengers of two different chemokines, RANTES and MIP-1β, respectively, that share a common receptor, CCR-5.
Figure 4.
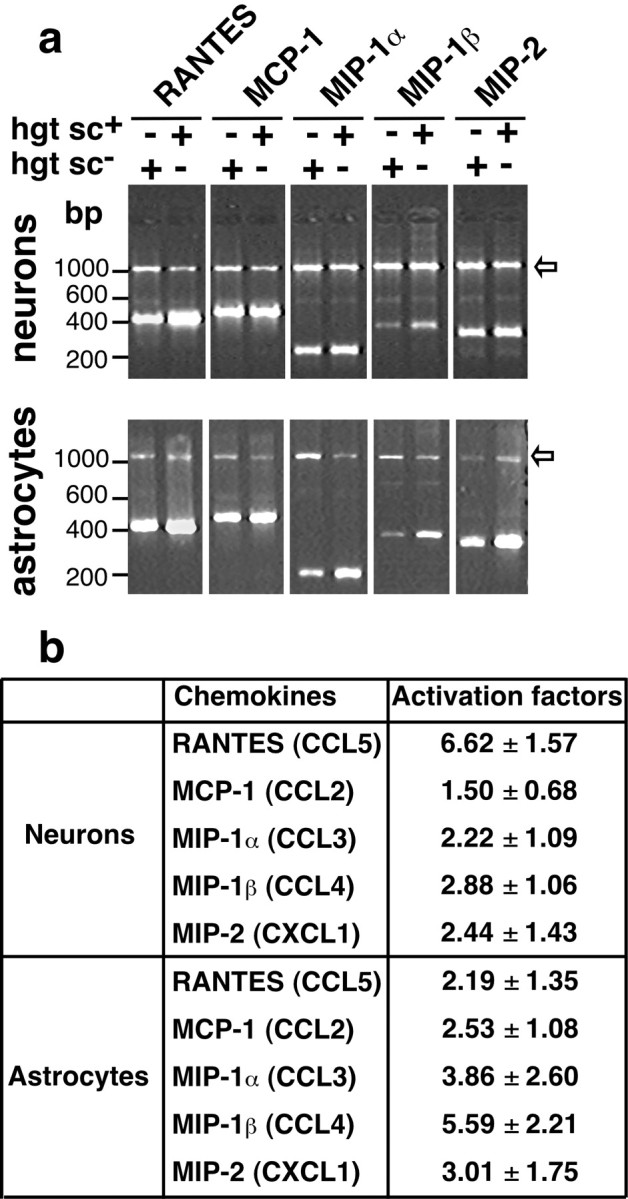
Chemokine expression profile in both hgtsc-- and hgtsc+-stimulated primary cultures of mouse neurons and astrocytes. a, Representative reverse transcription (RT)-PCR profile of chemokine activation in neurons or astrocytes. Mouse primary cultures of neurons or astrocytes were incubated in the presence of 5 μl of hgtsc- or hgtsc+. After 6 hr of incubation, mRNAs of the different conditions were purified and reverse transcribed. RT-PCR experiments were performed as described in Table 1 on 1 μg of total cDNA in the presence of appropriate primers for chemokines and GAPDH in the same assay. PCR products were analyzed on 2% agarose gels stained with SYBR Green I. Sizes of PCR products are, respectively, 431 bp for RANTES, 480 bp for MCP-1, 237 bp for MIP-1α, 367 bp for MIP-1β, 320 bp for MIP-2, and 1174 bp for GAPDH. Arrows indicate the PCR products of GAPDH mRNA. b, Semiquantitative profile of mRNA expression in neurons or astrocytes. RT-PCR gels for the genes indicated were quantified by densitometry using NIH Image software, with normalization to GAPDH mRNA levels. Results are expressed in terms of fold increase from hgtsc+-stimulated compared with hgtsc--stimulated cells and represent the means ± SD of data from four independent experiments. The systematic names of chemokines are in parentheses.
To confirm the importance of RANTES in neuron-induced microglia recruitment, in vitro cell migration assays were performed in the presence of increasing concentrations of an anti-RANTES monoclonal antibody. The addition of anti-RANTES antibody in the Boyden bottom chamber, in which mouse cortical neurons were seeded, resulted in a concentration-dependent decrease of the N11 microglial cell migration (Fig. 5a). An approximate 30% decrease in the rate of migration was obtained when the incubations were performed with 20 μg/ml anti-RANTES antibody. No significant cell migration change was measured when neurons were incubated with similar concentrations of an unrelated rat IgG. We further assayed the CCR-5 antagonist TAK-779 according to the same in vitro procedure. TAK-779 provoked a decrease of the microglia attraction rate in a dose-dependent manner. In the presence of saturated concentration of TAK-779 (40 μm), the migration rate was reduced by a twofold factor arguing for the involvement of CCR-5 (Fig. 5b). Table 2 summarizes the percentage of decrease of N11 and primary microglial cell migration induced by hgtsc+ in the presence of 40 μm TAK-779 in different coculture conditions. In every case, TAK-779 reduced significantly the rate of cell migration. Note that the decreased rates of migration induced by TAK-779 were lower when the experiments were performed with mouse microglia cells compared with the N11 cell line. This result could be explained by the heterogeneity of microglial cells in primary cultures. These experiments strongly suggest that CCR-5 plays an important role in the microglia directional migration induced by chemokines from neurons and astrocytes on PrPsc stimulation.
Figure 5.

Anti-RANTES antibody and CCR-5 antagonist TAK-779 inhibit microglia migration. a, Four-day-old cortical neurons were incubated with 5 μl of vehicle (white bar), hgtsc- (gray bar), or hgtsc+ (black bars) for 24 hr at 37°C. Migration experiments were initiated by plating N11 cells on a 8 μm pore size insert for 6 hr in the presence of the indicated concentrations of anti-RANTES monoclonal antibody or unrelated rat IgG. b, The experiment was performed as described above in the presence of the indicated concentrations of TAK-779 instead of antibodies. Results are expressed as the percentage of migration rate obtained with 5 μl of hgtsc+ alone (0). Histograms represent the means of data ± SD from three independent experiments. For each histogram, statistical significance was calculated compared with the preceding concentration of anti-RANTES antibody (a) or TAK-779 (b) (**p < 0.01; *p < 0.05; n.s., not significant).
Table 2.
Effect of TAK-779 on the microglial cell migration induced by hgtsc+
|
Bottom chamber |
Top chamber |
Decrease of microglial cell migration (%) |
|---|---|---|
| Neurons | N11 cells | 52.5 ± 8.3* |
| Astrocytes | N11 cells | 37.7 ± 1.2* |
| Neurons | Primary microglial cells | 28.1 ± 3.5** |
| Astrocytes |
Primary microglial cells |
33.6 ± 4.2**
|
Microglial cell migration induced by 5 μl of hgtsc+ decreased in the presence of 40 μm TAK-779. Results are expressed as the percentage of inhibition of the migration rate. Statistical significance: **p < 0.01; *p < 0.05.
In vitro neurotoxicity of microglia on primary cultured neurons
The chemokine-induced migration of microglia brings it to the contact of PrPsc. The following experiments were performed to understand how this contact could lead to neurotoxicity. Neuronal cell death is one of the main hallmarks of TSEs; however, precise molecular mechanisms of neurotoxicity remain to be elucidated. In vitro experiments indicate that microglia activation may play a pivotal role in neuronal damage (Brown et al., 1996). NO- and microglia-derived neurotoxins have been implicated in a range of neuropathological conditions such as Alzheimer's disease (Giulian et al., 1995). Thus, we investigated the effect of hgtsc+ on microglial cell activation by the measurement of nitrite concentration. When microglial cells were incubated with hgtsc+, the nitrite concentration in the culture medium increased with a threefold factor compared with hgtsc- incubation (Fig. 6a). The nitrite production capacity of N11 microglial cells was verified by incubating cells with a mixture of bacterial endotoxin LPS and IFNγ. The latter treatment induced an eightfold increase of nitrite concentration compared with the control condition. It was of interest to further characterize the neurotoxicity induced by PrPsc-activated microglia. Therefore, mouse cortical neurons were exposed to N11 microglial cell medium preconditioned with hgtsc- or hgtsc+. Neuronal DNA fragmentation analysis and 4′,6-diamidino-2-phenylindole (DAPI)-positive neuron examination were then performed. Mouse neurons treated with the hgtsc+ preconditioned microglia medium showed a ladder pattern of DNA degradation with bands corresponding to multiples of 180-200 bp (Fig. 6b). DNA laddering observed either in PBS-treated neurons or in neurons treated with the hgtsc- preconditioned microglia medium was barely noticeable. Moreover, when neurons were incubated with the hgtsc+ preconditioned microglia medium, the number of DAPI-positive neurons was significantly higher than during a hgtsc- preconditioned medium incubation (Fig. 6c). Together, these experiments clearly demonstrate that activated microglial cells exert their neurotoxic effects via an apoptotic cell death pathway in the absence of direct microglia-neurons contact. Note that we cannot totally rule out the possibility that neurotoxicity could be attributable, at least in part, to the presence of small, nonsedimentable oligomers of PrPsc in the microglia-conditioned medium. Interestingly, using the assays described above, no cytotoxity induced by hgtsc+ microglia-conditioned medium was observed on astrocyte cultures (data not shown).
Figure 6.

Microglia activation and neurotoxicity induced by PrPsc preconditioned microglia medium. a, Confluent N11 microglial cells were incubated with 5 μl of PBS (white bars), hgtsc- (gray bar), or hgtsc+ (black bar) for 12 hr. Then, nitrite concentration was measured using the Griess reagent and compared with the standard curve. Each histogram is the mean of triplicate determinations from six independent experiments ± SD. b, Representative gel analysis of 10 μg of neuronal DNA from untreated (C) or neurons treated with preconditioned microglia medium as described above. Sizes of the markers are expressed in base pairs (bp) on the left. C, DAPI staining of 4-d-old neurons treated with hgtsc- or hgtsc+ preconditioned microglia culture medium. Phase-contrast micrographs of representative microscopic fields are shown. Magnification, 40×. Arrows indicate apoptotic nuclei.
Discussion
To our knowledge, the present study is the first demonstration of the complementary response of neurons and astrocytes to prion infection leading to microglial cell recruitment within few days. The recruitment of microglia in the vicinity of PrPsc aggregates and their subsequent activation soon after the infection and long before the onset of the disease may contribute to both neuronal damage and pathogenesis. In our attempt to characterize the molecular and cellular events leading to microglial cell recruitment, we showed that PrPsc-stimulated neurons and astrocytes induced chemotactism by upregulation of chemokine expression. Additional investigations will be necessary to understand how PrPsc aggregates may influence the expression levels of glial and neuronal chemokines. However, we clearly demonstrate that PrPsc triggers the recruitment of microglial cells by interacting with both types of cells. In support of this point, Hetz et al. (2003) have recently demonstrated that purified PrPsc triggers apoptosis of neuroblastoma cells by inducing elevation of the intracellular calcium concentration and caspase-12 activation. Although we do not provide evidence for direct binding between PrPsc and cell surface, our experiments suggest the existence of functional interactions that might induce transduction signal pathways. Chemokines are widely expressed in the adult CNS, and their expression is regulated in response to stressful and pathological situations. They have been involved in the manifestation of various brain disorders such as Alzheimer's disease (Streit et al., 2001).
Because retina is an integral part of the CNS, formed by association of specialized neurons, astrocytes, and microglia, it is a well-suited model to study the cellular events occurring during TSEs. Retina is highly susceptible to infection and disease caused by prions (Fraser, 1982). Moreover, the eye is a natural route for scrapie infection, particularly in sheep, and cornea transplants could cause iatrogenic CJDs. The number of microglial cells may increase in response to pathological situations in the CNS through in situ proliferation (Sedgwick et al., 1998) or by recruitment of monocytes from the blood circulation (Giulian et al., 1989). Nevertheless, little is known concerning the origin of retinal microglia. Interestingly, during the time course of our experiments, we noted a large number of microglia located close to the optic nerve (Fig. 1e). This observation supports the hypothesis of a massive recruitment of microglia to retina trafficking via the optic nerve rather than an in situ cell proliferation. The spatiotemporal sequence of events such as microglia activation and recruitment remains to be put in regard to other pathological changes like PrPsc deposition. Our data support the idea that the recruitment of microglial cells may be one of the early steps of the pathogenesis and precedes neuronal loss. The involvement of microglia in neuronal cell death is well documented in a variety of CNS diseases, among them TSEs (for review, see Brown, 2001; Heppner et al., 2001). In the present study, the conditioned medium transfer paradigm was helpful to characterize the molecular mechanisms of neurotoxicity (Fig. 6). First, we showed that neurotoxicity could occur in the absence of direct contact between PrPsc and neurons. Second, PrPsc-stimulated microglia exerted its neurotoxic properties, at least in part, via soluble factors, in the absence of cell-cell contact. In agreement with our results, the expression of inducible NO synthase has been reported in scrapie-infected brains (Ju et al., 1998). Thus, our results suggest that mechanisms of neurotoxicity are set up immediately after the first contact between PrPsc aggregates and microglial cells. Retinal ganglion cells have been shown to degenerate after intraocular injection of a mouse scrapie strain, whereas neuronal cells present in the inner nuclear layer of the retina are more resistant (Russelakis-Carneiro et al., 1999). In agreement with this study, we show the presence of a number of microglial cells in the ganglion cell layer of the retina after hgtsc+ injection, suggesting that microglia cells are spatially coincident with neuronal loss. However, microglial cells may act as the primary cytotoxic cells and as a secondary activation response to the progressive neurodegeneration (Rezaie and Lantos, 2001). It is likely that the widespread neuronal degeneration also induce microglial activation, which in turn may further contribute to toxicity by releasing cytokines. The inflammatory expression profiles of microglia from CJD-affected brain contrast with that of healthy brain (Baker and Manuelidis, 2003). Alternatively, microglia might spread TSE disease and could be involved in neuroinvasion from retina to the CNS. Because microglia from infected brain display high levels of infectivity, it has been proposed that activated microglia may be a replication site for prions (Baker et al., 1999). Although the uptake of the P106-126 fragment has been shown in vitro (McHattie et al., 1999), the phagocytosis of PrPsc aggregates by microglia remains a poorly documented issue. The presence of amoeboid microglial cells in the phagocytic stage, observed in PrPsc-inoculated retina, might support this hypothesis. Moreover, gene expression studies have revealed the high level of cathepsin S expression in scrapie-affected brains. Cathepsin S, a member of the family of lysosomal proteases, is involved in the degradation of extracellular matrix and, thus, could play an important role in the clearance of proteinaceous debris and aggregates (Dandoy-Dron et al., 1998; Baker et al., 1999). Considering the possible phagocytosis properties of microglial cells, their recruitment around PrPsc aggregates could also constitute a survival strategy for neurons and astrocytes.
Interestingly, on PrPsc stimulation, both neurons and astrocytes preferentially synthesize chemokines acting through the same receptor, CCR-5. Supported by our in vivo and in vitro investigations, CCR-5 might constitute a novel therapeutic target for TSEs. It could be of interest to limit inflammatory components characterized by microglial activation and recruitment that appear long before the onset of TSEs. In conclusion, this study highlights the importance of the cross-talk of microglial cells with other brain cells and contributes to a better understanding of cellular interactions that take place during the development of TSEs.
Footnotes
This work was supported in part by Action Concertée Incitative-Jeunes Chercheurs. We thank Dr. Sylvain Lehmann for supplying the scrapie-infected neuroblastoma cell line. We also thank Dr. E. H. Hughes (University of Bristol, Bristol, UK) for advice on immunohistology procedures and Jean-Daniel Barde for animal care. We are grateful to Drs. N. Daude, F. Checler, and J. P. Vincent for advice and helpful comments on this manuscript. TAK-779 was obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Correspondence should be addressed to Dr. Joëlle Chabry, Institut de Pharmacologie Moléculaire et Cellulaire, Unité Mixte de Recherche 6097, Centre National de la Recherche Scientifique 660, route des lucioles, 06560 Valbonne, France. E-mail: chabry@ipmc.cnrs.fr.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240620-08$15.00/0
References
- Baba M, Nishimura O, Kanzaki N, Okamoto M, Sawada H, Iizawa Y, Shiraishi M, Aramaki Y, Okonogi K, Ogawa Y, Meguro K, Fujino M (1999) A small-molecule, nonpeptide CCR5 antagonist with highly potent and selective anti-HIV-1 activity. Proc Natl Acad Sci USA 96: 5698-5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CA, Manuelidis L (2003) Unique inflammatory RNA profiles of microglia in Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA 100: 675-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CA, Lu ZY, Zaitsev I, Manuelidis L (1999) Microglial activation varies in different models of Creutzfeldt-Jakob disease. J Virol 73: 5089-5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betmouni S, Perry VH, Gordon JL (1996) Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience 74: 1-5. [DOI] [PubMed] [Google Scholar]
- Brown DR (2001) Microglia and prion disease. Microsc Res Tech 54: 71-80. [DOI] [PubMed] [Google Scholar]
- Brown DR, Schmidt B, Kretzschmar HA (1996) Role of microglia and host prion protein in neurotoxicity of a prion protein fragment. Nature 380: 345-347. [DOI] [PubMed] [Google Scholar]
- Chabry J, Checler F, Vincent JP, Mazella J (1990) Colocalization of neurotensin receptors and of the neurotensin-degrading enzyme endopeptidase 24-16 in primary cultures of neurons. J Neurosci 10: 3916-3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabry J, Ratsimanohatra C, Sponne I, Elena PP, Vincent JP, Pillot T (2003) In vivo and in vitro neurotoxicity of the human prion protein (PrP) fragment P118-135 independently of PrP expression. J Neurosci 23: 462-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi W (1987) Single step method of RNA isolation by acid guanidium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156-159. [DOI] [PubMed] [Google Scholar]
- Dandoy-Dron F, Guillo F, Benboudjema L, Deslys JP, Lasmezas C, Dormont D, Tovey MG, Dron M (1998) Gene expression in scrapie. Cloning of a new scrapie-responsive gene and the identification of increased levels of seven other mRNA transcripts. J Biol Chem 273: 7691-7697. [DOI] [PubMed] [Google Scholar]
- Ettaiche M, Pichot R, Vincent JP, Chabry J (2000) In vivo cytotoxicity of the prion protein fragment 106-126. J Biol Chem 275: 36487-36490. [DOI] [PubMed] [Google Scholar]
- Fraser H (1982) Neuronal spread of scrapie agent and targeting of lesions within the retino-tectal pathway. Nature 295: 149-150. [DOI] [PubMed] [Google Scholar]
- Giese A, Brown DR, Groschup MH, Feldmann C, Haist I, Kretzschmar HA (1998) Role of microglia in neuronal cell death in prion disease. Brain Pathol 8: 449-457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Chen J, Ngeman J, George J, Noponen M (1989) The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J Neurosci 9: 4416-4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giulian D, Haverkamp LJ, Li J, Karshin WL, Yu J, Tom D, Li X, Kirkpatrick JB (1995) Senile plaques stimulate microglia to release a neurotoxin found in Alzheimer brain. Neurochem Int 27: 119-137. [DOI] [PubMed] [Google Scholar]
- Guiroy DC, Wakayama I, Liberski PP, Gajdusek DC (1994) Relationship of microglia and scrapie amyloid-immunoreactive plaques in kuru, Creutzfeldt-Jakob disease and Gerstmann-Straussler syndrome. Acta Neuropathol (Berl) 87: 526-530. [DOI] [PubMed] [Google Scholar]
- Hemmer K, Fransen L, Vanderstichele H, Vanmechelen E, Heuschling P (2001) An in vitro model for the study of microglia-induced neurodegeneration: involvement of nitric oxide and tumor necrosis factor-α. Neurochem Int 38: 557-565. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Prinz M, Aguzzi A (2001) Pathogenesis of prion diseases: possible implications of microglial cells. Prog Brain Res 132: 737-750. [DOI] [PubMed] [Google Scholar]
- Hetz C, Russelakis-Carniero M, Maundrell K, Castilla J, Soto C (2003) Caspase-12 and endoplasmic reticulum stress mediate neurotoxicity of pathological prion protein. EMBO J 22: 5435-5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone M, Gearing AJ, Miller KM (1999) A central role for astrocytes in the inflammatory response to beta-amyloid; chemokines, cytokines and reactive oxygen species are produced. J Neuroimmunol 93: 182-193. [DOI] [PubMed] [Google Scholar]
- Ju WK, Park KJ, Choi EK, Kim J, Carp RI, Wisniewski HM, Kim YS (1998) Expression of inducible nitric oxide synthase in the brains of scrapie-infected mice. J Neurovirol 4: 445-450. [DOI] [PubMed] [Google Scholar]
- Le Y, Yazawa H, Gong W, Yu Z, Ferrans VJ, Murphy PM, Wang JM (2001) The neurotoxic prion peptide fragment PrP(106-126) is a chemotactic agonist for the G protein-coupled receptor formyl peptide receptor-like 1. J Immunol 166: 1448-1451. [DOI] [PubMed] [Google Scholar]
- McHattie SJ, Brown DR, Bird MM (1999) Cellular uptake of the prion protein fragment PrP106-126 in vitro. J Neurocytol 28: 149-159. [DOI] [PubMed] [Google Scholar]
- Merril CR, Goldman D, Sedman SA, Ebert MH (1981) Ultrasensitive stain for proteins in polyacrylamide gels shows regional variation in cerebrospinal fluid proteins. Science 211: 1437-1438. [DOI] [PubMed] [Google Scholar]
- Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J, Casanova D, Milhavet O, Lehmann S (2000) Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J Virol 74: 320-325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE (1984) Purification and structural studies of a major scrapie prion protein. Cell 38: 127-134. [DOI] [PubMed] [Google Scholar]
- Race RE, Priola SA, Bessen RA, Ernst D, Dockter J, Rall GF, Mucke L, Chesebro B, Oldstone MB (1995) Neuron-specific expression of a hamster prion protein minigene in transgenic mice induces susceptibility to hamster scrapie agent. Neuron 15: 1183-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raeber AJ, Race RE, Brandner S, Priola SA, Sailer A, Bessen RA, Mucke L, Manson J, Aguzzi A, Oldstone MB, Weissmann C, Chesebro B (1997) Astrocyte-specific expression of hamster prion protein (PrP) renders PrP knockout mice susceptible to hamster scrapie. EMBO J 16: 6057-6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaie P, Lantos PL (2001) Microglia and the pathogenesis of spongiform encephalopathies. Brain Res Brain Res Rev 35: 55-72. [DOI] [PubMed] [Google Scholar]
- Righi M, Mori L, De Libero G, Sironi M, Biondi A, Mantovani A, Donini SD, Ricciardi-Castagnoli P (1989) Monokine production by microglial cell clones. Eur J Immunol 19: 1443-1448. [DOI] [PubMed] [Google Scholar]
- Russelakis-Carneiro M, Betmouni S, Perry VH (1999) Inflammatory response and retinal ganglion cell degeneration following intraocular injection of ME7. Neuropathol Appl Neurobiol 25: 196-206. [DOI] [PubMed] [Google Scholar]
- Sailer A, Bueler H, Fischer M, Aguzzi A, Weissmann C (1994) No propagation of prions in mice devoid of PrP. Cell 77: 967-968. [DOI] [PubMed] [Google Scholar]
- Sedgwick J, Ford A, Foulcher E, Airriess R (1998) Central nervous system microglial cell activation and proliferation follows direct interaction with tissue-infiltrating T cell blasts. J Immunol 160: 5320-5330. [PubMed] [Google Scholar]
- Streit WJ, Conde JR, Harrison JK (2001) Chemokines and Alzheimer's disease. Neurobiol Aging 22: 909-913. [DOI] [PubMed] [Google Scholar]
- Tran PB, Miller RJ (2003) Chemokine receptors: signposts to brain development and disease. Nat Rev Neurosci 4: 444-455. [DOI] [PubMed] [Google Scholar]
- Wilkinson PC (1998) Assays of leukocyte locomotion and chemotaxis. J Immunol Methods 216: 139-153. [DOI] [PubMed] [Google Scholar]
- Williams A, Lucassen PJ, Ritchie D, Bruce M (1997) PrP deposition, microglial activation, and neuronal apoptosis in murine scrapie. Exp Neurol 144: 433-438. [DOI] [PubMed] [Google Scholar]
- Zlotnik A, Yoshie O (2000) Chemokines: a new classification system and their role in immunity. Immunity 12: 121-127. [DOI] [PubMed] [Google Scholar]