

Abstract
The survival of neurons is maintained primarily by neurotrophic factors that suppress the apoptotic program. Axotomy or removal of peripheral targets causes neuronal cell death, but the mechanisms involved in the induction of this type of cell death remain poorly understood. Here, we show that DP5/Harakiri, a Bcl-2 homology domain 3-only member of the Bcl-2 family, is induced in motoneurons after transection of the hypoglossal nerve in mice and in sympathetic neurons after nerve growth factor (NGF) withdrawal. To assess the role of DP5 in neuronal cell death, mutant mice deficient in DP5 were generated by gene targeting. DP5–/– mice were viable and exhibited normal postnatal development. Notably, motoneurons from DP5–/– mice were highly protected from cell death induced by resection of the hypoglossal nerve compared with motoneurons from DP5+/+ littermate mice. In addition, deficiency of DP5 in superior cervical ganglia (SCG) neurons resulted in delayed neuronal cell death triggered by NGF withdrawal. Analysis of SCG neurons from DP5–/– mice revealed increased preservation of mitochondrial membrane potential and reduced activation of caspase-3 compared with neurons from wild-type mice. These results indicate that DP5 plays an important role in neuronal cell death induced by axotomy and NGF deprivation through the regulation of mitochondrial function and caspase-3 activation.
Keywords: BH3-only, Bcl-2, DP5, HRK, neuronal cell death, NGF
Introduction
Cell death plays an essential role during embryonic development and tissue homeostasis in multicellular organisms (Baehrecke, 2002). The cell-death mechanism is genetically controlled by a evolutionarily conserved program that is activated in the dying cell and serves to eliminate unnecessary and damaged cells (Strasser et al., 2000; Baehrecke, 2002). In the vertebrate nervous system, 20–80% of all neurons produced die during embryogenesis and early postnatal development (Oppenheim, 1991). Neuronal cell death appears to occur through competition for trophic factors derived from target tissues, which may serve to select the proper set of neuronal connections (Oppenheim, 1991). Removal of factors such as nerve growth factor (NGF) in vivo and in vitro induces apoptosis in populations of neurons that depend on the trophic factor for survival (Oppenheim, 1991; Deshmukh and Johnson, 1997). Similarly, axotomy or removal of peripheral targets causes neuronal cell death (Oppenheim, 1996; Goldberg and Barres, 2000).
Induction of apoptosis requires new gene expression in certain models of neuronal cell death, including that triggered by NGF deprivation (Oppenheim, 1991; Deshmukh and Johnson, 1997). NGF withdrawal in neonatal sympathetic neurons induces the expression of bim and dp5/harakiri, two proapoptotic Bcl-2 homology domain 3 (BH3)-only members of the bcl-2 family (Imaizumi et al., 1997; Putcha et al., 2001). Recent studies have shown that bim deletion conferred transient and partial protection against apoptosis triggered by NGF or K+ withdrawal in susceptible populations of neurons (Putcha et al., 2001). These results suggest that BIM has a redundant function in mediating the death of neurons induced by NGF deprivation. Dp5/harakiri was originally identified when screening for genes induced after NGF withdrawal in rat sympathetic neurons and for binding partners of prosurvival Bcl-XL (Imaizumi et al., 1997; Inohara et al., 1997). Studies from several laboratories have revealed that BH3-only proteins such as BIM and DP5/HRK function upstream of BAX and BAK to induce neuronal cell death (Bouillet and Strasser, 2002). BAX is essential for the death of sympathetic neurons induced by NGF withdrawal and the release of cytochrome c from the mitochondria, an event that mediates activation of caspases through apoptotic protease-activating factor-1 (Deckwerth et al., 1996; Putcha et al., 1999). The death of motoneurons triggered by transection of peripheral nerves has also been associated with the induction of gene expression (Tanabe et al., 1999; Honma et al., 2002). Here, we report that DP5 contributes to neuronal apoptosis triggered by NFG withdrawal and mediates the death of motoneurons induced by resection of the hypoglossal nerve.
Materials and Methods
Generation of DP5-deficient mice. DP5-null mice were generated by targeted disruption of dp5 through homologous recombination using a targeting vector designed to replace the single coding exon of dp5 with a neomycin-resistant cassette. The pTV targeting vector (Suzuki et al., 2002) was linearized with PacI and electroporated into 129/SV embryonic stem (ES) cells. Clones resistant to G418 and ganciclovir were selected and screened for homologous recombination by Southern blot and PCR analysis. Targeted ES clones were injected into C57BL/6J blastocysts. The resulting chimeric mice were backcrossed to C57BL/6J mice to generate DP5+/– heterozygous mice. Interbreeding of heterozygous mice was performed to generate homozygous DP5-deficient mice.
Genotype analysis. Genomic DNA was extracted from the tails of mice, and the dp5 genotype was analyzed by PCR analysis. For PCR amplification, the following primers were used: sense primer, 5′-GGAGCTGGAGCAAAACTGGATTGG-3′; antisense primer Neo2, 5′-GCAGGAGCAAGGTGAGATGACAGGAGAT-3′; and internal antisense primer, 5′-GACAGCCTCCGCACCTGTCAGCC-3′. The cycle parameters were 5 min at 95°C, 1 min at 95°C, 20 sec at 63°C, and 1 min at 72°C for 35 cycles, followed by 72°C for 10 min. PCR products were analyzed by agarose gel electrophoresis. Southern blot analysis was performed using a 0.4 kb genomic 32P-labeled probe (see Fig. 1 A).
Figure 1.

Generation of DP5-deficient mice. A, Schematic representation of the genomic dp5 locus (top), the gene-targeting construct (middle), and the targeted dp5 locus (bottom). DP5-null mice were generated by targeted disruption of dp5 through homologous recombination. The targeting vector (Tg Construct) was designed to replace the single coding exon of dp5 with a neomycin-resistant cassette. The probe used for Southern blot analysis is indicated. Nucleotide primers for PCR screening (arrowheads) are shown. S, SpeI; H, HindIII; N, NotI; B, BamHI; X, XbaI. B, Genomic tail DNA was digested with SpeI, and blots were hybridized with the probe indicated in A. Two mice of the indicated genotype are shown. C, Detection of normal and targeted alleles in genomic DNA by PCR analysis. D, Expression of dp5 transcripts in various tissues from DP5+/+ and DP5–/– mice by RT-PCR analysis. WT, Wild type; hsv-TK, herpes simplex virus-thymidine kinase; Bone M., bone marrow.
Cell culture and cell labeling. Superior cervical ganglia (SCG) neurons from 1-d-old mice were isolated and cultured as described previously (Imaizumi et al., 1997). The collected cells were plated on collagencoated dishes in DMEM containing 10% fetal calf serum (FCS), 2 mm glutamine, 20 μm fluorodeoxyuridine, 20 μm uridine, and 50 ng/ml NGF (Promega, Madison, WI). The neurons were maintained for 5 d in the presence of NGF before being subjected to reverse transcriptase (RT)-PCR or cell-death assay. NGF withdrawal was performed by changing the medium to DMEM and 10% FCS lacking NGF and containing anti-NGF antibodies (CIDtech Research, Cambridge, Canada), as reported previously (Imaizumi et al., 1997). To monitor mitochondrial membrane potential, cells that were deprived of NGF were loaded with 20 nm tetramethylrhodamine methylester (TMRM) (Molecular Probes, Eugene, OR) in PBS for 20 min at 37°C. To assess the expression of activated caspase-3, SCG neurons were stained at the indicated times with rabbit anti-active caspase-3 antibody (Promega), followed by incubation with FITC-conjugated secondary antibody.
RT-PCR assay. Aliquots of 3 μg of total RNA purified from plated SCG cultures or 10 μg from mouse tissues were reverse-transcribed with Moloney murine leukemia virus reverse transcriptase (Invitrogen, San Diego, CA) and subjected to PCR amplification. The oligonucleotide primers used for dp5, bim, neurofilament-M (NF-M), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) cDNAs were as follows: dp5-5′, 5′-AGACCCAGCCCGGACCGAGCAA-3′; dp5-3′, 5′-ATAGCACTGGGGTGGCTCTC-3′; bim-5′, 5′-TCAGGAGGAACCTGAAGATCTG-3′; bim-3′, 5′-TCAATGCCTTCTCCATACCAGAC-3′; NF-M-5′, 5′-GTGGTCACTAATGGCTTAGATGAG-3′; NF-M-3′, 5′-GTTGAGACCAGCTTCTCCTCAAAG-3′; GAPDH-5′, 5′-AGCTGTGGCGTGATGGCCGTGGGG-3′; and GAPDH-3′, 5′-GAGGGAGATGCTCAGTGTTGGGGC-3′, respectively. Cycle parameters were 1 min at 95°C, 1 min at 60°C, and 30 sec at 72°C for 25–30 cycles, followed by 72°C for 5 min. PCR products were analyzed by PAGE. Control experiments were performed to determine the range of PCR cycles over which amplification efficiency remained constant. Quantification was performed as reported previously (Honma et al., 2002).
Hypoglossal nerve injury model. Eight-week-old DP5–/– and DP5+/+ littermate mice were anesthetized with pentobarbital and put in the supine position. The right hypoglossal nerve of each mouse was then cut with scissors. For in situ hybridization, the operated and uninjured sides of the hypoglossal nuclei were dissected 7 d after surgery. For quantification of cell numbers after axotomy, mice were killed 35 d after the surgery, brains were collected, and 18 μm sections were taken through the hypoglossal nuclear region and subjected to thionine staining. The number of motor neurons (neurons >20 μm in diameter) on both control and operated sides was counted using 24 consecutive sections per mouse, and the percentage of surviving neurons within the injured and control sides was calculated as described previously (Honma et al., 2002).
Northern blotting and in situ hybridization. Northern blot analysis was performed using a 32P-labeled 844 bp DP5 cDNA fragment as a probe (Imaizumi et al., 1997). For in situ hybridization, animals were decapitated and brains were removed and quickly frozen in powdered dry ice. In situ hybridization was performed as described previously (Kiryu et al., 1995).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay. Fresh-frozen sections of mouse brains (n = 3 for each developmental stage analyzed) were fixed with 4% formaldehyde for 15 min and permeabilized with 0.5% (v/v) Triton X-100 in PBS for 10 min at room temperature. After washing and incubating in equilibration buffer (Promega), the terminal deoxynucleotidyl transferase (TdT) reaction mix containing fluorescein-12-deoxyUTP was applied, and the sections were incubated for 60 min at 37°C and washed in PBS. Sections were examined under a fluorescence microscope.
Results
Generation of mice deficient in DP5/HRK
To assess the role of DP5 in neuronal apoptosis, we generated DP5-deficient mice by homologous recombination of embryonic stem cells. A gene-targeting vector was constructed to replace the single coding exon of DP5 with a neomycin-resistant cassette (Fig. 1A). Homologous recombination was confirmed by Southern blot (Fig. 1B) and PCR analyses (Fig. 1C). The absence of DP5 expression was verified in homozygous mutant animals by RT-PCR (Fig. 1D) and Northern blot analyses (data not shown). Intercrosses of DP5+/– heterozygous mice produced DP5-deficient mice at the expected Mendelian ratio. DP5-deficient mice were fertile, showed no gross abnormalities, and appeared normal. Histological examination of tissue sections from 8-week-old DP5–/– brains showed no apparent abnormalities compared with DP5+/+ littermates (data not shown).
Absence of DP5 delays neuronal cell death induced by NGF deprivation
DP5 and BIM are induced in cultured SCG neurons from neonatal rats after NGF withdrawal (Imaizumi et al., 1997; Putcha et al., 2001). To assess whether these proapoptotic BH3-only genes are similarly upregulated in mouse SCG neurons, we examined the expression of dp5 and bim by semiquantitative RT-PCR analysis. In mouse SCG neurons, the expression levels of both dp5 and bim mRNAs were undetectable or low in the presence of NGF (Fig. 2A). By 15 hr of NGF withdrawal, dp5 was induced ∼14-fold, and bim was induced threefold, whereas the expression levels of neurofilament-M mRNA remained unchanged compared with neurons cultured in the presence of NGF (Fig. 2A and data not shown). To determine whether DP5 plays a role in neuronal cell death, we compared the survival of SCG neurons from DP5+/+, DP5+/–, and DP5–/– mice after NGF withdrawal. By 24 hr of NGF deprivation, neurons from wild-type mice underwent significant cell death, which increased to >60% after 36 hr of NGF deprivation. At 36 hr, ∼65% of the wild-type sympathetic neurons were dead, whereas only ∼45% of DP5-deficient neurons underwent apoptosis (Fig. 2B) (p < 0.01; Student's t test; n = 9–14). The protective effect of DP5 against apoptosis was transient in that the great majority of neurons from DP5+/+, DP5+/–, and DP5–/– mice were dead by 48 hr of NGF deprivation (Fig. 2B). Thus, DP5 deletion delays but does not prevent neuronal cell death triggered by NGF withdrawal. The slight and transient protective effect observed with DP5 deficiency is similar to that reported for BIM (Putcha et al., 2001), suggesting functional redundancy among proapoptotic BH3-only Bcl-2 family members in the induction of neuronal apoptosis associated with NGF withdrawal.
Figure 2.

DP5 deletion delays neuronal cell death induced by NGF deprivation. A, RT-PCR analysis ofdp5 and bim expression in SCG neurons from 1-d-old mice in the presence of 50 ng/ml NGF (+) and 15 hr after NGF withdrawal (–). Expression of NF-M was used as a control. B, SCG neurons from the indicated mice were maintained in NGF for 5 d, and then neuronal survival was determined in the absence of NGF at the times indicated. Results are shown as means ± SD (n = 9–14). Symbols denote statistically significant differences between DP5+/+ and DP5–/– neurons. *p < 0.05; **p < 0.01; Student's t test. C, Primary SCG neurons prepared from DP5+/+ and DP5–/– mice were loaded with TMRM, the retention of which is proportional to mitochondrial transmembrane potential. Results are presented as the mean percentages of TMRM-positive cells ± SD from 10 fields counted in three independent experiments. *p < 0.05 and **p < 0.01 compared with wild-type; Student's t test. D, Expression of active caspase-3 was determined after NGF deprivation in SCG neurons using rabbit anti-active caspase-3 antibody. Results are presented as the mean percentages of TMRM-positive cells ± SD from 10 fields counted in three independent experiments. *p < 0.05 and **p < 0.01 compared with wild-type; Student's t test.
Absence of DP5 promotes loss of mitochondrial membrane potential and activation of caspase-3 in SCG neurons
BH3-only proteins have been proposed to act upstream of the mitochondria to regulate caspase activation. To determine whether DP5 regulates the mitochondrial function, primary SCG neurons from DP5+/+ and DP5–/– mice were deprived of NGF and loaded with tetramethylrhodamine ethyl ester perchlorate (TMRE), a potentiometric dye that accumulates within polarized mitochondria, from which it is released on depolarization (Loew et al., 1993). In the absence of NGF deprivation, the great majority of SCG neurons from mutant and wild-type mice were labeled with TMRM (Fig. 2C). By 24 hr of NGF withdrawal, ∼50% of the SCG neurons from DP5+/+ mice were stained by TMRE, which decreased to <30% after 36 hr of NGF deprivation. TMRE labeling was significantly reduced in SCG neurons from DP5–/– mice compared with neurons from wild-type mice (Fig. 2C). Conversely, expression of activated caspase-3 was higher in SCG neurons from DP5+/+ mice than in neurons from DP5–/– mice after NGF deprivation (Fig. 2D). Thus, the absence of DP5 protects SCG neurons from the loss of mitochondrial transmembrane potential and inhibits the expression of activated caspase-3 induced by NGF deprivation.
DP5 is induced in the hypoglossal nucleus after axotomy
The death of motoneurons triggered by axotomy is thought to result from the deprivation of target-derived survival factors (Oppenheim, 1996; Goldberg and Barres, 2000). To examine the role of DP5 in axotomy-induced motoneuron cell death, we used the hypoglossal nerve injury model (Honma et al., 2002). In this model, motoneurons of the hypoglossal nucleus from neonatal animals undergo rapid cell death after hypoglossal nerve transection, whereas adult mice demonstrate delayed induction of cell death (H. Kiyama, unpublished observations). The expression level of dp5 was low or undetectable in the uninjured hypoglossal nucleus as assessed by in situ hybridization; however, dp5 expression in adult neurons was clearly induced 7 d after axotomy (Fig. 3A). Similar to the induction of dp5, the expression of bim mRNA was also increased in motoneurons of the injured side, but the intensity of the induced signal appeared relatively weak compared with uninjured neurons (Fig. 3A).
Figure 3.
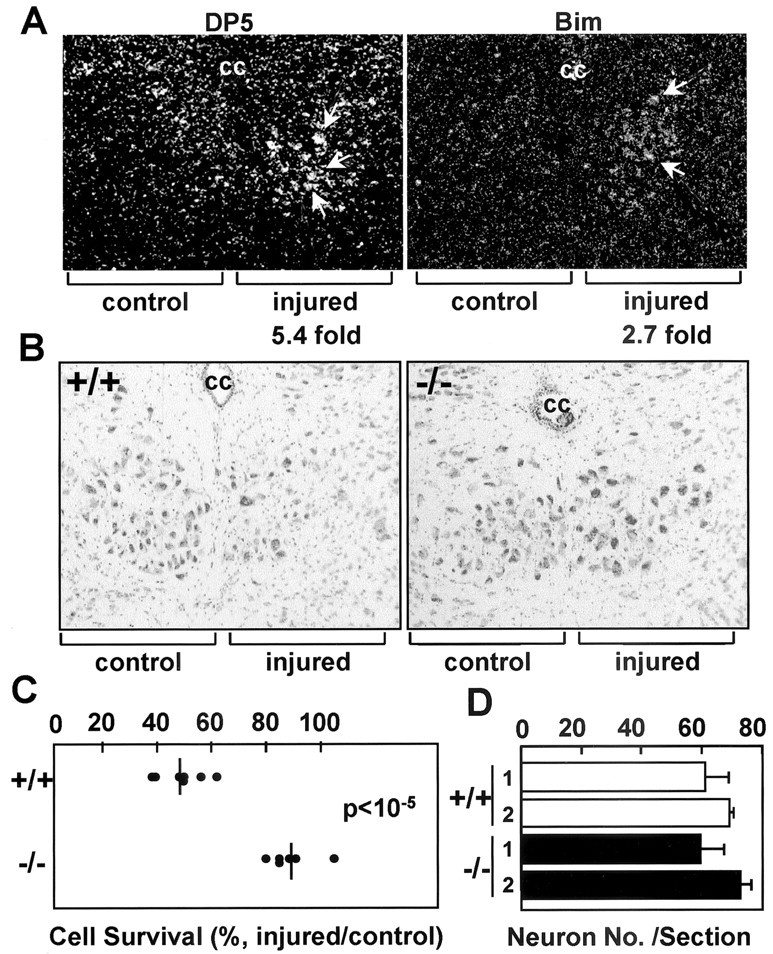
DP5 deletion protects motoneurons from axotomy-induced cell death. A, In situ hybridization of hypoglossal nuclei from 8-week-old mice with dp5- and bim-labeled probes 7 d after hypoglossal nerve transection. Injured and uninjured (control) sides are shown. The fold induction of dp5 and bim in injured neurons (compared with the uninjured side) calculated by image scanning is indicated. B, Coronal sections of the hypoglossal nuclei from DP5+/+ and DP5–/– mice 35 d after axotomy. The injured and control (contralateral) sides are shown. Motoneurons were stained with thionine. cc, Central canal. C, Survival of motoneurons from DP5+/+ and DP5–/– mice 35 d after hypoglossal nerve transection. The number of neurons in the injured and uninjured hypoglossal nuclei was determined by counting seven different sections per animal. Results represent the number of motoneurons in each axotomized nucleus as a percentage of the number in its contralateral (uninjured) nucleus. The DP5+/+ (n = 7) and DP5–/– (n = 6) mice were significantly different (p < 10–5; Student's t test). D, Number of neurons in hypoglossal nuclei (uninjured side) from 8-week-old DP5+/+ and DP5–/– mice. Values (number of neurons) represent the means ± SD derived from seven sections per animal. Results derived from two DP5+/+ and two DP5–/– mice are shown.
DP5 deficiency protects motoneurons from axotomy-induced cell death
We subsequently examined whether DP5 deficiency affects neuronal cell death triggered by transection of the hypoglossal nerve. In these experiments, the nerve was transected unilaterally in 8-week-old DP5+/+ and DP5–/– mice, and the number of hypoglossal nucleus neurons in the injured and uninjured control (contralateral) sides was determined by thionine staining. By 35 d after axotomy, the number of neurons was reduced ∼50% in wild-type mice compared with the uninjured side of the same mice (Fig. 3B,C). In contrast, the motoneurons from DP5-deficient mice were significantly protected from cell death, in that the number of neurons was reduced only ∼10% after transection of the hypoglossal nerve (Fig. 3B,C) (p < 10–5; Student's t test). As a control, we determined the number of neurons in the uninjured hypoglossal nucleus of 8-week-old wild-type and DP5-deficient mice. The number of neurons was similar in DP5+/+ and DP5–/– mice (Fig. 3D). We also assessed the percentage of motor neurons undergoing apoptosis at embryonic day 18 (E18) and at postnatal day 0 (P0) and P7 in the hypoglossal nuclei of DP5+/+ and DP5–/– mice. We found undetectable or very low levels of neuronal apoptosis as detected by TdT-mediated biotinylated UTP nick end labeling in the hypoglossal nucleus at all developmental stages examined, and no significant differences were found between DP5+/+ and DP5–/– mice (data not shown). Thus, although DP5 deficiency protects neurons from axotomy-induced cell death, developmental apoptosis and neuronal cell number in the hypoglossal nucleus are not affected by DP5 deletion.
Discussion
The partial and transient protection afforded by DP5 deficiency against cell death induced by NGF deprivation is consistent with a redundant function of DP5 and BIM during NGF deprivation, because BIM deficiency also delays this type of neuronal cell death (Putcha et al., 2001). Such an interpretation is compatible with our finding that there is reduced but not a total loss of caspase-3 activation in SCG neurons from DP5–/– mice compared with wild-type mice after NGF deprivation. In this model of neuronal cell death, SCG neurons from BAX-deficient mice are completely protected from cell death and caspase activation (Deckwerth et al., 1996; Putcha et al., 1999). Thus, the results suggest that both HRK and BIM play an important but redundant role upstream of BAX during neuronal cell death induced by NGF deprivation. Analysis of mice lacking both DP5 and BIM should provide insight in this regard.
The mechanism involved in neuronal cell death induced by axotomy is poorly understood. Delivery of neurotrophic factors can rescue motoneurons from axotomy-induced cell death (Oppenheim, 1996), suggesting that these survival factors block the induction of the cell-death program. We show that DP5 is upregulated in neurons after transection of the hypoglossal nerve and that DP5 deficiency protected motoneurons from axotomy-induced cell death. Because deficiency of DP5 greatly inhibits the death of motoneurons, the results suggest that neurotrophic factors promote cell survival at least in part by repressing the induction of DP5. Nonetheless, our results do not rule out that factors other than DP5 also play a role in the death of axotomized motoneurons, because a small but reproducible reduction in neuronal survival was observed after axotomy in DP5-deficient mice. Furthermore, it is possible that DP5 deletion delays but does not prevent hypoglossal neuronal cell death. We observed a more robust effect of DP5 deficiency in the axotomy model than in the culture NGF model of neuronal cell death. This could reflect differences in the expression levels of DP5 and/or other apoptosis regulatory genes in motoneurons versus SCG neurons and/or differences between the cell-death pathway activated through axotomy and NGF deprivation. We found no evidence for a role of DP5 in naturally occurring cell death in the hypoglossal nucleus. Pyknotic cells with features of apoptosis can be detected at E17–E19 in the mouse hypoglossal nucleus, and Bax deficiency delays this neuronal cell death (Sun et al., 2003). Thus, it is possible that Bim and/or another BH3-only protein could function upstream of Bax to regulate developmental neuronal apoptosis in the hypoglossal nucleus. However, the ultimate number of postnatal neurons in the hypoglossal nucleus is not affected by the deficiency of Bax, caspase-3, or caspase-9 (Oppenheim et al., 2001; Sun et al., 2003), indicating that neuronal cell death proceeds in the absence of these factors.
Proapoptotic BH3 proteins appear to function upstream of the mitochondria linking death stimuli to BAX and BAK (Bouillet and Strasser, 2002), because a deficiency of BAX (Deckwerth et al., 1996) and overexpression of Bcl-2 and Bcl-XL (Farlie et al., 1995; Blomer et al., 1998; Parsadanian et al., 1998) are known to protect neurons from axotomy-induced cell death. Consistent with this, we found that the absence of DP5 inhibited the loss of mitochondrial membrane potential and activation of caspase-3 in SCG neurons after NGF deprivation. DP5 may promote neuronal cell death by interacting with and inhibiting prosurvival Bcl-2 family members such as Bcl-2 and Bcl-XL. Such inhibition would allow proapoptotic Bax to oligomerize and to insert into the outer mitochondria, an event leading to mitochondrial dysfunction and caspase activation. In an alternative model, BH3-only proteins such as DP5 and BIM may activate BAX directly or indirectly without a need for repressing prosurvival Bcl-2 family members. Notably, the human DP5 ortholog HRK is induced in the spinal neurons of patients with amyotrophic lateral sclerosis (ALS) (Shinoe et al., 2001). Future studies will determine whether DP5/HRK plays a role in neuronal cell death associated with ALS in mouse models of this disease.
Footnotes
This work was supported by grants from the National Institutes of Health (G.N.), Toray Sciences Foundation, and Japan Society for the Promotion of Science (K.I., H.K.), and by postdoctoral fellowships from the Spanish Ministry of Science and Education (A.B., V.G.). We thank Peter Lucas for critical review of this manuscript.
Correspondence should be addressed to Dr. Gabriel Nuñez, University of Michigan Medical School, 1500 East Medical Center Drive, 4219 Cancer Center and Geriatric Center, Ann Arbor, MI 48109. E-mail: bclx@umich.edu.
A. Benito's present address: Unidad de Genética Molecular, Hospital Universitario Marqués de Valdecilla, 39008 Santander, Spain.
DOI:10.1523/JNEUROSCI.5101-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243721-05$15.00/0
K.I., A.B., and S.K.-S. contributed equally to this work.
References
- Baehrecke EH (2002) How death shapes life during development. Nat Rev Mol Cell Biol 3: 779–787. [DOI] [PubMed] [Google Scholar]
- Blomer U, Kafri T, Randolph-Moore L, Verma IM, Gage FH (1998) Bcl-xL protects adult septal cholinergic neurons from axotomized cell death. Proc Natl Acad Sci USA 95: 2603–2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillet P, Strasser A (2002) BH3-only proteins: evolutionarily conserved proapoptotic Bcl-2 family members essential for initiating programmed cell death. J Cell Sci 115: 1567–1574. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson Jr EM, Snider WD, Korsmeyer SJ (1996) BAX is required for neuronal death after trophic factor deprivation and during development. Neuron 17: 401–411. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Johnson Jr EM (1997) Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol Pharmacol 51: 897–906. [DOI] [PubMed] [Google Scholar]
- Farlie PG, Dringen R, Rees SM, Kannourakis G, Bernard O (1995) bcl-2 transgene expression can protect neurons against developmental and induced cell death. Proc Natl Acad Sci USA 92: 4397–4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JL, Barres BA (2000) The relationship between neuronal survival and regeneration. Annu Rev Neurosci 23: 579–612. [DOI] [PubMed] [Google Scholar]
- Honma M, Namikawa K, Mansur K, Iwata T, Mori N, Iizuka H, Kiyama H (2002) Developmental alteration of nerve injury induced glial cell line-derived neurotrophic factor (GDNF) receptor expression is crucial for the determination of injured motoneuron fate. J Neurochem 82: 961–975. [DOI] [PubMed] [Google Scholar]
- Imaizumi K, Tsuda M, Imai Y, Wanaka A, Takagi T, Tohyama M (1997) Molecular cloning of a novel polypeptide, DP5, induced during programmed neuronal death. J Biol Chem 272: 18842–18848. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ding L, Chen S, Nunez G (1997) harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L). EMBO J 16: 1686–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiryu S, Morita N, Ohno K, Maeno H, Kiyama H (1995) Regulation of mRNA expression involved in Ras and PKA signal pathways during rat hypoglossal nerve regeneration. Brain Res Mol Brain Res 29: 147–156. [DOI] [PubMed] [Google Scholar]
- Loew LM, Tuft RA, Carrington W, Fay FS (1993) Imaging in five dimensions: time-dependent membrane potentials in individual mitochondria. Biophys J 65: 2396–2407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheim RW (1991) Cell death during development of the nervous system. Annu Rev Neurosci 14: 453–501. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW (1996) Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron 17: 195–197. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Flavell RA, Vinsant S, Prevette D, Kuan CY, Rakic P (2001) Programmed cell death of developing mammalian neurons after genetic deletion of caspases. J Neurosci 21: 4752–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsadanian AS, Cheng Y, Keller-Peck CR, Holtzman DM, Snider WD (1998) Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci 18: 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson Jr EM (1999) BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci 19: 7476–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM (2001) Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron 29: 615–628. [DOI] [PubMed] [Google Scholar]
- Shinoe T, Wanaka A, Nikaido T, Kanazawa K, Shimizu J, Imaizumi K, Kanazawa I (2001) Upregulation of the pro-apoptotic BH3-only peptide harakiri in spinal neurons of amyotrophic lateral sclerosis patients. Neurosci Lett 313: 153–157. [DOI] [PubMed] [Google Scholar]
- Strasser A, O'Connor L, Dixit VM (2000) Apoptosis signaling. Annu Rev Biochem 69: 217–245. [DOI] [PubMed] [Google Scholar]
- Sun W, Gould TW, Vinsant S, Prevette D, Oppenheim RW (2003) Neuromuscular development after the prevention of naturally occurring neuronal death by Bax deletion. J Neurosci 23: 7298–7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y Miura H, Tanemura A, Kobayashi K, Kondoh G, Sano S, Ozawa K, Inui S, Nakata A, Takagi T, Tohyama M, Yoshikawa K, Itami S (2002) Targeted disruption of LIG-1 gene results in psoriasiform epidermal hyperplasia. FEBS Lett 521: 67–71. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Nakagomi S, Kiryu-Seo S, Namikawa K, Imai Y, Ochi T, Tohyama M, Kiyama H (1999) Expressed-sequence-tag approach to identify differentially expressed genes following peripheral nerve axotomy. Brain Res Mol Brain Res 64: 34–40. [DOI] [PubMed] [Google Scholar]