

Abstract
At many central synapses, endocannabinoids released by postsynaptic cells inhibit neurotransmitter release by activating presynaptic cannabinoid receptors. The mechanisms underlying this important means of synaptic regulation are not fully understood. It has been shown at several synapses that endocannabinoids inhibit neurotransmitter release by reducing calcium influx into presynaptic terminals. One hypothesis maintains that endocannabinoids indirectly reduce calcium influx by modulating potassium channels and narrowing the presynaptic action potential. An alternative hypothesis is that endocannabinoids directly and selectively inhibit N-type calcium channels in presynaptic terminals. Here we test these hypotheses at the granule cell to Purkinje cell synapse in cerebellar brain slices. By monitoring optically the presynaptic calcium influx (Cainflux) and measuring the EPSC amplitudes, we found that cannabinoid-mediated inhibition arises solely from reduced presynaptic Cainflux. Next we found that cannabinoid receptor activation does not affect the time course of presynaptic calcium entry, indicating that the reduced Cainflux reflects inhibition of presynaptic calcium channels. Finally, we assessed the classes of presynaptic calcium channels inhibited by cannabinoid receptor activation via peptide calcium channel antagonists. Previous studies established that N-type, P/Q-type, and R-type calcium channels are all present in granule cell presynaptic boutons. We found that cannabinoid activation reduced Cainflux through N-type, P/Q-type, and R-type calcium channels to 29, 60, and 55% of control, respectively. Thus, rather than narrowing the presynaptic action potential or exclusively modulating N-type calcium channels, CB1 receptor activation inhibits synaptic transmission by modulating all classes of calcium channels present in the presynaptic terminal of the granule cell to Purkinje cell synapse.
Keywords: calcium channels, cannabinoids, Purkinje cell, DSI, DSE, cerebellum
Introduction
The endocannabinoid signaling system allows neurons to modulate transiently the strength of their inputs by inhibiting synaptic transmission (Kreitzer and Regehr, 2001a; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Endocannabinoids such as anandamide and 2-arachidonylglycerol (2-AG) (Devane et al., 1992; Di Marzo et al., 1994; Stella et al., 1997) are released from the postsynaptic neuron. They activate presynaptic G-protein-coupled cannabinoid receptors, leading to a transient inhibition of excitatory and inhibitory synaptic transmission (Kreitzer and Regehr, 2001a,b; Maejima et al., 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001; Diana et al., 2002; Yoshida et al., 2002). Cannabinoid receptors are distributed widely throughout the brain, and such retrograde signaling has been observed at numerous synapses (Elphick and Egertova, 2001; Kreitzer and Regehr, 2002; Wilson and Nicoll, 2002).
A reduction in presynaptic calcium influx (Cainflux) leading to a decrease in neurotransmitter release has been implicated in cannabinoid-mediated synaptic modulation, but it is not clear how Cainflux is reduced. In cell culture and expression systems it has been shown that activation of CB1 receptors can modulate many types of channels, including several classes of potassium and calcium channels (Ameri, 1999; Alger, 2002), presenting many potential targets for physiological cannabinoid receptor activation. One possibility is that potassium channel modulation shortens the duration of the action potential and thereby indirectly reduces Cainflux. The primary evidence for this hypothesis is that potassium channel antagonists such as 4-aminopyridine (4-AP) and barium eliminate or decrease the inhibition following cannabinoid receptor activation (Alger et al., 1996; Daniel and Crepel, 2001; Robbe et al., 2001; Diana and Marty, 2003). However, cannabinoids do not appear to affect extracellular recordings of the presynaptic volley (Levenes et al., 1998; Takahashi and Linden, 2000), which are sensitive to changes in the presynaptic waveform (Sabatini and Regehr, 1997), making it unclear how modulation of potassium channels could contribute to decreased Cainflux.
Another proposed mechanism is direct modulation of presynaptic calcium channels (Sullivan, 1999; Hoffman and Lupica, 2000; Kreitzer and Regehr, 2001a; Alger, 2002). Specifically, several recent studies have suggested that N-type calcium channels play a privileged role in cannabinoid-mediated inhibition (Alger, 2002). For example, cannabinoid-mediated modulation of hippocampal inhibitory synapses is not affected by blockers of P/Q-type calcium channels but is eliminated by N-type calcium channel antagonists (Lenz et al., 1998; Wilson et al., 2001). Similarly, cannabinoid-mediated inhibition of excitatory synapses onto both striatal neurons (Huang et al., 2001) and trigeminal caudal neurons (Liang et al., 2004) is eliminated primarily by pretreatment with N-type calcium channel antagonists. These results suggest that selective regulation of N-type calcium channels may be a general mechanism for endocannabinoid-mediated retrograde inhibition.
We tested these hypotheses by determining the mechanism responsible for cannabinoid-mediated inhibition of transmission at the granule cell to Purkinje cell synapse. This synapse is well suited to these studies because it is highly sensitive to retrograde signaling by endocannabinoids (Kreitzer and Regehr, 2001a) and because the calcium channels responsible for transmitter release are known. Transmission at this synapse is mediated by three pharmacologically separable classes of calcium channels: N-type, P/Q-type, and R-type (Mintz et al., 1995; Dittman and Regehr, 1996; Sabatini and Regehr, 1997). We found that cannabinoid receptor activation inhibited neurotransmitter release by reducing Cainflux. The time course of calcium entry was unaltered, indicating that narrowing of the presynaptic action potential did not contribute to reduced Cainflux. Moreover, we found that, rather than selectively modulating N-type calcium channels, CB1 receptor activation inhibited all classes of calcium channels present in the presynaptic terminal.
Materials and Methods
Transverse slices (300 μm thick) were cut from the cerebellar vermis of 12- to 17-d-old Sprague Dawley rats in an ice-cold sucrose solution containing (in mm): 79 NaCl, 23 NaHCO3, 68 sucrose, 2.3 KCl, 1.1 NaH2PO4, 6.4 MgCl2, and 0.5 CaCl2 bubbled with 95% O2/5% CO2. After incubation for 20 min at 32°C the slices were transferred to a saline solution containing (in mm): 125 NaCl, 26 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 1 MgCl2, 2 CaCl2, and 25 glucose, bubbled with 95% O2/5% CO2 at 32°C for an additional 20-40 min; they subsequently were maintained at room temperature. Experiments were performed at room temperature in the presence of bicuculline (20 μm) to block inhibitory currents mediated by GABAA receptors. AM251 and methanandamide were purchased from Tocris Cookson (Ellisville, MO). ω-Conotoxin GVIA and ω-agatoxin IVA were purchased from Bachem (King of Prussia, PA), Alamone (Jerusalem, Israel), and Peptides International (Louisville, KY). All other chemicals were purchased from Sigma/RBI (St. Louis, MO). All procedures involving animals were approved by the Harvard Medical Area Standing Committee on Animals.
Electrophysiological recordings. Whole-cell voltage-clamp recordings of visualized Purkinje cells were obtained as previously described (Regehr and Mintz, 1994) by using an Axopatch 200B (Axon Instruments, Foster City, CA). Glass electrodes (1-2 MΩ) were filled with an internal solution containing (in mm): 35 CsF, 100 CsCl, 10 EGTA, 10 HEPES, 0.1 D600, pH 7.3, 300 mOsm. Synaptic currents were monitored at a holding potential of - 40 mV. Access resistance and leak currents were monitored continuously, and the experiment was terminated if either changed significantly.
Ca2+ imaging. In separate experiments parallel fibers were loaded with magnesium green AM or mag-fura-5 AM (Molecular Probes, Eugene, OR) as previously described (Regehr, 2000) and stimulated with an extracellular glass electrode placed in the molecular layer. For measurements using magnesium green, fluorescence intensity was measured with a photodiode on an upright microscope with a 470DF40 excitation filter (Zeiss, Thornwood, NY) and a 515DCLP dichroic and a 530LP emission filter (Omega Optical, Brattleboro, VT). For measurements using magfura-5, a 380DF15 excitation filter, a 435DRLP dichroic filter, and a 455LP emission filter (Omega Optical) were used.
Field potential recordings. To measure the presynaptic volleys, we placed an electrode (2-3 MΩ) filled with external solution in the molecular layer, ∼100 μm from the location being monitored optically and away from the stimulation site. In addition to 20 μm bicuculline, 5 μm NBQX was added to the external solution to prevent contamination of the signal by synaptic currents.
Data acquisition and analysis. All signals were digitized with a 16-bit analog-to-digital converter (Instrutech, Great Neck, NY) and Pulse Control software (Herrington and Bookman, 1995). EPSCs were filtered at 1 kHz and digitized at 5 kHz. Presynaptic volleys were filtered at 5 kHz and digitized at 50 kHz. Photodiode currents were filtered digitally at 300 Hz and digitized at 5 kHz and or were filtered at 2 kHz (Frequency Devices, Haverhill, MA) and digitized at 50 kHz. The Ca2+-dependent fluorescent signals were expressed as the change in fluorescence divided by the unstimulated fluorescence (ΔF/F). All analysis was performed by using custom macros written in IgorPro (Wavemetrics, Lake Oswego, OR). Averages are given as the mean ± SEM.
Results
We first examined whether a reduction in presynaptic Cainflux accounts for the cannabinoid-mediated inhibition of excitatory synaptic transmission at the granule cell to Purkinje cell synapse. Parallel fibers were stimulated extracellularly, and the resulting EPSCs were measured in Purkinje cells in whole-cell voltage-clamp mode. In separate experiments presynaptic Cainflux was quantified with the low-affinity calcium indicator, magnesium green, which provides a linear measure of changes in presynaptic Cainflux (Regehr, 2000). The cannabinoid receptor agonist WIN55,212-2 (5 μm) reduced both the EPSC amplitude and presynaptic Cainflux. A representative experiment shows the effect of WIN55,212-2 on EPSCs (Fig. 1A). On average, the EPSC was suppressed to 12 ± 4% of control (n = 4). WIN55,212-2 also decreased Cainflux (Fig. 1B). On average, peak Cainflux was decreased to 42 ± 3% of control (n = 6).
Figure 1.

Cannabinoid receptor activation attenuates parallel fiber EPSCs by decreasing presynaptic Cainflux. EPSCs (A, E) and presynaptic calcium transients (B-D, F) evoked by single stimulations of the parallel fiber pathway are shown. The cannabinoid receptor agonist WIN55,212-2 (5 μm) greatly reduced the amplitudes of both EPSCs (A) and presynaptic calcium transients (B). C, D, The depth of the loaded parallel fibers had a large effect on the time course of inhibition after the bath application of WIN55,212-2. C, The loaded fibers were ∼5 μm from the surface of the slice; in D they were ∼80 μm deep in the tissue. Methanandamide (5 μm), a partial cannabinoid receptor agonist, modestly reduced parallel fiber EPSCs (E) and presynaptic calcium transients (F). G, The peak EPSC amplitude is plotted as a function of peak presynaptic Cainflux in the presence of WIN55,212-2 and methanandamide. The power law relationship between presynaptic Cainflux and EPSC amplitude determined in previous studies (Eq. 1; n = 2.5-3.5) is plotted in gray for the purpose of comparison. A fit to Equation 1 for the methanandamide and WIN55,212-2 points yielded n = 3.0 (thick line). This fit was determined by using the logarithmic form of the function weighted by the SDs of the EPSCs measured in methanandamide and WIN55,212-2 and constrained to pass through the (100, 100) point.
We next determined the relationship between EPSC amplitude and Cainflux during cannabinoid modulation. We first evaluated the feasibility of determining a dose-response curve for the effects of WIN55,212-2 on both EPSC amplitude and Cainflux.We were, however, concerned that the highly lipophilic property shared by endocannabinoid receptor agonists and antagonists would interfere with their penetration into the slice preparation. We therefore took advantage of the predictable anatomy of parallel fibers in the transverse cerebellar slice to test the effectiveness of low concentrations of WIN55,212-2 in the slice. We loaded bands of parallel fibers with magnesium green and used optical methods to determine the depth of the labeled fibers. We then monitored Cainflux in the presence of varying concentrations of the agonist WIN55,212-2. In Figure 1C, parallel fibers running along the very surface of the slice were loaded with magnesium green. Within minutes, 0.1 μm WIN55,212-2 substantially decreased Cainflux. Increasing concentrations of agonist had little additional effect on Cainflux. However, when fibers 80 μm from the surface of the slice were loaded, 0.1 μm WIN55,212-2 had no discernible effect after 20 min (Fig. 1D). Even in the presence of 10 μm WIN55,212-2, almost 40 min were required to reach a stable baseline despite the nanomolar affinity of WIN55,212-2 for cannabinoid receptors. These results demonstrate that it is impractical to perform a dose-response curve because it is difficult to control the concentrations of lipophilic agonists such as WIN55,212-2 within a brain slice.
Therefore, rather than using low concentrations of a full agonist such as WIN55,212-2, we used saturating concentrations of methanandamide, a partial agonist of the CB1 receptor (Pertwee, 1999). Methanandamide (5 μm) reduced both the peak EPSC (Fig. 1E; to 32 ± 5% of control; n = 5) and presynaptic Cainflux (Fig. 1F; to 74 ± 1% of control; n = 4), but to a lesser extent than WIN55,212-2.
The relationship between EPSC amplitude and Cainflux allowed us to determine whether modulation by cannabinoids arises solely from changes in Cainflux. Previous studies have shown that, when Cainflux is altered at this synapse, the relationship between the amplitude of the EPSC and Cainflux is approximated by the expression:
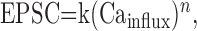 |
1 |
where k is a constant and n has been determined experimentally to be between 2.5 and 3.5 (Mintz et al., 1995; Dittman and Regehr, 1996; Sabatini and Regehr, 1997; Xu-Friedman and Regehr, 2000). This relationship is indicated by the shaded region of Figure 1G. If presynaptic modulation does not simply reduce Cainflux, the relationship deviates significantly from this line (Dittman and Regehr, 1996). In the extreme in which decreased Cainflux was not involved, reductions in EPSC amplitude are not accompanied by change in Cainflux (Chen and Regehr, 1997). This corresponds to a vertical line, with Cainflux = 100%. If the cannabinoid-mediated modulation at this synapse arises solely from alterations in Cainflux, the relationship between Cainflux and release measured in the presence of cannabinoid receptor agonists should fall within this shaded region (Dittman and Regehr, 1996). We found this to be the case, because the relationship between EPSC amplitude and Cainflux is well approximated by Equation 1, with n = 3.0 (Fig. 1G, thick line). Because the cannabinoid-mediated changes in EPSC amplitude can be accounted for by the decrease in Cainflux (Fig. 1G), these results suggest that cannabinoids inhibit parallel fiber to Purkinje cell synapses entirely by reducing presynaptic Cainflux.
Cannabinoids do not alter the presynaptic waveform
Previous studies have shown that presynaptic Cainflux can be reduced either by alterations in the waveform of the action potential or by direct modulation of presynaptic calcium channels. Subtle changes in the presynaptic action potential can lead to large changes in presynaptic Cainflux (Augustine, 1990; Byrne and Kandel, 1996; Sabatini and Regehr, 1997; Qian and Saggau, 1999; Zucker and Regehr, 2002). This suggests that modulation of potassium channels involved in spike repolarization can regulate synaptic strength. Alternatively, activation of G-protein-coupled receptors can modulate presynaptic calcium channels directly without affecting the presynaptic waveform (Zucker and Regehr, 2002).
Recent studies have suggested that the cannabinoid-mediated decrease in presynaptic Cainflux arises indirectly from modulation of presynaptic potassium channels (Alger et al., 1996; Daniel and Crepel, 2001; Varma et al., 2002; Diana and Marty, 2003). Studies showing that the potassium channel antagonist 4-AP (1 mm) prevents CB1-mediated inhibition of Cainflux present the strongest evidence for this hypothesis. However, no obvious change in the presynaptic volley, a sensitive means of detecting changes in the presynaptic waveform, has been identified in response to cannabinoid receptor agonists. This calls into question the role of changes in the presynaptic action potential in cannabinoid-mediated inhibition of synaptic transmission (Lenz et al., 1998; Takahashi and Linden, 2000; Daniel and Crepel, 2001).
To clarify the role of potassium channels in the cannabinoid-mediated decrease in presynaptic Cainflux, we first reexamined the effect of 4-AP on the parallel fiber to Purkinje cell synapse to address several concerns. First, 4-AP itself may affect the presynaptic waveform profoundly and alter calcium signaling in the presynaptic terminal. Under these conditions it may be difficult to examine the mechanisms of cannabinoid action. Second, the original study assessed Cainflux evoked by five stimuli at 100 Hz. It is not clear whether parallel fibers can follow this frequency of stimulation faithfully in the presence of 4-AP.
We first assessed the effect of 1 mm 4-AP on the timing and amplitude of presynaptic Cainflux by using the low-affinity calcium-sensitive indicator mag-fura-5 (dissociation constant of 28 μm for calcium). 4-AP increased calcium levels to >25 times that observed in control conditions (Fig. 2A). In addition, 4-AP greatly slowed the time course of the calcium signal. The pronounced difference in the time courses of the calcium signal under control conditions and in the presence of 4-AP is underscored by comparing the normalized calcium transients evoked under these two conditions (Fig. 2B). Summary data from five experiments are shown in Figure 2C. The time to peak (which includes >1 msec for the evoked presynaptic action potential to reach the recording site) increased from 2.3 ± 0.4 msec in control conditions to 174 ± 12 msec in the presence of 4-AP (n = 5).
Figure 2.

The potassium channel antagonist 4-AP profoundly alters presynaptic calcium signaling. A, Representative traces of presynaptic calcium signals evoked by single stimuli quantified with the calcium-sensitive indicator mag-fura-5 are shown on the same scale under control conditions and in the presence of 1 mm 4-AP. B, Representative traces of presynaptic calcium signals were normalized to illustrate the difference in the time course of the signals. C, The amplitude (top) and time course (bottom) of calcium signals were averaged for five experiments. The presynaptic calcium signals (D-F) and the presynaptic volleys (G-I) evoked by one and two stimuli were quantified also. Representative traces of the calcium signal evoked by one (thick traces) and two (thin traces) stimuli are shown under control conditions (D) and in the presence of 1 mm 4-AP (E). Stimulation times in D and E are indicated by vertical bars below the traces. F, These results are summarized for five experiments. G, Representative traces of the presynaptic volleys under control conditions (top) and in the presence of 4-AP (bottom) are shown. H, The first presynaptic volleys under control conditions (thick trace) and in the presence of 4-AP (thin trace) are shown. I, The presynaptic volley amplitudes were normalized to the values measured before the application of 4-AP. The amplitudes of the first (open squares) and second (open circles) presynaptic volleys were averaged for five experiments.
The slow rise time of the calcium signal indicates that Cainflux persisted for >100 msec in the presence of 4-AP. Furthermore, it suggests that repolarization of the presynaptic action potential might not be complete for >100 msec. To test this hypothesis, we examined the calcium signals evoked by one and two stimuli in control conditions and in the presence of 4-AP. In control conditions the incremental increase in fluorescence was similar for a single stimulus and the second of two stimuli (ΔF2/ΔF1 ≈ 1; Fig. 2D). However, in the presence of 4-AP the second stimulus generated only a very slight additional increase in fluorescence (ΔF2/ΔF1 ≈ 0.07; Fig. 2E). These data are summarized in Figure 2F (n = 5). This small incremental increase did not arise from saturation of the calcium indicator, because we used a calcium indicator with a low calcium sensitivity.
Further evidence that 4-AP profoundly alters the presynaptic waveform is suggested by the effect of 4-AP on the presynaptic volley. Under control conditions two stimuli separated by 20 msec evoked presynaptic volleys with the triphasic structure characteristic of a propagating action potential, although the volley activated by the second stimulus was slightly smaller in amplitude (Fig. 2G, top). In the presence of 4-AP the amplitude of the first volley was reduced, and the second volley was virtually eliminated (Fig. 2G, bottom). The effects of 4-AP on the amplitude of the first and second volley are summarized in Figure 2I (n = 4). In addition, 4-AP profoundly altered the shape of the first presynaptic volley (Fig. 2H).
Taken together, these data indicate that, in the presence of 1 mm 4-AP, calcium signaling in the presynaptic terminal differs markedly from control conditions. The magnitude of the presynaptic Cainflux is increased by >25-fold and persists for ∼85 times longer than under control conditions. Furthermore, in 4-AP a second stimulus no longer evokes a presynaptic volley nor causes a significant incremental increase in Cainflux. These findings suggest that 4-AP prevents spike repolarization for >100 msec. Because the effects of 4-AP on cannabinoid modulation also have been examined in the presence of low external calcium (Cae), we also characterized the presynaptic calcium signals evoked in 0.1 mm Cae. We found that in 0.1 mm Cae the time to peak of the presynaptic calcium signal also was delayed greatly (89 ± 10 msec; n = 4). These effects of 4-AP on presynaptic calcium signaling make it difficult to use 4-AP to examine the mechanism of cannabinoid action.
We therefore took a different approach to assess whether CB1 receptor activation decreased presynaptic Cainflux by altering the presynaptic action potential. Previously, we showed that, under appropriate experimental conditions, the time course of presynaptic Cainflux is highly sensitive to changes in the presynaptic waveform. Furthermore, the derivative of the presynaptic ΔF/F signals evoked by a single stimulus provides a good measure of the time course of presynaptic Cainflux (Sabatini and Regehr, 1997, 1998). Therefore, if the large decrease in Cainflux after CB1 receptor activation arises from more rapid action potential repolarization and a narrowing of the presynaptic action potential, there should be a substantial effect on the time course of Cainflux.
Presynaptic calcium transients were measured with the fluorescent calcium indicator magnesium green while the presynaptic volleys were monitored. The effects of WIN55,212-2, the cannabinoid receptor antagonist AM251, and the potassium channel antagonist tetraethylammonium (TEA) then were assessed. TEA was used because it provides a means of broadening the presynaptic action potential in a more controlled manner than is possible with 4-AP (Sabatini and Regehr, 1997). In the representative experiment shown in Figure 3A, WIN55,212-2 reduced Cainflux to 48% of control. AM251 reversed this inhibition. Application of TEA (100 μm) increased Cainflux to 133% of control. The presynaptic volley was unaffected by CB1 receptor activation (Fig. 3B, middle), whereas TEA altered the volley consistent with an effect on the presynaptic waveform (Fig. 3B, right).
Figure 3.
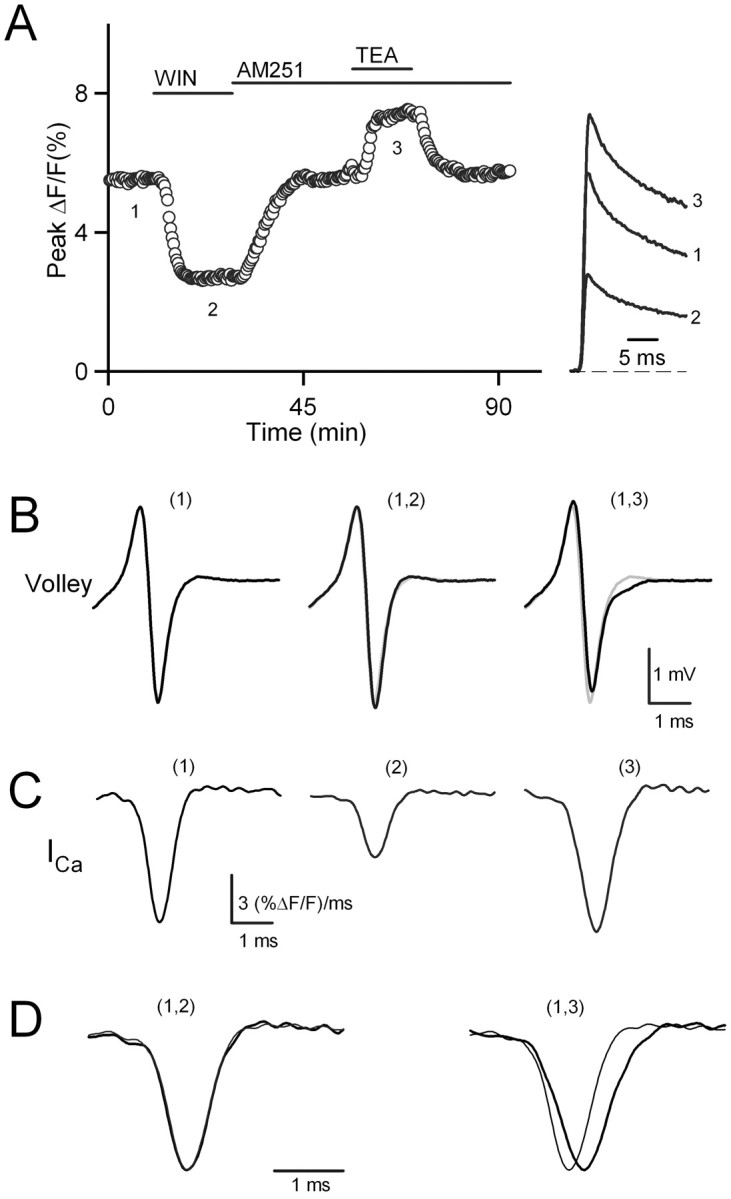
Activation of CB1 receptors does not affect the time course of presynaptic calcium influx. Fluorescence changes evoked by single stimuli were measured with the calcium-sensitive indicator magnesium green and a photodiode with a response time of 3 kHz. A, The effects of WIN55,212-2, AM251, and TEA on both the peak ΔF/F signal (left) and on the time course measured during the indicated times (right) are shown for a representative experiment. In addition, the presynaptic volley was monitored (B) at the times indicated in A (left). B, The presynaptic volley is shown under control conditions (left) and in the presence of WIN55,212-2 (middle, black trace) or TEA (right, black trace) as compared with the volley under control conditions (middle and right, gray traces). C, The derivative of the fluorescence transient indicates the time course of presynaptic calcium influx corresponding to ICa under control conditions (left) and in the presence of WIN55,212-2 (middle) or TEA (right). D, The time courses of presynaptic calcium influx measured in the presence of WIN55,212-2 (left, bold trace) and in the presence of TEA (right, bold trace) are normalized and compared with control traces (left and right, thin traces).
The derivative of the fluorescence transients provides a measure of the presynaptic calcium current (ICa). CB1 receptor activation reduced the amplitude of ICa without affecting the time course of Cainflux (Fig. 3C). On the other hand, TEA increased the total Cainflux primarily by broadening the time course of the presynaptic calcium current without affecting the peak of ICa (Fig. 3C). Comparing the normalized calcium currents under control conditions and in the presence of WIN55,212-2 highlights the lack of any effect of CB1 receptor activation on the time course of Cainflux (Fig. 3D, left). The alteration in the time course of Cainflux in TEA is clear (Fig. 3D, right). The average width at half-maximum (w1/2) of ICa was 658 ± 15 μsec under control conditions, 655 ± 21 μsec in the presence of WIN55,212-2, and 776 ± 23 μsec in the presence of TEA (n = 4). Thus, although WIN55,212-2 had a larger effect on total Cainflux than did TEA (45 ± 4% of control compared with 127 ± 3% for TEA; n = 4), activation of the CB1 receptor did not alter the half-width of ICa significantly (Student's t test, p > 0.5), whereas spike broadening with TEA did (Student's t test, p < 0.01).
These findings argue against CB1 receptor activation altering Cainflux by affecting the presynaptic action potential. Although the effect of TEA on Cainflux was smaller than that of WIN55,212-2, TEA clearly affected the presynaptic volley and the time course of presynaptic calcium entry, whereas WIN55,212-2 did not. Thus we conclude that the cannabinoid-mediated reduction in presynaptic Cainflux does not arise from potassium channel modulation. Rather, the decrease in Cainflux reflects direct modulation of calcium channels.
Cannabinoids modulate multiple classes of calcium channels
We next examined the classes of presynaptic calcium channels modulated by cannabinoid receptor activation. The three types of calcium channels contributing to neurotransmitter release at the parallel fiber to Purkinje cell synapse are separable pharmacologically: N-type calcium channels correspond to CaV2.2 and are blocked selectively by ω-conotoxin GVIA (CgTx), and P/Q-type channels that correspond to CaV2.1 are blocked selectively by ω-agatoxin IVA (Aga-IVA); the remaining fraction is resistant to both of these toxins (Mintz et al., 1995; Ertel et al., 2000). We refer to this component that is resistant to organic blockers of L-type calcium channels and these peptide toxins as R-type (Randall and Tsien, 1995; Catterall et al., 2003). It is likely that this component is mediated primarily by CaV2.3.
Calcium channel modulation was not restricted to a single class of channels. First we blocked N-type calcium channels with 500 nm CgTx and assessed the effect of WIN55,212-2 on the remaining fraction (Fig. 4A). Cannabinoid receptor activation inhibited Cainflux even when N-type calcium channels were blocked, demonstrating that the modulation of calcium channels by cannabinoid receptor activation is not restricted to N-type channels. CgTx alone blocked 25 ± 2% of the peak Cainflux. Coapplication of WIN55,212-2 blocked an additional 38 ± 3% of the peak Cainflux under control conditions (n = 3). WIN55,212-2 also reduced Cainflux when P/Q-type calcium channels were blocked with 200 nm Aga-IVA (Fig. 4B), indicating that calcium channel modulation similarly is not restricted to P/Q-type channels. Aga-IVA alone blocked 47 ± 3% of the peak Cainflux. Coapplication of WIN55,212-2 blocked an additional 31 ± 2% of the peak Cainflux under control conditions (n = 3). Finally, CgTx and Aga-IVA were coapplied to block N-type and P/Q-type calcium channels simultaneously to assess the effect of WIN55,212-2 on the remaining component mediated by R-type calcium channels. The R-type component clearly was inhibited by activation of cannabinoid receptors (Fig. 4C). The combination of CgTx and Aga-IVA reduced peak Cainflux by 77 ± 2%. Coapplication of WIN55,212-2 blocked an additional 11 ± 1% of the peak Cainflux under control conditions (n = 4).
Figure 4.
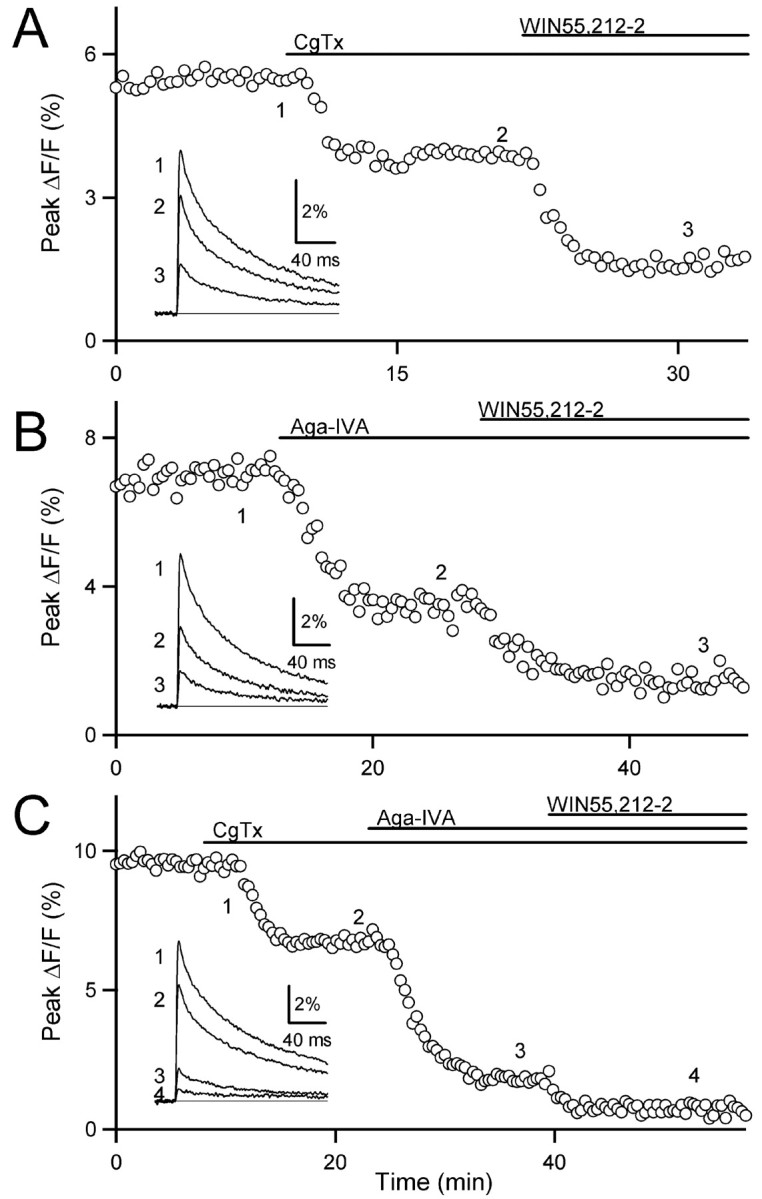
CB1 receptor activation modulates three types of calcium channel at the parallel fiber to Purkinje cell synapse. A-C, Representative experiments showing the effects of the cannabinoid receptor agonist WIN55,212-2 (5 μm), the N-type calcium channel blocker ω-conotoxin GVIA (CgTx; 500 nm), and the P/Q-type calcium channel blocker ω-agatoxin IVA (Aga-IVA; 200 nm) on presynaptic calcium transients. A, With CgTx-sensitive channels blocked, application of WIN55,212-2 further decreased Cainflux. B, Likewise, with Aga-IVA-sensitive channels blocked, application of WIN55,212-2 further decreased Cainflux. C, After the coapplication of CgTx and Aga-IVA, the application of WIN55,212-2 further inhibited Cainflux through the toxin-insensitive channels.
N-type calcium channels are most sensitive to cannabinoid modulation
To determine the percentage of each calcium channel type modulated by cannabinoid receptor activation, we first applied WIN55,212-2 and then determined the effects of CgTx and Aga-IVA on Cainflux. As shown in a representative experiment, the application of CgTx and Aga-IVA still reduced Cainflux even in the presence of this cannabinoid receptor agonist (Fig. 5).
Figure 5.

CB1 receptor activation inhibits a subset of each class of calcium channel. After the application of WIN55,212-2, application of CgTx and Aga-IVA each further reduced presynaptic Cainflux.
Using this approach, we quantified the contribution of each type of calcium channel to Cainflux in the presence of WIN55,212-2. Expressed as a percentage of total Cainflux in control conditions, the Cainflux was 8 ± 1, 28 ± 3, and 13 ± 1% through N-, P/Q-, and R-type calcium channels, respectively (Fig. 6A, black bars; n = 5). In these experiments the sum of the three contributions was 49%, the total Cainflux in WIN55,212-2 relative to control. These results are compared with the fraction of Cainflux through each channel type in control conditions (Fig. 6A, open bars). In control conditions the Cainflux was 27 ± 1% (n = 7), 48 ± 2% (n = 7), and 23 ± 3% (n = 4) for N-, P/Q-, and R-type calcium channels, respectively.
Figure 6.

Activation of cannabinoid receptors differentially affects three components of presynaptic calcium influx. A, The percentage contribution of each calcium channel type to Cainflux in parallel fiber synapses in control conditions (open bars) and in the presence of 5 μm WIN55,212-2 (filled bars). Contributions are expressed as the percentage of total Cainflux in control conditions. B, The percentage of inhibition by WIN55,212-2 of each type of voltage-gated calcium channel calculated as described in Results from A.
We then used these experiments to determine the effect of cannabinoid receptor activation on each component of presynaptic Cainflux (Fig. 6). Expressed in terms of the total Cainflux in control conditions, the contribution of N-type dropped from 27 ± 1% in control conditions to 8 ± 1% in the presence of WIN55,212-2. Thus WIN55,212-2 reduced calcium entry through N-type calcium channels by 71 ± 8% (100%-8%/27%) (Fig. 6B). Similar measurements indicated that at this synapse WIN55,212-2 reduced calcium entry through P/Q-type calcium channels by 40 ± 8% and through R-type channels by 45 ± 13% (Fig. 6B). A comparison of the effect of WIN55,212-2 on the different types of calcium channels revealed that, although there was no significant difference in the modulation of P/Q- and R-type calcium channels, N-type channels were inhibited to a greater extent than were either P/Q-type or R-type calcium channels (p < 0.05 for both, Student's t test). Therefore, although N-type channels are modulated most strongly by cannabinoid receptor activation, all three calcium channel types found at this synapse are inhibited strongly.
Discussion
In this study we have clarified the mechanism by which cannabinoids modulate synaptic transmission at the parallel fiber to Purkinje cell synapse. We tested two current hypotheses and found that cannabinoids inhibited transmission neither by modulating potassium channels and narrowing the presynaptic action potential nor by selectively modulating N-type calcium channels. Rather, cannabinoid-mediated inhibition of synaptic transmission reflected presynaptic modulation of all three calcium channel types that underlie transmitter release at this synapse.
Cannabinoids reduce presynaptic calcium entry
We found that generating dose-response curves with the highly lipophilic cannabinoid receptor agonists and antagonists was impractical in cerebellar brain slices. Optical methods revealed that WIN55,212-2, a widely used cannabinoid agonist, acted slowly and incompletely even at synapses located <100 μm within the slice. These data underscore the limitations of using cannabinoid agonists and antagonists in physiological studies when the depth of the synapses being studied is difficult to control or unknown. These difficulties prevented us from completing a traditional dose-response curve with WIN55,212-2 to determine the relationship between presynaptic Cainflux and peak EPSC amplitude. Instead, we quantified the effects on Cainflux and the EPSC amplitude of a partial and a full agonist of CB1 receptors. This approach promises to be a generally useful strategy for studying the effects of cannabinoids in brain slices. This approach revealed that the relationship between Cainflux and the EPSC amplitude was consistent with cannabinoids decreasing the EPSC amplitude exclusively by inhibiting presynaptic Cainflux (Fig. 1G).
The observation that cannabinoids inhibit transmission at the parallel fiber to Purkinje cell synapse solely by reducing presynaptic calcium entry is consistent with previous studies. First, these studies showed that cannabinoid receptor agonists do not alter the frequency or amplitude of miniature EPSCs at the parallel fiber to Purkinje cell synapse, suggesting that cannabinoid receptor activation does not affect presynaptic processes downstream from calcium entry or postsynaptic AMPA receptor sensitivity (Levenes et al., 1998; Takahashi and Linden, 2000). Second, by monitoring presynaptic Cainflux with high affinity indicators, they demonstrated that cannabinoids reduce Cainflux, although the extent of the reduction could not be quantified (Daniel and Crepel, 2001). Finally, they suggested that cannabinoids acted, at least in part, by modulating presynaptic calcium channels, consistent with our results (Levenes et al., 1998; Takahashi and Linden, 2000).
Cannabinoids do not narrow the presynaptic action potential
Previous studies have shown that 1 mm 4-AP prevents cannabinoid-mediated inhibition of synaptic transmission of parallel fibers (Daniel and Crepel, 2001). These results gave rise to the hypothesis that cannabinoid receptor activation modulates 4-AP-sensitive potassium channels, thereby narrowing the presynaptic action potential and indirectly reducing presynaptic Cainflux. Several of our experimental findings argue against this hypothesis. First, in 1 mm 4-AP the Cainflux increased by >25-fold and persisted for ∼85 times longer than under control conditions. Furthermore, in 4-AP a second stimulus was unable to evoke a presynaptic volley. Together these findings suggest that 1 mm 4-AP may prolong spike repolarization for 100 msec, dramatically altering calcium signaling in the presynaptic terminal. Thus our findings suggest that experiments using high concentrations of potassium channel antagonists to study synaptic transmission are difficult to interpret. Second, by measuring the time course of presynaptic calcium entry, we found that cannabinoids do not cause a narrowing of the presynaptic action potential. These findings are consistent with our results and those of previous studies showing that the PF presynaptic volley is unaffected by cannabinoids (Levenes et al., 1998; Takahashi and Linden, 2000). Third, we showed that presynaptic calcium channel types are modulated differentially (see below), which is inconsistent with inhibition arising exclusively from changes in the presynaptic waveform (Sabatini and Regehr, 1997). Finally, we considered the possibility that potassium channel modulation occurs in concert with calcium channel modulation to offset any waveform changes that arise from calcium channel modulation (as might occur if calcium-activated potassium conductances contributed to spike repolarization at this synapse). This seems unlikely because changes in presynaptic calcium entry do not change the presynaptic waveform significantly at this synapse (Sabatini and Regehr, 1997). Thus multiple lines of evidence argue that modulation of presynaptic potassium channels does not contribute significantly to cannabinoid-mediated inhibition of synaptic transmission at this synapse.
Cannabinoids modulate multiple types of calcium channels at this synapse
We found that cannabinoids modulate all three classes of calcium channels mediating transmitter release at PF terminals. We found that N-type channels are modulated more strongly by cannabinoids than either P/Q-type or R-type channels. However, because calcium flowing through N-type channels accounts for only ∼30% of presynaptic Cainflux at PF terminals, modulation of P/Q- and R-type channels mediates the bulk of cannabinoid-mediated changes in presynaptic Cainflux at this synapse. In addition, because P/Q-type calcium channels are particularly effective at evoking release at this synapse (Mintz et al., 1995), modulation of P/Q-type calcium channels likely accounts for most of the cannabinoid-mediated inhibition of the EPSC.
The modulation of N-type, P/Q-type, and R-type calcium channels is consistent with studies in culture and expression systems demonstrating that a variety of voltage-gated calcium channels can be modulated by cannabinoids (Ameri, 1999; Alger, 2002). However, our results differ from those in hippocampal, striatal, and trigeminal neurons, in which blocking presynaptic N-type calcium channels eliminates the effects of cannabinoids (Huang et al., 2001; Wilson et al., 2001; Liang et al., 2004). The difference between these studies and our results simply may reflect the types of channels present in the presynaptic terminals under investigation rather than any selectivity of the cannabinoid signaling system. For example, although three types of calcium channels mediate transmission at parallel fibers, only N-type calcium channels mediate transmission for the class of hippocampal interneurons that express cannabinoid receptors (Katona et al., 1999; Tsou et al., 1999; Wilson et al., 2001; Alger, 2002). Our findings indicate that the hypothesis that cannabinoids exclusively modulate N-type calcium channels in presynaptic terminals does not hold in general. In the specific cases in which it does hold, this specificity may reflect the modulation of the only type of calcium channel present in the presynaptic bouton.
Footnotes
This work was supported by National Institutes of Health Grants RO1 NS 32405 and RO1 NS 44396. We thank M. Beierlein, D. Blitz, S. Brenowitz, K. Foster, A. Kreitzer, and M. Xu-Friedman for comments on this manuscript.
Correspondence should be addressed to Wade Regehr, Goldenson 308, Department of Neurobiology, Harvard Medical School, 220 Longwood Avenue, Boston, MA 02115. E-mail: wade_regehr@hms.harvard.edu.
S. P. Brown's present address: Department of Comparative Medicine, Stanford University School of Medicine, Edwards Building R314, 300 Pasteur Drive, Stanford, CA 94305-5342.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245623-09$15.00/0
References
- Alger BE (2002) Retrograde signaling in the regulation of synaptic transmission: focus on endocannabinoids. Prog Neurobiol 68: 247-286. [DOI] [PubMed] [Google Scholar]
- Alger BE, Pitler TA, Wagner JJ, Martin LA, Morishita W, Kirov SA, Lenz RA (1996) Retrograde signaling in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J Physiol (Lond) 496[Pt 1]: 197-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameri A (1999) The effects of cannabinoids on the brain. Prog Neurobiol 58: 315-348. [DOI] [PubMed] [Google Scholar]
- Augustine GJ (1990) Regulation of transmitter release at the squid giant synapse by presynaptic delayed rectifier potassium current. J Physiol (Lond) 431: 343-364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JH, Kandel ER (1996) Presynaptic facilitation revisited: state and time dependence. J Neurosci 16: 425-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Striessnig J, Snutch TP, Perez-Reyes E (2003) International Union of Pharmacology. XL. Compendium of voltage-gated ion channels: calcium channels. Pharmacol Rev 55: 579-581. [DOI] [PubMed] [Google Scholar]
- Chen C, Regehr WG (1997) The mechanism of cAMP-mediated enhancement at a cerebellar synapse. J Neurosci 17: 8687-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel H, Crepel F (2001) Control of Ca2+ influx by cannabinoid and metabotropic glutamate receptors in rat cerebellar cortex requires K+ channels. J Physiol (Lond) 537: 793-800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R (1992) Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science 258: 1946-1949. [DOI] [PubMed] [Google Scholar]
- Diana MA, Marty A (2003) Characterization of depolarization-induced suppression of inhibition using paired interneuron-Purkinje cell recordings. J Neurosci 23: 5906-5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A (2002) Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci 22: 200-208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D (1994) Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature 372: 686-691. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG (1996) Contributions of calcium-dependent and calcium-independent mechanisms to presynaptic inhibition at a cerebellar synapse. J Neurosci 16: 1623-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elphick MR, Egertova M (2001) The neurobiology and evolution of cannabinoid signaling. Philos Trans R Soc Lond B Biol Sci 356: 381-408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA (2000) Nomenclature of voltage-gated calcium channels. Neuron 25: 533-535. [DOI] [PubMed] [Google Scholar]
- Herrington J, Bookman RJ (1995) Pulse control v4.5: IGOR XOPs for patch-clamp data acquisition. Miami: University of Miami.
- Hoffman AF, Lupica CR (2000) Mechanisms of cannabinoid inhibition of GABAA synaptic transmission in the hippocampus. J Neurosci 20: 2470-2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CC, Lo SW, Hsu KS (2001) Presynaptic mechanisms underlying cannabinoid inhibition of excitatory synaptic transmission in rat striatal neurons. J Physiol (Lond) 532: 731-748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Sperlagh B, Sik A, Kafalvi A, Vizi ES, Mackie K, Freund TF (1999) Presynaptically located CB1 cannabinoid receptors regulate GABA release from axon terminals of specific hippocampal interneurons. J Neurosci 19: 4544-4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001a) Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron 29: 717-727. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2001b) Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci 21: RC174(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG (2002) Retrograde signaling by endocannabinoids. Curr Opin Neurobiol 12: 324-330. [DOI] [PubMed] [Google Scholar]
- Lenz RA, Wagner JJ, Alger BE (1998) N- and L-type calcium channel involvement in depolarization-induced suppression of inhibition in rat hippocampal CA1 cells. J Physiol (Lond) 512: 61-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenes C, Daniel H, Soubrie P, Crepel F (1998) Cannabinoids decrease excitatory synaptic transmission and impair long-term depression in rat cerebellar Purkinje cells. J Physiol (Lond) 510: 876-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang YC, Huang CC, Hsu KS, Takahashi T (2004) Cannabinoid-induced presynaptic inhibition at the primary afferent trigeminal synapse of juvenile rat brainstem slices. J Physiol (Lond) 555: 85-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maejima T, Ohno-Shosaku T, Kano M (2001) Endogenous cannabinoid as a retrograde messenger from depolarized postsynaptic neurons to presynaptic terminals. Neurosci Res 40: 205-210. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG (1995) Calcium control of transmitter release at a cerebellar synapse. Neuron 15: 675-688. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M (2001) Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron 29: 729-738. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (1999) Pharmacology of cannabinoid receptor ligands. Curr Med Chem 6: 635-664. [PubMed] [Google Scholar]
- Qian J, Saggau P (1999) Modulation of transmitter release by action potential duration at the hippocampal CA3-CA1 synapse. J Neurophysiol 81: 288-298. [DOI] [PubMed] [Google Scholar]
- Randall A, Tsien RW (1995) Pharamcological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. J Neurosci 15: 2995-3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG (2000) Monitoring presynaptic calcium dynamics with membrane-permeant indicators. In: Imaging neurons: a laboratory manual (Yuste R, Lanni F, Konnerth A, eds), pp 37.1-37.11. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Regehr WG, Mintz IM (1994) Participation of multiple calcium channel types in transmission at single climbing fiber to Purkinje cell synapses. Neuron 12: 605-613. [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ (2001) Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci 21: 109-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG (1997) Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. J Neurosci 17: 3425-3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG (1998) Optical measurement of presynaptic calcium currents. Biophys J 74: 1549-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella N, Schweitzer P, Piomelli D (1997) A second endogenous cannabinoid that modulates long-term potentiation. Nature 388: 773-778. [DOI] [PubMed] [Google Scholar]
- Sullivan JM (1999) Mechanisms of cannabinoid receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol 82: 1286-1294. [DOI] [PubMed] [Google Scholar]
- Takahashi KA, Linden DJ (2000) Cannabinoid receptor modulation of synapses received by cerebellar Purkinje cells. J Neurophysiol 83: 1167-1180. [DOI] [PubMed] [Google Scholar]
- Tsou K, Mackie K, Sanudo-Pena MC, Walker JM (1999) Cannabinoid CB1 receptors are localized primarily on cholecystokinin-containing GABAergic interneurons in the rat hippocampal formation. Neuroscience 93: 969-975. [DOI] [PubMed] [Google Scholar]
- Varma N, Brager D, Morishita W, Lenz RA, London B, Alger B (2002) Presynaptic factors in the regulation of DSI expression in hippocampus. Neuropharmacology 43: 550-562. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2001) Endogenous cannabinoids mediate retrograde signaling at hippocampal synapses. Nature 410: 588-592. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296: 678-682. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Kunos G, Nicoll RA (2001) Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron 31: 453-462. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG (2000) Probing fundamental aspects of synaptic transmission with strontium. J Neurosci 20: 4414-4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Hashimoto K, Zimmer A, Maejima T, Araishi K, Kano M (2002) The cannabinoid CB1 receptor mediates retrograde signals for depolarization-induced suppression of inhibition in cerebellar Purkinje cells. J Neurosci 22: 1690-1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]