

Abstract
PKC plays a critical role in competitive activity-dependent synapse modification at the neuromuscular synapse in vitro and in vivo. This action involves a reduction of the strength of inactive inputs to muscle cells that are activated by other inputs. A decrease of postsynaptic responsiveness and a loss of postsynaptic acetyl choline receptors account for the heterosynaptic loss in vitro. The loss is not seen in preparations in which PKC has been blocked pharmacologically. Here, we show that the loss does not occur in in vitro preparations made from animals genetically modified to lack the theta isoform of PKC. Synapse elimination in the newborn period in vivo is delayed but is eventually expressed in knock-out animals. PKC-dependent synapse reduction is suppressed in heterologous cultures combining normal nerve and PKC theta-deficient muscle, as might be expected from the postsynaptic locus of the changes that underlie the activity-dependent plasticity. Preparations in which PKC theta-deficient neurons innervated normal muscle also exhibited a marked deficit in PKC-deficient synapse reduction. The presynaptic action of PKC theta implied by this observation is blocked by TTX, and we propose that activity-related synapse strengthening is decreased by presynaptic PKC theta. Thus, PKC theta in both presynaptic and postsynaptic elements plays a critical role in activity-dependent synapse modulation and loss. We provide a model for activity-dependent synapse loss incorporating these findings.
Keywords: PKC theta, knock-out, synapse elimination, neuromuscular junction, in vivo, in vitro
Introduction
Neurons and synapses typically are generated in excessive, redundant numbers initially and subsequently undergo a reduction in numbers and complexity. This “pruning” is regulated by the functional activity of the circuits and results in more effective organization of the circuits. Neurotrophins and proteases have been implicated in this process (O'Brien et al., 1978; Cabelli et al., 1997). Such selective activity-dependent modification of synaptic strength constitutes a major mechanism for the epigenetic regulation of the development of synaptic circuits (Shatz, 1990; Fields and Nelson, 1992). Synaptic pruning has been studied extensively at the neuromuscular junction (NMJ) at which major neonatal activity-dependent modification of synaptic connections occurs (Brown et al., 1976; Thompson, 1985). Alterations in both postsynaptic receptor stability and neural structures contribute to the elimination process (Sanes and Lichtman, 1999).
We have used both in vivo and in vitro preparations in explicating some of the cell biological mechanisms underlying this synapse elimination (Jia et al., 1999; Lanuza et al., 2000, 2001, 2002). Serine protein kinases [both protein kinase A (PKA) and PKC] play a role in the use-dependent synapse modification, with PKC activation resulting in a decrease in synaptic strength, whereas PKA action opposes or reverses the PKC effect (Li et al., 2001a, 2002). The involvement of PKC in the synapse elimination process was also observed in vivo but was seen only during the first two-thirds of the 2–3 week postnatal period during which synapse elimination occurs in vivo (Lanuza et al., 2001). It appeared that our cultured preparations correspond to a relatively early stage of the normal developmental process, because synapse strength was always affected by PKC activation throughout the 3–5 week period that we maintained them in vitro.
PKC comprises a family of related isoforms with different properties and tissue distributions (Tanaka and Nishizuka, 1994; Newton, 1995). A mouse strain in which the PKC theta gene has been inactivated has become available (Sun et al., 2000). This isoform has been shown to be one of the forms strongly expressed in skeletal muscle and is under a degree of neural control (Hilgenberg and Miles, 1995; Hilgenberg et al., 1996). We examined whether PKC-dependent synaptic plasticity was expressed in animals lacking the PKC theta isoform. Muscle, nerve, or glia cells from mutant animals can be grown with wild-type (WT) cultures of the other cell types in our three-compartment tissue culture system that permitted us to examine in which cell type PKC theta was required for synapse reduction to be expressed.
Here, we report that activity-dependent synapse elimination was severely compromised in cultured preparations in which PKC theta was not expressed in muscle cells. More surprisingly, marked suppression of synapse elimination was also seen in preparations in which only the neurons were deficient in PKC theta. Morphological and pharmacological studies in vivo have shown that normally occurring synapse elimination requires PKC action, but this is true only during the first 6–10 d postnatal. We present experiments on mice genetically deficient in PKC theta that confirm this. A preliminary report of some of this work has been published previously (Li et al., 2001b).
Materials and Methods
PKC theta-deficient animals. Founders for the PKC theta-deficient animals were kindly supplied by Dr. Hua Gu (National Institute for Allergy and Infectious Diseases, Bethesda, MD) and were developed initially in Dr. Dan Littman's laboratory (Dr. Littman, Molecular Pathogenesis Program, Skirball Institute of Biomolecular Medicine, New York University School of Medicine, New York, NY) (Sun et al., 2000). The mutant animals were produced from the C57Bl/J6 line, which is the same as that used for our WT preparations. The PKC theta-deficient strain had been backcrossed at least five times with the wild-type C57Bl strain (Dr. Littman, personal communication).
Western blot analysis. Muscles from the WT and PKC theta-deficient [knock-out (KO)] animals were homogenized using a high-speed homogenizer (overhead stirrer, VWR International, Clarksburg, MD) in lysis buffer containing 150 mm NaCl, 20 mm Tris-HCl, pH 7.5, 2 mm EGTA, and 5 mm EDTA supplemented with 1% Triton X-100, 1 mm PMSF, 30 μg/ml aprotinin, 50 mm NaF, 25 μg/ml leupeptin, and 1 mm sodium orthovanadate. In vitro cultures were washed twice with ice-cold Dulbecco's PBS, scraped, and extracted for 30 min on ice in the same lysis buffer as above. Clarification of the extracts was achieved by centrifugation for 20 min in the microcentrifuge at 10,000 rpm and 4°C. The protein concentrations were determined using the BCA protein assay kit (Pierce, Rockford, IL). Equivalent protein samples were subjected to NuPAGE 4–12% gradient Bis-Tris gel electrophoresis (Invitrogen, San Diego, CA) and transferred to polyvinylidene difluoride membrane (Millipore, Bedford, MA) by electroblotting. After blocking in TBS with 5% nonfat milk and 0.1% Tween 20, the membranes were incubated with monoclonal antibodies specific to PKC theta and PKC α (BD Transduction Labs), followed by incubation with HRP-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). In some experiments, specific antibodies against the myosin heavy chain were used (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA). Labeled protein bands were identified with ECL (Amersham Biosciences, Arlington Heights, IL).
Chambers and tissue culture. The tissue culture preparations involved three-compartment chambers with two populations of neurons from the ventral half of the mouse spinal cord in combination with skeletal myotubes and cerebral background (CB) or glia and were essentially as described in a previous study (for details of chamber fabrication and culture methodology, see Jia et al., 1999). Neurons from the ventral half of the spinal cord were grown in the side chambers and skeletal muscle fibers in the center chamber. The neurites from the side chambers extending into the central chamber formed synapses with the muscle cells in the center chamber. The cultures were maintained in a 37°C, 10% CO2 incubator.
The twitch assay. A platinum electrode was placed in each side chamber, and the ground electrode was in the center chamber. A stimulus delivered to axons from either side compartment elicited a contraction in some myotubes. This response in a given myotube was scored as a synaptic response. The percentage of these initially responsive myotubes that became unresponsive after an experimental or control period was taken as the functional synapse loss, and this was used as an index of a decrease of synaptic efficacy (for details of this assay, see Jia et al., 1999). (N = the total number of synaptic responses counted; n = total number of culture dishes tested.) In some experiments, chronic extracellular neuronal stimulation was performed. The stimulation pattern used in experiments was: 5 V biphasic (each pulse was 0.2 msec in duration, with a separation of 5 msec) electrical stimulation at a burst of 30 Hz, 2 sec duration, every 10 sec. Low-frequency stimuli (0.3 Hz) exert a strong synapse-preserving effect on the stimulated input. The description of stimulus pattern is complicated by the fact that even single stimuli may evoke repetitive responses in the muscle, presumably because of repetitive firing in the neural circuits in the side chambers.
Intracellular recording. Intracellular recordings were performed at room temperature in culture medium containing 5% horse serum. The pH was balanced with CO2. Electrodes filled with 4 m potassium acetate solution had resistances of ∼100 MΩ. Monosynaptic end plate potentials (EPPs) were evoked by extracellular stimulation of the axons from one side compartment. Data were collected through an interface (Instrutech) and analyzed on a G3 Mac computer with software developed by Synergy Research Inc. The muscle sodium channel blocker μ-conotoxin (1 μm; BACHEM Bioscience, Torrence, CA) was used to diminish the muscle action potential during intracellular recordings.
In vivo methods. Experiments were performed on the Levator auris longus (LAL) muscle of C57Bl and PKC theta KO neonatal mice. Mice and pups obtained from timed pregnancies were used in the study. The day of birth was designated postnatal day (P) 0. As in previous experiments (Lanuza et al., 2001, 2002), we minimized variability in our measurements with careful control of the timing of conception. Furthermore, we used only pups whose weights were within 5% of the mean for a given day after conception. These precautions resulted in synapse maturation data with very low variance. Animals were cared for in accordance with national guidelines for the humane treatment of laboratory animals.
Immunocytochemistry and morphological analysis. Whole mounts of LAL muscles were processed for immunocytochemistry to simultaneously detect acetyl choline receptors (AChRs) and axons using rhodamine-conjugated α-bungarotoxin (r-aBTX) and antibodies against the 200 kDa neurofilament protein and synaptophysin, as described previously (Lanuza et al., 2001). NMJs were viewed with a fluorescent Reichert microscope or imaged with a confocal laser-scanning microscope (Leica TCS NT), and three-dimensional reconstructions were generated. The number of axons per endplate was counted, and the “percentage of polyneuronal innervation” that represents all fibers that receive two or more axons and the “average number of axonal connections” were calculated. All NMJs visible in their entirety were scored, a minimum of 94 per muscle. At each age and condition examined, a minimum of six muscles was studied. Differences between groups were tested using a t test or U test (Mann–Whitney) with the Komogorov–Smirnov test to compare mean values. The χ2 or Fisher's test was applied to compare percentages. The criterion for statistical significance was p < 0.05. The preparations were scored, and counting was performed by a person without knowledge of the age or type of animals.
For the in vitro preparations, AChR aggregates were labeled with r-αBTX at a concentration of 1 μg/ml for 30 min. After fixation with 2% paraformaldehyde, the preparations were permeabilized with Triton X-100 and blocked with normal goat serum and incubated with mouse anti-neurofilament (1:1000 dilution) and mouse anti-synaptophysin (1:50 dilution) at 4°C overnight. Next day, after washing, the preparations were incubated with 1:500 FITC-conjugated goat anti-mouse IgG for 2 hr at room temperature.
Results
We confirmed that the mouse strain we used was deficient in the expression of PKC theta by means of Western blot analysis of skeletal muscle, spinal cord tissue, and glia cells in conjunction with isoform-specific antibodies. Figure 1A shows the robust expression of PKC theta, as expected, in WT muscle and the absence of expression of the theta isoform, but normal expression of the α isoform (PKC α), of PKC in muscle tissue from the mutant animals (Fig. 1B). Expression of the theta isoform of PKC in the ventral spinal cord could be detected as well as in cortical background (glia) cells (Fig. 1C) from the WT animals but not from the mutant animals.
Figure 1.

A, PKC theta does not exist in PKC theta –/– mice, whereas PKC α is well expressed. Western blots for muscle tissues and cultured myotubes from PKC theta KO and WT mice were probed with a monoclonal antibody to PKC theta. B, Western blots in A were stripped and reprobed with a monoclonal antibody to PKCα. C, Western blots for PKC theta from tissues: WT ventral spinal cord (VH), whole spinal cord (SC) from KO animals, and CB or glia cells from WT and PKC theta KO animals. Twenty micrograms of total protein were loaded on each lane in A and B, 10 μg for spinal cord tissue and 1 μg for the glia cells.
There was no difference between WT and KO animals in the size of the muscle fibers in vivo (Table 1), and no constant, significant difference in weight between the KO and WT animals was detectable at any point during the development of the animals. We measured by Western blots the amount of a developmental marker, myosin heavy chain, and found that levels in WT and KO muscles were not significantly different at P6 or P7. Except for the difference in the rate of maturation (see below), the NMJs were morphologically indistinguishable both in vitro and in vivo (Fig. 2a,b). In WT and KO preparations in vitro, we quantitated the area of postsynaptic AChR aggregates, and there was no significant difference. The resting membrane potential and spontaneous electrical activity (neurogenic plus myogenic) were not detectably different between the two in vitro. The mean size of evoked EPPs was also closely similar (Table 1). Thus, general development of the nerve–muscle system did not seem to differ between the WT and KO animals.
Table 1.
Muscle fiber and synaptic properties of WT and PKC-theta KO preparations
|
|
Wild type (n) |
PKC theta KO (n) |
|---|---|---|
| In vivo | ||
| Muscle fiber diameter (μm), P7 | 7.77 ± 0.81 (3 muscles, 120 each) | 7.74 ± 0.71 (3 muscles, 120 each) |
| In vitro | ||
| EPP amplitude (mV) | 5.87 ± 1.01(7) | 5.82 ± 0.99(15) |
| Spontaneous activity (spikes/sec) | 1.01 ± 0.58(7) | 1.28 ± 0.39(9) |
| RMP (mV) |
-51.6 ± 3.16(13) |
-53.8 ± 2.26(21) |
Figure 2.
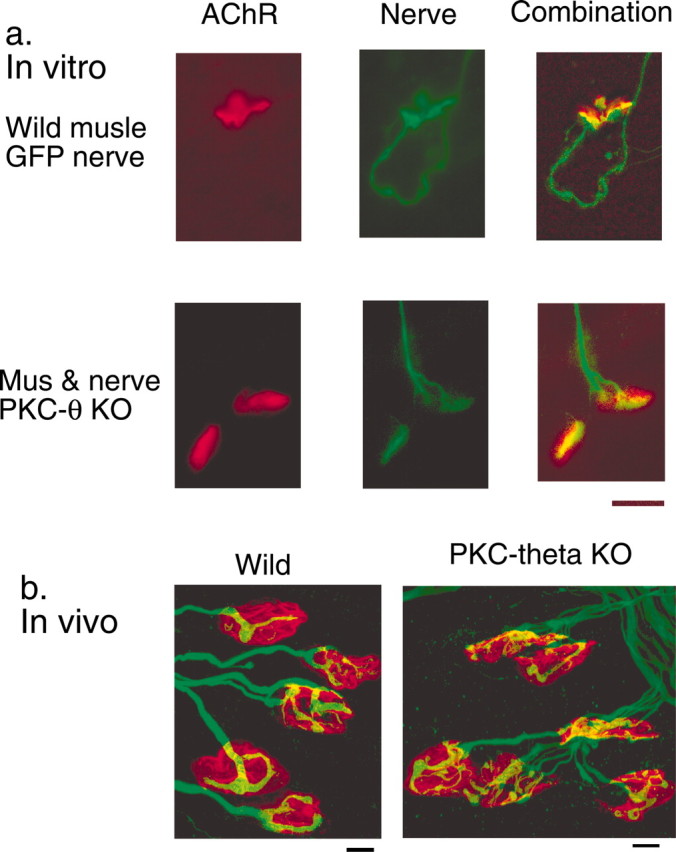
Neuromuscular contacts. a, Neuromuscular contacts from preparations in vitro. Normal: the neurites are visualized because of their expression of green fluorescent protein, and the AChR are stained with r-aBGTX. PKC theta KO: nerve and muscle are deficient in PKC theta; the neurites are stained green with anti-synaptophysin and anti-neurofilament protein antibodies, as described in Materials and Methods, and the AChR are stained red with r-aBGTX. In both sets of pictures, the left images are of the receptor, the middle images are of the neurites, and the right images combine the two. b, Microphotographs of WT and PKC theta KO NMJs in vivo, stained for the AChR (red) and axons/synaptic terminals (green), as described in Materials and Methods. Scale bars, 10 μm.
PKC theta is required for heterosynaptic activity-dependent and phorbol ester-elicited synapse reduction
Hebbian activity-dependent synapse reduction is robustly expressed in our culture system (Jia et al., 1999; Li et al., 2002). Thus, when one input to myotubes innervated by each of the two populations of motoneurons in our tissue culture system was stimulated, the stimulated input showed a significant but relatively small decrease (Fig. 3, Wild Stim Side), whereas the other, nonstimulated synapse was diminished by >50% (significantly more than the stimulated input) (Li et al., 2001a) (Fig. 3, Wild Unstim Side). In sharp contrast to this result, the response of neither the stimulated nor the unstimulated input is decreased by unilateral stimulation in preparations made from the PKC theta –/– animals as shown in Figure 3 (KO, Stim Side and Unstim Side). When six synaptic pairs were studied in this way, there was not a statistically significant change produced in the KO preparations. WT preparations with no stimulation were not different from the stimulated KO preparations (p > 0.2; data not shown). The changes in efficacy in the stimulated inputs were significantly different (p < 0.03) in the WT and KO preparations. The difference between unstimulated inputs in unilaterally stimulated WT and KO preparations was significant at p < 0.002. Thus, the loss in synaptic efficacy of both stimulated and unstimulated inputs produced by unilateral stimulation in WT preparations is completely absent in the PKC theta KO preparations.
Figure 3.
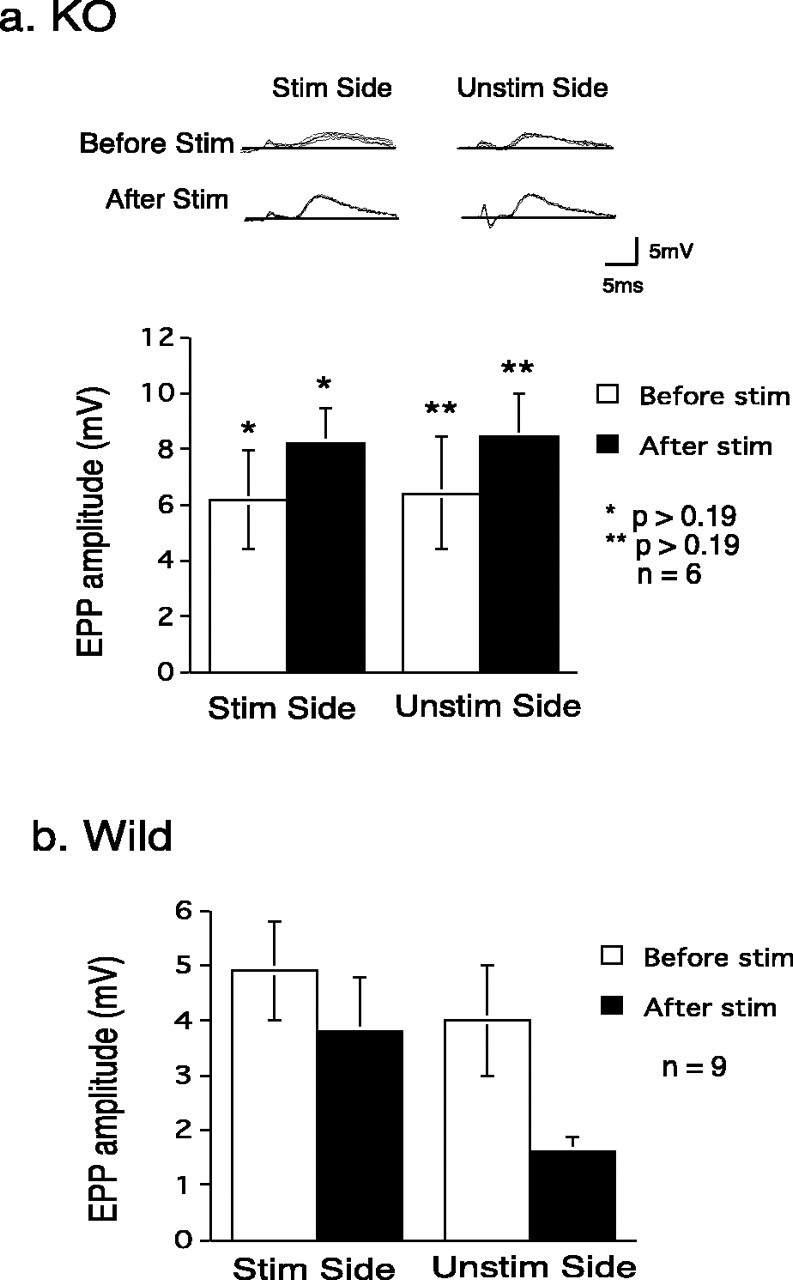
Intracellular microelectrode recordings from PKC theta –/– animals. a, KO. The top panel shows examples of EPPs evoked from the stimulated and unstimulated sides before and after a 2 hr period of stimuli (30 Hz for 2 sec every 10 sec) delivered to the “stimulated” side. Note the lack of decrement in the responses elicited from either the stimulated or unstimulated side. The bottom panel shows a summary of results from six experiments, like that shown above. There is a nonsignificant trend for both responses to increase. b, Wild type [from Li et al. (2002) for comparison purposes]. This work with preparations from WT animals consistently showed a significant decrease in the unstimulated input in this type of experiment (Right bars, WT, Unstim; from Li et al., 2001, 2002). This change is highly significantly different from that in the unstimulated input in the KO preparations. There was less loss from the stimulated input (left bars), but there is significantly more loss from the WT stimulated side than from the KO stimulated side. In this and subsequent figures, except as noted, the error bars represent ±SEM.
Previous work showed that this selective loss was mediated by PKC and that PKC activation by phorbol ester application could be used as a surrogate for electrical activation of the synapses (Lanuza et al., 2000), but this work gave no information as to which specific isoform of PKC was involved in the synapse elimination process. To address this issue, we have compared the response to the phorbol ester using the twitch assay in preparations made from WT animals with the responses from mutant animals lacking the specific theta isoform of PKC. As shown in Figure 4, bars 1–3, PMA produced no significant loss of synaptic efficacy in the preparations made from the mutant animals in contrast to the extensive loss seen in WT preparations, showing that the theta isoform is necessary for the expression of the PKC-dependent synapse loss.
Figure 4.

Effect of PMA on synapse efficacy in heterologous cultures. The twitch assay (see Materials and Methods) was used to evaluate changes in the synaptic strength of synaptic connections in the chambers over a 2 hr period under different conditions. In the absence of treatment, connections in the PKC theta –/– preparations were quite stable (data not shown), as was the case for WT preparations (bar 1). Under treatment with 100 μm PMA there was a substantial loss (∼70%) in WT synaptic connections (bar 2), whereas there was no significant loss of connections with the same treatment in preparations made from the PKC theta –/– animals (bar 3). Results using the twitch assay from heterologous cultures composed of tissues from either WT or KO animals in different combinations are shown in bars 4–7. All preparations received the same treatment of 100 nm PMA for 2 hr. Preparations with either muscle cells or neurons (or both) from PKC theta –/– animals did not show significant synapse loss with PMA treatment (bars 4–6), whereas preparations in which only the glia cell component was from the KO animal (bar 7) showed loss equivalent to that with all WT tissue (bar 2).
The cellular basis of phorbol ester induced synapse reduction
Our previous experiments indicated that the PKC-mediated mechanism for activity-dependent synapse downregulation involves a postsynaptic site of action and an increased loss of AChR from the myotube surface membrane (Lanuza et al., 2000). We wished to explore further the question of the locus of action of the PKC in the multi-compartment experimental system. The neuronal, glial, and muscle components of the system were prepared and plated separately so that preparations could be made that contain the different cell types coming from animals of different genetic makeup. Using the twitch assay, we explored the functional properties of these heterologous preparations (Fig. 4). All combinations of WT and mutant neurons, CB cells, and muscle cells were examined. The origin of the glial background cells did not affect the results. When normal nerve and muscle were combined with mutant glia, a normal degree of synapse loss was produced by PMA treatment (Fig. 4, bar 7). The presence of PKC theta in the muscle cells was necessary for PMA-induced synapse loss, however, because even with normal nerve and glia in the preparation, with deficient muscle cells there was no significant synapse loss with PMA treatment (Fig. 4, bar 5). This finding is fully compatible with our previous interpretation that the action of PKC was primarily, if not exclusively, on the postsynaptic muscle cell. It also indicates that the theta isoform is the form mediating synapse reduction. More surprising was the finding that in a preparation composed of normal muscle and normal glia with PKC theta-deficient neurons, there was a markedly reduced loss of synaptic connections in response to PMA treatment compared with the loss in all WT preparations (Fig. 4, bar 6). This indicates that PKC theta in the nerve cell plays some role crucial to the process of PMA-induced synapse elimination. Thus, the data in Figure 4 shows that PKC theta activity in both nerve and muscle (but not in glia) is essential to this PKC-dependent plasticity.
This conclusion is borne out by the results of experiments in which synaptic efficacy was evaluated with intracellular recordings of EPPs before and after PMA treatment in the different types of heterologous culture preparations. The results of some experiments of this sort are shown in Figure 5 (top). In preparations made from WT animals and, as expected, evoked EPPs were markedly reduced as a result of PMA treatment. If PKC theta KO-derived nerve or muscle were used, however, there was little, if any, reduction in EPP amplitude after PMA treatment. The results of several such experiments are shown in Fig. 5 (bottom). There was no effect of PMA treatment in preparations in which the muscle cells are from PKC theta-deficient animals (with WT nerve and glia). The same was true when PKC theta-deficient nerve cells were used in conjunction with WT muscle and glia; there was no decrease in the EPP under PMA treatment. It may be noted that there was no significant difference in the amplitude of EPPs produced in normal muscles by WT or PKC theta KO neurons. As shown in Figure 1, PKC theta is strongly expressed in skeletal muscle and in ventral spinal cord and glia as well. In the KO animals, there was no expression of PKC theta in the ventral spinal cord or even in whole spinal cord tissue (Fig. 1C). In our hands, immunocytochemical methods coupled with confocal microscopy has shown PKC theta expression in WT motor axons (data not shown).
Figure 5.
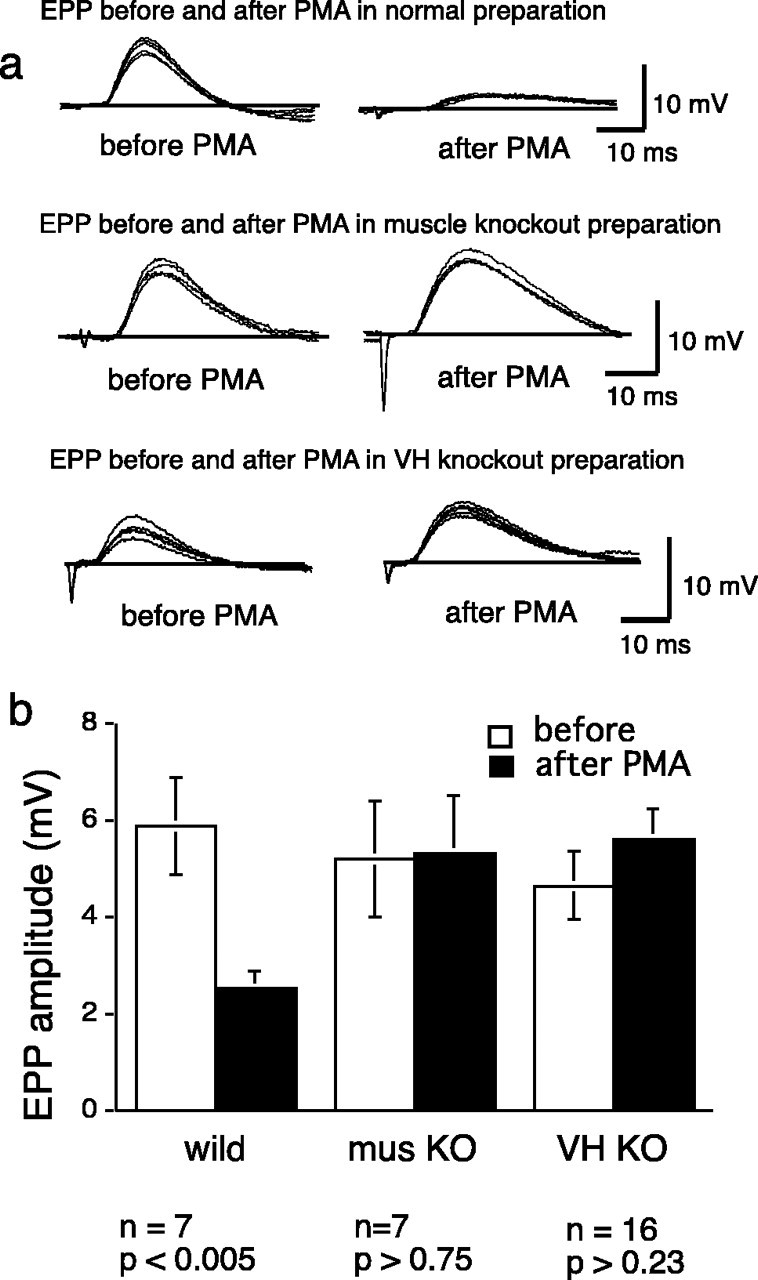
EPP amplitude in heterologous cultures, analogous experiments to those shown in Figure 4, but with intracellular recordings as the test for synaptic efficacy. a, Top traces, A preparation made from WT animals shows marked reduction in EPP amplitude after PMA treatment. Middle traces, A preparation made with normal neurons and glia but with KO muscle cells shows no loss of synapse strength after PMA. Bottom traces, A preparation made with normal muscle and glia but KO neurons also fails to show synapse decrement with PMA. b, Data from several experiments like those shown in a are summarized, confirming the lack of PMA effect in preparations in which either the nerve or the muscle cells are from KO animals.
In cultured preparations made from heterozygous animals, the degree of synapse loss produced by PMA was intermediate between that seen with preparations from WT and KO animals. Loss in wild type and KO was 75.6 ± 4% and 6.1 ± 2%, respectively, whereas in the heterozygous preparations loss was 46.9 ± 2%. This loss in the heterozygous preparations was significantly different from the wild type and KO at p < 0.001.
Despite the similarity in evoked and spontaneous synaptic activity in WT and PKC theta KO cultures (Table 1), we sought to determine the role of spontaneous electrical activity in determining the response of the preparations to phorbol ester stimulation. To this end, we treated heterologous cultures with PMA in the presence of 10–7 m TTX to block all electrical activity. The results are shown in Figure 6. As noted above, PMA treatment is without effect in cultures made from KO muscle and normal nerve, and this is true also in the presence of TTX treatment. In contrast, in cultures made of KO nerve and WT muscle, there is no effect of PMA in the absence of TTX, but in the presence of TTX PMA does produce a major decrement of synaptic efficacy. This result will be discussed below.
Figure 6.

The effect of activity on PMA action in different heterologous culture combinations as revealed by blocking activity with TTX. EPPs were measured in myotubes before treatment (open bars) and again, in the same myotubes, after 2 hr of treatment (filled bars) combining 100 nm PMA and 10-7 m TTX. The preparations were washed with normal growth medium after the treatment and before the second EPP measurement. Note that TTX treatment restores the sensitivity to PMA treatment in the cultures prepared from KO neurons and normal muscle (bars on the right). This is not true for the cultures made from KO muscle and normal nerve. The data labeled Mus KO + TTX were from preparations made with muscle cells from KO animals and nerve and CB from WT animals. Ventral horn (VH) KO preparations were made with VH neurons from KO animals and muscle and CB from WT animals.
Role of PKC theta in vivo
We had found that the process of developmental synapse elimination in the rodent in vivo could be strongly influenced by procedures that blocked or enhanced PKC activity (Lanuza et al., 2001). To complement the pharmacological studies and determine the specific PKC isoform involved in the elimination process in vivo, we looked for abnormalities in the synapse elimination process that might be characteristic of the PKC theta KO animals.
Previous work has documented the decrease in polyneuronal innervation that occurs postnatally in the rodent, as well as the postnatal changes in postsynaptic AChR distribution at the LAL NMJ (Lanuza et al., 2001, 2002). Also, we identified a PKC-dependent period between P4 and P8 in the synapse elimination period and a PKC-independent period from day 8 to adulthood (Lanuza et al., 2001). In the present study, we found that mice lacking the PKC theta isoform showed an initial delay of synapse elimination although, finally, the NMJs become monoinnervated. Either the percentage of junctions with more than one axon or the mean number of axons per junction can be used in following the process of synapse elimination. The average number of axons innervating a neuromuscular synapse falls from three to nearly one over the first 14 d postnatal (Fig. 7). The time at which the halfway point of loss (two axons per synapse) occurs is delayed from 5.5 in normal animals to 9.0 d in the KO animals. By P8, when synapse loss had begun in the PKC theta-deficient muscles, there was a significant difference in the percentage of polyneuronal innervation (50.2 ± 3.25 in the normal muscle vs 74.9 ± 1.88 in the KO muscles; p < 0.001). By P14, the percentage of polyneuronal innervation was not different between the two types of animals (∼96% of the NMJs had become monoinnervated in both). This difference in the rate of loss in polyneuronal innervation was not accompanied by a general deficit in development as indicated by the weight of the developing fetuses. At day 7, the weight of the WT and KO animals were within 8% of one another (the KO heavier). As noted, the diameters of muscle fibers in WT and KO animals were within 5% of one another at the time during development of maximal differences in polyinnervation, and the levels of a muscle marker, myosin heavy chain, were not different at either day 6 or 7 postnatal. We could not detect any morphological differences between synapses of the WT and KO animals in vivo or in vitro (Fig. 2), except for their maturation rate (i.e., degree of polyinnervation and AChR distribution) (Lanuza et al., 2002; M.A. Lanuza, unpublished results).
Figure 7.

The time course of synapse elimination in normal and PKC theta –/– mice in vivo, as measured by the average number of axons contacting the NMJs as a function of days postnatal. Data were calculated as mean ± SEM, although the SEM bars are smaller than the symbols on the graph. The arrows show the times at which the halfway point of loss (two axons per synapse) occurred. The LAL preparations were removed from postnatal pups at the ages indicated and stained with a-bunagrotoxiin and antibodies against neurofilament protein. The number of axons innervating at least 94 end plates were counted and the average number of axons per end plate calculated (for details, see Materials and Methods).
Discussion
The present results using a genetic deletion of a specific PKC isoform confirms previous data implicating PKC in the process of synapse elimination and shows that the theta isoform is essential for normal expression of the elimination process in vitro. The Hebbian synaptic plasticity (Hebb, 1949) expressed in our system in WT cultures is manifested as a substantial (>50%) decrease in the efficacy of unstimulated inputs to myotubes that are activated by other stimulated inputs, and this heterosynaptic loss is completely prevented by the specific PKC blocker calphostin C (Jia et al., 1999). This heterosynaptic synapse loss also does not occur in preparations made from PKC theta-deficient animals. Thus, the kinase isoform is essential for Hebbian, stimulus-specific, activity-dependent synapse downregulation in vitro. When the responses of stimulated inputs before and after activation were compared in WT and KO preparations, the PKC-mediated effect of activation on efficacy was also revealed. The stimulated input was not decreased in the KO in contrast to the modest reduction that occurred in the wild type.
Synapse efficacy was downregulated by direct PKC activation with phorbol ester application in WT preparations (Lanuza et al., 2000; Li et al., 2001a). Previous results showed that the PKC-mediated effects involved postsynaptic AChRs; PKC activation served to decrease the number of postsynaptic receptors with a consequent decrease in synaptic efficacy. This was reflected in a decrease of unitary or quantal synaptic response, with no detectable change in the presynaptically determined number of released quanta (Li et al., 2002). The postsynaptic locus of action for the theta isoform of the PKC is confirmed in the present study by the heterologous culture experiment pairing normal neurons and glia cells with PKC theta-deficient muscle cells. When this combination was stimulated with PMA for a time and at a dosage that produces reliable synapse decrement in WT cultures, there was no significant effect on synapse efficacy (Figs. 4, 5). The theta isoform must be expressed in the muscle cells for general activation of PKC to result in reduction of synapse efficacy. This result was not affected by the state of activity of the preparation; TTX-treated preparations with KO muscle also were insensitive to PMA.
The heterologous preparations can be made in any combination to examine whether PKC theta was required in either neurons or glia for the Hebbian process to be expressed. Absence of PKC theta in glia cells did not interfere with the synapse reduction process, but preparations with PKC theta-deficient neurons failed to demonstrate the Hebbian plasticity (Fig. 4) even when combined with normal muscle and glia. This result was confirmed with intracellular recordings of EPPs in which the reduction of EPPs produced by the phorbol ester activator of PKC seen in preparations made with cells from WT animals is not seen in preparations made with nerve from PKC theta-deficient animals (Fig. 5). Furthermore, if the activity of these WT muscle/KO nerve preparations was suppressed with TTX, PMA treatment did produce synapse downregulation. These observations are not compatible with an exclusively postsynaptic model of synapse regulation. Our previous observations (Lanuza et al. 2000) and those of Balice-Gordon and Lichtman (1994) did involve predominantly or exclusively postsynaptic mechanisms. In particular, loss of AChR was shown to be involved in synapse loss.
The observation that synapse modulation is dependent on neural PKC theta suggests that neural PKC theta either produces postsynaptic receptor loss or that neural PKC theta blocks a process that maintains the synapse (and AChR). The latter seems more plausible, and we hypothesize that PKC theta in the nerve terminal acts to decrease release of a molecule, perhaps a cotransmitter such as calcitonin gene-related peptide (CGRP), that activates postsynaptic PKA and hence stabilizes the AChR and the synapse. This stabilizing cotransmitter would be released in an activity-dependent manner and would, therefore, not be released in the TTX-treated preparations. The lack of synapse (AChR) stabilization in these activity-blocked preparations would render them vulnerable to PMA activation of postsynaptic PKC theta. Thus, one simple hypothesis would be that the presynaptic PKC action blocks the release of the transmitter that mediates the positive effect of activity. This would occur, for instance, if PKC acted presynaptically to block CGRP release.
Heterozygous preparations show a degree of synapse loss with PMA treatment that is midway between that of WT and KO preparations. This may indicate the kinase activity is limiting for the expression of synapse loss. The expression of the kinase is reduced (but not eliminated) in the heterozygous animals (Pfeifhofer et al., 2003; our unpublished results), and preparations from such animals might be expected to show a corresponding decrease in synapse reduction. This result is in correspondence to the observations of Sun et al. (2000), who found that the stimulation of lymphocyte proliferation by appropriate antibodies was suppressed in PKC theta KO animals and the effect in heterozygous animals is intermediate between WT and KO animals.
The several synapse-related processes that we hypothesize based on the present and previous work can be summarized as: (1) Activation of the AChR results in the stimulation of PKC (Kim et al., 2002) and perhaps PKA. (2) Activation of peptidergic (e.g., CGRP) receptors locally activates PKA. As noted in previous publications (Li et al., 2002; Nelson et al., 2003), this localization of PKA would be attributable to interactions of the kinase with anchoring proteins (AKAPs) (Cantrell et al., 1999; Sik et al., 2000). (3) PKA phosphorylates the AChR (or other critical scaffold protein) so that the receptor is stabilized in the synaptic membrane. (4) PKC activation acts against the PKA effect to destabilize the receptor. It acts generally at both activated synapses and also at other inactive synapses on activated myotubes. (5) Presynaptic PKC activation acts to block release of CGRP or other peptides. This serves to diminish the activity-related positive effect of PKA. We hypothesize that CGRP mediates this step because CGRP is known to be present in motor neurons (Piehl et al., 1993), to be released from cholinergic neurons (Miles et al., 1989; Csillik et al., 1992), and to induce PKA activation in muscle with a consequent AChR phosphorylation (Miles et al., 1989). We have shown that CGRP stabilizes synaptic efficacy at the NMJ (Li et al., 2001a).
These mechanisms may be operative at synapses other than the NMJ. Both excitatory and inhibitory receptors are targets of various kinases, including PKA and PKC (Blackstone et al., 1994; Vaello et al., 1994; Tingley et al., 1997; Swope et al., 1999), and the phosphorylation reactions can affect receptor function and stability. Presynaptic actions of the kinases are well documented in a number of preparations (Malenka et al., 1986; D'Angelo et al., 1992; Ghirardi et al., 1992). In the latter work, PKC activation at the NMJ resulted in an increase in transmitter (ACh) release, in contrast to the hypothesized decrease of a cotransmitter (CGRP) augmenting postsynaptic responsiveness in the present work.
We concluded from previous work in vivo that PKC was involved in a stage of the synapse elimination process that occurs normally during the first 2 weeks of postnatal life, but that the process was not dependent on the kinase after approximately day 10 postnatal (Lanuza et al., 2001). After that time, PKC-independent mechanisms take over. The present data are compatible with this interpretation in that the process of synapse elimination is significantly slowed (there is a 3.5 d delay in the time of 50% loss of connections) during the first week or 10 d of postnatal life in PKC theta KO animals, but the process goes to completion by a month or so postnatal, even in the absence of PKC theta expression. The delay in synapse elimination is not attributable to a general slowing of muscle development, because other markers of muscle development (e.g., fiber diameter and myosin heavy chain) were not different for the WT and KO animals at the time during development when the difference in synapse elimination was maximal.
Two major conclusions thus arise from the present work: (1) PKC theta is involved in the synapse elimination process both in vitro and in vivo, but other mechanisms are also involved and can eventually produce mononeuronal innervation of the muscle cells in vivo even in the absence of the theta isoform of the PKC; and (2) the actions of the kinases are complex and involve interactions in both the presynaptic and postsynaptic elements. It will be of great interest to understand the nature of these interactions.
Footnotes
M.A.L. was supported in part by grants from Fondo de Investigaciones Sanitarias de la Seguridad Social (Spain) (2PI02/0448 and 1326 UO7 E20 N2001BEAI-49). The monoclonal antibody against myosin heavy chain developed by Dr. D. A. Fishman was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the National Institute of Child Health and Human Development and maintained at the Department of Biological Sciences and the University of Iowa (Iowa City, IA).
Correspondence should be addressed to Dr. Min-Xu Li, Section on Neurobiology, Laboratory of Developmental Neurobiology, National Institute of Child Health and Human Development, National Institutes of Health, Building 49, Room 5A38, Bethesda, MD 20892. E-mail:liminxu@mail.nih.gov.
DOI:10.1523/JNEUROSCI.3930-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243762-08$15.00/0
References
- Balice-Gordon RJ, Lichtman JW (1994) Long-term synapse loss induced by focal blockade of postsynaptic receptors. Nature 372: 519–524. [DOI] [PubMed] [Google Scholar]
- Blackstone C, Murphy T, Moss S, Baraban J, Huganir R (1994) Cyclic AMP and synaptic activity-dependent phosphorylation of AMPA-preferring glutamate receptors. J Neurosci 14: 7585–7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Jansen JK, Van Essen D (1976) Polyneuronal innervation of skeletal muscle in new-born rats and its elimination during maturation. J Physiol (Lond) 261: 387–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelli RJ, Shelton DL, Segal RA, Shatz CJ (1997) Blockade of endogenous ligands of trkB inhibits formation of ocular dominance columns. Neuron 19: 63–76. [DOI] [PubMed] [Google Scholar]
- Cantrell A, Tibbs V, Westenbroek R, Scheuer T, Catterall WA (1999) Dopaminergic modulation of voltage-gated Na+ current in rat hippocampal neurons requires anchoring of cAMP-dependent protein kinase. J Neurosci 19: RC21(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csillik B, Knyihar-Csillik E, Kreutzberg GW, Tajti L, Kereszturi A, Kovacs T (1992) Calcitonin gene-related peptide is released from cholinergic synapses. Ann NY Acad Sci 657: 466–468. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Rossi P, Tanzi F, Taglietti V (1992) Protein kinase C facilitation of acetylcholine release at the rat neuromuscular junction. Eur J Neurosci 4: 823–831. [DOI] [PubMed] [Google Scholar]
- Fields RD, Nelson PG (1992) Activity-dependent development of the vertebrate nervous system. Int Rev Neurobiol 34: 133–214. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Braha O, Hochner B, Montarolo P, Kandel E, Dale N (1992) Roles of PKA and PKC in facilitation of evoked and spontaneous transmitter release at depressed and nondepressed synapses in Aplysia sensory neurons. Neuron 9: 479–489. [DOI] [PubMed] [Google Scholar]
- Hebb DO (1949) The organization of behavior. New York: Wiley.
- Hilgenberg L, Miles K (1995) Developmental regulation of a protein kinase C isoform localized in the neuromuscular junction. J Cell Sci 108: 51–61. [DOI] [PubMed] [Google Scholar]
- Hilgenberg L, Yearwood S, Milstein S, Miles K (1996) Neural influence on protein kinase C isoform expression in skeletal muscle. J Neurosci 16: 4994–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M, Li MX, Dunlap V, Nelson PG (1999) The thrombin receptor mediates functional activity-dependent neuromuscular synapse reduction via protein kinase C activation in vitro. J Neurobiol 38: 369–381. [PubMed] [Google Scholar]
- Kim S, Bondeva T, Nelson PG (2002) Activation of protein kinase C isozymes in primary mouse myotubes by carbachol. Brain Res Dev Brain Res 137: 13–21. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Li MX, Jia M, Kim S, Davenport R, Dunlap V, Nelson PG (2000) Protein kinase C mediated changes in synaptic efficacy at the neuromuscular junction in vitro: the role of postsynaptic acetylcholine receptors. J Neurosci Res 61: 616–625. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Garcia N, Santafe M, Nelson PG, Fenoll-Brunet MR, Tomas J (2001) Pertussis toxin-sensitive G-protein and protein kinase C activity are involved in normal synapse elimination in the neonatal rat muscle. J Neurosci Res 63: 330–340. [DOI] [PubMed] [Google Scholar]
- Lanuza MA, Garcia N, Santafe M, Gonzalez CM, Alonso I, Nelson PG, Tomas J (2002) Pre- and postsynaptic maturation of the neuromuscular junction during neonatal synapse elimination depends on protein kinase C. J Neurosci Res 67: 607–617. [DOI] [PubMed] [Google Scholar]
- Li MX, Jia M, Jiang H, Dunlap V, Nelson PG (2001a) Opposing actions of protein kinase A and C mediate Hebbian synaptic plasticity. Nat Neurosci 4: 871–872. [DOI] [PubMed] [Google Scholar]
- Li MX, Jia M, Yang LX, Dunlap V, Nelson PG (2001b) The role of PKC theta in synapse loss in neuro-muscular junction in vitro. Soc Neurosci Abstr 27: 799.3. [Google Scholar]
- Li MX, Jia M, Yang LX, Dunlap V, Nelson PG (2002) Pre- and postsynaptic mechanisms in Hebbian activity-dependent synapse modification. J Neurobiol 52: 241–250. [DOI] [PubMed] [Google Scholar]
- Malenka R, Madison D, Nicoll R (1986) Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature 321: 175–177. [DOI] [PubMed] [Google Scholar]
- Miles K, Greengard P, Huganir RL (1989) Calcitonin gene-related peptide regulates phosphorylation of the nicotinic acetylcholine receptor in rat myotubes. Neuron 2: 1517–1524. [DOI] [PubMed] [Google Scholar]
- Nelson PG, Jia M, Li MX (2003) Protein kinases and Hebbian function. The Neurosientist 9: 110–116. [DOI] [PubMed] [Google Scholar]
- Newton A (1995) Protein kinase C: structure, function, and regulation. J Biol Chem 270: 28495–28498. [DOI] [PubMed] [Google Scholar]
- O'Brien RA, Ostberg AJ, Vrbova G (1978) Observations on the elimination of polyneuronal innervation in developing mammalian skeletal muscle. J Physiol (Lond) 282: 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifhofer C, Kofler K, Gruber T, Tabrizi NG, Lutz C, Maly K, Leitges M, Baier G (2003) Protein kinase C theta affects Ca2+ mobilization and NFAT cell activation in primary mouse T cells. J Exp Med 197: 1525–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehl F, Arvidsson U, Hokfelt T, Cullheim S (1993) Calcitonin gene-related peptide-like immunoreactivity in motoneuron pools innervating different hind limb muscles in the rat. Exp Brain Res 96: 291–303. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389–442. [DOI] [PubMed] [Google Scholar]
- Shatz CJ (1990) Impulse activity and the patterning of connections during CNS development. Neuron 5: 745–756. [DOI] [PubMed] [Google Scholar]
- Sik A, Gulacsi A, Lai Y, Doyle W, Pacia S, Mody I, Freund T (2000) Localization of the A kinase anchoring protein AKAP79 in the human hippocampus. Eur J Neurosci 12: 1155–1164. [DOI] [PubMed] [Google Scholar]
- Sun Z, Arendt C, Ellmeier W, Schaeffer E, Sunshine M, Gandhi L, Annes J, Petrzilka D, Kupfer A, Schwartzberg P, Littman D (2000) PKC-theta is required for TCR-induced NF-kappaB activation in mature but not immature T lymphocytes. Nature 404: 402–407. [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Raymond LA, Huganir RL (1999) Regulation of ligandgated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res 33: 49–78. [DOI] [PubMed] [Google Scholar]
- Tanaka C, Nishizuka Y (1994) The protein kinase C family for neuronal signaling. Annu Rev Neurosci 17: 551–567. [DOI] [PubMed] [Google Scholar]
- Thompson WJ (1985) Activity and synapse elimination at the neuromuscular junction. Cell Mol Neurobiol 5: 167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tingley WG, Ehlers MD, Kameyama K, Doherty C, Ptak JB, Riley CT, Huganir RL (1997) Characterization of protein kinase A and protein kinase C phosphorylation of the N-methyl-D-aspartate receptor NR1 subunit using phosphorylation site-specific antibodies. J Biol Chem 272: 5157–5166. [DOI] [PubMed] [Google Scholar]
- Vaello ML, Ruiz-Gomez A, Lerma J, Mayor Jr F (1994) Modulation of inhibitory glycine receptors by phosphorylation by protein kinase C and cAMP-dependent protein kinase. J Biol Chem 269: 2002–2008. [PubMed] [Google Scholar]