

Abstract
Axonal degeneration is the major cause of permanent neurological disability in individuals with inherited diseases of myelin. Axonal and neuronal changes that precede axonal degeneration, however, are not well characterized. We show here that dysmyelinated lower motor neurons retract and regenerate dysfunctional presynaptic terminals, leading to severe neurological disability before axonal degeneration. In addition, dysmyelination led to a decreased synaptic quantal content, an indicator of synaptic dysfunction. The amplitude and rise time of miniature endplate potentials were also increased, but these changes were primarily consistent with an increase in the passive membrane properties of the transgenic muscle fibers. Maintenance of synaptic connections should be considered as a therapeutic target for slowing progression of neurological disability in primary diseases of myelin.
Keywords: myelin, axon, acetylcholine receptor, dysmyelination, axonal sprouting, neuromuscular junction
Introduction
Axons are myelinated by Schwann cells in the PNS and by oligodendrocytes in the CNS. Point mutations, duplications, and deletions in genes encoding proteins specific for or enriched in these cells cause dysmyelination and neurological disability (Scherer, 1999). Although dysmyelination can significantly slow nerve conduction velocities, irreversible and progressive neurological disabilities in most inherited diseases of myelin are caused by axonal degeneration. Axonal pathology and axonal degeneration have been documented morphologically in many inherited and acquired diseases of myelin (Trapp et al., 1998, 1999; Scherer, 1999; Garbern et al., 2002). The timing of clinical disease onset and rate of disease progression depend on the mutation and the severity of dysmyelination. Individuals with congenital hypomyelination or Déjérine-Sottas syndrome have an early onset and often fatal clinical course. Dysmyelination, neurological disability, and axonal degeneration in many forms of Charcot-Marie Tooth disease do not manifest until adulthood (Dyck et al., 1989; Yin et al., 1998; Lappe-Siefke et al., 2003), indicating that long-term survival of axons depends on glial-derived support after myelination is complete. Degeneration of chronically dysmyelinated axons, therefore, is a major cause of permanent neurological disability associated with inherited diseases of myelin (Scherer, 1999).
Little is known about the molecular mechanisms responsible for axonal loss in inherited myelin diseases, and no effective therapies have been established. Axonal pathologies that precede axonal degeneration in animal models of dysmyelination include atrophy, swelling, altered axonal cytoskeleton, reduced axonal transport (de Waegh et al., 1992; Griffiths et al., 1998; Yin et al., 1998; Frei et al., 1999; Sahenk et al., 1999; Sancho et al., 1999), and misregulation of gene expression by the dysmyelinated neuron (Brady et al., 1999). As a consequence of these axonal pathologies, it seems likely that synaptic connections of dysmyelinated neurons are altered before axonal degeneration, but this has not been investigated.
We described recently peripheral nerve dysmyelination in several lines of transgenic mice that overexpress P0 protein (P0tg), the major structural protein of PNS myelin (Wrabetz et al., 2000). Overexpressed P0 protein prevented spiral and longitudinal myelin growth (Yin et al., 2000) and caused neurological disability, which correlated with transgene copy number and degree of dysmyelination. These mice resemble and serve as an animal model of the human peripheral neuropathies Déjérine-Sottas syndrome and congenital hypomyelination (Warner et al., 1996; Wrabetz et al., 2000). The transgene was not expressed by oligodendrocytes, and the CNS was appropriately myelinated. To better understand the pathogenesis of neurological disability during chronic dysmyelination, we analyzed the synaptic connections of lower motor neurons (LMNs) in Ptg0 mice. We report here that PNS dysmyelination leads to structural and functional changes at neuromuscular junctions (NMJs) and severe neurological disability.
Materials and Methods
P0-overexpressing mice. The transgene consisted of all introns and exons of the mouse P0 gene, 6 kb of the promoter, and the natural polyadenylation signal. Generation and characterization of mice expressing this transgene have been described in detail previously (Wrabetz et al., 2000). This report focused on the line (Tg 80.2, referred to here as Ptg0) with the highest transgene copy number and most severe peripheral neuropathy. In addition, a lower expressing line (Tg 80.4) was examined and crossed with P0-null mice (Wrabetz et al., 2000) for rescue experiments.
Behavior testing. Body weight, running speed, and grip strength were assessed in Ptg0 and wild-type (WT) littermates at postnatal day 30 (P30) (Ptg0, n = 6; WT, n = 6), P60 (Ptg0, n = 4; WT, n = 6), and P90 (Ptg0, n = 3; WT, n = 5) using methods described in detail Mitsumoto et al. (1994). Forelimb paw grip strength was measured using a digital force transducer (Chatillion DFIS-2; AMETEK, Paoli, PA). The maximal grip strength of five trials was recorded. Running time is defined as the shortest time (the best of three trials) to run 75 cm and is calculated in centimeters per second. The test–retest reliability of these assessment techniques has been established previously (Mitsumoto et al., 1994).
Electromyography. Electromyography (EMG) was performed on anesthetized Ptg0 and WT littermates at P37 (n = 6), P72 (n = 4), and P111 (n = 2). The muscles examined include the following: gluteus maximus (proximal hindlimb extensor), gastrocnemius (distal hindlimb flexor), triceps (proximal forelimb extensor), and forepaw flexors (distal forelimb flexors). Spontaneous electrical activity (fibrillation potentials) was acquired with a TECA Sapphire EMG using a concentric 25 gauge electrode, inserted into the middle of the muscle belly for each muscle examined. Fibrillation potentials (50 μV or greater in amplitude) were quantified per second.
Confocal analysis of neuromuscular junctions. A minimum of six gluteus maximus and soleus muscles were removed from WT and Ptg0 mice at P15, P45, and P90. The muscles were fixed and permeabilized as described previously (Son and Thompson, 1995). Specimens were labeled for postsynaptic acetylcholine receptors (AChRs) with fluorescently conjugated α-bungarotoxin (Molecular Probes, Eugene, OR) and immunostained for axons with a combination of neurofilament (SMI31; Sternberger Monoclonals, Baltimore, MD) and synaptophysin (Sigma, St. Louis, MO) antibodies. Additional sections were tripled labeled for axons, acetylcholine receptors, and P0 protein. Primary antibodies were detected using FITC and Texas Red-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Biotinylated α-bungarotoxin was detected using Cy5-avidin (Jackson ImmunoResearch). Sections were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and examined on a Leica (Heidelberg, Germany) confocal microscope (TCS-NT system). Either Leica TCS-NT acquisition software or Scion (Frederick, MD) Image software was used to reconstruct z-series images into maximum intensity projections. Images were prepared for publication using Photoshop 7 (Adobe Systems, San Jose, CA) software.
The relationship between terminal axons and bungarotoxin staining was investigated in gluteus muscles using whole-mount preparations. The numbers of NMJs analyzed from a minimum of six WT and Ptg0 muscles at each age were as follows: at P15, 329 WT and 224 Ptg0; at P45, 200 WT and 222 Ptg0; and at P90, 292 WT and 263 Ptg0. The junctions were divided into three categories: those normally innervated; those in which axons were separated from bungarotoxin staining (partially denervated); and those junctions without axons (totally denervated). Axonal sprouting was quantified by determining the percentage of NMJs that either extended or received an axonal sprout. The numbers of NMJs analyzed for sprouts were as follows: at P15, 83 WT and 142 Ptg0; at P45, 110 WT and 223 Ptg0; and at P90, 106 WT and 275 Ptg0. Data were analyzed by Student's t test. Similar analysis was performed on an additional line of P0-overexpressing mice (Tg 80.4; n = 59 NMJs) and Tg 80.4 mice bred to P0-null mice (n = 80 NMJs from three mice).
Nerve transection. Four WT and four Ptg0 mice (P30) were anesthetized, and their left sciatic nerves were transected at the sciatic notch. Both soleus muscles were removed 48 hr later, immersion fixed, immunostained for axons and postsynaptic specializations, and examined by confocal microscopy as described above.
Electrophysiological analyses of neuromuscular synaptic transmission. Two WT and six Ptg0 mice at P60 were anesthetized by intraperitoneal injection of 0.05 cc of a mixture of 17.4 mg/ml ketamine and 2.6 mg/ml xylazine (Phoenix Pharmaceuticals, St. Joseph, MO), and the soleus muscle and its innervation were dissected under oxygenated (95% O2, 5% CO2) Rees' Ringer's solution (Rees, 1978) (110 mm NaCl, 5 mm KCl, 1 mm MgCl2,25mm NaHCO3,2mm CaCl2,11mm glucose, 0.3 mm glutamate, 0.4 mm glutamine, 5 mm BES [N, N -bis(2-hydroxyethyl)-2-aminoethanesulfonic acid] buffer, 0.434 μm cocarboxylase, and 36 μm choline chloride). Muscles were pinned in a Sylgard-lined Petri dish and superfused with oxygenated Ringer's solution, and the muscle nerve was placed into a suction electrode.
Physiological analyses were performed as described by Kopp et al. (2000). Briefly, to determine whether nerve-evoked muscle contractions were present, the nerve was stimulated with 0.1–0.2 msec duration rectangular pulses at 0.5–1 Hz. The stimulus voltage was adjusted from 0.1 to 10 V, and muscle contractions were visually monitored through a dissecting microscope. To determine whether muscle fibers were electrically excitable, muscle fibers were placed into a suction electrode and were stimulated with 0.5 msec duration rectangular pulses delivered at 0.5–1 Hz. The stimulus voltage was adjusted over a range from 0.5 to 10 V, and muscle contractions were visually monitored.
To determine whether spontaneous and evoked neurotransmitter release was present, skeletal muscle fibers were cut at each end to prevent muscle contractions attributable to muscle fiber depolarization (Glavinovic, 1979; Ribchester et al., 1994). Intracellular recordings were performed using glass microelectrodes filled with 3 m KCl (30–570 MΩ resistance). All experiments were performed at room temperature. A total of 43 junctions from six Ptg0 mice and 25 junctions from five wild-type littermates were studied.
Electrical potentials were amplified using an Axoprobe 1A amplifier (Axon Instruments, Union City, CA), low-pass filtered at 1 kHz, and digitized at 10 kHz using an analog-to-digital converter (DigiData; Axon Instruments) and interactive software (Axoscope; Axon Instruments). Muscle fiber resting membrane potential was continuously monitored, and only fibers with resting potentials more hyperpolarized than –40 mV and in which the resting potential did not change by >5 mV during the course of the experiment were studied further. This was the minimum resting potential that allowed a sufficient signal-to-noise ratio to detect miniature endplate potentials (mepps). Muscle fiber input resistance was calculated as voltage–current after injection of a 100 msec hyperpolarizing current pulse. mepps were recorded for 10–20 min and analyzed using interactive software (Mini Analysis; Synaptosoft, Decatur GA). To characterize nerve-evoked endplate potentials, the stimulation voltage to the soleus muscle nerve was varied from 0.01 to 2.00 V. Quantal content was determined under conditions of reduced Ca2+ (1 mm CaCl) and elevated Mg22+(10 mm MgCl2) using the method of failures (Del Castillo and Katz, 1954; Kopp et al., 2000) or expressed as the ratio of mean endplate potential amplitude/mean mepp amplitude. The epp amplitudes were corrected for nonlinear summation (McLachlan and Martin, 1981) using an f value of 0.8. The quantal content, i.e., the number of ACh quanta released after a single nerve impulse, was calculated at each NMJ by dividing the mean corrected epp amplitude by the mean mepp amplitude. (McLachlan and Martin, 1981). Statistical analyses were performed using Microsoft (Seattle, WA) Excel software. Data are presented as mean ± SEM.
FM1-43 labeling of synaptic vesicle recycling. Postnatal day 60 WT (n = 6) and Ptg0 (n = 10) mice were anesthetized, and the soleus muscle and nerves were dissected under cold oxygenated (95% O2, 5% CO2) mammalian Ringer's solution and were pinned into a Sylgard-lined Petri dish. Muscles were placed in 5 μm FM1-43 (Molecular Probes) in oxygenated Ringer's solution at room temperature. The nerve was placed into a suction electrode and was stimulated with 5 sec trains of 0.1 msec duration pulses delivered at 30 Hz, repeated every 20 sec for 5 min. The stimulus intensity used was the minimum that reliably caused muscle contraction, typically 0.1–0.5 V. After stimulation, muscles were rinsed with 10 min washes in oxygenated Ringer's solution and 2 min washes in Ringer's solution with 1 mm ADVASEP-7 (Biotium, Hayward, CA) over a 1 hr period. After FM1-43 labeling, AChRs were labeled by incubation in 10 μg/ml rhodamine-conjugated α-bungarotoxin in Ringer's solution for 4 min. Muscles were mounted onto glass slides and coverslipped. Muscles were mounted onto glass slides and coverslipped. NMJs (Ptg0, 22 junctions; WT, 10 junctions) were imaged using a laser scanning confocal microscope (Leica TCS 4D system). The percentage of AChR staining apposed by FM1-43 was determined using interactive software (MetaMorph; Universal Imaging Corporation, West Chester, PA).
Quantification of AChR subunit mRNA. Three gluteus maximus muscles from P15, P30, and P90 WT and Ptg0 mice were removed. RNA was isolated using the Total RNA isolation kit (Promega, Madison, WI), quantified using standard spectrophotometric measurements and run on an Agilent (Palo Alto, CA) 2100 Bioanalyzer to ensure RNA quality. RNA (devoid of DNA) was reverse transcribed and amplified using gene-specific primers for AChR α subunit (GenBank accession number M17640; forward, GAGGACCACCGTGAGATTGT; reverse, AATCGACCCATTGCTGTTTC), β subunit (GenBank accession number M14537; forward, AGCCTGAACGAGAAGGATGA; reverse, AGCAGTGATGCGGAGAGAAT), and γ subunit (GenBank accession number X03818; forward, GTGTCTTCGAGGTG GCTCTC; reverse, TCTGGGATTGGAAGATGAGG). PCR fragments were subsequently ligated to the T7 polymerase promoter sequence (Lig-n-scribe kit; Ambion, Austin, TX) and PCR amplified. Five micrograms of total RNA were used for Northern analysis using the Northern Max kit (Ambion) protocols. Radiolabeled ([32P]UTP) antisense RNA, transcribed using T7 polymerase (Maxiscript kit; Ambion) was used as probe. Hybridized membranes were exposed to Eastman Kodak (Rochester, NY) X-Omat films for 1–2 d. Bands were quantified using the Scion Image software and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels. After detection, membranes were stripped by boiling in 0.1% SDS solution and reprobed (in the order of α subunit, γ subunit, followed by β subunit).
Electron microscopy. Three WT and mutant mice were perfused at P15 and P90 with 2.5% glutaraldehyde, 4% paraformaldehyde, and 0.08 m Sorensen's buffer. The gluteus maximus muscle and L4 and L5 ventral roots were removed and processed to Epon embedding resin. NMJs were identified in 1-μm-thick sections and then sectioned for electron microscopic analysis. The relationship between axons, postsynaptic specializations, and Schwann cells was investigated in electron micrographs from 18 WT NMJs and 16 Ptg0 NMJs. Three of the Ptg0 NMJs were examined in serial sections. Montages of the entire left and right L4 and L5 ventral roots from three P90 WT and Ptg0 mice were generated from electron micrographs. Total axons were quantified in the montages. The data were analyzed using Student's t test.
Results
Neurological disability correlates with muscle fibrillation and atrophy but not LMN degeneration
Several lines of mice that overexpress P0 protein were generated (Wrabetz et al., 2000). The present study focuses on the most severely affected line (Tg 80.2). A progressive decline in motor performance accompanied reduced body weight and muscle atrophy (Fig. 1A). Running speed in Ptg0 was reduced by 50% at P30 and 80% at P60, and the mice were non-ambulatory at P90. Grip strength was reduced by 20% at P30, 68% at P60, and 75% at P90. Body weight was reduced at P30, when pelvic girdle muscle atrophy appears, and decreased at subsequent time points, paralleling more generalized muscle atrophy. EMG of girdle, hindlimb, and forelimb muscles was performed to test for muscle denervation. Fibrillation potentials (Fig. 1B, inset), an indicator of muscle denervation, were graded quantitatively in WT and Ptg0 mice at P37, P72, and P111 (Fig. 1B). Fibrillation potentials were not detected in WT muscles. At P37, fibrillations were prominent in the pelvic girdle (gluteus) muscles, minimal in distal hindlimb flexor (gastrocnemius) muscles, and absent in forelimb distal flexors. By P72, fibrillations were present in all muscles examined but were most prominent in distal hindlimb muscles. At P111, fibrillation potentials persisted in girdle and hindlimb muscles and became more prominent in forelimb muscles. These data support a progressive denervation of Ptg0 musculature that parallels the proximal-to-distal gradient of muscle atrophy.
Figure 1.

P0 overexpression causes severe neurological phenotypes and muscle fibrillation but not proximal motor axon degeneration. A, Running speed, grip strength, and body weight decrease progressively in Ptg0 mice between P30 and P90. Values represent means and SEM for mice tested at P30 (Ptg0, n = 6; WT, n = 6), P60 (Ptg0, n = 4; WT, n = 6), and P90 (Ptg0, n = 3; WT, n = 5). B, Ptg0 mice display spontaneous fibrillations of muscle (inset) that appearing irdle and proximal muscles before distal muscles. C–E, Comparison of toluidine blue-stained Epon sections of ventral roots from P90 wild-type (C) and Ptg0 (D) mice. Schwann cells ensheath but fail to myelinate motor axons in the mutant mice. The number of motor axons in L4 and L5 ventral roots are identical in wild-type and Ptg0 mice (E). Scale bars, 10 μm. Grip strength was normalized to body weight. Table and histograms show mean and SEM. *p < 0.05;†p < 0.001;‡p < 0.0001; t tests of Ptg0 versus WT.
To determine whether degeneration of LMNs or their proximal axons contribute to the muscle denervation, axons in the L4 and L5 ventral roots from P90 WT (Fig. 1C) and Ptg0 (Fig. 1D) mice were quantified by electron microscopy (Fig. 1E). Despite the severe dysmyelination in ventral roots of Ptg0 mice, the number of axons was identical to WT. Degeneration of LMNs or their proximal axons, therefore, do not contribute to the neurological deficits in Ptg0 mice.
Terminal axonal withdrawal and sprouting at neuromuscular junctions
During normal development, two or more motor axons initially innervate individual NMJs (Sanes and Lichtman, 1999; Lichtman and Colman, 2000). By the end of the second postnatal week, however, most NMJs are innervated by single axons. We compared innervation of the gluteus maximus and soleus muscles at P15, P45, and P90 in Ptg0 and WT mice by confocal analysis of tissue immunostained for terminal axons with neurofilament and synaptophysin antibodies and for AChRs with bungarotoxin. Changes were significant and similar in the gluteus maximus and soleus muscles; quantitative data are presented for the gluteus. At P15, NMJs in both mouse strains (Fig. 2A,B) were innervated by single axons, and >98% of the postsynaptic AChR clusters in WT mice were appropriately apposed by axonal terminals (Fig. 2C). In contrast to WT NMJs (Fig. 2D), axonal terminals in Ptg0 NMJs often ended in small ovoids (Fig. 2E, arrows), and large regions of the postsynaptic specialization were unopposed by axons (Fig. 2F, asterisks), leaving NMJs partially denervated. Twenty-five percent of the NMJs in P15 Ptg0 gluteus maximus were partially denervated (Table 1). The preterminal axons, however, did not degenerate or form axonal retraction bulbs, the stereotypic appearance of preterminal axonal withdrawal at NMJs (Balice-Gordon et al., 1993). On the basis of these observations, NMJs in Ptg0 mice form normally during development. However, synaptic connections between terminal axons and muscle fibers begin to break down by P15.
Figure 2.

Dysmyelinated PNS axons form but do not maintain normal NMJs. A, B, At P15, single axons (green) innervate NMJs (red) in both WT (A) and Ptg0 (B) mice. C, NMJs in WT mice are normal at P30 (C) and P90. D–F, P15 WT axons branch normally within NMJs (D). Intrajuctional axons in P15 Ptg0 mice often ended in small ovoids (E, arrows), leaving areas of postsynaptic AChRs devoid of axons (F, asterisks). AChR staining appeared normal in D and E and was omitted to demonstrate axonal differences. Terminal axon retraction and sprouting increased as the Ptg0 mice aged (F, arrowheads). At P90, postsynaptic AChR clusters (G, H, red) became fragmented and punctate, and the resultant small AChR aggregates were often innervated by individual terminal axonal branches (G–I, green). Interjunctional sprouts (G–I, arrowheads) become more prominent as the mutant mice age. For quantification, see Table 1. A–H are from gluteus; I is from soleus. Scale bars: A, B, 25 μm; C–E, 10 μm; F, 2 μm; G–I, 5 μm.
Table 1.
Terminal axon retraction and sprouting increase with age at neuromuscular junctions in P0tg mice and can be rescued by crossbreeding with P0-null mice
|
Ages |
Strain |
% Partial denervation |
% Complete denervation |
% NMJ with axon sprouts |
|---|---|---|---|---|
| P15 | WT | 1.7 ± 0.7 | 0 ± 0 | 1.0 ± 1.0 |
| P0tg | 25.2 ± 0.5*** | 0.8 ± 0.5 | 3.9 ± 2.0 | |
| P45 | WT | 0.8 ± 0.4 | 0 ± 0 | 3.1 ± 0.3 |
| P0tg | 72.0 ± 1.1*** | 7.3 ± 0.5*** | 63.7 ± 9.9* | |
| P90 | WT | 1.5 ± 0.5 | 0 ± 0 | 3.2 ± 0.7 |
| P0tg | 89.5 ± 0.6*** | 7.2 ± 0.2*** | 90.5 ± 0.3** | |
| Tg 80.4 | 31.3 ± 2.3** | 2.6 ± 1.0 | 14.9 ± 1.3** | |
|
|
P0R |
1.2 ± 0.4 |
0 ± 0 |
3.0 ± 0.2 |
Three animals were examined, both left and right muscles, at each age per strain, except Tg 80.4, in which two animals were analyzed.
p < 0.005; **p < 0.001; ***p < 0.0001; t test for transgenic versus WT.
Terminal axonal withdrawal from NMJs increased as the mutant mice aged (Fig. 2G). By P45, 72% of the NMJs examined were partially denervated, and 7% were totally denervated (Table 1). At P90, only 3% of the mutant NMJs appeared normal, 89% were partially denervated, and 7% were entirely devoid of presynaptic nerve terminals and were thus denervated. In addition, the normal pretzel-like shape of AChR clusters (Fig. 2C) became punctate and discontinuous (Fig. 2F–I), and the terminal axons extended multiple intrajunctional branches to the many small discrete AChR aggregates at NMJs in both the gluteus maximus (Fig. 2H) and soleus (Fig. 2I) muscles. Axonal sprouts also extended to neighboring NMJs (Fig. 2F–I). In muscles from P15–P90 WT mice, <3% of NMJs extended and/or received interjunctional axonal sprouts. In Ptg0 mice, however, these percentages were significantly increased at both P45 (45%) and P90 (89%). Although dysmyelinated axons cannot maintain normal NMJs, they retain the ability to sprout to neighboring NMJs.
The relationship between synaptic abnormalities and axonal degeneration was investigated by examining NMJ innervation after transection of P30 WT and P0tg sciatic nerves. Whereas bungarotoxin staining was unaffected, axonal terminals were eliminated by 48 hr after transection in both strains of mice (data not shown). Synaptic withdrawal and sprouting at the NMJ, therefore, precedes axonal degeneration and is not a response of axons disconnected from their somas.
Loss of functional nerve terminals and changes in neuromuscular synaptic transmission in Ptg0 mice
To determine whether the Ptg0 transgenic mouse NMJs with dramatically altered presynaptic and postsynaptic specializations are capable of releasing neurotransmitter, the ability of junctions to internalize the fluorescent dye FM1-43 was evaluated in the soleus muscle from 2-month-old Ptg0 and WT mice. The soleus muscle was selected for this analysis because of the ease of dissection without muscle or nerve damage. Nerves were stimulated in short trains of pulses delivered at high frequency in Ringer's solution containing FM1-43 (Cochilla et al., 1999), and neuromuscular junctions were analyzed with confocal microscopy.
In 10 of 10 NMJs examined in P60 WT muscles, an average of 92% of the postsynaptic AChR cluster area was occupied by FM1-43-stained presynaptic nerve terminals (Fig. 3A). However, in NMJs in the soleus muscle of P60 Ptg0 mice, an average of <40% of the postsynaptic AChR cluster area was occupied by FM1-43-stained nerve terminals (Fig. 3B,C), and 5 of 22 junctions examined were entirely devoid of FM1-43-stained nerve terminals. These data are consistent with the partial denervation observed after immunostaining. This observation supports widespread loss of functional nerve terminals in the soleus muscles from mice overexpressing P0.
Figure 3.
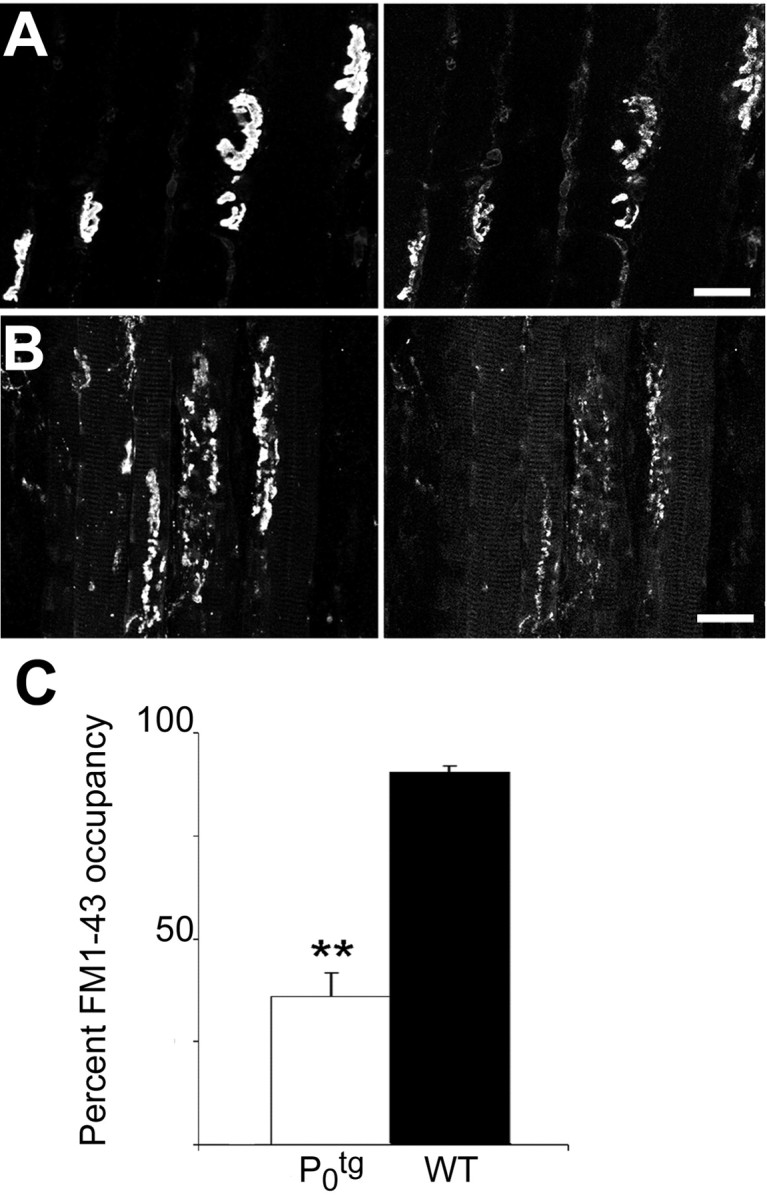
FM1-43 labeling demonstrates defects in synaptic vesicle recycling and loss of functional nerve terminals in Ptg0 NMJs. A, In 10 of 10 NMJs in the soleus muscle of P 60 WT mice, >95% of the postsynaptic AChR cluster area (left) was occupied in its entirety by FM1-43-stained presynaptic nerve terminals (right). B, In 17 of 22 NMJs in the soleus muscle of P60 Ptg0 mice, a portion of the postsynaptic AChR cluster area was occupied by FM1-43-stained nerve terminals. C, Average NMJ area occupied by FM1-43 labeling for soleus muscles from P60 Ptg0 mice (white) was reduced to 60% compared with WT mice (black). Error bars indicate mean ± SEM. t test indicated that Ptg0 was significantly different from WT; **p < 0.0001. Scale bars, 25 μm.
To determine how the loss of functional nerve terminals in Ptg0 mice suggested by FM1-43 staining affected neuromuscular synaptic transmission, the soleus muscle was dissected, and spontaneous and nerve-evoked synaptic potentials were analyzed after intracellular recording using standard methods. Spontaneous mepps were recorded for 5–10 min. In the soleus muscle from 2-month-old mice, the frequency of spontaneous mepps was similar between Ptg0 and WT mice (Fig. 4A,B), suggesting that spontaneous release of neurotransmitter quanta is relatively unaffected. However, mepp amplitude was increased 2.3-fold (p < 0.001; Student's t test), and rise time was increased 1.9-fold (p < 0.005) compared with WT mice (Fig. 4C–E).
Figure 4.
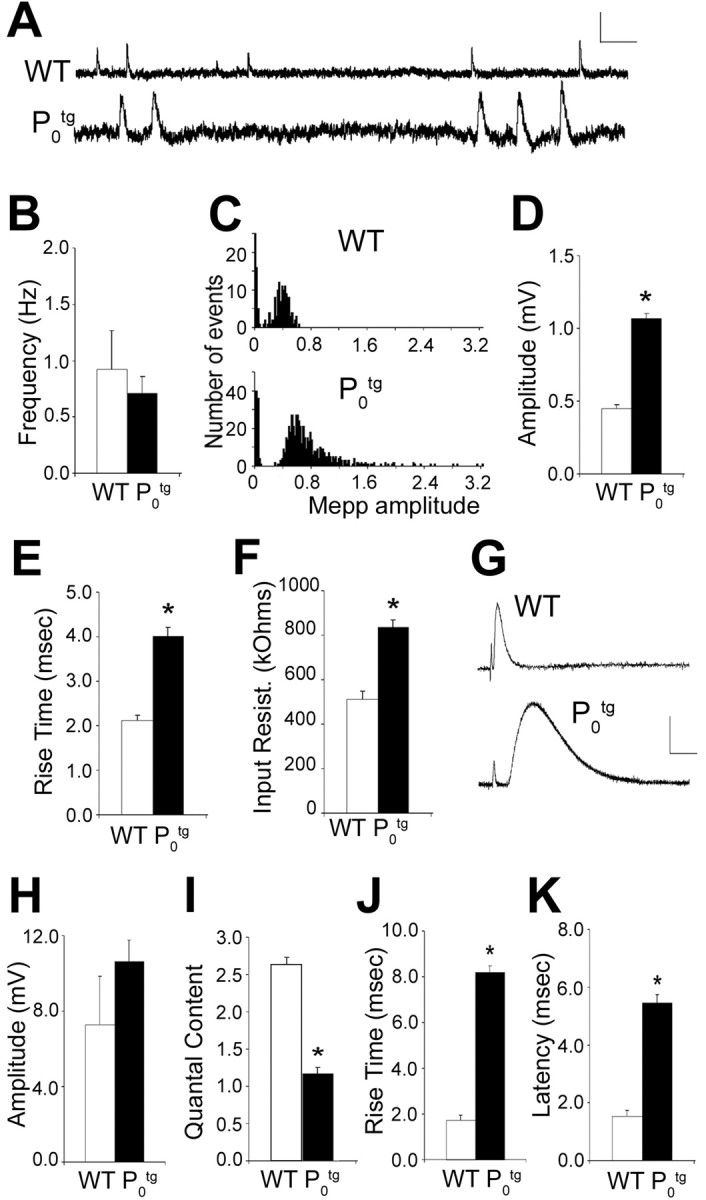
Spontaneous and evoked synaptic transmission in the soleus muscle from Ptg0 and WT mice. A, Spontaneous mepps from a soleus muscle fiber from P60 Ptg0 and WT mice. mepp frequency (B) is not significantly different between WT (white) and Ptg0 (black) mice. C, Distribution of the amplitude of all events from muscle fibers shown in A. Distributions are not significantly different; Komolgorov–Smirnov test; p = 0.425. In each histogram, the first peak is the distribution of baseline noise in the recordings. mepp amplitude (D), rise time (E), and inputresistance (F) were increased in soleus muscle fibers from Ptg0 (blackbars) compared with WT (white bars) mice.G, Nerve-evoked end plate potentials from a soleus muscle fiber from Ptg0 and WT mice. The vertical deflection at the left of each trace is an artifact of nerve stimulation. Note the increased latency from on set of nerve stimulation to on set of eppins oleus muscle from mice overexpressing P0. H–K, epp amplitude (H) was not significantly different between WT and Ptg0, but quantal content was reduced (I), and epp rise time (J) and latency (K) were increased in the soleus muscle of Ptg0 compared with WT mice. Values are shown as mean ± SEM. *p < 0.005, significant difference between transgenic and WT junctions; Student's t test. Calibration: A, 0.5 mV, 100 msec; G, 5 mV, 10 msec.
The increase in mepp size might be attributable to muscle fiber atrophy and thus reflect an increase in muscle fiber input resistance as has been observed after denervation, inactivity, and as a consequence of neuromuscular disease (Tonge, 1974; Weinstein, 1980; Roy et al., 2000). Measurements of muscle fiber input resistance showed that input resistance was greater in Ptg0 soleus than in WT muscle fibers (Fig. 4F) (839 ± 38 kΩ in Ptg0; 513 ± 77 kΩ in WT; p < 0.001; Student's t test). This is consistent with the significant muscle fiber atrophy measured by muscle fiber cross-sectional area reported previously in Ptg0 muscles (Wrabetz et al., 2000). These differences in input resistance could account at least in part for the differences in mepp amplitude.
To characterize nerve-evoked neurotransmitter release, the nerve innervating the soleus muscle was stimulated and recorded. The quantal content, i.e., the number of ACh quanta released after a single nerve impulse, was calculated as the mean epp amplitude, corrected for nonlinear summation (McLachlan and Martin, 1981), divided by the mean mepp amplitude. In the soleus muscle from 2-month-old mice, epp amplitude was similar between Ptg0 and WT (Fig. 4G,H). However, quantal content was significantly reduced in junctions from Ptg0 compared with wild-type soleus muscle (Fig. 4I). Moreover, in Ptg0 muscles but not WT littermates, a small percentage of junctions in Ptg0 muscles (<5%) had significantly reduced epp amplitude. In addition, no evoked epps or spontaneous mepps were observed in ∼10% of junctions (4 of 43) from Ptg0 soleus muscle, suggesting that these muscle fibers were completely denervated. These observations are consistent with the percentage of junctions that were partially or completely denervated after FM1-43 labeling (see above) and suggest that synaptic transmission is reduced in Ptg0 mice.
The increased mepp amplitude and rise time observed in the soleus muscle of Ptg0 mice might also be consistent with a change in the expression of postsynaptic AChR subunits, as has been observed after denervation and reinnervation (Gu and Hall, 1988; Sanes and Lichtman, 1999). To evaluate this possibility, levels of α, β, and γ AChR mRNA were compared in WT and Ptg0 gluteus and soleus at P15, P30, and P90 by Northern blot analysis (Fig. 5). AChR subunit mRNA expression was similarly altered in Ptg0 gluteus and soleus: data shown are from the gluteus. Levels of the α AChR subunit mRNA were significantly increased in Ptg0 muscle at P30 (sixfold) and P90 (fourfold). The β subunit was increased twofold at P30 and was unchanged at P90. The embryonic γ subunit was abundant in WT newborn muscle (Fig. 5), barely detectable at P30 in the Ptg0 muscle, and was not detectable at other time points in either WT or Ptg0 muscle total RNA extracts. These data suggest that the changes observed in mepp amplitude may reflect a change in postsynaptic AChR expression in addition to a change in muscle fiber input resistance.
Figure 5.

Dysmyelination alters muscle acetylcholine receptor mRNA levels. α and β, but not γ, acetylcholine receptor mRNAs are increased in Ptg0 gluteus maximus muscle. Northern blot comparisons of α, β, and γ AChR mRNAs from wild-type (W) and P0-overexpressing (P) mice. The α subunit is increased at P30 (6-fold) and P90 (4-fold). The β subunit was increased by twofold at P30 but was unchanged at P90. The γ subunit is prominent at birth but undetectable in wild type and Ptg0 controls at P15, P30, or P90. mRNA levels were normalized to GAPDH (G) levels.
In the soleus muscle, epp rise time was significantly greater than that observed in WT mice (Fig. 4G,J), as was observed for mepps. One possible explanation is that the muscle fiber membrane time constant is altered in Ptg0 mice as a consequence of atrophy. Alternatively, the density of AChRs may be decreased in muscles from Ptg0 mice, because a slowing of spontaneous and evoked epps has been reported under circumstances in which AChR density is low (Katz and Miledi, 1973; Albuquerque et al., 1976; Betz and Osborne, 1977; Kidokoro, 1980; Colman et al., 1997). A third possibility is that there is an increased separation between nerve terminals and postsynaptic AChRs at junctions in Ptg0 mice, as would be expected to occur if nerve terminals were retracting. A slowing of mepp and epp rise times has been suggested to be attributable to the increased time that would be required for acetylcholine molecules to bind to unoccupied AChRs (Katz and Miledi, 1973; Betz and Osborne, 1977; Kidokoro, 1980). In addition to the slowing of epp and mepp rise time, the latency from the stimulus artifact to epp onset was delayed by more than fourfold in the soleus muscle of Ptg0 mice compared with WT (Fig. 4K). This is consistent with the dysmyelination and slowing of axonal conduction velocity observed after P0 overexpression (Wrabetz et al., 2000). Together, these data suggest that neuromuscular synaptic function is impaired in the soleus muscle of Ptg0 mice, consistent with partial denervation and reinnervation by terminal sprouts (Fig. 2).
NMJ denervation correlates with dysmyelination
When NMJs were examined in P90 gluteus maximus from another P0-overexpressing line (Tg 80.4) with less severe dysmyelination and neurological disability, 32% were partially denervated (Table 1). The neurological phenotype and dysmyelination in the Tg 80.4 line was rescued by breeding to P0-null mice (Wrabetz et al., 2000). We investigated whether the NMJ abnormalities in Tg 80.4 mice were also rescued by the P0-null mice. In gluteus maximus from P90 Tg 80.4 × P0-null mice (P0R), muscle fibrillations were absent, and 99% of the NMJs were appropriately innervated (Table 1). These rescue experiments provide compelling evidence that dysmyelination is the primary cause of NMJ denervation in P0-overexpressing mice.
Terminal Schwann cells do not play a primary role in NMJ denervation
Confocal studies were performed to determine whether the Schwann cells that cap Ptg0 NMJs play a primary role in NMJ denervation. In WT and Ptg0 mice, detectable P0 immunoreactivity terminated 10–30 μm before the entry of the axon into the NMJ (Fig. 6A,B). In WT mice, P0-expressing cells near NMJs formed myelin sheaths that closely apposed axons (Fig. 6A). In the mutant mice, P0-expressing Schwann cell perikarya (Fig. 6B) were larger and covered shorter axonal segments than myelinating Schwann cells in WT mice. P0 protein was not detected by confocal microscopy in terminal Schwann cells of the NMJ in either WT or Ptg0 mice. To extend these light microscopic observations, we compared the ultrastructure of NMJs from P15 WT (Fig. 6C) and Ptg0 (Fig. 6D) gluteus maximus muscles. Sixty-five percent of NMJs from P15 Ptg0 mice (10 of 16 NMJs) were indistinguishable from WT NMJs (Fig. 6C). The axon terminal was appropriately apposed to postsynaptic specializations, and Schwann cells capped the surface of the axon terminal. Six of the 16 Ptg0 NMJs examined by electron microscopy contained degenerating presynaptic terminals (Fig. 6D), identified by an abundance of synaptic vesicles (Fig. 6D, inset). Regions of the postsynaptic membrane were unapposed by axons or Schwann cells (Fig. 6D). Terminal Schwann cells or their processes were not detected between Ptg0 presynaptic and postsynaptic specializations. These observations support the conclusion from confocal imaging that terminal Schwann cells are not playing a primary role in separating presynaptic and postsynaptic components of the NMJ. The ultrastructure of NMJs in P90 Ptg0 gluteus was significantly altered (Fig. 6E). Large expanses of postsynaptic membranes were unapposed by terminal axons. Terminal axons of varying diameters (Fig. 6E) were present and ensheathed by Schwann cells.
Figure 6.
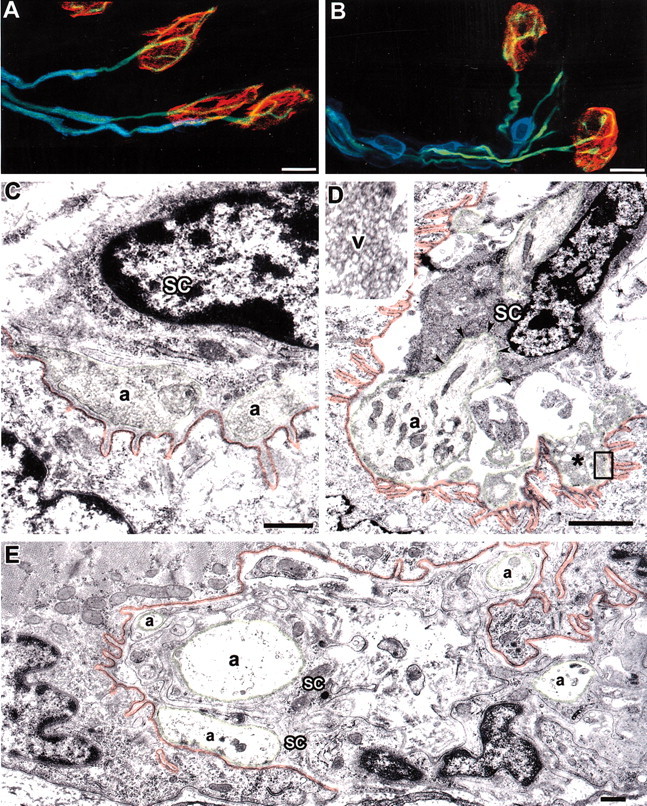
Dysmyelination causes axonal withdrawal from the NMJ. A,B, P0 protein is not detected in terminal Schwann cells of the NMJ. Triple labeling for P0 (blue), axons (green), and postsynaptic specializations (red) in WT (A) and mutant (B) NMJs detected P0 in Schwann cells along the preterminal axon but not in terminal Schwann cells that “cap” the NMJ. C, D, In electron micrographs of WT NMJs (C), terminal Schwann cells (sc) cap the NMJ. In partially denervated NMJs from P 15 Ptg0 muscle (D), terminal Schwann cells surround the axon (a, green arrowheads) as it enters the NMJ but did not separate presynaptic and postsynaptic (red) specializations. Degenerating axonal profiles (*, inset) were detected in some P15 Ptg0 NMJs and contained numerous vesicles. E, In electron micrographs of P90 Ptg0 NMJs, multiple axons (a, green) were present. The majority of post synaptic muscle membranes (red) were not apposed by axons terminals. Some axons were < 1 μm india meter and may represent regenerated sprouts.S chwann cells surrounded these axons and provided a terminal cap (E). Scale bars: A, B,10 μm; C–E,1 μm.
Discussion
Here we describe a mouse model in which chronically dysmyelinated lower motor neurons retract and regenerate dysfunctional synaptic terminals. These synaptic changes precede the axonal degeneration that becomes prominent by 6 months of age and that reduces the lifespan of the mice by ∼50% (Wrabetz et al., 2000). NMJs of Ptg0 LMNs formed normally during early postnatal development and were unchanged in P90 P0-overexpressing mice crossed to P0-null mice. Terminal axonal withdrawal and NMJ dysfunction in Ptg0 mice, therefore, are pathological consequences of chronic dysmyelination. Maintenance of synaptic connectivity should thus be considered as a therapeutic target in primary diseases of myelin.
Schwann cells have essential roles in axonal migration, axonal maturation, and axonal survival. Alterations in Ptg0 Schwann cell function, therefore, could cause neurological disability by several mechanisms. Although peripheral myelin does not form, axons in Ptg0 peripheral nerves are ensheathed by Schwann cells (Yin et al., 2000). This Schwann cell ensheathment promotes maturation of axons, including appropriate muscle innervation and normal distribution of AChRs on the surface of muscle. Molecular requirements for formation of the NMJ include axonally derived agrin, which orchestrates the postsynaptic distribution and organization of acetylcholine receptors by interacting with MuSK (muscle-specific receptor tyrosine kinase) (McMahan, 1990; DeChiara et al., 1996). Our studies indicate that these molecular events are intact in Ptg0 mice. We describe a sequence of axonal pathologies in Ptg0 nerves in which an early downstream effect of dysmyelination was partial withdrawal of axon terminals from the NMJs. The preterminal axons initially retained one-to-one relationships with NMJs and generated intrajunctional sprouts that project to AChR clusters at NMJs. These structural changes have functional consequences, as evidenced by a decrease in quantal content in Ptg0 compared with WT mice. Other changes in mepp amplitude and rise time likely reflect an increase in muscle fiber input resistance as a result of atrophy, whereas the fourfold increase in epp latency reflect dysmyelination and slowing of axonal conduction velocity observed after P0 overexpression (Wrabetz et al., 2000). After sciatic nerve transection, distal axons and their terminals at NMJs rapidly degenerated in both WT and Ptg0 mice. Synaptic changes in Ptg0 NMJs, therefore, precede axonal degeneration. As the mice aged, intrajunctional and interjunctional sprouts increased to the point at which 96% of NMJs examined at P90 were abnormal. Many junctions in P90 Ptg0 muscle attained the appearance of CNS synaptic terminal arbors, with multiple sprouts contacting punctate clusters of AChRs. Chronic dysmyelination not only altered the distribution of AChRs within individual junctions but also increased the expression of mRNA encoding the α and β subunit of the AChR at P30–P90. This increase is consistent with an increase in extrajunctional AChR expression, similar to that observed after denervation or inactivity (Fambrough, 1974; Berg and Hall, 1975; Blondet et al., 1989).
A number of studies support the existence of unidentified molecules or signals that are required to maintain axon–muscle contact once NMJs are formed (Lichtman and Colman, 2000). The separation of maintenance of axon–muscle contact from NMJ formation and terminal axon sprouting described here supports this hypothesis and raises the possibility that chronic dysmyelination preferentially alters the unidentified signals that maintain NMJs. Altered axonal cytoskeleton and impaired axonal transport precede axonal degeneration in mice with a variety of mutations in myelin protein genes (de Waegh et al., 1992; Griffiths et al., 1998; Yin et al., 1998; Lappe-Siefke et al., 2003). It is possible, therefore, that dysmyelination in Ptg0 mice impairs antegrade transport of LMN-derived molecules needed for maintenance of the NMJ. This may initiate a response by the muscle that eventually implements terminal axon sprouting. As the Ptg0 mice age, progressive muscle atrophy, reduced muscle fibrillations (Fig. 1B), and the lack of muscle fiber grouping (Wrabetz et al., 2000) support conduction block as an additional determinant of the sprouting phenotype. Synaptic stripping by terminal Schwann cells has been described in laminin β2-deficient mice (Noakes et al., 1995) and after exogenous application of neuregulin (Trachtenberg and Thompson, 1997) but, on the basis of confocal and EM analysis, did not appear to contribute to the denervation of Ptg0 NMJs.
Denervation of the NMJs did not conform to a general dying-back distal axonopathy (Fig. 1B), because the longest fibers were not affected first. Our data do support, however, a mechanism of distal axonopathy that affects proximal muscles before distal muscles. The progression of proximal-to-distal muscle phenotype and of predominately slow muscle implicates muscle usage as a contributor to the NMJ phenotype in Ptg0 mice. This mechanism has also been suggested to contribute to NMJ dysfunction in hereditary canine spinal muscular atrophy (HCSMA), a neurodegenerative disease affecting LMNs (Sack et al., 1984; Blazej et al., 1998). Physiological and morphological NMJ phenotypes, however, are dramatically dissimilar in Ptg0 mice and in HCSMA dogs, which exhibit motor unit tetanic failure before structural and functional alterations in NMJs are apparent (Balice-Gordon et al., 2000; Rich et al., 2002).
Mechanisms of neuronal degeneration are highly compartmentalized (Gillingwater and Ribchester, 2001). Injury or deprivation of growth factors can lead to apoptotic death of the neuronal cell body, followed by rapid degeneration of dendrites, axons, and synaptic terminals (Pettmann and Henderson, 1998). Axonal transection results in rapid degeneration of the isolated distal axon by a caspase-independent mechanism (Raff et al., 2002). Rapid axonal degeneration after transection is delayed in mice with the Wlds mutation (Brown et al., 1992). Whether this mutation, which involves a proteosomal protein, would protect against the NMJ changes in Ptg0 mice remains to be determined. We described here significant alteration in the synaptic connections of chronically dysmyelinated neurons that precedes axonal degeneration. Our data have important implications regarding the progression of neurological disability in primary myelin diseases because it provides proof of principle for gradual muscle deafferentiation by chronically dysmyelinated or demyelinated neurons. Gradual and progressive neuronal diaschsis may contribute to the continuous, irreversible neurological disability that is common in inherited myelin disorders (Scherer, 1999; Garbern et al., 2002) and secondary progressive stages of multiple sclerosis (Trapp et al., 1999; Herndon, 2002). Maintenance of synaptic connectivity should be considered as a therapeutic target in inherited and acquired diseases of myelin.
Footnotes
This work was supported by National Institutes of Health Grants NS38186 (B.D.T.), NS38517 (R.J.B.-G.), and NS41319 and NS45630 (M.L.F., L.W.) and Telethon Italy Grant GGP030074 (L.W.). We thank Victoria Pickett for editorial assistance.
Correspondence should be addressed to Dr. Bruce D. Trapp, Department of Neurosciences/NC30, Lerner Research Institute, Cleveland Clinic Foundation, 9500 Euclid Avenue, Cleveland, OH 44195. E-mail: trappb@ccf.org.
DOI:10.1523/JNEUROSCI.4617-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243890-09$15.00/0
References
- Albuquerque EX, Rash JE, Mayer RF, Satterfield JR (1976) An electrophysiological and morphological study of the neuromuscular junction in patients with myasthenia gravis. Exp Neurol 51: 536–563. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Chua CK, Nelson CC, Lichtman JW (1993) Gradual loss of synaptic cartels precedes axon withdrawal at developing neuromuscular junctions. Neuron 11: 801–815. [DOI] [PubMed] [Google Scholar]
- Balice-Gordon RJ, Smith DB, Goldman J, Cork LC, Shirley A, Cope TC, Pinter MJ (2000) Functional motor unit failure precedes neuromuscular degeneration in canine motor neuron disease. Ann Neurol 47: 596–605. [PubMed] [Google Scholar]
- Berg DK, Hall ZW (1975) Loss of alpha-bungarotoxin from junctional and extrajunctional acetylcholine receptors in rat diaphragm muscle in vivo and in organ culture. J Physiol (Lond) 252: 771–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz W, Osborne M (1977) Effects of innervation on acetylcholine sensitivity of developing muscle in vitro. J Physiol (Lond) 270: 75–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazej RG, Mellersh CS, Cork LC, Ostrander EA (1998) Hereditary canine spinal muscular atrophy is phenotypically similar but molecularly distinct from human spinal muscular atrophy. J Hered 89: 531–537. [DOI] [PubMed] [Google Scholar]
- Blondet B, Rieger F, Verdiere-Sahugue M (1989) Activity-independent modulation of acetylcholine receptor levels in rat skeletal muscle following neonatal denervation. Neurosci Lett 102: 273–278. [DOI] [PubMed] [Google Scholar]
- Brady ST, Witt AS, Kirkpatrick LL, de Waegh SM, Readhead C, Tu PH, Lee VM (1999) Formation of compact myelin is required for maturation of the axonal cytoskeleton. J Neurosci 19: 7278–7288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MC, Lunn ER, Perry VH (1992) Consequences of slow Wallerian degeneration for regenerating motor and sensory axons. J Neurobiol 23: 521–536. [DOI] [PubMed] [Google Scholar]
- Cochilla AJ, Angleson JK, Betz WJ (1999) Monitoring secretory membrane with FM1-43 fluorescence. Annu Rev Neurosci 22: 1–10. [DOI] [PubMed] [Google Scholar]
- Colman H, Nabekura J, Lichtman JW (1997) Alterations in synaptic strength preceding axon withdrawal. Science 275: 356–361. [DOI] [PubMed] [Google Scholar]
- de Waegh SM, Lee VM, Brady ST (1992) Local modulation of neurofilament phosphorylation, axonal caliber, and slow axonal transport by myelinating Schwann cells. Cell 68: 451–463. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD (1996) The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell 85: 501–512. [DOI] [PubMed] [Google Scholar]
- Del Castillo J, Katz B (1954) Quantal components of the end-plate potential. J Physiol (Lond) 124: 560–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyck PJ, Karnes JL, Lambert EH (1989) Longitudinal study of neuropathic deficits and nerve conduction abnormalities in hereditary motor and sensory neuropathy type 1. Neurology 39: 1302–1308. [DOI] [PubMed] [Google Scholar]
- Fambrough DM (1974) Acetylcholine receptors. Revised estimates of extrajunctional receptor density in denervated rat diaphragm. J Gen Physiol 64: 468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frei R, Motzing S, Kinkelin I, Schachner M, Koltzenburg M, Martini R (1999) Loss of distal axons and sensory Merkel cells and features indicative of muscle denervation in hindlimbs of P0-deficient mice. J Neurosci 19: 6058–6067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbern JY, Yool DA, Moore GJ, Wilds IB, Faulk MW, Klugmann M, Nave KA, Sistermans EA, van der Knaap MS, Bird TD, Shy ME, Kamholz JA, Griffiths IR (2002) Patients lacking the major CNS myelin protein, proteolipid protein 1, develop length-dependent axonal degeneration in the absence of demyelination and inflammation. Brain 125: 551–561. [DOI] [PubMed] [Google Scholar]
- Gillingwater TH, Ribchester RR (2001) Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J Physiol (Lond) 534: 627–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glavinovic MI (1979) Voltage clamping of unparalysed cut rat diaphragm for study of transmitter release. J Physiol (Lond) 290: 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths I, Klugmann M, Anderson T, Yool D, Thomson C, Schwab MH, Schneider A, Zimmermann F, McCulloch M, Nadon N, Nave K-A (1998) Axonal swellings and degeneration in mice lacking the major proteolipid of myelin. Science 280: 1610–1613. [DOI] [PubMed] [Google Scholar]
- Gu Y, Hall ZW (1988) Immunological evidence for a change in subunits of the acetylcholine receptor in developing and denervated rat muscle. Neuron 1: 117–125. [DOI] [PubMed] [Google Scholar]
- Herndon RM (2002) Medical hypothesis: why secondary progressive multiple sclerosis is a relentlessly progressive illness. Arch Neurol 59: 301–304. [DOI] [PubMed] [Google Scholar]
- Katz B, Miledi R (1973) The binding of acetylcholine to receptors and its removal from the synaptic cleft. J Physiol (Lond) 231: 549–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidokoro Y (1980) Developmental changes of spontaneous synaptic potential properties in the rat neuromuscular contact formed in culture. Dev Biol 78: 231–241. [DOI] [PubMed] [Google Scholar]
- Kopp DM, Perkel DJ, Balice-Gordon RJ (2000a) Disparity in neurotransmitter release probability among competing inputs during neuromuscular synapse elimination. J Neurosci 20: 8771–8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33: 366–374. [DOI] [PubMed] [Google Scholar]
- Lichtman JW, Colman H (2000) Synapse elimination and indelible memory. Neuron 25: 269–278. [DOI] [PubMed] [Google Scholar]
- McLachlan EM, Martin AR (1981) Non-linear summation of end-plate potentials in the frog and mouse. J Physiol (Lond) 311: 307–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahan UJ (1990) The agrin hypothesis. Cold Spring Harb Symp Quant Biol 55: 407–418. [DOI] [PubMed] [Google Scholar]
- Mitsumoto H, Ikeda K, Holmlund T, Greene T, Cedarbaum JM, Wong V, Lindsay RM (1994) The effects of ciliary neurotrophic factor on motor dysfunction in wobbler mouse motor neuron disease. Ann Neurol 36: 142–148. [DOI] [PubMed] [Google Scholar]
- Noakes PG, Gautam M, Mudd J, Sanes JR, Merlie JP (1995) Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature 374: 258–262. [DOI] [PubMed] [Google Scholar]
- Pettmann B, Henderson CE (1998) Neuronal cell death. Neuron 20: 633–647. [DOI] [PubMed] [Google Scholar]
- Raff MC, Whitmore AV, Finn JT (2002) Axonal self-destruction and neurodegeneration. Science 296: 868–871. [DOI] [PubMed] [Google Scholar]
- Rees D (1978) A non-phosphate-buffered physiological saline for in vitro electrophysiological studies on the mammalian neuromuscular junction. J Physiol (Lond) 278: 8P–9P. [PubMed] [Google Scholar]
- Ribchester RR, Mao F, Betz WJ (1994) Optical measurements of activity-dependent membrane recycling in motor nerve terminals of mammalian skeletal muscle. Proc R Soc Lond B Biol Sci 255: 61–66. [DOI] [PubMed] [Google Scholar]
- Rich MM, Wang X, Cope TC, Pinter MJ (2002) Reduced neuromuscular quantal content with normal synaptic release time course and depression in canine motor neuron disease. J Neurophysiol 88: 3305–3314. [DOI] [PubMed] [Google Scholar]
- Roy RR, Zhong H, Monti RJ, Vallance KA, Kim JA, Edgerton VR (2000) Mechanical properties and fiber type composition of chronically inactive muscles. J Gravit Physiol 7: 103–104. [PubMed] [Google Scholar]
- Sack Jr GH, Cork LC, Morris JM, Griffin JW, Price DL (1984) Autosomal dominant inheritance of hereditary canine spinal muscular atrophy. Ann Neurol 15: 369–373. [DOI] [PubMed] [Google Scholar]
- Sahenk Z, Chen L, Mendell JR (1999) Effects of PMP22 duplication and deletions on the axonal cytoskeleton. Ann Neurol 45: 16–24. [DOI] [PubMed] [Google Scholar]
- Sancho S, Magyar JP, Aguzzi A, Suter U (1999) Distal axonopathy in peripheral nerves of PMP22-mutant mice. Brain 122: 1563–1577. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389–442. [DOI] [PubMed] [Google Scholar]
- Scherer S (1999) Axonal pathology in demyelinating diseases. Ann Neurol 45: 6–7. [DOI] [PubMed] [Google Scholar]
- Son YJ, Thompson WJ (1995) Schwann cell processes guide regeneration of peripheral axons. Neuron 14: 125–132. [DOI] [PubMed] [Google Scholar]
- Tonge DA (1974) Chronic effects of botulinum toxin on neuromuscular transmission and sensitivity to acetylcholine in slow and fast skeletal muscle of the mouse. J Physiol (Lond) 241: 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Thompson WJ (1997) Nerve terminal withdrawal from rat neuromuscular junctions induced by neuregulin and Schwann cells. J Neurosci 17: 6243–6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338: 278–285. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Ransohoff RM, Fisher E, Rudick RA (1999) Neurodegeneration in multiple sclerosis: relationship to neurological disability. The Neuroscientist 5: 48–57. [Google Scholar]
- Warner LE, Hilz MJ, Appel SH, Killian JM, Kolodny EH, Karpati G, Carpenter S, Watters GV, Wheeler C, Witt D, Bodell A, Nelis E, Van Broeckhoven C, Lupski JR (1996) Clinical phenotypes of different MPZ (P0) mutations may include Charcot-Marie-Tooth type 1B, Dejerine-Sottas, and congenital hypomyelination. Neuron 17: 451–460. [DOI] [PubMed] [Google Scholar]
- Weinstein SP (1980) A comparative electrophysiological study of motor end-plate diseased skeletal muscle in the mouse. J Physiol (Lond) 307: 453–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrabetz L, Feltri ML, Quattrini A, Imperiale D, Previtali S, D'antonio M, Martini R, Yin X, Trapp BD, Zhou L, Chiu SY, Messing A (2000) P0 glycoprotein overexpression causes congenital hypomyelination of peripheral nerves. J Cell Biol 148: 1021–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Crawford TO, Griffin JW, Tu P-H, Lee VMY, Li C, Roder J, Trapp BD (1998) Myelin-associated glycoprotein is a myelin signal that modulates the caliber of myelinated axons. J Neurosci 18: 1953–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Kidd GJ, Wrabetz L, Feltri ML, Messing A, Trapp BD (2000) Schwann cell myelination requires timely and precise targeting of P0 protein. J Cell Biol 148: 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]