

Abstract
Choline acetyltransferase (ChAT), the enzyme that synthesizes the neurotransmitter acetylcholine (ACh), is thought to be present in kinetic excess in cholinergic neurons. The rate-limiting factor in ACh production is the provision of choline to ChAT. Cholinergic neurons are relatively unique in their expression of the choline transporter 1 (CHT1), which exhibits high-affinity for choline and catalyzes its uptake from the extracellular space to the neuron. Multiple lines of evidence indicate that the activity of CHT1 is a key determinant of choline supply for ACh synthesis. We examined the interaction of ChAT and ChT activity using mice heterozygous for a null mutation in the Chat gene (Chat+/-). In these mice, brain ChAT activity was reduced by 40-50% relative to the wild type, but brain ACh levels as well as ACh content and depolarization-evoked ACh release in hippocampal slices were normal. However, the amount of choline taken up by CHT1 and ACh synthesized de novo from choline transported by CHT1 in hippocampal slices, as well as levels of CHT1 mRNA in the septum and CHT1 protein in several regions of the CNS, were 50-100% higher in Chat+/- than in Chat+/+ mice. Thus, haploinsufficiency of ChAT leads to an increased expression of CHT1. Increased ChT activity may compensate for the reduced ChAT activity in Chat+/- mice, contributing to the maintenance of apparently normal cholinergic function as reflected by normal performance of these mice in several behavioral assays.
Keywords: acetylcholine, behavior, choline acetyltransferase, choline transporter, congenital myasthenic syndrome with episodic apnea, cholinergic
Introduction
The neurotransmitter acetylcholine (ACh) is required for the central and peripheral control of movement, autonomic nervous system function, regulation of sleep, and multiple cognitive processes including timing, attention, learning, and memory. Cholinergic neurons express three proteins with combined activities that ensure efficient synthesis, storage, and release of ACh: (1) the sodium-dependent high-affinity choline transporter (ChT) catalyzes the uptake of choline from the extracellular space to the neuronal cytoplasm; (2) choline acetyltransferase (ChAT) synthesizes ACh, using choline and acetyl-CoA as substrates; and (3) the vesicular acetylcholine transporter (VAChT) packages the cytoplasmically formed ACh into secretory synaptic vesicles. Studies using biochemical and pharmacological approaches indicate that cholinergic nerve terminals may contain ChAT activity in kinetic excess of what is needed to support normal synthesis of ACh (for review, see Blusztajn and Wurtman, 1983), and two recent observations in vivo are consistent with this notion. In humans, polymorphism of the CHAT gene underlies the etiology of one form of presynaptic congenital myasthenic syndrome (CMS) designated as CMS with episodic apnea (CMS-EA) (Ohno et al., 2001; Byring et al., 2002). In this disease, the affected individuals inherit two abnormal CHAT alleles. Most of the different mutant alleles encode proteins characterized by lower affinity for one or both of the two ChAT substrates. One variant is a frame-shift mutation generating a true null allele and thus an inactive enzyme. Heterozygote parents of the CMS-EA patients are asymptomatic (Ohno et al., 2001). Similarly, whereas Chat null mice are stillborn and display developmental abnormalities because of the total lack of ACh, Chat+/- mice appear to be grossly normal and are fertile (Misgeld et al., 2002; Brandon et al., 2003). Together, these data suggest that a loss of as much as one-half of normal ChAT activity is well tolerated. However, because the rate of ACh synthesis could be diminished when ChAT activity is reduced, it is possible that a lowered rate would compromise the normal functioning of cholinergic neurons and that a compensatory change might be required to offset the loss of ChAT activity and maintain normal levels of ACh. Because the rate-limiting factor in the synthesis of ACh is thought to be the supply of its precursor, choline (for review, see Blusztajn and Wurtman, 1983), such a compensatory mechanism might entail an increased delivery of choline to ChAT. Choline is an essential nutrient for mammals, including humans (Food and Nutrition Board, 1998), and consequently most of the choline used for ACh synthesis derives from the diet, whereas a smaller fraction is synthesized de novo (Blusztajn and Wurtman, 1983) predominantly by the liver (Walkey et al., 1997; Watkins et al., 2003) but additionally in situ by brain cells (Blusztajn and Wurtman, 1981; Lakher and Wurtman, 1987), including cholinergic neurons (Blusztajn et al., 1987). The diet-derived and endogenously synthesized circulating choline enters the brain via a blood-brain barrier transporter (Pardridge and Oldendorf, 1977; Klein et al., 1990). Once in the extracellular space of the brain, choline can be taken up by all cells and used as a precursor of structural membrane phospholipids such as phosphatidylcholine and sphingomyelin and uniquely by cholinergic neurons for the conversion to ACh (for review, see Blusztajn and Wurtman, 1983). Phosphatidylcholine in these neurons also comprises a storage pool of choline, and the hydrolysis of phosphatidylcholine catalyzed by phospholipase D liberates choline from this reservoir for the conversion to ACh by ChAT (Lee et al., 1993; Zhao et al., 2001). In cholinergic nerve terminals, however, most of the choline for ACh is taken up from the extracellular space by ChT. In a variety of experimental models, the rates of ACh synthesis correlate well with the activity of ChT, and choline transport catalyzed by ChT can be rapidly stimulated by increased neuronal activity (Murrin and Kuhar, 1976; Collier and Ilson, 1977; Polak et al., 1977; Sherman et al., 1978), indicating that this process responds to an increased demand for choline when ACh synthesis and release are high. The latter appears to be mediated by a recruitment of ChT molecules from a synaptic vesicular pool to the plasma membrane resulting in an increased maximal transport rate (Vmax) of choline transport without a change in the substrate concentration at which the transport rate is one-half Vmax (Kt) (Ferguson et al., 2003). Moreover, in the brain, almost all ChAT-expressing neurons also express ChT, indicating that ChT is a key component of the cholinergic phenotype (Misawa et al., 2001; Kus et al., 2003). ChT exhibits high affinity for choline (Kt, ∼0.5-5 μm), and its activity is potently inhibited by hemicholinium-3 (HC-3) (Yamamura and Snyder, 1973). These properties have been explored in functional studies of this protein (Dowdall and Simon, 1973; Haga and Noda, 1973; Kuhar et al., 1973; Apparsundaram et al., 2000; Okuda et al., 2000). The gene encoding the neuronal ChT has been cloned recently and designated CHT1 (Apparsundaram et al., 2000; Okuda et al., 2000), permitting the use of molecular biological techniques and generation of antibodies for the assessment of its tissue levels and distribution. Here, we tested the hypothesis that ChT may be upregulated when ChAT activity is reduced using the heterozygous Chat+/- mouse model. We found that in hippocampal slices from Chat+/- mice, ChT activity and protein levels are upregulated by 50-100% relative to slices from control mice and that CHT1 mRNA levels are similarly increased in the septum, a brain region containing the cell bodies of cholinergic neurons that project to the hippocampus. Presumably as a result of this upregulation, total ACh and depolarization-evoked ACh release are normal. Moreover, no behavioral abnormalities were observed in the Chat+/- mice.
Materials and Methods
Mice
All mice used in these experiments were offspring between two and seven generations removed from the original founders carrying the Chat null allele, Chattm1Fhg (Brandon et al., 2003). All animal procedures were performed in accordance with the statutes of the Institutional Animal Care and Use Committees of the Salk Institute and Boston University.
ChAT and AChE activity
Adult Chat+/- mice and age- and sex-matched wild-type control (Chat+/+) mice were killed, and the brains were rapidly removed and dissected on ice. Dissected frontal and parietal neocortex, hippocampus, olfactory bulbs, striatum, and medial septum were frozen at -80°C until assayed. Tissue was sonicated in 400 μl of 50 mm phosphate buffer, pH 7.4. Tissue homogenate was diluted as needed so that results would fall in the linear response range of each respective assay. This dilution factor varied between region and assay type. ChAT activity was measured by incorporation of [14C]acetyl-coenzyme A into [14C]ACh (Fonnum, 1975) as described previously (Pizzo et al., 1999). AChE activity was assayed by measuring the hydrolysis of [14C]ACh into [14C]acetate (Fonnum, 1975). The protein content of the samples was determined by the method of Lowry et al. (1951). ChAT activity is expressed as nanomoles of ACh formed per milligram of tissue per minute. AChE activity is expressed as micromoles ACh hydrolyzed per milligram of protein per minute.
Tissue ACh content
Experiment 1. Mice were pretreated with 0.32 mg/kg physostigmine 30 min before they were killed to maximally inhibit endogenous cholinesterase activity. Mice were killed by decapitation, and the brains were rapidly removed and chilled. Specific brain regions were rapidly dissected and denatured with 100 μl of 10% trichloroacetic acid (TCA) with brief sonication, enabling extraction of choline and ACh. The samples were centrifuged, and the precipitate was retained for protein analysis. The supernatant fluid was further purified by centrifugation through a 5000 kDa molecular weight cutoff filter (Millipore, Bedford, MA). To remove the TCA and lipids, the supernatant was subjected to four sequential extractions with water-saturated ethyl ether. The final ACh extract was stored at -80°C. ACh content was determined by HPLC using a commercial kit from Bioanalytical Systems (West Lafayette, IN). The protein precipitate was solubilized using Solvable (Packard Instrument Company, Meriden, CT), and protein content was determined using the method of Lowry et al. (1951).
Experiment 2. Mice were deeply anesthetized with CO2 and killed by decapitation. Brains were rapidly removed, and cortex and striatum were dissected. ACh and choline were extracted from brain cerebral cortex and striatum by preparing a 2% tissue homogenate in a mixture of methanol: 1N formic acid (1:0.1 by volume) and centrifuging to obtain a protein pellet. The supernatant fluid was then extracted with two volumes of chloroform and one volume of water. The samples were vortex mixed and centrifuged for 10 min at 2000 rpm. The aqueous phase was collected, dried under a vacuum, and reconstituted in 200 μl of water. ACh and choline contents were determined by HPLC using a commercial kit from BAS. Protein content of the extracts was determined by the method of Smith et al. (1985).
ACh synthesis from choline transported by ChT
Mice were anesthetized with CO2 and then decapitated. Each brain was removed rapidly, and the hippocampus was dissected, placed on ice, and cut into 400 μm transverse slices using a McIlwain tissue chopper (Mickle Laboratory Engineering, Guildford, Surrey, UK). The slices were incubated in tissue strainers (five slices per strainer) at 37°C for 1 hr in a physiological salt solution, NaPSS, pH 7.4 (145 mm NaCl, 5 mm KCl, 1 mm CaCl2, 0.75 mm MgSO4,10mm glucose, and 10 mm HEPES) with 15 μm neostigmine (Sigma, St. Louis, MO). During this period, strainers were transferred to fresh buffer every 15 min, allowing the released molecules to be removed so that they did not interfere with the remaining part of the experiment. Each strainer was incubated for 10 min in a well containing NaPSS, 1 μm [3H]choline (3 μCi/ml; Du Pont, Boston, MA), and 15 μm neostigmine (Sigma), either with or without 10 μm HC-3 (Sigma). Slices were then washed with ice-cold NaPSS. ACh and choline were extracted with 600 μl of methanol, processed as described above for experiment 2, and their mass was determined by HPLC. After separation by HPLC, the ACh and choline fractions as well as a baseline sample were collected and their radioactivities determined by liquid scintillation spectrophotometry. To obtain a measure of [3H]ACh and [3H]choline derived from choline specifically transported by ChT, the amount of radioactivity in [3H]choline and [3H]ACh fractions in the presence of HC-3 was subtracted from that measured in the absence of HC-3. The mass of choline taken up by ChT, and of newly synthesized ACh, was calculated by dividing the radioactivity (in disintegrations per minute) of the choline and ACh fraction by the specific radioactivity of the [3H]choline precursor added to the medium. To determine the amount of choline taken up by ChT and of newly synthesized ACh as a fraction of the total intracellular choline and ACh, respectively, the mass of choline taken up by ChT and of newly formed ACh, calculated as described above, was divided by the total choline and ACh mass, respectively, as determined by HPLC. These calculations permitted the determination of the degree of enrichment of the total choline pool with that taken up by ChT and of the ACh pool with newly synthesized ACh and allowed normalization of results from three separate experiments.
Potassium-evoked ACh release
Hippocampal slices were prepared as described above. Five slices were transferred into each cell strainer and were allowed to equilibrate for 1 hr at 37°C in NaPSS with 15 μm neostigmine. Slices were then incubated at 37°C for 5 min periods sequentially in a normal (NaPSS) or depolarizing (KPSS: 105 mm NaCl, 45 mm KCl, 1 mm CaCl2, 0.75 mm MgSO4, 10 mm glucose, and 10 mm HEPES) solution with 15 μm neostigmine as follows: NaPSS, KPSS, NaPSS, KPSS, NaPSS, KPSS, NaPSS, KPSS. The medium from each 5 min incubation was collected and centrifuged to remove debris, and ACh and choline contents were determined by HPLC as described above. ACh and choline were also extracted from the slices and quantified by HPLC as described above.
Reverse transcriptase PCR
Mice were anesthetized by CO2 inhalation and decapitated. Each brain was removed rapidly, and the septum was dissected. The RNA was extracted using phenol/chloroform method (Chomczynski and Sacchi, 1987) and precipitated once with isopropanol and then with ethanol. RNA was redissolved in DEPC-treated water, and its quantity was determined using RiboGreen RNA quantitation reagent and kit (Molecular Probes, Eugene, OR) and a Victor3 multilabel plate reader (PerkinElmer Life Sciences, Emeryville, CA). The RNAs were used for reverse transcriptase (RT)-PCR using Superscript One-Step RT-PCR with Platinum Taq (Invitrogen, San Diego, CA). First strand cDNA synthesis was performed with 20 ng of total RNA, oligo dT primer, and reverse transcriptase at 48°C (45 min). Primers used for PCR include glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (BD Biosciences, San Jose, CA) and CHT1 (Forward: CGGGGAACCATTGAATTCGTTGAAGTCTAC; reverse: GGGGCAAGCTTCCACTTTCAAATAGAGATACT). PCR was performed using Platinum TaqDNA polymerase with a denaturing step for 2 min at 94°C followed by 32 cycles of 45 sec at 94°C, 1 min at 55°C, and 2 min at 72°C and terminated by an elongation step at 72°C for 7 min. Under these conditions, the amount of the CHT1 PCR product was proportional to the amount of the RNA analyzed (data not shown). PCR products were analyzed by electrophoresis on a 10% polyacrylamide gel, stained with ethidium bromide, and visualized with the Kodak (Rochester, NY) Image Station 440. Band intensities were quantified using Kodak ID software.
Western blot analysis
For Western blot analysis, whole tissue extracts were prepared by adding lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 1% Nonidet NP-40, 10% glycerol, 2 mm 4-(2-aminoethyl)-benzenesulfonyl fluoride, 1 μg/ml leupeptin, 2 μg/ml aprotinin, 2 μg/ml pepstatin) to a frozen sample of a CNS region or hippocampal slice, gently sonicating, incubating for 15 min on ice, and briefly centrifuging to clear. The extracts were normalized for total protein and subjected to SDS-PAGE. After transfer of protein to an Immoblin P membrane (Millipore), the membrane was blocked with 5% nonfat dry milk in 1× Tris-buffered saline (TBS) containing 0.1% Tween 20 for 1 hr and then probed with affinity-purified rabbit anti-CHT1 polyclonal antibody AB5966 (1:1000; Chemicon, Temecula, CA) overnight. The antibody/antigen complexes on the membranes were detected using a peroxidase-conjugated anti-rabbit IgG (1:5000) and visualized using the enhanced chemiluminescence method (Western Lightning; PerkinElmer Life Sciences) and a Kodak Image Station 440. Digitized images of immunoblots were quantified using Kodak ID software.
Behavioral analyses
All testing was performed during the light phase of the 12 hr light/dark cycle. Mice tested were males, 6-12 months of age. All experiments were performed using age-matched littermate controls. Two of the tested cohorts were of a mixed genetic background (on average, 50% C57BL/6, 50% 129S4; referred to herein as 50/50 background), and two cohorts were of a uniform 129S4 genetic background.
Activity chamber. Activity was monitored over the course of 60 min in a square 1 × 1 m Plexiglas chamber using an automatic infrared photobeam interruption system (San Diego Instruments, San Diego, CA) in a darkened room. The chamber was cleaned with 10% ethanol between each session. Results were analyzed by repeated measures ANOVA.
Rotarod. All mice were naive to the task before any testing. Mice were placed on the apparatus (Rotamex 4/8; Columbus Instruments, Columbus, OH) while it was idling at 5 rpm. In the first set of experiments, rotation was accelerated from 5 to 65 rpm over the course of 3 min, and mice were tested on three trials per day, with an intertrial interval of ∼2 min for five sequential days. Latency to fall was automatically recorded; if a mouse remained on the rod for an entire trial, it received the maximum score. In the endurance version of the task, rotation was increased from 5 to 15 rpm over the course of 60 min, and mice were tested only one time each. During a 60 min trial, mice were allowed to fall off up to a total of 10 times, and immediately after each fall were placed back on the rotarod. Time of each of the (up to) 10 falls was recorded by hand. Results were analyzed by repeated measures ANOVA.
Water maze.
The water maze used was 1.3 m in diameter, and the platform was 10 × 10 cm, set 25 cm from the wall of the pool and 1 cm below the surface of 26°C opaque water. All trials were recorded with an automatic tracking system (San Diego Instruments). Before training, each mouse was placed on the platform for 30 sec. With the first group of mice, visible platform training (two trials per day for 2 d) was performed before hidden platform training, but this test was not included with subsequent groups. Hidden platform training (two trials per day) was initiated from each of four quadrants in a balanced pseudorandom order, with the mouse facing the wall. Mice were allowed 60 sec to find and climb onto the platform; if they did not find it in 60 sec, they were placed gently on the platform by the experimenter. Mice rested on the platform for 10 sec between trials. Mice were trained for 8 d, and in the reversal version of the paradigm, the platform location was shifted to the opposite quadrant for six more days of training. Probe trials were conducted with the platform removed at the following time points: 4 hr after training on days 4, 8, 11, and 14, and 23 hr after training on days 8 and 14. Variables measured during training trials included latency to find platform, swim distance to platform, swim speed, and average distance from mouse to platform. Additional measures for probe trials included time spent in each quadrant, swim distance in each quadrant, and number of platform location crossings. Results were analyzed by ANOVA.
Results
ChAT activity and AChE activity in Chat heterozygous adults
Similar to our observations with heterozygous embryos (Brandon et al., 2003), compared with wild-type littermates, adult mice carrying one null allele of the Chat gene have a highly significant, 40-50% reduction in ChAT activity in several brain structures. These structures include the cortex, hippocampus, septum, striatum, and olfactory bulb (Fig. 1a). In contrast to the dramatic depletion in ChAT activity found in the Chat+/- mice, no difference in AChE activity was observed between heterozygous and wild-type mice (Fig. 1b).
Figure 1.
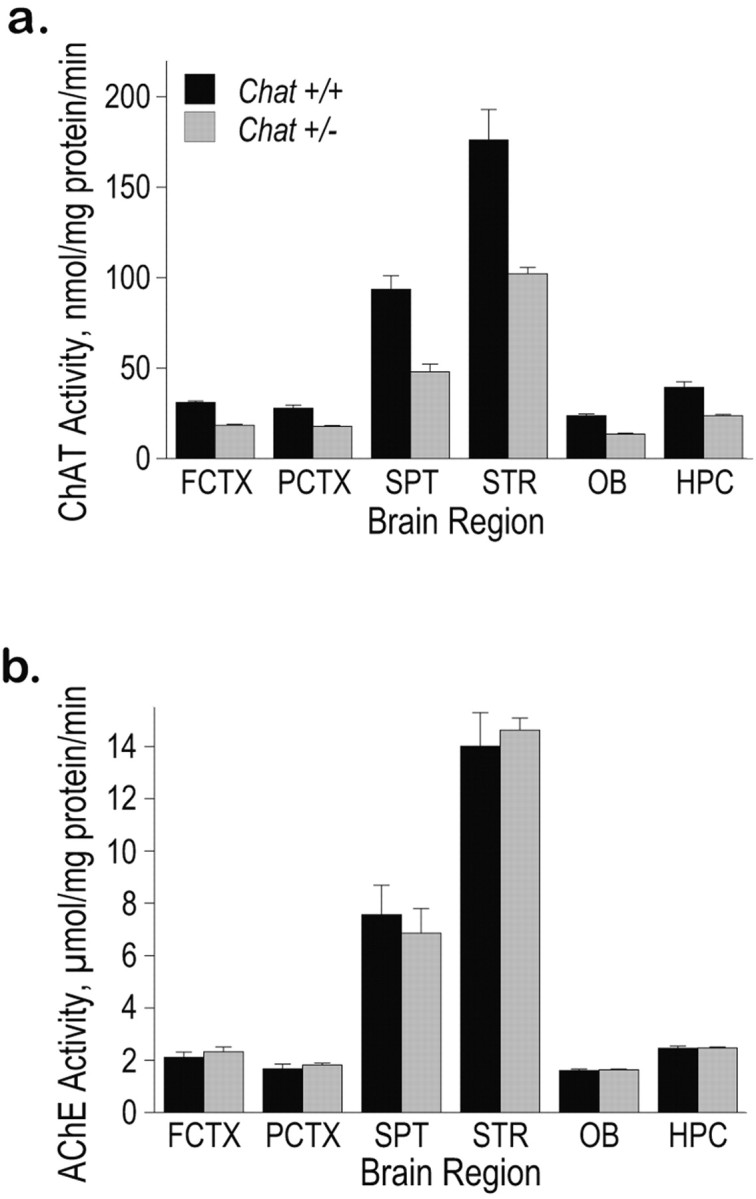
ChAT and AChE activity in Chat heterozygotes. a, ChAT activity is reduced in Chat+/- brain regions. In each of several regions of the brain (FCTX, frontal cortex; PCTX, parietal cortex; SPT, septum; STR, striatum; OB, olfactory bulb; HPC, hippocampus), ChAT activity (nmol/mg of protein/min) was reduced by almost half (FCTX, 41%; PCTX, 36%; HPC, 40%; SPT, 49%; STR, 42%; OB, 43%). Error bars represent SEM. Significant differences were observed between Chat+/+ mice and Chat+/- mice in all brain regions examined (FCTX, p < 0.0001; PCTX, p < 0.0005; HPC, p < 0.001; SPT, p < 0.001; STR, p < 0.005; OB, p < 0.0001). b, AChE activity in various brain regions of Chat+/+ and Chat+/- mice. No significant effect of genotype was found in any brain region.
Behavioral analyses of Chat heterozygous mice
The Chat+/- mice offer a unique opportunity to assess the behavioral effects of a 40-50% reduction in ChAT activity. We hypothesized that behavioral effects could arise from ACh deficiency in any number of the various cholinergic systems, including those required for either motor or cognitive functions, and therefore mice were tested in a battery of behavioral assays.
Testing activity in a novel open field could reveal differences in motor function (overall activity), learning (decreased exploration over the course of each session), or anxiety (thigmotaxis; central versus peripheral activity). Mice of mixed 50/50 genetic background (on average 50% C57BL/6, 50% 129S4) were tested in a 1 hr session, and no differences were observed in overall activity or in the extinction curves that reflect the decreased exploration that occurs as time in the chamber increases (Fig. 2a). Subsequently, heterozygous and wild-type littermates of a uniform genetic background (100% 129S4) were tested to reduce potential variability because of the mixed background and increase the likelihood of detecting a subtle difference between genotypes. The 129S4 mice were tested in 1 hr sessions on two sequential days, and again, no differences between the genotypes were detected (p > 0.05; data not shown). Furthermore, in neither of these experiments were differences observed between genotypes on central activity only or peripheral activity only (all p > 0.05; data not shown). Chat heterozygotes were compared with wild-type littermates in an accelerating rotarod paradigm. This test could reveal differences in motor function, motor learning, or fear-motivated behavior. As shown in Figure 2b, Chat+/- mice of the 50/50 background showed no difference from control littermates when tested three times per day for five sequential days. Mice of the 129S4 background also showed no difference between genotypes in this paradigm (p > 0.05; data not shown). We hypothesized that the reduced ChAT activity in heterozygotes might only manifest as a defect under conditions of great demand for ACh synthesis, as might occur if synapses were required to release ACh repeatedly over an extended period. Therefore, we also tested Chat+/- mice in an “endurance” version of the rotarod. In this paradigm, naive mice were placed on the rotarod at 5 rpm, and speed was accelerated to 15 rpm over the course of 1 hr. The latency to each of the first 10 falls was measured, and no differences between genotypes were detected (p > 0.05; data not shown).
Figure 2.

Behavioral analyses revealed no defects in Chat heterozygotes. a-d, Chat+/+ (closed circles) and Chat+/- (open squares) mice were tested in the activity chamber (a), the accelerating rotarod (b), either learning or relearning curves in the training phase of the water maze (c; arrow represents platform reversal to opposite quadrant), and recall in the probe trial of the water maze (d; time spent in each quadrant from the trial 4 hr after training on day 8 is shown). OPP, Opposite quadrant; AR, adjacent right quadrant; TARGET, correct quadrant; AL, adjacent left quadrant. Error bars represent SEM. ANOVA revealed no significant main effects of genotype in any of the behavioral tests, including the activity chamber (F(1,25) = 0.03; p = 0.87), accelerating rotarod (F(1,26) < 0.01; p = 0.99), learning and relearning in the training phase of the water maze (F(1,32) < 0.05, p = 0.94 for learning; F(1,32) = 0.27, p = 0.61 for relearning), or probe trial of the water maze (F(1,32) = 0.41; p = 0.53) experiments shown here.
Last, several cholinergic systems in the brain are thought to be involved in cognition. Therefore, the Chat+/- mice were tested in the water maze, which is a test that requires motor function, spatial navigation, learning, and recall. Chat heterozygotes of the 50/50 background showed no difference from control littermates in any measure on the visual platform version of the task (all p > 0.05; data not shown) or in hidden platform training using two trials per day over 8 d (Fig. 2c). To assess the ability of the mice to replace a previous solution with a new correct solution to the task, we tested Chat+/- mice in reversal learning as well. Again, no differences were observed between genotypes on any measure, including latency to find the platform, distance swam to platform, or swim speed. Probe trials, wherein the hidden platform is removed and the animal's swim pattern is recorded, were conducted 4 and 24 hr after day 4 training, day 8 training, and day 14 “retraining,” and no significant differences were found in time spent or distance swam in correct quadrant, platform location crossings, or mean distance from the platform location during the trial (typical trial shown in Fig. 2d). This training and retraining protocol was repeated with an additional cohort of mice of the 129S4 background, and again, no differences were observed in any of the measures (all p > 0.05; data not shown). In summary, the Chat heterozygotes were found to be normal in both learning and performance of motor and nonmotor tasks, even under relatively challenging conditions.
The lack of behavioral defects in these mice despite testing several independent groups of mice in a wide array of tests, including rather demanding versions of these tests, was striking. How does one reconcile the consistent reduction in ChAT activity, but normal behavioral abilities, of the Chat heterozygotes? One obvious possibility is that in normal mice, ChAT is simply expressed in at least twofold excess of the amount needed biologically with 50% of normal activity being entirely sufficient for cholinergic function. Alternatively, mechanisms may exist or may have arisen in these mice to maintain normal releasable amounts of ACh in the context of reduced ChAT activity. To determine whether the reduction in ChAT activity reduced the amount of total ACh, we measured ACh levels in several brain regions.
Tissue ACh content and ACh release
ACh content in the cortex and striatum was measured in Chat+/+ and Chat+/- mice in two independent experiments. There was no significant difference in the ACh content between the two groups in any brain region (Fig. 3a). Therefore, Chat+/- mice are able to maintain normal levels of intracellular ACh despite reduced activity of ChAT.
Figure 3.
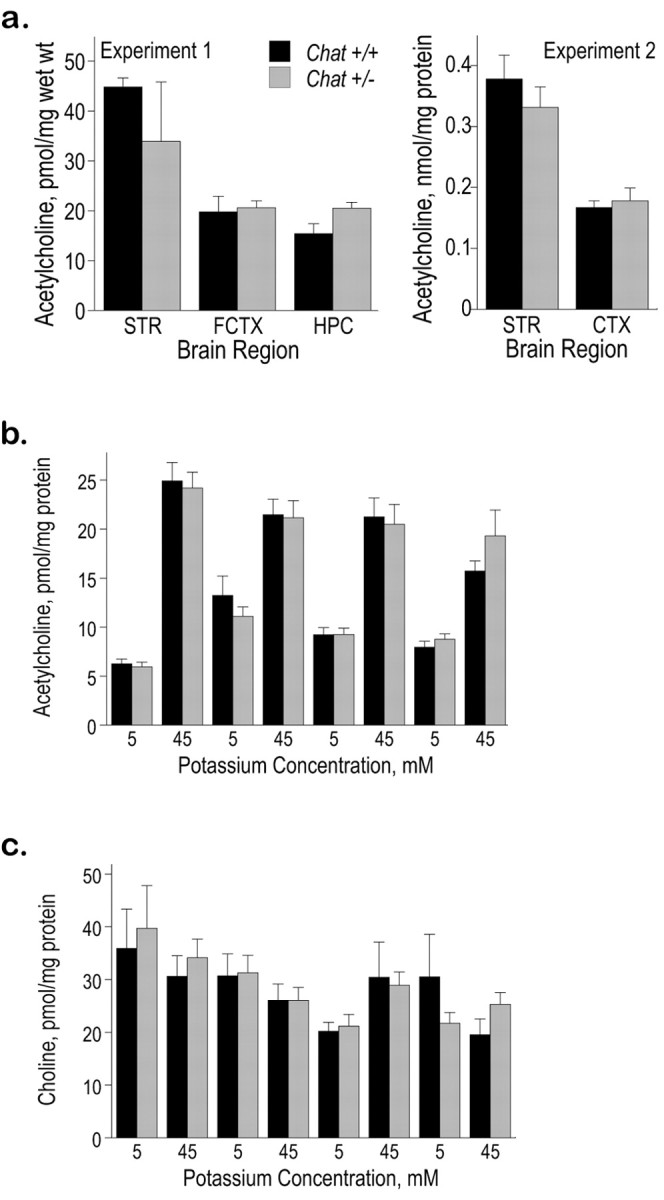
Normal levels of ACh in Chat heterozygotes. a, Tissue ACh content in the striatum, hippocampus, and cortex of Chat+/+ and Chat+/- mice. Two separate methods were used to assay ACh content using HPLC as described in Materials and Methods. In both experiments, there were no significant differences between the two groups of mice in any of the brain regions assayed. STR, Striatum; FCTX, frontal cortex; HPC, hippocampus; CTX, cortex. b, c, Potassium-evoked ACh and choline release in Chat+/+ and Chat+/- mice. Hippocampal slices were incubated in each solution for 5 min, and ACh (b) and choline (c) levels in the medium were measured using HPLC as described in Materials and Methods. No significant differences in ACh or choline release were observed between the groups.
ACh was released from hippocampal slices at rest, and this basal release increased fourfold to fivefold during incubation in a depolarizing medium (Fig. 3b). Hippocampal slices from Chat+/- mice were capable of sustaining this high rate of ACh release during four periods of depolarization. Choline was also released into the medium, but this release was unaffected by either depolarization or the animal's genotype (Fig. 3c). This apparently normal sustained ACh release observed in vitro is consistent with the ability of Chat+/- mice to maintain normal cholinergic function.
Choline transport and ACh synthesis
The amounts of choline taken up by ChT and of ACh derived from this choline was determined by incubating hippocampal slices with [3H] choline (1 μm for 10 min) in the presence or absence of a ChT inhibitor, HC-3 (10 μm), and calculating the HC-3-sensitive enrichment of the endogenous pools of these compounds with the labeled [3H]choline. In Chat+/- mice, the enrichment of the free-choline pool with choline taken up by ChT was 110% higher than in the wild-type animals (Fig. 4a). Moreover, the enrichment of the ACh pool was 60% higher in the Chat+/- mice than in the controls (Fig. 4a). These results indicate that ChT activity is strikingly increased in Chat+/- mice.
Figure 4.
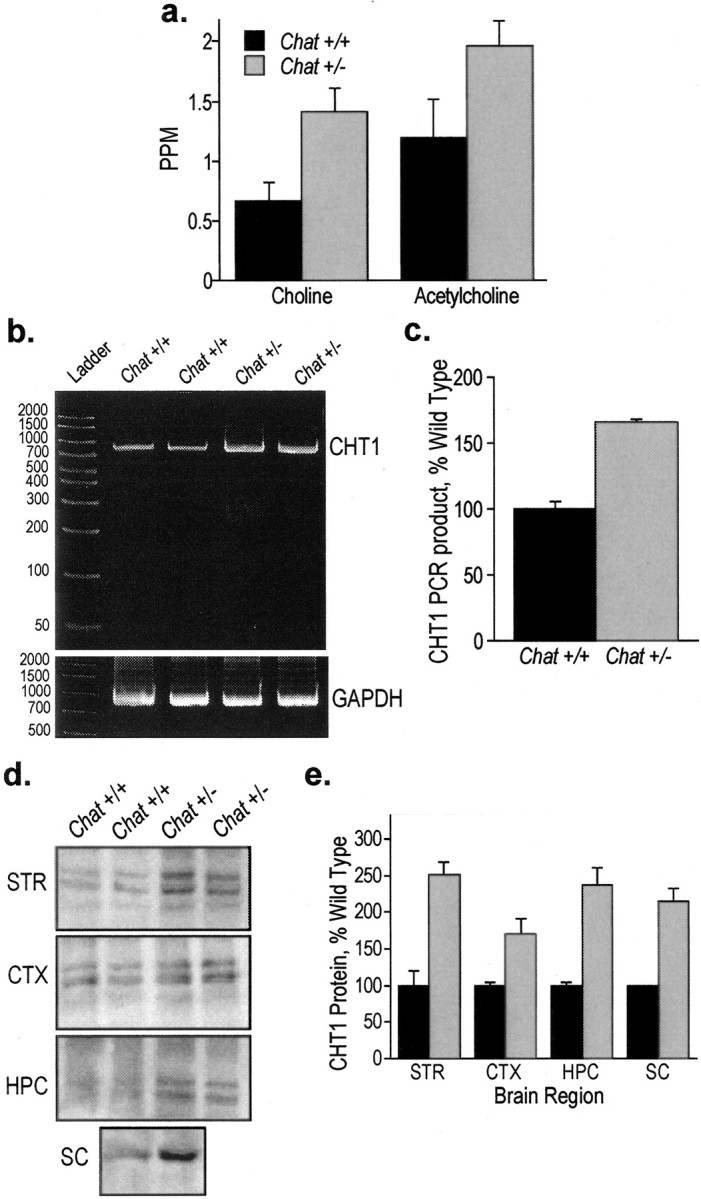
Increased ChT activity, mRNA, and protein in Chat heterozygotes. a, [3H]Choline uptake was measured in the presence and absence of HC-3, an inhibitor of ChT activity, as described in Materials and Methods. The amount of ChT-transported choline was calculated by subtracting the amount of intracellular [3H]choline in the presence of HC-3 from the amount of [3H]choline in the absence of HC-3. The [3H]choline constituted a higher fraction of the HC-3-sensitive choline level in the Chat+/- mice compared with the wild-type mice (p < 0.05). ACh synthesized from [3H]choline was also measured in the presence and absence of HC-3. The amount of ACh synthesized from ChT-transported choline was calculated by subtracting the amount of ACh synthesized in the presence of HC-3 from the amount of ACh produced in the absence of HC-3. The newly made ACh constituted a higher fraction of the HC-3-sensitive ACh pool in the Chat+/- mice than it did in the wild-type littermates (p < 0.05). b, RT-PCR of CHT1 mRNA in Chat+/+ and Chat+/- mice. Septal RNA was used for RT-PCR of CHT1 and GAPDH. As expected, the PCR product from the amplification of CHT1 was present as an 840 bp band. The PCR product from the amplification of GAPDH was present as a 983 bp band. c, CHT1 product levels were quantified by Kodak Image Station Software and normalized to GAPDH product levels. The results are presented as a percentage of wild-type control ± range. d, CHT1 protein levels in Chat+/+ and Chat+/- mice. CHT1 protein levels in several CNS structures were measured by Western blot analysis. Chat+/- mice have an approximately twofold increase in CHT1 protein levels in each of the brain regions compared with Chat+/+ mice. e, CHT1 levels were quantified by Kodak Image Station Software and are presented as a percentage of wild-type control ± range. STR, Striatum; CTX, cerebral cortex; HPC, hippocampus; SC, spinal cord.
CHT1 mRNA and protein levels
To determine whether these results are attributable to the increased expression of CHT1 in the cholinergic neurons of the septohippocampal pathway, CHT1 mRNA was measured in the septum, where the cell bodies reside, and CHT1 protein was measured in the hippocampus, which contains their terminals. Using RT-PCR, we detected a single 840 base pair PCR product as expected. The amount of the CHT1 mRNA was ∼70% higher in the septum from Chat+/- mice compared with Chat+/+ animals (Fig. 4b,c).
CHT1 protein levels were assessed by Western blot analysis. In our hands, CHT1 appears as a single band of ∼63 kDa in the spinal cord and as a doublet of ∼66 and 73 kDa in the brain (Fig. 4d). This pattern is consistent with previous reports (Misawa et al., 2001; Ferguson et al., 2003). Similar results were obtained with two additional anti-CHT1 antibodies (data not shown). As shown in Figure 4, d and e, CHT1 levels were approximately twofold higher in the hippocampus of Chat+/- mice compared with Chat+/+ mice. These data are in good agreement with the activity measurements reported above (Fig. 4a). Similar changes in CHT1 protein levels were observed in striatum, cerebral cortex, and spinal cord (Fig. 4d,e).
Discussion
Mice that express a single active Chat allele, and thus have approximately one-half of the wild-type ChAT activity in the CNS, do not exhibit behavioral defects, contain normal amounts of ACh in the brain, and release normal amounts of ACh in vitro. These data indicate that a 50% reduction in ChAT activity is well tolerated. Therefore, the following question arises: how much of a decline in ChAT activity is needed to reduce ACh levels and manifest a behavioral defect? In mice treated with the ChAT inhibitor 4-(1-naphtylvinyl)pyridine (NVP) (250 mg/kg), a 60% drop in ChAT activity resulted in a 20% reduction of brain ACh levels after swimming stress (Krell and Goldberg, 1975), whereas in an analogous experiment in rats, NVP (200 mg/kg) caused a reduction in brain ACh levels by 90% (Budai et al., 1986). In rats treated with the ChAT inhibitor BW813U, a 60% reduction of ChAT activity did not impair performance on a previously learned spatial task (the rate of acquisition was not tested) (Wenk et al., 1986). Clinical evidence indicates that the threshold beyond which reduced ChAT activity causes functional impairments is quite close to 50%. One of the patients with CMS-EA studied by Ohno et al. (2001) was a compound CHAT heterozygote with one null allele and one allele encoding a CHAT protein with a V506L substitution. This patient was symptomatic even though the activity of the V506L CHAT isoform was not severely reduced.
One way that normal ACh production could be sustained in the presence of reduced amounts of Chat protein might be via an increased affinity of the enzyme for one or both of its substrates, acetyl-CoA and choline, or by a mechanism that increases the supply of these substrates to the enzyme without alterations in its kinetic properties. Little is known about in vivo regulation of the affinity of ChAT for its substrates. Polymorphism in the CHAT gene results in the production of CHAT proteins that, when expressed in bacteria, exhibit wide-ranging affinities for its substrates and a range of Vmax values (Ohno et al., 2001). However, no information is available about the regulation of these properties in vivo. Wild-type CHAT expressed in bacteria or in human cells exhibits an increased Vmax when phosphorylated by protein kinase C (PKC) (Dobransky et al., 2000, 2001), suggesting that activation of PKC in neurons may increase ACh production.
An increased supply of acetyl-CoA to ChAT could also accelerate ACh synthesis. However, given that this compound is used by all cells for energy generation and fatty acid synthesis, it has been difficult to study the regulation of acetyl-CoA production specifically in cholinergic neurons and to identify intraneuronal pools of this compound used for ACh synthesis (for review, see Jope, 1979; Tucek, 1990). Increased availability of glucose, a metabolic precursor of acetyl-CoA, increases acetyl-CoA levels and stimulates ACh synthesis (Ricny and Tucek, 1980; Dolezal and Tucek, 1982). However, little is known about physiological mechanisms that may increase acetyl-CoA synthesis in cholinergic neurons, although the enzyme that generates acetyl-CoA in the cytoplasm of these cells, ATP-citrate lyase, is more highly expressed in these neurons than in other cells of the CNS (Tomaszewicz et al., 2003; Beigneux et al., 2004). It remains to be determined whether the level of expression or activity of ATP-citrate lyase is regulated by the demand for acetyl-CoA in cholinergic neurons.
The principal finding reported here is that ChT is dramatically upregulated in Chat heterozygotes. Therefore, a 50% loss of normal ChAT activity produces an apparently adaptive response that increases the supply of choline to ChAT and thereby accelerates ACh synthesis. This kind of adaptation indicates the existence of a mechanism that detects a drop in ChAT activity. It is likely that this hypothetical mechanism monitors the demand for ACh (e.g., the cholinergic neuron firing rate or presynaptic receptor activity) rather than ChAT activity per se. Indeed increased neuronal activity acutely stimulates ChT function (Murrin and Kuhar, 1976; Collier and Ilson, 1977; Polak et al., 1977; Sherman et al., 1978; Marchbanks, 1982), and the reliability of this phenomenon has formed the basis for the proposal that measurements of ChT activity in vitro can be used as an index of the in vivo firing rate of cholinergic neurons (Simon, et al., 1976). In the current study, the increased ChT activity in Chat+/- mice correlated with the amount of CHT1 mRNA and protein, suggesting that in addition to rapid ChT activation reported previously (Murrin and Kuhar, 1976; Collier and Ilson, 1977; Polak et al., 1977; Sherman et al., 1978; Marchbanks, 1982), ChT may also be regulated at the level of transcription. If this is the case, then CHT1 may be a member of a large set of genes with transcription that is regulated by neuronal activity (West et al., 2001).
Behavioral and physiological data indicate that cholinergic neurotransmission is relatively normal in Chat+/- mice and in people expressing a single copy of a mutant CHAT allele. Our data suggest that this phenotype may be attributable to, at least in part, a compensatory increase of choline transport and imply that cholinergic neurotransmission in humans and animals with reduced ChAT activity would become vulnerable to defects in CHT1 function. A mutant CHT1 allele occurring with a 6% frequency among Ashkenazi Jews has been discovered recently (Okuda et al., 2002). The mutant allele encodes an amino acid substitution in the CHT1 protein (I89V) that results in a 40-50% reduced Vmax of choline transport compared with the wild type when measured in a cell culture expression system. However, no phenotype associated with this polymorphism has been described to date.
In summary, the Chat+/- mouse model demonstrates that a 50% loss of ChAT activity causes an induction of CHT1 mRNA, and an increase in CHT1 protein abundance, and ChT activity. ChT induction increases the provision of choline to ChAT and thus may constitute a compensatory mechanism that contributes to the maintenance of normal cholinergic neurotransmission in Chat+/- mice.
Footnotes
This study was supported by National Institutes of Health (NIH) Grants AG09525, AG17533, NS42793, and NS44238. T.M. was supported by NIH Training Grant AG00115. E.P.B. was supported by a grant from the John Douglas French Alzheimer's Foundation. We thank Mary L. Gage and Rebecca Cole for helpful comments on this manuscript.
Correspondence should be addressed to Jan Krzysztof Blusztajn, Boston University School of Medicine, 715 Albany Street, L804, Boston, MA 02118. E-mail: jbluszta@bu.edu.
E. P. Brandon's present address: Ceregene, 9381 Judicial Drive, San Diego, CA 92121.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245459-08$15.00/0
E.P.B. and T.M. contributed equally to this work.
References
- Apparsundaram S, Ferguson SM, George Jr AL, Blakely RD (2000) Molecular cloning of a human, Hemicholinium-3-sensitive choline transporter. Biochem Biophys Res Commun 276: 862-867. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Kosinski C, Gavino B, Horton JD, Skarnes WC, Young SG (2004) ATP-citrate lyase deficiency in the mouse. J Biol Chem 279: 9557-9564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blusztajn JK, Wurtman RJ (1981) Choline biosynthesis by a preparation enriched in synaptosomes from rat brain. Nature 290: 417-418. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK, Wurtman RJ (1983) Choline and cholinergic neurons. Science 221: 614-620. [DOI] [PubMed] [Google Scholar]
- Blusztajn JK, Liscovitch M, Richardson UI (1987) Synthesis of acetylcholine from choline derived from phosphatidylcholine in a human neuronal cell line. Proc Natl Acad Sci USA 84: 5474-5477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Lin WC, D'Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KF (2003) Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci 23: 539-549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budai D, Ricny J, Kasa P, Tucek S (1986) 4-(1-Naphthylvinyl)pyridine decreases brain acetylcholine in vivo, but does not alter the level of acetyl-CoA. J Neurochem 46: 990-992. [DOI] [PubMed] [Google Scholar]
- Byring RF, Pihko H, Tsujino A, Shen XM, Gustafsson B, Hackman P, Ohno K, Engel AG, Udd B (2002) Congenital myasthenic syndrome associated with episodic apnea and sudden infant death. Neuromuscul Disord 12: 548-553. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156-159. [DOI] [PubMed] [Google Scholar]
- Collier B, Ilson D (1977) The effect of preganglionic nerve stimulation on the accumulation of certain analogues of choline by a sympathetic ganglion. J Physiol (Lond) 264: 489-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobransky T, Davis WL, Xiao GH, Rylett RJ (2000) Expression, purification and characterization of recombinant human choline acetyltransferase: phosphorylation of the enzyme regulates catalytic activity. Biochem J 349: 141-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobransky T, Davis WL, Rylett RJ (2001) Functional characterization of phosphorylation of 69-kDa human choline acetyltransferase at serine 440 by protein kinase C. J Biol Chem 276: 22244-22250. [DOI] [PubMed] [Google Scholar]
- Dolezal V, Tucek S (1982) Effects of choline and glucose on atropine-induced alterations of acetylcholine synthesis and content in the caudate nuclei of rats. Brain Res 240: 285-293. [DOI] [PubMed] [Google Scholar]
- Dowdall MJ, Simon EJ (1973) Comparative studies on synaptosomes: up-take of (N-Me-3H)choline by synaptosomes from squid optic lobes. J Neurochem 21: 969-982. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Savchenko V, Apparsundaram S, Zwick M, Wright J, Heilman CJ, Yi H, Levey AI, Blakely RD (2003) Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J Neurosci 23: 9697-9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F (1975) A rapid radiochemical method for the determination of choline acetyltransferase. J Neurochem 24: 407-409. [DOI] [PubMed] [Google Scholar]
- Food and Nutrition Board. Institute of Medicine (1998) Dietary reference intakes for thiamin, riboflavin, niacin, vitamin B6, folate, vitamin B12, panthotenic acid, biotin, and choline. Washington, DC: National Academy. [PubMed]
- Haga T, Noda H (1973) Choline uptake systems of rat brain synaptosomes. Biochim Biophys Acta 291: 564-575. [DOI] [PubMed] [Google Scholar]
- Jope RS (1979) High affinity choline transport and acetylCoA production in brain and their roles in the regulation of acetylcholine synthesis. Brain Res Brain Res Rev 1: 313-344. [DOI] [PubMed] [Google Scholar]
- Klein J, Köppen A, Löffelholz K (1990) Small rises in plasma choline reverse the negative arteriovenous difference of brain choline. J Neurochem 55: 1231-1236. [DOI] [PubMed] [Google Scholar]
- Krell RD, Goldberg AM (1975) Effect of choline acetyltransferase inhibitors on mouse and guinea-pig brain choline and acetylcholine. Biochem Pharmacol 24: 391-396. [DOI] [PubMed] [Google Scholar]
- Kuhar MJ, Sethy VH, Roth RH, Aghajanian GK (1973) Choline: selective accumulation by central cholinergic neurons. J Neurochem 20: 581-593. [DOI] [PubMed] [Google Scholar]
- Kus L, Borys E, Ping Chu Y, Ferguson SM, Blakely RD, Emborg ME, Kordower JH, Levey AI, Mufson EJ (2003) Distribution of high affinity cho-line transporter immunoreactivity in the primate central nervous system. J Comp Neurol 463: 341-357. [DOI] [PubMed] [Google Scholar]
- Lakher M, Wurtman RJ (1987) In vivo synthesis of phosphatidylcholine in rat brain via the phospholipid methylation pathway. Brain Res 419: 131-140. [DOI] [PubMed] [Google Scholar]
- Lee H-C, Fellenz-Maloney M-P, Liscovitch M, Blusztajn JK (1993) Phospholipase D-catalyzed hydrolysis of phosphatidylcholine provides the choline precursor for acetylcholine synthesis in a human neuronal cell line. Proc Natl Acad Sci USA 90: 10086-10090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193: 265-275. [PubMed] [Google Scholar]
- Marchbanks RM (1982) The activation of presynaptic choline uptake by acetylcholine release. J Physiol (Paris) 78: 373-378. [PubMed] [Google Scholar]
- Misawa H, Nakata K, Matsuura J, Nagao M, Okuda T, Haga T (2001) Distribution of the high-affinity choline transporter in the central nervous system of the rat. Neuroscience 105: 87-98. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR (2002) Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron 36: 635-648. [DOI] [PubMed] [Google Scholar]
- Murrin LC, Kuhar MJ (1976) Activation of high-affinity choline uptake in vitro by depolarizing agents. Mol Pharmacol 12: 1082-1090. [PubMed] [Google Scholar]
- Ohno K, Tsujino A, Brengman JM, Harper CM, Bajzer Z, Udd B, Beyring R, Robb S, Kirkham FJ, Engel AG (2001) Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci USA 98: 2017-2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I (2000) Identification and characterization of the high-affinity choline transporter. Nat Neurosci 3: 120-125. [DOI] [PubMed] [Google Scholar]
- Okuda T, Okamura M, Kaitsuka C, Haga T, Gurwitz D (2002) Single nucleotide polymorphism of the human high-affinity choline transporter alters transport rate. J Biol Chem 277: 45315-45322. [DOI] [PubMed] [Google Scholar]
- Pardridge WM, Oldendorf WH (1977) Transport of metabolic substrates through the blood-brain barrier. J Neurochem 28: 5-12. [DOI] [PubMed] [Google Scholar]
- Pizzo DP, Waite JJ, Thal LJ, Winkler J (1999) Intraparenchymal infusions of 192 IgG-saporin: development of a method for selective and discrete lesioning of cholinergic basal forebrain nuclei. J Neurosci Methods 91: 9-19. [DOI] [PubMed] [Google Scholar]
- Polak RL, Molenaar PC, van Gelder M (1977) Acetylcholine metabolism and choline uptake in cortical slices. J Neurochem 29: 477-485. [DOI] [PubMed] [Google Scholar]
- Ricny J, Tucek S (1980) Relation between the content of acetyl-coenzyme A and acetylcholine in brain slices. Biochem J 188: 683-688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman KA, Zigmond MJ, Hanin I (1978) High affinity choline uptake in striatum and hippocampus: differential effects of treatments which release acetylcholine. Life Sci 23: 1863-1870. [DOI] [PubMed] [Google Scholar]
- Simon JR, Atweh S, Kuhar MJ (1976) Sodium-dependent high affinity cho-line uptake: a regulatory step in the synthesis of acetylcholine. J Neurochem 26: 909-922. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC (1985) Measurement of protein using bicinchoninic acid. Anal Biochem 150: 76-85. [DOI] [PubMed] [Google Scholar]; [Erratum (1987) 163: 279]. [Google Scholar]
- Tomaszewicz M, Rossner S, Schliebs R, Cwikowska J, Szutowicz A (2003) Changes in cortical acetyl-CoA metabolism after selective basal forebrain cholinergic degeneration by 192IgG-saporin. J Neurochem 87: 318-324. [DOI] [PubMed] [Google Scholar]
- Tucek S (1990) The synthesis of acetylcholine: twenty years of progress. Prog Brain Res 84: 467-477. [PubMed] [Google Scholar]
- Walkey CJ, Donohue LR, Bronson R, Agellon LB, Vance DE (1997) Disruption of the murine gene encoding phosphatidylethanolamine N-methyltransferase. Proc Natl Acad Sci USA 94: 12880-12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SM, Zhu X, Zeisel SH (2003) Phosphatidylethanolamine-N-methyltransferase activity and dietary choline regulate liver-plasma lipid flux and essential fatty acid metabolism in mice. J Nutr 133: 3386-3391. [DOI] [PubMed] [Google Scholar]
- Wenk G, Sweeney J, Hughey D, Carson J, Olton D (1986) Cholinergic function and memory: extensive inhibition of choline acetyltransferase fails to impair radial maze performance in rats. Pharmacol Biochem Behav 25: 521-526. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME (2001) Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA 98: 11024-11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamura HI, Snyder SH (1973) High affinity transport of choline into synaptosomes of rat brain. J Neurochem 21: 1355-1374. [DOI] [PubMed] [Google Scholar]
- Zhao D, Frohman MA, Blusztajn JK (2001) Generation of choline for acetylcholine synthesis by phospholipase D isoforms. BMC Neurosci 2: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]