

Abstract
To investigate lipid rafts as a site where amyloid β protein (Aβ) oligomers might accumulate and cause toxicity in Alzheimer's disease (AD), we analyzed Aβ in the Tg2576 transgenic mouse model of AD. Aβ was highly concentrated in lipid rafts, which comprise a small fraction of brain volume but contain 27% of brain Aβ42 and 24% of Aβ40 in young mice. In the Tg2576 model, memory impairment begins at 6 months before amyloid plaques are visible. Here we show that Aβ dimers appear in lipid rafts at 6 months and that raft Aβ, which is primarily dimeric, rapidly accumulates reaching levels >500× those in young mice by 24–28 months. A similar large accumulation of dimeric Aβ was observed in lipid rafts from AD brain. In contrast to extracellular amyloid fibrils, which are SDS-insoluble, virtually all Aβ in lipid rafts is SDS soluble. Coupled with recent studies showing that synthetic and naturally occurring Aβ oligomers can inhibit hippocampal long-term potentiation, the in vivo age-dependent accumulation of SDS-soluble Aβ dimers in lipid rafts at the time when memory impairment begins in Tg2576 mice provides strong evidence linking Aβ oligomers to memory impairment. After dimeric Aβ began to accumulate in lipid rafts of the Tg2576 brain, apolipoprotein E (ApoE) and then phosphorylated tau accumulated. A similar increase in ApoE and a large increase in phosphorylated tau was observed in lipid rafts from AD brain. These findings suggest that lipid rafts may be an important site for interaction between dimeric Aβ, ApoE, and tau.
Keywords: lipid rafts, amyloid β protein, Alzheimer's disease, ApoE, tau, Tg2576 mouse model
Introduction
There is compelling evidence that amyloid β protein (Aβ) aggregation is an essential early event in Alzheimer's disease (AD) pathogenesis (Selkoe, 2001, 2002). Growing evidence suggests that the pathogenic effects of Aβ aggregation are mediated, in large part, by small diffusible Aβ oligomers formed during Aβ fibrillogenesis (Lambert et al., 1998; Lue et al., 1999; McLean et al., 1999; Hsia et al., 1999; Mucke et al., 2000) (for review, see Walsh et al., 2002). Lipid rafts are dynamic assemblies of cholesterol and sphingolipids (for review, see Simons and Toomre, 2000) that play an important role in signal transduction, and they have also been implicated in cell adhesion and lipid/protein sorting (Sargiacomo et al., 1993; Simons and Ikonen, 1997; Simons and Toomre, 2000). Because lipid rafts resist solubilization in cold, nonionic detergents and have low buoyant density, they are most commonly isolated as buoyant, detergent-resistant membranes (DRMs) using sucrose density gradient centrifugation (Simons and Ikonen, 1997; Brown and London, 1998; Hooper, 1999). Using this approach, Aβ has been identified in lipid rafts from cultured cells and from human and rodent brain (Lee et al., 1998; Morishima-Kawashima and Ihara, 1998; Oshima et al., 2001; Wahrle et al., 2002). Presenilin 1 (PS-1), β secretase (BACE1, Asp-2, memapsin-1), the amyloid β protein precursor (APP), and CTFβ, the Aβ-bearing C-terminal fragment produced by β secretase, have also been identified in DRMs (Lee et al., 1998; Morishima-Kawashima and Ihara, 1998; Oshima et al., 2001; Riddell et al., 2001; Wahrle et al., 2002). On this basis, we reasoned that Aβ oligomers might form and accumulate in lipid rafts, where they would be well positioned to cause toxicity by interfering with signal transduction.
To test this hypothesis, we analyzed the effect of aging on lipid raft Aβ in the brains of Tg2576 mice. In Tg2576 mice, which overexpress human APP695 with the “Swedish” mutation (Hsiao et al., 1996), memory deficits appear at 6 months (Westerman et al., 2002), whereas amyloid plaques first appear at 8 months (Kawarabayashi et al., 2001). Our results show that Aβ dimers appear in lipid rafts at 6 months and that raft Aβ, which is primarily dimeric, rapidly accumulates, reaching levels >500× those in young mice by 24–28 months. A similar large accumulation of dimeric Aβ was observed in lipid rafts from AD, as compared with control brain. Significantly, the dimeric Aβ in lipid rafts from AD or aging Tg2576 brains did not require formic acid for solubilization. Coupled with recent studies showing that synthetic and naturally occurring Aβ oligomers can inhibit hippocampal long-term potentiation (Lambert et al., 1998; Walsh et al., 2002), the age-dependent appearance of Aβ dimers in lipid rafts at the time when memory impairment begins in the Tg2576 model of AD provides strong evidence implicating diffusible Aβ oligomers in AD pathogenesis.
To determine if lipid rafts might also be involved in the formation of neurofibrillary tangles, we analyzed lipid rafts from the brains of aged Tg2576 mice and patients with AD for phosphorylated tau, the major constituent of neurofibrillary tangles. Remarkably, this analysis showed increasing levels of phosphorylated tau in the brains of very old Tg2576 mice and high levels of phosphorylated tau in lipid rafts from AD brain. Apolipoprotein E (ApoE), which is known to be involved in AD, was also increased in lipid rafts from the brains of aged Tg2576 mouse and AD brain.
Materials and Methods
Materials. Thirty-three Tg2576 mice [3 (n = 6), 4 (n = 2), 5 (n = 3), 6 (n = 2),7(n = 2),8(n = 3),9(n = 3), 10 (n = 3), 11 (n = 3), 12 (n = 2), 18 (n = 1), 20 (n = 1), 24 (n = 1), and 28 (n = 1) months old] and six nontransgenic littermates [3 (n = 4), 9 (n = 1), and 28 (n = 1) months old] were examined. Six brains of AD (mean 77 ± 10 years old), two brains of pathological aging (69 and 81 years old), and two normal control brains with only a few senile plaques (55 and 88 years old) were investigated. Pathological aging indicates cognitively normal subjects who have many senile plaques in the brain (Dickson et al., 1995), suggesting a preclinical or very early phase of AD.
Antibodies. The following antibodies were used: BAN-50 (anti-Aβ1–16) (Suzuki et al., 1994), BNT-77 (anti-Aβ11–28) (Shinkai et al., 1997), BA-27 (anti-Aβ1–40) (Suzuki et al., 1994), BC-05 (anti-Aβ35–43) (Suzuki et al., 1994), and 4G8 (anti-Aβ17–24; Senetek PLC, Napa, CA); 3160 (anti-Aβ1–40) for immunoprecipitation (Kawarabayashi et al., 2001); O443 [antibody to C-terminal 20 amino acid residues of APP, a gift from K. Sambamurti (Medical College of South Carolina, Charleston, SC) (Pinnix et al., 2001)]; Z31preA [antibody to 577–596 of APP695, which corresponds to just N-terminal side of β-cleavage (Kawarabayashi et al., 1991)]; anti-flotillin antibody (Transduction Laboratories, Lexington, KY) as a lipid raft marker (Bickel et al., 1997), anti-calnexin antibody (StressGen Biotechnologies, Victoria, British Columbia, Canada) as an endoplasmic reticulum (ER) marker; anti-mouse ApoE antibody [a gift from N. Yamada (University of Tokyo, Tokyo, Japan) (Shimano et al., 1991)], anti-human ApoE antibody (Chemicon, Temecula, CA); PHF-1 to phosphorylated tau at pSer396/pSer404 sites (Greenberg and Davies, 1990), and WKS46 as a phosphorylation-independent antibody against tau 359–370 [a gift from S. H. Yen (Mayo Clinic Jacksonville) (Kenessey et al., 1997)]; antibody to N terminus of mouse PS1 (N1) and antibody to loop of mouse PS-1 (L1) (gifts from T. Koothan); anti-BACE-1 antibody (rb206; Amersham Biosciences, Peapack, NJ), and anti-neprilysin antibody (CD10; Novocastra Laboratories, Newcastle, UK). Cholera toxin B subunit conjugated with peroxidase (Sigma, St. Louis, MO) was used to detect GM1 ganglioside (Parton, 1994).
Preparation of lipid rafts. Brains were minced with a razor blade and homogenized on ice in MBS-T buffer with protease inhibitors (50 mm MES, 150 mm NaCl, pH 6.5, 1% Triton X-100, 5 μg/ml leupeptin, 5 μg/ml aprotinin, and 2 μg/ml pepstatin A) at 200 mg brain/ml MBS-T using 20 strokes of a Dounce homogenizer. The homogenate was sonicated for 5 sec, five times at level 10 with an XL-2000 Microson Ultrasonic Cell Disruptor (Misonix Inc., Farmingdale, NY), and then centrifuged to remove nuclei and cell debris at 2000 × g for 10 min at 4°C. The pellet was re-extracted twice by sonicating the pellet with 25% of the original volume of buffer and centrifugation under the same conditions. The three supernatants that had been extracted from the low-speed Triton pellet (fraction 14) were pooled to form a Triton homogenate (fraction 13) that was fractionated by sucrose gradient centrifugation. Sucrose in distilled water (1.5 ml, 80%) and 1.5 ml of fraction 13 were mixed, put into a 12 ml ultracentrifuge tube, and overlayed with 5 ml of 38% sucrose and 3 ml of 5% sucrose. The sucrose gradient was centrifuged at 100,000 × g for 19 hr at 4°C in an SW41 rotor (Beckman Instruments, Palo Alto, CA). Starting from the top, 1 ml of sample was aspirated, and the samples were named as fractions 1–11 with the pellet designated fraction 12 (high speed gradient pellet).
Aβ ELISA. Twenty-five microliters of 20% SDS in distilled water were added to 225 μl of each fraction to make a 2% SDS concentration. Each sample was then sonicated for 5 sec six times and centrifuged at 100,000 × g for 1 hr at 20°C. The supernatant contained SDS-soluble Aβ (AβSDS). To analyze SDS-insoluble fibrillar Aβ that could be extracted with formic acid (AβFA), each pellet was sonicated with 225 μl of 70% FA and then centrifuged at 100,000 × g for 1 hr at 4°C (Harigaya et al., 1995). Both SDS and FA extracted fractions were measured by ELISA using BNT-77/BA-27 for Aβ40 and BNT-77/BC-05 for Aβ42 (Suzuki et al., 1994; Shinkai et al., 1997). The concentration of Aβ in each fraction was adjusted to per gram of original wet weight of the brain. After normality testing, one-way ANOVA and unpaired t test with Welch's correction were performed for statistical analysis (SPSS, version 11; SPSS, Chicago, IL).
Western blot analysis. Sucrose gradient fractions 1–11 were separated on 10–20% or 16% Tricine SDS gels (Novex, Wadsworth, OH) and electrotransferred to an Immobilon P membrane (Millipore, Bedford, MA) at 100 V for 1.5 hr. Membranes were labeled with primary antibodies overnight at 4°C and incubated with horseradish peroxidase-linked secondary antibodies (Amersham Biosciences, Buckingham, UK) for 1 hr. The signal was detected using Supersignal (Pierce, Rockford, IL). To detect Aβ dimers in the AD brain, each fraction was directly applied to the gel. To detect Aβ dimers in Tg2576, immunoprecipitation was performed as described below.
Immunoprecipitation. Each fraction was mixed with one-ninth volume of 20% SDS in distilled water. Each 2% SDS fraction was sonicated for 5 sec, five times and diluted 40-fold with radioimmunoprecipitation assay (RIPA) buffer (150 mm NaCl, 1% Triton X-100, 0.5% cholic acid, 0.1% SDS, and 50 mm Tris, pH 8) containing protease inhibitors. The samples were then immunoprecipitated overnight with 10 μl of protein G-agarose that had been incubated with 2 μg of 3160. The samples were washed three times with RIPA buffer, one time with TBS, and used for Western blotting.
Electron microscopy and immunoelectron microscopy. Lipid rafts (fraction 4), the high-speed gradient pellet (fraction 12), and the low-speed Triton pellet (fraction 14) were centrifuged at 100,000 × g for 5 min at 4°C. Respective pellets were fixed in 1.5% glutaraldehyde in 0.1 m cacodylate buffer at pH 7.4 containing 1% CaCl2, then postfixed in OsO4, dehydrated in ethanol, infiltrated, and embedded in Epon 812 (Polysciences, Warrington, PA). Ultrathin sections were stained with uranyl and lead. For post-embedding immunogold labeling, pellets were fixed in 2% paraformaldehyde in 0.1 m phosphate buffer, dehydrated in up to 90% ethanol, infiltrated, and embedded in LR White resin (Polysciences). Thin sections collected on nickel grids were incubated overnight with primary antibodies at 4°C, followed by secondary antibodies conjugated with 5 or 10 nm gold particles. They were briefly stained with uranyl acetate and lead formate.
Results
Characterization of lipid rafts
Lipid rafts were isolated conventionally as buoyant DRMs (Simons and Toomre, 2000). Brains from 3-month-old Tg2576 mice were homogenized on ice in MBS-T (MBS with 1% Triton X-100 and protease inhibitors). The postnuclear supernatants were then fractionated on sucrose gradients, and the 11 gradient fractions were analyzed by immunoblotting (Fig. 1A). Flotillin and GM-1 ganglioside, two markers for lipid rafts in brain, were found almost exclusively in fraction 4, indicating that lipid rafts float into this fraction during centrifugation (Fig. 1A, note that longer exposures showed some staining for flotillin and GM-1 ganglioside in fractions 3 and 5). Transmembrane proteins like calnexin, which is a good marker for the endoplasmic reticulum that is excluded from lipid rafts, were found only in fractions 8–11 as expected (Fig. 1A). Electron microscopic (EM) examination of fraction 4 showed vesicular structures of various sizes as previously reported (Lee et al., 1998) (Fig. 1B).
Figure 1.

Characterization of lipid rafts from 3-month-old Tg2576 mouse brains. A, Western blotting of sucrose gradient centrifuge fractions. Lanes 1–11 show the 11 fractions from the sucrose gradient. The lipid raft markers, flotillin and GM1 ganglioside, localized in fraction 4. The ER marker, calnexin, localized in fractions 8–11. APP, CTF of APP, BACE1, PS1-N, PS1-C, neprilysin, and ApoE localized in lipid rafts as well as in fractions 8–11. B, Fraction 4 was composed of vesicular structures of various sizes by EM. Scale bar, 600 nm. C, D, Levels of Aβ42SDS (C) and Aβ40SDS (D) in fractions 1–11 from 3-month-old Tg2576 brains (n = 6). E, Immunoblotting of Aβ in lipid rafts from 3-month-old transgenic brains. Synthetic Aβ1–40 (a, 2,000 fmol; b, 500 fmol; c, 125 fmol), fraction 4 of 3-month-old transgenic brains (d, 0.5 ml; e, 2 ml; f, 2 ml) were immunoprecipitated with 3160 (anti-Aβ1–40). Proteins were separated on 16% tricine gels, transferred to polyvinylidene difluoride membrane, and detected with BAN-50 (anti-Aβ1–16, lanes a–e) or 4G8 (anti-Aβ17–24, lane f). The arrowhead identifies CTFβ, and the arrow identifies Aβ monomers.
Full-length APP, APP CTFs, the putative γ secretase (N- and C-fragments of PS1) and ApoE were all detected not only in fractions 8–11 at the bottom of the gradient (which contain buffer-soluble extracellular and intracellular proteins as well as proteins released from organelle membranes by Triton) but also in the lipid rafts in fraction 4. β secretase (BACE1) and the recently identified Aβ degrading enzyme neprilysin were also present both in the lipid raft and Triton-soluble fractions. Thus, our evaluation of molecules known to be involved in Aβ generation (APP, APP CTFs, PS1-N/PS1-C, BACE1), degradation (neprilysin), and aggregation (ApoE) showed that they are all present in the lipid raft fraction.
Aβ42 and Aβ40 are found in the lipid rafts
The Aβ in sucrose fractions was analyzed by sandwich ELISA as described in Materials and Methods. When sucrose fractions were analyzed directly, the ELISA signals from all eleven fractions were weak (data not shown). When SDS was added to a concentration of 2% before dilution and analysis, there was a substantial increase in signal, and Aβ42 (Fig. 1C) and Aβ40 (Fig. 1D) were readily detected. This effect presumably occurs because SDS dissociates Aβ from binding proteins that mask detection by sandwich ELISA.
Remarkably, fraction 4 from 3 month Tg2576 brains contained more Aβ42 (Fig. 1C) and Aβ40 (Fig. 1D) than any other fraction. Overall, the lipid rafts (fractions 3–5) contained 27% (2.3 ± 0.30 pmol/gm) of all Aβ42 and 24% (6.2 ± 0.9 pmol/gm) of all Aβ40 in the brains of young Tg2576 mice. Fraction 4 alone contained 19% (1.6 ± 0.2 pmol/gm) of all Aβ42 and 20% (5.3 ± 0.9 pmol/gm) of all Aβ40.
To be certain that the sandwich ELISAs were detecting Aβ in fraction 4, we immunoprecipitated this fraction with an affinity-purified polyclonal antibody to Aβ. The immunoprecipitate was then analyzed on immunoblots labeled with BAN-50, which detects Aβ1–16, or with 4G8, which detects Aβ17–24. For standardization, synthetic Aβ in amounts ranging from 125 to 2000 fmol was immunoprecipitated in identical manner in parallel. The results of this analysis are shown in Figure 1E. Both BAN-50 (lanes d, e) and 4G8 (lane f) labeled Aβ immunoprecipitated from fraction 4, and most of the Aβ detected by both antibodies was the 4 kDa monomeric form. In 0.5 ml of fraction 4 (lane d), we found ∼500–700 fmol of Aβ (lane d vs increasing synthetic Aβ in lanes a-c) and ∼4 times that amount in 2 ml (lane e). This corresponds to some 5–7 pmol of Aβ/g in young Tg2576 brain, a result that is in excellent agreement with our analysis by sandwich ELISA. The ∼11 kDa Aβ-bearing CTF in the lipid rafts (fraction 4) was also immunoprecipitated by the anti-Aβ antibody (Fig. 1E).
Aβ accumulates with aging in the lipid rafts of Tg2576 mouse brain
In young Tg2576 brain and in control human or mouse brain, only 5% of total Aβ is soluble Aβ that can be extracted in the absence of any detergent, and there is essentially no fibrillar Aβ that requires formic acid for solubilization (Kawarabayashi et al., 2001). In Tg2576 brain, aggregation begins at 6–8 months as formic acid requiring Aβ (AβFA) appears. Aβ then accumulates exponentially reaching levels like those seen in AD brain by 18–24 months (Kawarabayashi et al., 2001).
Figure 2 shows the changes in SDS-soluble or formic acid requiring Aβ42 (Fig. 2A) and Aβ40 (Fig. 2B) that occur in the brains of Tg2576 mice with aging. The Aβ measured in each fraction at each age is expressed in terms of the SDS-soluble Aβ (AβSDS) in that fraction at 3–5 months (Aβ in fraction at specified age/AβSDS in fraction at 3–5 months). As Tg2576 mice aged from 6–10 months, SDS-insoluble Aβ that required formic acid for solubilization (AβFA) appeared in the brain and began to accumulate as expected from our previous work (Kawarabayashi et al., 2001). During this early period, AβFA was detected exclusively in the low-speed Triton and high-speed gradient pellets (fraction 12 + 14), that are summed and shown in Figure 2, Ae and Be (AβFA: open columns) for Aβ42 and Aβ40, respectively. Because AβFA appeared and began to accumulate in pellets, SDS-soluble Aβ began to accumulate in lipid rafts (fractions 3–5). The increasing amount of Aβ42SDS in lipid rafts as mice aged from 6 to 10 months is evident in Figure 2Aa. Aβ40SDS also accumulated in lipid rafts from 6–10 months (Fig. 2Ba), but to a lesser degree than Aβ42SDS. Remarkably, the Triton-soluble Aβ in fractions 8–11 showed no change in the 6–10 month period.
Figure 2.
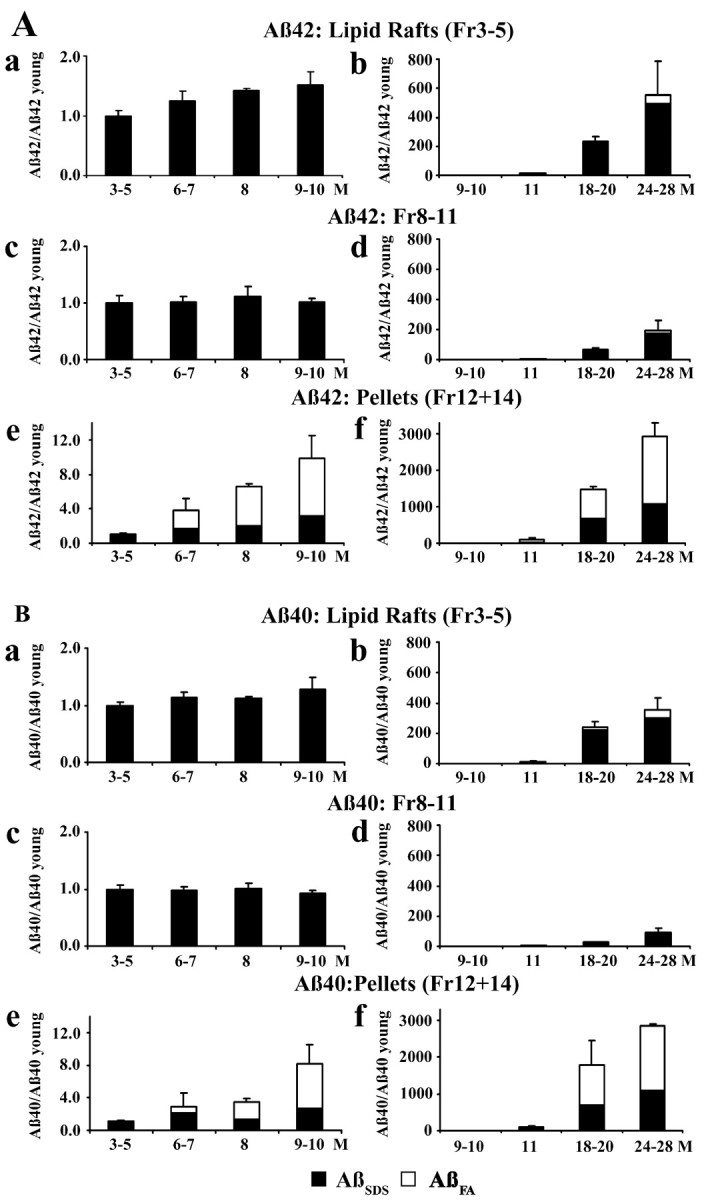
Age-dependent changes in Aβ42 (A) and Aβ40 (B) in Tg2576 mouse brain. The figures on the left show brain Aβ in 3- to 10-month-old mice; those on the right show brain Aβ in 9- to 28-month-old mice. Fractions shown are lipid raft Aβ (fractions 3–5; a, b), Triton-soluble Aβ (fractions 8–11; c, d), and pellet Aβ (fractions 12 + 14; e, f). The Aβ measured in each fraction at each age is expressed in terms of the SDS-soluble Aβ (AβSDS) in that fraction at 3–5 months (Aβ in fraction at specified age/AβSDS in fraction at 3–5 months). SDS-soluble Aβ (AβSDS) is shown in filled columns; SDS-insoluble Aβ that was solubilized in formic acid (AβFA) is shown in open columns.
Between 10 and 28 months, senile plaques similar in appearance and number to those in the AD brain develop in the brains of Tg2576 mice, and a massive amount of fibrillar Aβ is deposited extracellularly. During this period, the pellet fractions showed an enormous increase in Aβ42 (Fig. 2Af) and Aβ40 (Fig. 2Bf). Most of this was SDS-insoluble Aβ42 and Aβ40 that required formic acid for solubilization (AβFA: open columns), but there was also a substantial increase in SDS-soluble Aβ42 and Aβ40 (AβSDS: filled columns). In the 10–28 month period, the Aβ42 (Fig. 2Ab) in lipid rafts also increased enormously to >500× the normal level as did the Aβ40 in lipid rafts (Fig. 2Bb), which increased to >300× normal. Most of this increase was SDS-soluble Aβ42 and Aβ40 (AβSDS: filled columns), but some of the Aβ42 and Aβ40 in lipid rafts required formic acid for solubilization (AβFA: open columns). Because these enormous increases occurred in the pellet and lipid raft fractions, the Aβ42 in fractions 8–11 also increased (Fig. 2Ad) as did the Aβ40 in these fractions (Fig. 2Bd) but the increases, which were almost entirely caused by SDS-soluble Aβ, were much smaller.
Electron microscopic analysis of Tg2576 lipid raft and pellet fractions
Analysis of lipid rafts (fraction 4) from 28 month Tg2576 brain by immunoelectron microscopy (IEM) showed vesicles that were labeled by BAN-50 (Fig. 3a), which recognizes Aβ1–16. Vesicles were also labeled by BA-27 (Fig. 3b), which is specific for the C terminus of Aβ40, and BC-05 (Fig. 3c), which detects the C terminus of Aβ42. Consistent with our finding that there is some Aβ in lipid rafts of old mice that requires formic acid for solubilization, rare amyloid fibrils were observed in the lipid raft fraction from 28 month Tg2576 brain (data not shown). Because amyloid fibrils were not observed in the lipid raft fraction of young mice during the early phase of Aβ accumulation (data not shown) when all raft Aβ is SDS-soluble (Fig. 2), it may be that a few fibrils associated artifactually with rafts during homogenization in very old mice, which have large amounts of extracellular fibrillar amyloid. In the high-speed gradient pellet (fraction 12) from 28 month mice, there were many amyloid fibrils labeled by 3160 (Fig. 3d), a polyclonal anti-Aβ antibody, as expected from our observation that most of the Aβ in this pellet requires formic acid for solubilization. In the low-speed Triton pellet (fraction 14), dense amyloid cores from cored senile plaques and numerous amyloid fibrils were observed (Fig. 3e), again as expected because most of the Aβ in this fraction required formic acid for solubilization.
Figure 3.

Electron microscopy of Aβ42, Aβ40, and Aβ fibrils in the lipid raft fraction and pellets from 28-month-old Tg2576 brain. a–c, IEM showed that vesicular profiles in fraction 4 were labeled with anti-Aβ antibodies (a, BAN-50; b, BA-27; c, BC-05). No labeling was observed in control experiments in which anti-Aβ antibodies were omitted (data not shown). d, Many amyloid fibrils stained with 3160 coupled with 5 nm gold particles were present in fraction 12 (high speed gradient pellet). e, Amyloid cores were detected in fraction 14 (low-speed Triton pellet). Scale bar: a–d, 150 nm; e, 300.
Aβ is increased in the lipid rafts of Alzheimer's disease brain
The results of our analysis of human brain from controls, subjects with pathological aging, and patients with AD is shown in Figure 4. As expected, Aβ42 (Fig. 4C) and Aβ40 (Fig. 4F) showed marked increases in the pellet fraction from AD as compared with control and most of the Aβ in the pellets was SDS-insoluble Aβ that solubilized in formic acid. As in the Tg2576 model, Aβ42 (Fig. 4A) and Aβ40 (Fig. 4D) also increased markedly in lipid rafts from AD as compared with control brain, and there was some increase in fractions 8–11 (Fig. 4B,E).
Figure 4.

Aβ42 and Aβ40 in control, pathological aging (PA), and AD brains. The figures on the left show Aβ42, and those on the right Aβ40 in control (n = 2), pathologic aging (n = 2), and AD (n=6) brains. Fractions shown are lipid raft Aβ (fractions 3–5; A,D), Triton-soluble Aβ (fractions 8–11; B, E), and pellet Aβ (fractions 12 + 14; C, F). SDS-soluble Aβ (AβSDS) is shown in filled columns; SDS-insoluble Aβ that was solubilized in formic acid (AβFA) is shown in open columns. Standard error bars are not shown for pathologic aging and controls because only two brains were analyzed in each instance.
Subjects with pathological aging are cognitively normal but have substantial Aβ deposited in their brains primarily in diffuse plaques. In all fractions (Fig. 4A–F), the amount of Aβ40 and Aβ42 in brains from subjects with pathological aging was intermediate between the low level in control brain and the elevated level in AD brain. A striking difference between pathological aging and AD was the extremely low level of Aβ40 in the brains of subjects with pathological aging.
High levels of Aβ dimers accumulate in lipid rafts beginning at 6 months
Enya et al. (1999) have shown that Aβ dimers are not detected by the ELISAs we used to evaluate Aβ40 and Aβ42. For this reason, we evaluated lipid rafts from Tg2576 brains of all ages by immunoprecipitating Aβ from fraction 4 with polyclonal antibody 3160 followed by immunoblotting with 4G8, a monoclonal antibody that detects both monomers and dimers. The results of our analysis of all fractions from an 11 month Tg2576 brain are shown in Figure 5A. At this age, Aβ in the lipid rafts increased markedly but the Aβ in fractions 8–11 increased very little (Fig. 2). For this reason, Aβ was visible in the lipid rafts (fraction 4) at the exposure shown in Figure 5A but not in fractions 8–11. At this age, SDS-stable dimers were the major form of Aβ present in the lipid rafts, and they were not detected in any other fraction (Fig. 5A).
Figure 5.

Aβ dimers in lipid rafts from Tg2576 and human brains. A, Fractions 1–11 from 11-month-old Tg2576 brain. Ba, Fraction 4 from 4-, 8-, 10-, and 12-month-old Tg2576 brains. Bb, Fraction 4 from 5-, 6-, 7-, 8-, 10-, and 11-month-old Tg2576 brains labeled with 4G8. The Aβ in A and B was immunoprecipitated with polyclonal antibody 3160 and then immunoblotted with 4G8, a monoclonal antibody that detects both monomers and dimers. Dimers were the predominant form of Aβ detected in lipid rafts. Aβ dimers were detected at 6 months and progressively accumulated thereafter. C, Epitope mapping of fraction 4 from 17-month-old Tg2576 brain using anti-Aβ and anti-APP antibodies. Aβ was immunoprecipitated with 3160 and then immunoblotted with Z31preA (B), BAN-50 (C), 4G8 (D), BA-27 (E), BC-05 (F), and O443 (G). Lane A is synthetic Aβ40 (100 pmol) immunoprecipitated with 3160 and detected by BAN-50. The 8 kDa Aβ dimer and the 4 kDa Aβ monomer were detected by all anti-Aβ antibodies. BA-27 specifically detects the C terminus of Aβ40 and BC-05 is selective for the C terminus of Aβ42, so the labeling by these antibodies shows that the 8 kDa protein terminates at Aβ40 or at Aβ42 on the carboxylside. Z31preA, which recognizes the APP epitope just amino to Aβ failed to detect the 8 kDa band, showing that the 8 kDa band is dimeric Aβ and not an Aβ-bearing APP fragment that extends past Aβ on the amino side. The 8 kDa Aβ dimers were also not detected by anti-C terminus of APP (O443). Because 3160 recognizes the N-terminal region of Aβ, p3 was not detected with this immunoprecipitation. D, Fractions 1–11 from AD, pathological aging (PA), and control brains. The Aβ in the fractions shown in D was directly immunoblotted with 4G8.
Figure 5B shows the extraordinary increase that occurs in lipid raft dimers between 4 and 12 months. In these experiments, the volume of each fraction that was immunoprecipitated was considerably less than in the analysis (Fig. 1E) showing monomeric Aβ in 3 month mice (0.125 ml vs 0.50–2.0 ml). For this reason, monomeric Aβ was not detected at 4 months (Fig. 5Ba) or 5 months (Fig. 5Bb). In our initial experiment (Fig. 5Ba), which analyzed the brains of 4, 8, 10, and 12 month mice, dimers were clearly visible at 8 months and had become the dominant form. At 10 months dimers were intensely stained and monomers were clearly visible, and at 12 months dimers were even more intensely stained and monomer showed intense staining (Fig. 5Ba). Because the substantial change in lipid raft Aβ occurred between 4 and 10 months, there was no change in the 4G8-labled APP CTF in lipid rafts (Fig. 5Ba), and there was no change in flotillin, full-length APP, or APP CTFs labeled with a polyclonal antibody to the C terminus, as shown in Figure 6A. To evaluate changes in the 5–8 month interval, a second experiment was performed (Fig. 5B). In this experiment, which analyzed the brains of 5, 6, 7, 8, 10, and 11 month-old mice, dimers were absent at 5 months but were clearly visible at 6 months and increased steadily from 7 to 11 months (Fig. 5Bb). The results in Figure 2, which were obtained by ELISA, showed a significant increase in lipid raft Aβ42 between 4 and 10 months and Aβ40 also increased, but the magnitude of the large increase in dimeric lipid raft Aβ that occurs in this interval (Fig. 5B) is underestimated because the ELISAs used do not detect dimers well.
Figure 6.

Accumulation of ApoE and phosphorylated tau in lipid rafts. A, One microliter of fraction 4 at 3, 4, 5, 8, 12, 18, 24, and 28 months was analyzed by immunoblot with antibodies to flotillin, APP, ApoE, WKS46 (aphosphorylation-independent antibody against tau 359–370), and PHF-1 (an antibody to tau phosphorylated at pSer396/pSer404) sites. The amount of flotillin and APP did not change with aging. ApoE increased substantially beginning at 12 months. Total tau detected by WKS46 did not change with aging, whereas phosphorylated-tau stained by PHF-1 increased beginning at 18 months. Sucrose gradient centrifuge fractions (1–11) from AD and control brains were analyzed by immunoblot using anti-flotillin antibody (B), anti-human ApoE antibody (C), or PHF-1 (D). Note the accumulation of ApoE in lipid rafts (fraction 4) from AD as compared with control brains and the intensely labeled smear of phosphorylated tau in lipid rafts from AD but not control brain.
To establish that the ∼8 kDa protein that appears with aging in lipid rafts is Aβ dimers and not an 8 kDa Aβ-bearing APP fragment, epitope mapping was performed using lipid rafts from 17-month-old Tg2576 brain (Fig. 5C). The 8 kDa band as well as 4 kDa Aβ monomer was detected not only by BAN-50 (anti-Aβ1–16) and 4G8 (anti-Aβ17–28) but also BA-27 (which specifically detects the C terminus of Aβ40) and BC-05 (which is selective for the C terminus of Aβ42). Thus, the 8 kDa band is composed of Aβ that terminates at Aβ40 or at Aβ42. At 17 months, Aβ monomers were more abundant than dimers. That the 8 kDa band does not contain an Aβ fragment that extends beyond Aβ on the amino side was established with Z31preA antibody, which recognizes the APP epitope just amino to Aβ but failed to detect the 8 kDa band.
Analysis of fractions from AD brain showed intensely labeled dimers and faintly stained monomers in fraction 4 of AD brain (Fig. 5D) when there was no detectable staining of fractions 8–11, similar to what was observed in Tg2576. In pathological aging (PA), dimers and monomers were also observed in fraction 4, but there was no detectable Aβ in lipid rafts from control brain.
ApoE and phosphorylated tau accumulate in lipid rafts of aged Tg2576 and AD brain
Flotillin, APP, APP CTFs, ApoE, total tau, and phosphorylated tau were examined in lipid rafts (fraction 4) from 3–28 month Tg2576 mice (Fig. 6A). None of these proteins showed an age-dependent change except ApoE and phosphorylated tau, two proteins known to play an important role in AD. ApoE increased progressively beginning at 12 months. Phosphorylated tau, stained by PHF-1, increased progressively beginning at 18 months.
When lipid rafts and all other fractions were analyzed in AD as compared with control brain, flotillin was found exclusively in lipid rafts (fraction 4) and showed no change (Fig. 6B). ApoE (Fig. 6C) was found both in lipid rafts (fraction 4) and in the Triton-soluble fractions (8–11). In AD, ApoE was definitely increased in the lipid rafts, but there was no apparent increase in fractions 8–11. The change in phosphorylated tau, stained by PHF-1, was particularly striking (Fig. 6D) in lipid rafts from AD brain. Phosphorylated tau was undetectable in control brain, but there was an intensely stained smear of phosphorylated tau in the lipid rafts (fraction 4) from AD brain and only faint staining of phosphorylated tau in fractions 8–11.
Discussion
Lee et al. (1998) previously showed that full-length APP, APP CTFs that are the substrate for γ secretase, and the PS1 fragments now known to be important elements in the γ secretase complex are all present in lipid rafts along with Aβ. In this report, we confirm these findings and those of Riddell et al. (2001), who showed that β secretase (BACE1), the enzyme that produces CTFβ, is present in lipid rafts. In addition, we show that neprilysin, which has recently been shown to be involved in Aβ degradation (Iwata et al., 2000), is present in lipid rafts. In experiments performed collaboratively with T. Golde (Mayo Clinic Jacksonville) (Wahrle et al., 2002), we showed that most of the γ secretase activity in isolated membranes resides in lipid rafts, where processing of CTFβ to Aβ40, Aβ42, and CTFγ occurs in a cholesterol-dependent manner. Ehehalt et al. (2003) recently reported that lipid rafts are critically involved in regulating the β secretase cleavage that produces Aβ. Thus APP, CTFβ, γ secretase, β secretase, and cholesterol are all associated with lipid rafts in a functionally meaningful way, making it likely that lipid rafts play an important role in Aβ production. Our finding that neprilysin is associated with lipid rafts suggests that rafts may also be involved in Aβ degradation.
Aβ is a secreted protein, and it has long been known that the amyloid fibrils that accumulate in senile plaques have an extracellular location. The concentration of Aβ in CSF, the average Aβ concentration in the volume of the brain, and the estimated concentration of extracellular Aβ in the brain are, however, below the concentration needed for fibril formation in vitro, raising the question as to how fibril formation could be initiated in the extracellular space. Our analysis of Tg2576 brains shows that there is a marked accumulation of SDS-soluble, dimeric Aβ in lipid rafts as mice age. This increase begins at 6 months before Aβ deposition is visible histologically, and it closely parallels the appearance and accumulation of SDS-insoluble fibrillar Aβ in the pellets.
Although it is possible that the formation of extracellular, SDS-insoluble amyloid fibrils and the parallel accumulation of SDS-soluble Aβ dimers in lipid rafts occur independently, it is far more likely that there is a dynamic interaction between SDS-soluble raft Aβ and extracellular Aβ assemblies including mature SDS-insoluble fibrils. Our data show that in young mice >20% of brain Aβ is located in lipid rafts, which constitute a small fraction of total cellular volume [Sargiacomo et al. (1993) estimated that lipid rafts constitute only 0.4–0.8% of the plasma membrane in Madin-Darby canine kidney cells]. It is likely, therefore, that Aβ is at a much higher concentration in lipid rafts than in the extracellular space. Lipid rafts are enriched in cholesterol and GM-1 ganglioside, molecules that foster Aβ aggregation in vitro (Yanagisawa et al., 1995; Avdulov et al., 1997; Choo-Smith et al., 1997). In addition, Aβ has been found complexed to GM-1 ganglioside in AD brain (Yanagisawa et al., 1995), and EM analysis of AD brains has suggested that Aβ may start to deposit as a membrane-bound form on cell surface plasma membranes (Yamaguchi et al., 2000). Thus, it is reasonable to propose that Aβ aggregation may be initiated in lipid rafts and that the marked accumulation of SDS-stable dimers in lipid rafts between 6 and 10 months reflects the involvement of lipid rafts in this process. Because lipid rafts occupy the outer leaflet of the membrane, oligomers formed in the rafts might be expected to enter the extracellular space using the normal secretory pathway. This concept fits well with the observation that oligomers form intracellularly and are secreted from cultured cells overexpressing APP (Podlisny et al., 1995; Morishima-Kawashima and Ihara, 1998; Walsh et al., 2000, 2002), and it links well to AD because the secretion of oligomers is increased by the expression of AD-causing mutations (Xia et al., 1997). Once seeded, the extracellular space could be the preferred site for fibril growth because fibril growth is much less concentration-dependent than seed formation and because the mechanisms for degrading extracellular fibrils are likely to be less effective than those for degrading abnormal Aβ assemblies in rafts.
Importantly, our analysis of AD brains showed marked increases in lipid raft Aβ like those observed in the oldest Tg2576 brains analyzed. These findings in AD brain fit well with the previous work of Oshima et al. (2001), whose thorough investigation of Aβ in lipid raft fractions from human AD and control brain prompted them to conclude, as we have, that there is likely to be a dynamic relationship between lipid raft Aβ and the Aβ that accumulates extracellularly in AD brain.
We recently showed that the onset of memory loss in Tg2576 mice occurs at 6 months (Westerman et al., 2002). Because SDS-insoluble Aβ did not correlate with memory impairment when mice of all ages were examined, we hypothesized that memory deficits in Tg2576 mice were not caused by SDS-insoluble Aβ fibrils, but were caused instead by soluble assemblies of Aβ. Our current results show that the onset of memory loss coincides with the accumulation of SDS-soluble Aβ dimers in lipid rafts. Because SDS was used to solubilize this Aβ, the native structure of the Aβ assemblies and their interaction with other raft molecules is uncertain. It is nevertheless highly likely that the dimers observed are either toxic themselves or related to native Aβ assemblies that are toxic. The dimers observed are remarkably similar to oligomers released from cultured cells that have been shown to inhibit hippocampal long-term potentiation in vivo (Walsh et al., 2002), and in their native state they could be similar to the diffusible Aβ ligands formed in vitro by Lambert et al. (1998) that were shown to be neurotoxic and to inhibit long-term potentiation in cultured hippocampal slices. Because lipid rafts play an important role in signal transduction and other cellular functions, the Aβ accumulating in lipid rafts is well positioned to impair neuronal function in a way that could account for the memory loss observed in the Tg2576 model of AD.
It is well established that the ApoE4 allele is associated with increased risk for AD, and Bales et al. (1997) showed that amyloid fibrils do not form in the PDAPP model of AD when ApoE is absent, thereby establishing that ApoE plays an important role in the amyloid fibril formation that occurs in this animal model. Holtzman et al. (1999, 2000a,b) studied mice expressing only human ApoE isoforms and found that amyloid fibril formation is accelerated in mice expressing human ApoE4 as compared with other ApoE isoforms. Based on studies showing that ApoE isoforms can differentially affect amyloid fibril formation in vitro (Ma et al., 1994; Castano et al., 1995; Evans et al., 1995), it is likely that the effects of ApoE are mediated through a direct interaction between Aβ and ApoE. In this study, we show that ApoE accumulates progressively with aging in the lipid raft fraction from Tg2576 mice and that ApoE is elevated in the lipid fraction from AD as compared with control human brain. These findings suggest that lipid rafts may be an important site where ApoE and Aβ interact in a way that influences Aβ fibril formation. Additional evidence for this comes from recent experiments showing that lipid raft Aβ is decreased in mice that lack ApoE (Fagan et al., 2002).
One of the most remarkable findings of the current study is our observation that phosphorylated tau accumulates in the lipid raft fraction from very old Tg2576 mice and shows a striking elevation in lipid rafts from AD brain. This observation fits well with recent reports that Aβ can accelerate tangle formation in transgenic models (Gotz et al., 2001; Lewis et al., 2001), and it suggests that Aβ dimers in lipid rafts may be linked to the formation of paired helical filaments. The microtubule-associated protein tau interacts with the neuronal plasma membrane in addition to microtubules, and there is evidence that this interaction is influenced by phosphorylation of tau at sites that are modified in paired helical filaments (Brandt et al., 1995; Maas et al., 2000). Tau is also expressed in oligodendrocytes, and recent experiments suggest that recruitment of tau and tubulin to activated Fyn kinase in oligodendrocyte rafts is an important step in the initiation of myelination (Klein et al., 2002). Fyn kinase is also present in lipid rafts of mouse brain (Fagan et al., 2002). Thus, it is interesting to speculate that the remarkable levels of phosphorylated tau in lipid rafts from the AD brain may be attributable to an Aβ dimer-induced alteration in the interaction between kinases (or other signaling molecules) in lipid rafts and tau recruited to the cytoplasmic face of the neuronal plasma membrane.
Footnotes
This work was supported by National Institutes of Health Grant AG15453 (S.Y.), Grants-in Aid for Primary Amyloidosis Research Committee (S. Ikeda and T. Ishihara), surveys and research on special disease from the Ministry of Health, Labor, and Welfare of Japan, and by Grants-in-Aid for Scientific Research (B) (1437028), and Scientific Research on Priority Areas (C), Advanced Brain Science Project, from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We thank Daniel Sevlever for help in preparing lipid rafts, Shu-Hui Yen for tau antibody, Takaomi C. Saido for neprilysin antibody, Kumar Sambamurti for anti-APP antibody, Nobuhiro Yamada for anti-ApoE antibody, Thillai Koothan for anti-PS-1 antibody, and Blaze Birinyi and Yoshinori Kosugi for their technical assistance.
Correspondence should be addressed to Steven G. Younkin, Department of Neuroscience, Mayo Clinic, 4500 San Pablo Road, Jacksonville, FL. E-mail: younkin.steven@mayo.edu.
DOI:10.1523/JNEUROSCI.5543-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/243801-09$15.00/0
References
- Avdulov NA, Chochina SV, Igbavboa U, Warden CS, Vassiliev AV, Wood WG (1997) Lipid binding to amyloid β-peptide aggregates: preferential binding of cholesterol as compared with phosphatidylcholine and fatty acids. J Neurochem 69: 1746–1752. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, Paul SM (1997) Lack of apolipoprotein E dramatically reduces amyloid β-peptide deposition. Nat Genet 17: 263–264. [DOI] [PubMed] [Google Scholar]
- Bickel PE, Scherer PE, Schnitzer JE, Oh P, Lisanti MP, Lodish HF (1997) Flotillin and epidermal surface antigen define a new family of caveolae-associated integral membrane proteins. J Biol Chem 272: 13793–13802. [DOI] [PubMed] [Google Scholar]
- Brandt R, Leger J, Lee G (1995) Interaction of tau with the neural plasma membrane mediated by tau's amino-terminal projection domain. J Cell Biol 131: 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, London E (1998) Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol 14: 111–136. [DOI] [PubMed] [Google Scholar]
- Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C, Frangione B (1995) Fibrillogenesis in Alzheimer's disease of amyloid β peptides and apolipoprotein E. Biochem J 306: 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo-Smith LP, Garzon-Rodriguez W, Glabe CG, Surewicz WK (1997) Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J Biol Chem 272: 22987–22990. [DOI] [PubMed] [Google Scholar]
- Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P (1995) Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging 16: 285–298; discussion 298–304. [DOI] [PubMed] [Google Scholar]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K (2003) Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enya M, Morishima-Kawashima M, Yoshimura M, Shinkai Y, Kusui K, Khan K, Games D, Schenk D, Sugihara S, Yamaguchi H, Ihara Y (1999) Appearance of sodium dodecyl sulfate-stable amyloid β-protein (Aβ) dimer in the cortex during aging. Am J Pathol 154: 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans KC, Berger EP, Cho CG, Weisgraber KH, Lansbury PT Jr (1995) Apolipoprotein E is a kinetic but not a thermodynamic inhibitor of amyloid formation: implications for the pathogenesis and treatment of Alzheimer disease. Proc Natl Acad Sci USA 92: 763–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM (2002) Human and murine ApoE markedly alters Aβ metabolism before and after plaque formation in a mouse model of Alzheimer's disease. Neurobiol Dis 9: 305–318. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ42 fibrils. Science 293: 1491–1495. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P (1990) A preparation of Alzheimer paired helical filaments that displays distinct tau proteins by polyacrylamide gel electrophoresis. Proc Natl Acad Sci USA 87: 5827–5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harigaya Y, Shoji M, Kawarabayashi T, Kanai M, Nakamura T, Iizuka T, Igeta Y, Saido TC, Sahara N, Mori H, Hirai S (1995) Modified amyloid β protein ending at 42 or 40 with different solubility accumulates in the brain of Alzheimer's disease. Biochem Biophys Res Commun 211: 1015–1022. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Wu S, Bhat P, Parsadanian M, Fagan AM, Chang LK, Sun Y, Paul SM (1999) Expression of human apolipoprotein E reduces amyloid-β deposition in a mouse model of Alzheimer's disease. J Clin Invest 103: R15–R21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Ashe KH, Irizarry MC, Hyman BT (2000a) Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Ann Neurol 47: 739–747. [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM (2000b) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97: 2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper NM (1999) Detergent-insoluble glycosphingolipid/cholesterolrich membrane domains, lipid rafts and caveolae (review). Mol Membr Biol 16: 145–156. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96: 3228–3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99–102. [DOI] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC (2000) Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat Med 6: 143–150. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Shoji M, Harigaya Y, Yamaguchi H, Hirai S (1991) Expression of APP in the early stage of brain damage. Brain Res 563: 334–338. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid (β) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenessey A, Nacharaju P, Ko LW, Yen SH (1997) Degradation of tau by lysosomal enzyme cathepsin D: implication for Alzheimer neurofibrillary degeneration. J Neurochem 69: 2026–2038. [DOI] [PubMed] [Google Scholar]
- Klein C, Kramer EM, Cardine AM, Schraven B, Brandt R, Trotter J (2002) Process outgrowth of oligodendrocytes is promoted by interaction of fyn kinase with the cytoskeletal protein tau. J Neurosci 22: 698–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Aβ1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95: 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury PT Jr, Kosik KS (1998) A detergent-insoluble membrane compartment contains Aβ in vivo. Nat Med 4: 730–734. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293: 1487–1491. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999) Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155: 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer Jr HB, Das S, Potter H (1994) Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372: 92–94. [DOI] [PubMed] [Google Scholar]
- Maas T, Eidenmuller J, Brandt R (2000) Interaction of tau with the neural membrane cortex is regulated by phosphorylation at sites that are modified in paired helical filaments. J Biol Chem 275: 15733–15740. [DOI] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860–866. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Ihara Y (1998) The presence of amyloid β-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochemistry 37: 15247–15253. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of Aβ1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshima N, Morishima-Kawashima M, Yamaguchi H, Yoshimura M, Sugihara S, Khan K, Games D, Schenk D, Ihara Y (2001) Accumulation of amyloid β-protein in the low-density membrane domain accurately reflects the extent of β-amyloid deposition in the brain. Am J Pathol 158: 2209–2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG (1994) Ultrastructural localization of gangliosides; GM1 is concentrated in caveolae. J Histochem Cytochem 42: 155–166. [DOI] [PubMed] [Google Scholar]
- Pinnix I, Musunuru U, Tun H, Sridharan A, Golde T, Eckman C, Ziani-Cherif C, Onstead L, Sambamurti K (2001) A novel gamma-secretase assay based on detection of the putative C-terminal fragment-gamma of amyloid β protein precursor. J Biol Chem 276: 481–487. [DOI] [PubMed] [Google Scholar]
- Podlisny MB, Ostaszewski BL, Squazzo SL, Koo EH, Rydell RE, Teplow DB, Selkoe DJ (1995) Aggregation of secreted amyloid β-protein into sodium dodecyl sulfate-stable oligomers in cell culture. J Biol Chem 270: 9564–9570. [DOI] [PubMed] [Google Scholar]
- Riddell DR, Christie G, Hussain I, Dingwall C (2001) Compartmentalization of β-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr Biol 11: 1288–1293. [DOI] [PubMed] [Google Scholar]
- Sargiacomo M, Sudol M, Tang Z, Lisanti MP (1993) Signal transducing molecules and glycosyl-phosphatidylinositol-linked proteins form a caveolin-rich insoluble complex in MDCK cells. J Cell Biol 122: 789–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (2001) Alzheimer's disease: genes, proteins, and therapy. Physiol Rev 81: 741–766. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (2002) Deciphering the genesis and fate of amyloid β-protein yields novel therapies for Alzheimer disease. J Clin Invest 110: 1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimano H, Yamada N, Shimada M, Ohsawa N, Fukazawa C, Yazaki Y, Takaku F, Katsuki M (1991) Hepatic and renal expression of rat apolipoprotein E under control of the metallothionein promoter in transgenic mice. Biochim Biophys Acta 1090: 91–94. [DOI] [PubMed] [Google Scholar]
- Shinkai Y, Yoshimura M, Morishima-Kawashima M, Ito Y, Shimada H, Yanagisawa K, Ihara Y (1997) Amyloid β-protein deposition in the leptomeninges and cerebral cortex. Ann Neurol 42: 899–908. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387: 569–572. [DOI] [PubMed] [Google Scholar]
- Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1: 31–39. [DOI] [PubMed] [Google Scholar]
- Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L Jr, Eckman C, Golde TE, Younkin SG (1994) An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (β APP717) mutants. Science 264: 1336–1340. [DOI] [PubMed] [Google Scholar]
- Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE (2002) Cholesterol-dependent gamma-secretase activity in buoyant cholesterolrich membrane microdomains. Neurobiol Dis 9: 11–23. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ (2000) The oligomerization of amyloid β-protein begins intracellularly in cells derived from human brain. Biochemistry 39: 10831–10839. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Rowan MJ, Selkoe DJ (2002) Amyloid-β oligomers: their production, toxicity and therapeutic inhibition. Biochem Soc Trans 30: 552–557. [DOI] [PubMed] [Google Scholar]
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 22: 1858–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia W, Zhang J, Kholodenko D, Citron M, Podlisny MB, Teplow DB, Haass C, Seubert P, Koo EH, Selkoe DJ (1997) Enhanced production and oligomerization of the 42-residue amyloid β-protein by Chinese hamster ovary cells stably expressing mutant presenilins. J Biol Chem 272: 7977–7982. [DOI] [PubMed] [Google Scholar]
- Yamaguchi H, Maat-Schieman ML, van Duinen SG, Prins FA, Neeskens P, Natte R, Roos RA (2000) Amyloid β protein (Aβ) starts to deposit as plasma membrane-bound form in diffuse plaques of brains from hereditary cerebral hemorrhage with amyloidosis-Dutch type, Alzheimer disease and nondemented aged subjects. J Neuropathol Exp Neurol 59: 723–732. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Odaka A, Suzuki N, Ihara Y (1995) GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer's disease. Nat Med 1: 1062–1066. [DOI] [PubMed] [Google Scholar]