

Abstract
Human sensory neuron-specific G-protein-coupled receptors (SNSRs) are expressed solely in small diameter primary sensory neurons. This restricted expression pattern is of considerable therapeutic interest because small nociceptors transmit chronic pain messages. The neuronal function of human SNSRs is difficult to assess because rodent orthologs have yet to be clearly defined, and individual isoforms are found only in a small subset of primary sensory neurons. To circumvent this problem, we expressed human SNSR4 (hSNSR4; also known as Hs.mrgX1) in rat superior cervical ganglion (SCG), dorsal root ganglion (DRG), and hippocampal neurons using nuclear injection or recombinant adenoviruses and examined modulation of ion channels and neurotransmission using whole-cell patch-clamp techniques. BAM8-22 (a 15 amino acid C-terminal fragment of bovine adrenal medulla peptide 22), a peptide agonist derived from proenkephalin, inhibited high (but not low) voltage-activated Ca2+ current in both DRG and SCG neurons expressing hSNSR4, whereas no response was detected in control neurons. The Ca2+ current inhibition was concentration dependent and partially sensitive to Pertussis toxin (PTX) treatment. Additionally, the peptide was highly effective in modulating current arising from M-type K+ channels in SCG neurons expressing hSNSR4. In hippocampal neurons expressing hSNSR4, BAM8-22 induced presynaptic inhibition of transmission that was abolished after PTX treatment. Our data indicate that hSNSR4, when heterologously expressed in rat neurons, can be activated by an opioid-related peptide, couples to Gq/11-proteins as well as PTX-sensitive Gi/o-proteins, and modulates neuronal Ca2+ channels, K+ channels, and synaptic transmission.
Keywords: channel, sympathetic, dorsal root ganglion, nociception, pain, neuropeptide
Introduction
Painful stimuli are initially detected at the peripheral terminals of specialized primary sensory neurons called nociceptors. Thus, proteins that mediate action potential initiation, propagation, and neurotransmission in these neurons are potential therapeutic targets for the development of novel analgesics. Of particular interest are proteins that are expressed predominantly, if not exclusively, in primary sensory neurons. Such a restricted expression pattern would help limit the actions of selective agents to the peripheral nervous system and presumably avoid undesirable CNS side effects such as mental impairment and dependence. In this regard, several ion channels have been reported that are expressed primarily in small nociceptive neurons. These include a voltage-gated tetrodotoxin-insensitive Na+ channel (NaV1.8), an ATP-gated ion channel (P2X3 receptor), and a nonselective cationic channel (vanilloid receptor VR1) activated by capsaicin, noxious heat, and extracellular protons (Julius and Basbaum, 2001). In each case, genes encoding orthologous proteins have been identified from murine and human genomes, thus facilitating the construction of mouse knock-out models. Such models have been useful in evaluating the participation of the targeted gene in conveying painful stimuli to the CNS.
Recently, a large family of heterotrimeric G-protein-coupled receptors (GPCRs) was identified with an expression pattern restricted to primary sensory neurons using a subtractive strategy to identify a cDNA population specific to trkA-expressing neurons (Dong et al., 2001). The receptor family was termed mrg for Mas-related genes (Young et al., 1986), and numerous members were identified in the rat, mouse, and human genome. An independent search by Lembo et al. (2002) based on a rat orphan GPCR sequence identified six human genes, which they termed sensory neuron-specific G-protein-coupled receptors (SNSRs). Three of the genes found by the two groups appear to be identical: Hs.mrgX1 and human SNSR4 (hSNSR4); Hs.mrgX3 and hSNSR1; Hs.mrgX4 and hSNSR6. Surprisingly, clear orthologous relationships between interspecies subfamily members are not apparent (Choi and Lahn, 2003; Zylka et al., 2003), although the human genes share highest homology with the mrgA subfamily of murine genes.
Given the properties of these receptors, it will be challenging to delineate physiological functions. First, endogenous ligands have not been unequivocally identified, although interesting candidates are beginning to emerge (Dong et al., 2001; Bender et al., 2002; Han et al., 2002; Lembo et al., 2002; Robas et al., 2003). Second, these receptors appear to be expressed in a very small subset of nociceptors; thus, in the absence of a method of identifying expressing neurons, a very large number of neurons would need to be sampled to obtain meaningful results. Third, it is not clear whether receptor protein is expressed throughout the neuron or restricted to a specific compartment (e.g., nerve terminals). Finally, as noted above, the function of the human receptors may not be accurately inferred from studying rodent receptors in situ. To circumvent these problems, we heterologously expressed hSNSR4 in rat dorsal root ganglion (DRG), superior cervical ganglion (SCG), and hippocampal neurons using nuclear injection or recombinant adenoviruses and examined modulation of ion channels and neurotransmission using whole-cell patch-clamp techniques.
Materials and Methods
All experiments were approved by the Institutional Animal Care and Use Committee.
Cell cultures. Single SCG neurons were isolated from 10- to 14-week-old male Wistar rats as described previously (Ikeda, 2004). Briefly, after dissection, the ganglia were desheathed, cut into small pieces, and incubated with 0.7 mg/ml collagenase type D (Roche Molecular Biochemicals, Indianapolis, IN), 0.4 mg/ml trypsin (Worthington Biochemical, Lakewood, NJ), and 0.1 mg/ml DNase 1, type IV (Sigma, St. Louis, MO) in minimal essential medium (MEM) saturated with 5% CO2/95% O2 at 35°C for 1 hr. Thereafter, cells were dissociated by vigorous shaking of the flask. After centrifugation twice at 50 × g, the dispersed neurons were resuspended in MEM containing 10% fetal bovine serum, 1% glutamine, and 1% penicillin-streptomycin solution (all from Invitrogen, Carlsbad, CA). Neurons were then plated into polystyrene culture dishes coated with poly-l-lysine and stored in a humidified atmosphere containing 5% CO2 in air at 37°C. Single DRG neurons were isolated from 1- to 2-week-old Sprague Dawley rats using essentially the same procedure, except the collagenase concentration was reduced to 0.1 mg/ml. Hippocampal neurons were grown on collagen/poly-d-lysine microislands as reported previously (Bekkers and Stevens, 1991; Chen and Lambert, 2000). Briefly, hippocampi were dissected from newborn Sprague Dawley rats and digested with papain (∼25 U/ml) at 37°C for 1 hr. After dissociation, 5 × 104 cells were plated in 35 mm dishes that had been coated with 0.15% agarose and sprayed with a 1:4 (v/v) mixture of rat tail collagen (3.6 mg/ml) and poly-d-lysine (0.5 mg/ml). Where indicated, SCG and DRG neurons were incubated overnight with 500 ng/ml Pertussis toxin (PTX; List Biological, Campbell, CA), and hippocampal neurons were incubated with 100 ng/ml PTX for 24-72 hr before recording.
Intranuclear injection of SCG neurons. Because the coding region for hSNSR4 (GenBank accession number AF474990) contains no introns, the sequence was directly amplified from human genomic DNA (Promega, Madison, WI) with Pfu ultra polymerase (Stratagene, La Jolla, CA). The resulting PCR product was ligated into the multiple cloning region of pCI, a mammalian expression vector (Promega), at the EcoRI-EcoRV site, and the sequence was confirmed by automated DNA sequencing. The forward primer was designed such that the start codon was under the optimal context for translational initiation (Kozak, 1999). The hSNSR4 construct (pCI-hSNSR4; 0.1 μg/μl in 10 mm Tris, pH 8.0) was injected into the nucleus of SCG neurons 4-8 hr after cell isolation with an Eppendorf (Brinkmann Instruments, Westbury, NY) FemtoJet microinjector and 5171 micromanipulator (Ikeda, 2004). Neurons were coinjected with a plasmid, pEGFP-N1 (5 ng/μl; Clontech, Palo Alto, CA), encoding the enhanced green fluorescent protein (EGFP) cDNA to facilitate later identification of successfully injected cells. SCG neurons expressing EGFP were used for whole-cell patch-clamp recordings within 24 hr after injection.
Adenoviral vectors and infection of DRG and hippocampal neurons. Adenoviruses coexpressing EGFP and hSNSR4 were generated using the AdEasy system (He et al., 1998). Briefly, the XhoI-EcoRV fragment of hSNSR4 coding region in the above pCI-hSNSR4 vector was subcloned into the shuttle vector pAdTrack-CMV (kindly provided by T.-C. He, Johns Hopkins University, Baltimore, MD). The shuttle vector has a separate EGFP expression cassette that allows visualization of virus production and identification of infected cells (He et al., 1998). The resultant plasmid (pAdTrack-CMV-hSNSR4) was linearized with PmeI and electroporated into Escherichia coli BJ5183 cells that had been transformed with pAdEasy-1, an adenoviral backbone plasmid (Stratagene). Recombination between these two plasmids was confirmed by restriction analysis and sequencing. High-titer virus stocks were generated by transfecting PacI-linearized recombinant plasmids into human embryonic kidney (HEK) 293 cells. The crude lysate of virus-infected HEK 293 cells was used for experiments. The effective titer was calculated by counting the number of EGFP-positive cells in a 12-well plate ∼40 hr after infection. Control adenoviruses expressing EGFP alone were generated similarly, except the shuttle vector was directly used for recombination. DRG neurons were infected 6-12 hr after isolation by adding 3 × 108 infectious units (diluted in MEM) to each 35 mm dish followed by replacement of the culture medium after 12 hr. Approximately 20-30 hr after infection, infection efficiency was ∼100% as judged from EGFP fluorescence, and whole-cell recordings were performed at this time. Hippocampal neurons (10-14 d in vitro) were infected by adding 2-5 × 107 infectious units per dish. Approximately 1 hr before infection, a glutamate receptor antagonist, d-2-amino-5-phosphonovaleric acid (100-200 μm), was added to minimize toxicity. Recordings of infected hippocampal neurons were performed 30-60 hr after infection.
Electrophysiology. Whole-cell currents were recorded with a patch-clamp amplifier (Axopatch 200B; Axon Instruments, Union City, CA) at room temperature (21-24°C) using the conventional whole-cell variant of the patch-clamp technique (Hamill et al., 1981). For recording Ca2+ current (ICa), patch electrodes were pulled from borosilicate glass capillaries (Corning 7052; Garner Glass, Claremont, CA) on a P-97 Flaming-Brown micropipette puller (Sutter Instruments, Novato, CA), fire polished on a microforge, and filled with a solution containing (in mm): 120 N-methyl-d-glucamine, 20 tetraethylammonium (TEA)-OH, 10 HEPES, 11 EGTA, 1 CaCl2, 14 Tris-creatine phosphate, 4 MgATP, and 0.3 Na2GTP; pH was adjusted to 7.24 with methanesulfonic acid and HCl (final Cl- concentration of 20 mm). Pipette resistance ranged from 1 to 3 MΩ, yielding uncompensated series resistances of 2-6 MΩ. Series resistance compensation of 80% was used in all recordings. For recording ICa from SCG neurons, the external solution consisted of the following: 145 mm TEA methanesulfonate, 10 mm HEPES, 15 mm glucose, 10 mm CaCl2, and 300 nm tetrodotoxin; pH 7.35 and 320 mOsm/kg. For recording ICa from DRG neurons, the bathing solution CaCl2 was reduced to 5 mm, and the HEPES increased by an identical amount. Low voltage-activated (LVA) ICa from DRG neurons was evoked at -40 mV from a holding potential of -80 mV. High voltage-activated (HVA) ICa from DRG neurons was evoked at +10 mV. Because of the heterogeneity in DRG neuron ICa composition, calculation of receptor-mediated inhibition of the HVA current component was determined from neurons in which ICa(+10)/ICa(-40) ratio exceeded 1. This procedure helped exclude neurons in which a small HVA current component might be contaminated from residual LVA current. Voltage protocol generation and data acquisition were performed using custom data acquisition software on a Macintosh G4 computer (Apple Computers, Cupertino, CA) with an ITC-18 interface board (InstruTECH, Port Washington, NY). ICa traces were low-pass filtered at 2-5 kHz and digitized at 10 kHz. For M-current (IM) recordings, pipettes were pulled from borosilicate glass capillaries (1B150F-4; World Precision Instruments, Sarasota, FL) and filled with an internal solution that consisted of the following (in mm): 140 KCl, 0.1 EGTA, 10 HEPES, 4 MgATP, 0.1 Na2GTP, 0.1 Tris-creatine phosphate; pH adjusted to 7.25 with KOH, 300 mOsm/kg. The external solution contained the following: 150 mm NaCl, 2.5 mm KCl, 10 mm HEPES, 1 mm MgCl2,1mm CaCl2,15mm glucose, 300 nm tetrodotoxin; pH adjusted to 7.35 with NaOH, 325 mOsm/kg. IM traces were low-pass filtered at 1 kHz and digitized at 2 kHz.
For recording synaptic currents, hippocampal neurons were depolarized every 10 sec with pairs (50 msec interval) of depolarizing command pulses (2 msec, 60 mV) from a holding potential between -40 and -60 mV. The depolarizations evoked unclamped action currents and synaptic (autaptic) currents (Bekkers and Stevens, 1991). Series resistance (after compensation) was monitored during the recording, and cells were discarded if a significant increase occurred. Patch electrodes were pulled from borosilicate glass capillaries (TW150F-4; World Precision Instruments) and filled with a solution containing the following (in mm): 140 K-gluconate, 5 KCl, 0.2 EGTA, 10 HEPES, 3 MgATP, 0.3 Na2GTP; pH 7.25, 300 mOsm/kg. The external solution contained the following (in mm): 145 NaCl, 2.5 KCl, 10 HEPES, 10 glucose, 1.5 CaCl2, 2.5 MgCl2; pH adjusted to 7.35 with NaOH, 320 mOsm/kg. Application of drugs to the neuron under study was performed by positioning a silica tube (inner diameter, 200 μm) of a custom-designed gravity-fed microperfusion system at ∼100 μm from the cell. BAM8-22 (a 15 amino acid C-terminal fragment of bovine adrenal medulla peptide 22; human, bovine, rat, and mouse sequences are identical) was synthesized by Invitrogen and used at 3 μm unless otherwise indicated. The other drugs were purchased from Sigma unless otherwise stated. Numerical values are expressed as mean ± SEM. Statistical comparisons were made by using paired, unpaired Student's t test or ANOVA followed by Newman-Keuls test as appropriate. The differences were considered significant if p < 0.05.
Results
Ca2+ channels in rodent DRG neurons are not modulated by the peptide BAM8-22
Although the identities of endogenous ligands for hSNSRs are not known, a Ca2+ mobilization assay was used to identify a proenkephalin A gene product, BAM22, as a high-potency agonist for hSNSR3/4 (Lembo et al., 2002). BAM22 is a potent agonist at classical opioid receptor sub-types (e.g., μ, κ, δ) as well as SNSRs. However, a truncation product of BAM22, BAM8-22, lacks the characteristic Met-enkephalin motif YGGFM and is unable to activate classical opioid receptors while retaining high affinity for hSNSR4 (Lembo et al., 2002).
We thus exploited the apparent specificity (at least in regard to opioid receptors) of BAM8-22 to probe for native receptors coupling to Ca2+ channels in rodent sensory neurons. DRG neurons express many types of voltage-dependent Ca2+ channels (Fedulova et al., 1985; Kostyuk et al., 1988; Fox et al., 1987; Regan et al., 1991; Mintz et al., 1992; Yusaf et al., 2001), and as shown in Figure 1, the composition of ICa varies considerably from cell to cell. To separate the effects of BAM8-22 on ICa arising from LVA and HVA Ca2+ channels, a voltage protocol consisting of 30 msec test pulses to -40 mV and +10 mV evoked from a holding potential of -80 mV was used (Fig. 1A, bottom). The test pulses were separated by a 50 msec pulse to -60 mV, which inactivated the majority of LVA Ca2+ channels. The current evoked at -40 mV should arise primarily from LVA (i.e., T-type) Ca2+ channels because HVA Ca2+ channels are not usually activated at -40 mV. ICa recorded during the +10 mV test pulse is enriched in the HVA current component, although a small fraction may still arise from the LVA component.
Figure 1.

Heterologously expressed hSNSR4 couples to HVA but not LVA Ca2+ channels in rat DRG neurons. A, Representative whole-cell patch-clamp ICa traces acquired in the absence or presence of the peptide agonist BAM8-22 (BAM; 3 μm) from a control (Con; top) or an EGFP-expressing DRG neuron (middle). The voltage protocol used to evoke ICa for experiments described in Fig. 1 is illustrated at the bottom. The current evoked at -40 mV arises from LVA Ca2+ channels, whereas the current evoked at +10 mV (after an inactivating interpulse) arises primarily for HVA Ca2+ channels. B, Representative current traces (top) illustrating marked inhibition of ICa evoked (+10 mV) after application of BAM8-22 to a DRG neuron expressing hSNSR4. The neuron illustrated exhibited virtually no LVA Ca2+ channels. The time course of inhibition is shown at the bottom. C, Representative current traces (top) obtained in the absence or presence of BAM8-22 from an hSNSR4-expressing DRG neuron with a large component of LVA-type Ca2+ channels. The time course of inhibition is shown at the bottom. D, Summary bar graph comparing mean BAM8-22-induced inhibition of HVA (filled) and LVA (open) ICa under experimental conditions listed below each bar. HVA ICa amplitude was measured 10 msec from the start of the voltage step to +10 mV, whereas LVA ICa amplitude was measured 15 msec after initiation of the voltage step to -40 mV. Heterologous expression was accomplished using recombinant adenovirus.
As shown in Figure 1, A and D, application of BAM8-22 (3 μm) did not alter the amplitude of HVA or LVA ICa recorded from young rat DRG neurons (0.04 ± 0.98%, n = 10, and 4.5 ± 1.5%, n = 5, respectively). BAM8-22 (3 or 10 μm) also had no effect on HVA or LVA ICa recorded from adult mouse DRG neurons (data not shown; n = 23). The majority of neurons were small to medium in size with a mean capacitance of 27.8 pF (range, 13.9-63.8 pF). The lack of BAM8-22 effect is consistent with the idea that hSNSRs/mrgs are not orthologous to rodent members of the same gene family. It is also possible that neurons sensitive to BAM8-22 represent only a very small subset of all DRG neurons and thus were missed with the sample size used (41 neurons). Alternatively, it is possible that appropriate receptors may not be localized to neuronal soma, are altered by enzymatic treatment, or do not modulate ICa under the recording conditions used (see below).
Heterologously expressed hSNSR4 couples to high but not low voltage-activated Ca2+ channels in DRG neurons
Failure to detect a BAM8-22 response in DRG neurons prompted us to express hSNSR4 in rat DRG neurons using recombinant adenoviruses. Adenoviruses expressing EGFP alone or coexpressing EGFP and hSNSR4 were constructed using the AdEasy system (He et al., 1998). As shown in Figure 1, A and D, application of BAM8-22 (3 μm) did not alter the amplitude of the double test pulse-evoked ICa recorded from neurons expressing EGFP only (inhibition of HVA ICa was 1.0 ± 1.3%; n = 5), consistent with the lack of effect in uninfected neurons. In neurons coexpressing EGFP and hSNSR4 (Fig. 1B,C), application of BAM8-22 induced robust inhibition of HVA ICa (59.7 ± 5.2%; n = 16) (Fig. 1D). The inhibition was relatively rapid, reaching steady state within ∼30 sec and reversed completely ∼1 min after washout of the agonist. In contrast, as shown in Figure 1C, BAM8-22 apparently did not inhibit significantly LVA ICa recorded from neurons coexpressing hSNSR4 and EGFP (4.3 ± 0.9%; n = 5) (Fig. 1D).
A common form of fast HVA Ca2+ channel inhibition is mediated by Gβγ subunits released after activation of Gi/o-proteins (for review, see Ikeda and Dunlap, 1999; Dolphin, 2003; Elmslie, 2003). PTX has the ability to ADP-ribosylate a cysteine residue in the Gα subunit of Gi/o subfamily (four residues from the C terminus), thereby disrupting the coupling between these G-proteins and GPCRs. In neurons pretreated with PTX (500 ng/ml), the inhibition of ICa evoked at +10 mV by BAM8-22 was significantly reduced (23.1 ± 3.4%; n = 12; p < 0.0001) (Fig. 1D). These data suggest that hSNSR4, when heterologously expressed in rat DRG neurons, can modulate HVA but not LVA Ca2+ channels via both PTX-sensitive Gi/o and PTX-insensitive G-proteins.
The ability of hSNSR4 to modulate HVA Ca2+ channels is further illustrated by current-voltage (I-V) relationships obtained in the absence or presence of BAM8-22 (Fig. 2B,C). ICa was evoked by 70 msec test pulses to successively increase potentials (ranging from -120 to +80 mV) from a holding potential of -80 mV. ICa arising from LVA Ca2+ channels (T-type) displayed rapid, voltage-dependent inactivation, whereas ICa arising from HVA Ca2+ channels lacked rapid inactivation during a 70 msec step of depolarization. This is clearly demonstrated by representative records of ICa (evoked by a 70 msec test pulse to -10 mV from holding potential at -80 mV) recorded from neurons that had predominantly LVA versus HVA (Fig. 2A, left and right, respectively) current components. The I-V relationships shown in Figure 2, B and C, were determined from ICa amplitudes (same neuron as depicted in Fig. 1C) measured near the beginning and end of the test pulses (i.e., 15 and 62.5 msec from the beginning of the test pulse, respectively). Because LVA channels inactivate rapidly, measurements at the end of the test pulse enhance visual separation of current components arising from LVA and HVA channels. In Figure 2C, the selective inhibition of the HVA ICa component by BAM8-22 is clearly illustrated.
Figure 2.

Voltage dependence of BAM8-22-induced ICa inhibition in a rat DRG neuron heterologously expressing hSNSR4. A, Representative current traces comparing the time course of ICa inactivation in neurons displaying primarily LVA (left) or HVA (right) ICa. Voltage protocols used to evoke ICa are illustrated below. B, C, Superimposed current-voltage (I-V) relationships obtained before and during application of BAM8-22 (BAM; 3 μm). ICa was recorded from the same DRG neuron illustrated in Figure 1C. ICa was evoked by a 70 msec test pulse of successively increasing voltage from a holding potential of -80 mV. Current amplitudes were measured 15 msec (B) or 62.5 msec (C) after initiation of the test pulse. Con, Control.
Heterologously expressed hSNSR4 couples to voltage-dependent Ca2+ channels in SCG neurons
DRG neurons express multiple types of both LVA and HVA Ca2+ channels. Hence, isolating an ICa component arising from a single Ca2+ channel type can be difficult. Moreover, the heterogeneity of primary sensory neurons in regard to expression of ion channels and GPCRs also complicates analysis. To circumvent these problems, we opted to use adult rat SCG neurons as expression hosts, because the composition of ICa in these neurons is relatively homogeneous (primarily N-type Ca2+ channels), and the modulation pathways are well described. Expression of hSNSR4 was achieved by directly injecting cDNA coding for the receptor into the nuclei of acutely dissociated SCG neurons (Ikeda, 2004). Data in Figure 3 demonstrate that application of BAM8-22 to neurons expressing hSNSR4, but not control (i.e., uninjected) neurons, inhibited ICa evoked by a double-pulse voltage protocol (Fig. 3A, top). The protocol (Elmslie et al., 1990) consisted of an initial 20 msec test pulse to +10 mV (“prepulse”) followed by a 50 msec depolarizing conditioning pulse to +80 mV and a subsequent 20 msec test pulse to +10mV (“postpulse”). Before application of the agonist, the postpulse current amplitude usually exceeded the prepulse amplitude recorded from control (i.e., uninjected) or hSNSR4 cDNA-injected neurons as shown in Figure 3, A and B. This basal facilitation (defined as the ratio of postpulse to prepulse amplitude) has been shown previously to arise from tonic G-protein activation (Ikeda, 1991). Application of BAM8-22 (3 μm) resulted in robust decrease of ICa in neurons injected previously (18-24 hr) with hSNSR4 cDNA, whereas BAM8-22 at a higher concentration (10 μm) was ineffective in control neurons. The inhibition was rapid and completely reversible as shown in Figure 3B. In SCG neurons, ICa inhibition mediated by activation of GPCRs has been classified as either voltage dependent (VD) or voltage independent (VI). VD inhibition is mediated by Gβγ (Herlitze et al., 1996; Ikeda, 1996) and can be relieved by strong depolarization (Bean, 1989; Elmslie et al., 1990). For example, as shown in Figure 3A and summarized in Figure 3D, the inhibition of prepulse ICa in uninjected neurons after norepinephrine (NE; 10 μm) application was robust (67.8 ± 2.2%; n = 7), whereas postpulse ICa inhibition was much weaker (27.7 ± 2.3%; n = 7; p < 0.0001; paired t test). The mechanism of VI inhibition is more complex and less completely understood. Clearly, hSNSR4-mediated ICa inhibition exhibits VD characteristics, because inhibition of prepulse ICa amplitude by BAM8-22 (66.6 ± 2.6%; n = 17) was much stronger than postpulse amplitude inhibition (44.8 ± 3.4%; n = 17; p < 0.0001; paired t test). However, although NE and BAM8-22 induced virtually the same degree of prepulse ICa inhibition (67.8 ± 2.2%, n = 7, and 66.6 ± 2.6%, n = 17, respectively), postpulse ICa amplitude was differentially inhibited by NE and BAM8-22 (27.7 ± 2.3%, n = 7, vs 44.8 ± 3.4%, n = 17; p < 0.001; t test). The fact that BAM8-22 mediated ICa inhibition was only partially alleviated (compared with NE-mediated ICa inhibition) by the conditioning voltage step suggests that BAM8-22-mediated ICa inhibition arises from both VD and VI mechanisms.
Figure 3.
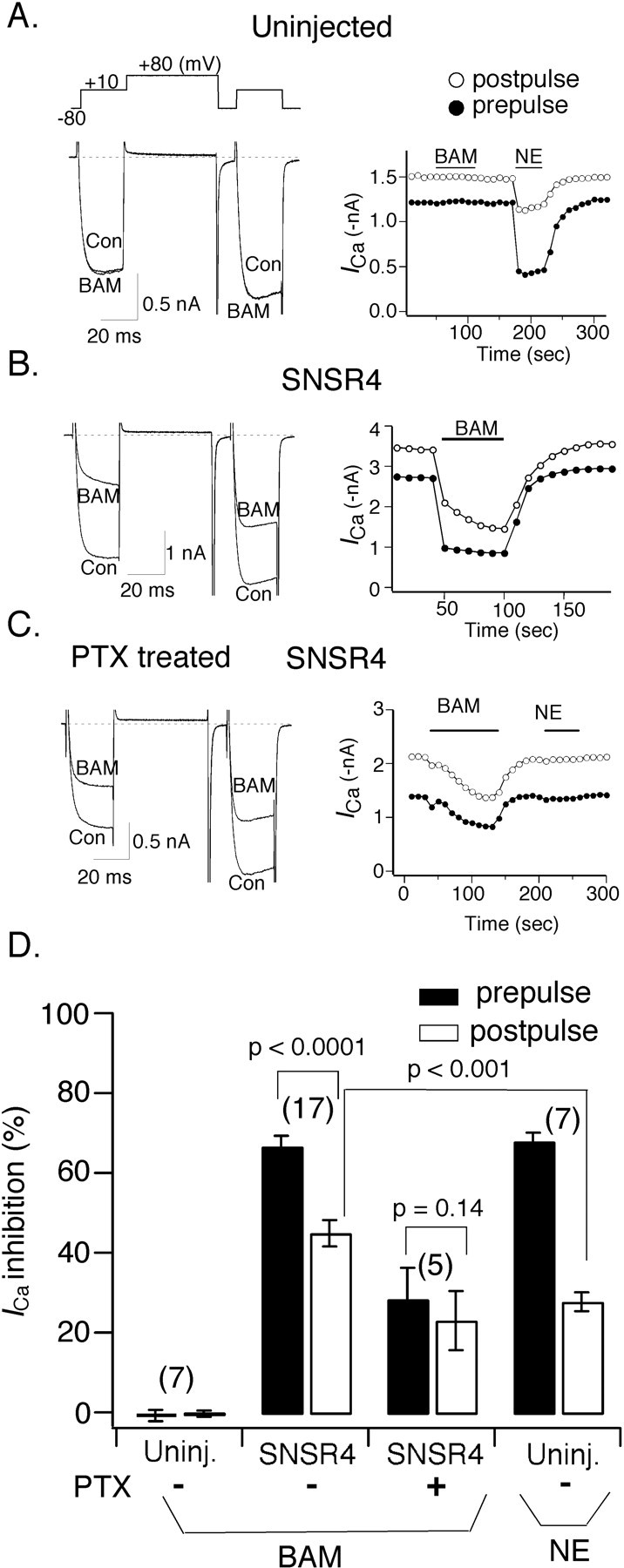
Heterologously expressed hSNSR4 couples to voltage-activated Ca2+ channels in rat SCG neurons. A-C, Comparison of BAM8-22 (BAM) and NE-induced ICa inhibition. Representative whole-cell ICa traces (left) and time course (right) obtained in the absence or presence of BAM8-22 recorded from a control (Con) SCG neuron (A), an SCG neuron injected previously with cDNA encoding hSNSR4 (B), and an SCG neuron injected with the cDNA encoding hSNSR4 and pretreated overnight with 500 ng/ml PTX (C). The voltage protocol shown above the current traces was repeated at 0.1 Hz. Filled and open circles represent ICa amplitude measured during the prepulse and postpulse, respectively. ICa amplitude was determined 10 msec after initiation of the test pulse. D, Bar graph summarizing mean (±SEM) percentage inhibition of prepulse (filled) and postpulse (open) evoked ICa by BAM8-22 or NE (10 μm). Experimental conditions are specified under each bar. BAM, 10 μm for control uninjected (Uninj.) neurons, 3 μm was used for the remaining experiments. The numbers of cells tested for each condition are shown in parentheses.
The VD component of ICa inhibition in SCG neurons is mediated by PTX-sensitive G-proteins
GPCRs that mediate VD ICa inhibition in SCG neurons usually couple to Ca2+ channels via PTX-sensitive heterotrimeric Gi/o-proteins (Ikeda and Dunlap, 1999). However, activation of some receptors linked to Gs-proteins such as vasoactive intestinal polypeptide (VIP) receptor can also result in VD ICa inhibition (Zhu and Ikeda, 1994). To differentiate these possibilities, we tested the effect of overnight pretreatment with PTX (0.5 μg/ml; 16-24 hr) on hSNSR4-mediated ICa inhibition. As shown in Figure 3C and summarized in Figure 3D, PTX pretreatment greatly attenuated BAM8-22-mediated ICa inhibition in neurons expressing hSNSR4. Interestingly, the onset of ICa inhibition after PTX treatment also was much slower, taking ∼2 min to achieve maximal inhibition. The mechanism responsible for this slow time course is unclear and deserves additional investigation. NE-mediated ICa inhibition was abolished in the same neurons establishing the activity of the toxin. The inhibition of ICa by BAM8-22 in neurons pretreated with PTX was VI, because inhibition of the prepulse amplitude (28.3 ± 7.9%; n = 5) was not significantly different from postpulse inhibition (22.9 ± 7.5%; n = 5; p = 0.14; paired t test). As mentioned previously, in the absence of PTX pretreatment, prepulse and postpulse inhibitions were much greater (66.6 ± 2.6 and 44.8 ± 3.8%, respectively).
The finding that BAM8-22-mediated ICa inhibition arises from both VD and VI mechanisms also was supported by examining the relationship between hSNSR4-mediated ICa inhibition and test potential (Fig. 4). I-V relationships were obtained in the absence or presence of 3 μm BAM8-22 (Fig. 4A). The effect of voltage on hSNSR4-mediated inhibition is illustrated by the bell-shaped relationship between ICa inhibition and test potential (Fig. 4B). Maximal inhibition by BAM8-22 in hSNSR4-expressing neurons occurs over a voltage range of 0-10 mV. This was similar to NE-mediated inhibition of ICa in control (uninjected) neurons (Fig. 4B, open circles). However, over the voltage range +30 to +60 mV, BAM8-22-mediated inhibition was much greater than NE-mediated inhibition. Specifically, at +42 mV, ICa inhibition by BAM8-22 in hSNSR4-expressing neurons was significantly greater (45.0 ± 3.0%; n = 7) than NE-mediated inhibition in control neurons (22.9 ± 2.8%; n = 5; p < 0.001; t test). Together, these data indicate that BAM8-22-mediated ICa inhibition in SCG neurons was comprised of two approximately equal components, VD inhibition mediated via PTX-sensitive Gi/o-proteins and VI inhibition mediated via PTX-insensitive G-proteins.
Figure 4.

Voltage and concentration dependence of ICa modulation mediated by activation of hSNSR4. A, ICa I-V relationships obtained in the absence (open circles) or presence (filled triangles) of BAM8-22 (BAM; 3 μm) from a neuron previously injected with cDNA encoding hSNSR4. B, Plot of mean (±SEM) percentage ICa inhibition versus test potential. The filled triangles represent data from neurons previously injected with hSNSR4 cDNA (n = 7) and exposed to BAM8-22 (3 μm). For comparison, data from uninjected neurons (n = 5; open circles) treated with NE (10 μm) are superimposed on the plot. C, Time courses of ICa inhibition produced by sequentially increasing concentration of BAM8-22. Data obtained from two separate neurons expressing hSNSR4 are illustrated. In general, three concentrations of agonist were tested for each neuron. D, Concentration-response curve for hSNSR4-mediated mean ICa inhibition. The EC50 for BAM8-22 was determined by fitting the data to the Hill equation using a nonlinear regression algorithm. Numbers of cells tested for each concentration of BAM8-22 are shown in parentheses.
Potency of BAM8-22 in hSNSR4-expressing SCG neurons
To derive the concentration-response curve, ICa was evoked every 10 sec using the voltage protocol depicted in Figure 3A. Figure 4C illustrates time courses of prepulse-evoked ICa inhibition after sequential application of increasing concentration of BAM8-22 recorded from SCG neurons expressing hSNSR4. Note that between agonist applications, ICa amplitude rapidly (∼30 sec) recovered often beyond the baseline amplitude. Such “over recovery” after agonist application has been reported previously (Kasai, 1991) for ICa inhibition, although the underlying mechanisms remain unclear. The EC50 value for BAM8-22 (0.60 μm) was estimated by fitting mean ICa inhibition data to the Hill equation (Fig. 4D) using nonlinear regression. In uninjected neurons, BAM8-22 at 10 μm had no effect on ICa amplitude (Fig. 3D).
Constitutive activity of hSNSR4 in SCG neurons
In uninjected SCG neurons, the amplitude of ICa evoked by the postpulse is usually slightly larger than that evoked by the prepulse. This basal facilitation has been attributed to tonic G-protein activation (Ikeda, 1991). As summarized in Figure 5, B and C, the basal facilitation ratio defined as the ratio of postpulse ICa amplitude to prepulse ICa amplitude in the absence of agonist ranged between 1.07 and 1.67 with a mean value of 1.30 ± 0.02 (n = 33) in uninjected neurons. During the course of these experiments, we noticed that a subset of hSNSR4-expressing neurons (19 of 65) exhibited a basal facilitation ratio that greatly exceeded that of the control group (p < 0.0001), ranging from 1.70 to 2.17 with a mean of 1.88 ± 0.04 (Fig. 5A,B). Additionally, as illustrated in Figure 5A, the activation kinetics of ICa evoked by the prepulse was much slower than that of uninjected neurons (Fig. 3A). Such kinetic slowing of current activation and strong VD facilitation are hallmarks of Gβγ-mediated inhibition of ICa in SCG neurons (Ikeda and Dunlap, 1999). The mean basal facilitation ratio derived from all hSNSR4-injected neurons (1.54 ± 0.04; n = 66) (Fig. 5B,C) was also significantly higher than that of uninjected neurons (p < 0.0001).
Figure 5.

Constitutive activity of hSNSR4 expressed in SCG neurons. A, Representative ICa trace (recorded from an SCG neuron injected with hSNSR4 cDNA) illustrating strong depolarization-induced relief of ICa inhibition in the absence of overt agonist. The voltage protocol for evoking ICa is shown below the trace. B, Basal (i.e., absence of agonist) ICa facilitation (i.e., the ratio of the postpulse to prepulse ICa amplitude measured at 10 msec after initiation of the test pulse) for individual SCG neurons. Neurons were uninjected (Uninj.; control), injected with hSNSR4 cDNA, or injected with hSNSR4 cDNA and treated overnight with PTX. C, Bar graph summarizing mean (±SEM) basal facilitation. Data are derived from B. Numbers of neurons tested are shown in parentheses.
Additional experiments were performed to shed light on the increased basal facilitation ratio seen in a subset of hSNSR4-injected neurons. Injection of EGFP cDNA alone did not significantly alter basal facilitation ratio (1.17 ± 0.03; n = 6; data not shown) when compared with uninjected neurons (p = 0.24). Thus, the injection procedure and heterologous expression per se did not alter mean basal facilitation ratio. Pretreatment of hSNSR4-injected neurons with PTX (0.5 μg/ml; 16-24 hr) reduced the mean basal facilitation ratio to 1.38 ± 0.04 (range, 1.19-1.68; n = 16) (Fig. 5B,C), a value comparable with that of uninjected cells (p = 0.27). Note that none of the PTX-treated hSNSR4-injected neurons or control neurons (including both uninjected and EGFP-expressing) had basal facilitation ratios >1.7, whereas 19 of 66 hSNSR4-injected cells (Fig. 5B) exhibited basal facilitation ratios greater than this value. The effect of PTX on basal facilitation ratio in hSNSR4-expressing neurons suggests that in some neurons, hSNSR4 was active in the absence of overt agonist.
Heterologously expressed hSNSR4 couples to M-type K+ channels in SCG neurons
To test the hypothesis that hSNSR4 couples to Gq/11-proteins, we examined the modulation of M-type K+ channel currents (IM) in SCG neurons. IM is a voltage-gated noninactivating K+ current (Brown and Adams, 1980) that influences neuronal excitability in both peripheral and central neurons. IM is modulated by GPCRs that couple to Gq/11 via a mechanism involving activation of phospholipase C (PLC) and breakdown of phosphatidylinositol 4,5-bisphosphate (Suh and Hille, 2002; Zhang et al., 2003).
Whole-cell IM from SCG neurons was recorded using pipette and external solutions that approximate physiological ion concentrations. Neurons were voltage clamped at a holding potential of -30 mV (which serves to inactivate other K+ channels found in SCG neurons) and periodically (0.1 Hz) stepped to -60 mV for 0.5 sec (Fig. 6A). IM amplitude was determined from the magnitude of the slow “tail current” resulting from M-channel deactivation during the voltage step to -60 mV. Application of BAM8-22 (3 μm) to uninjected neurons produced no detectable change in IM amplitude or time course (Fig. 6A). In neurons injected previously (18-24 hr) with hSNSR4 cDNA, application of BAM8-22 (3 μm) resulted in a relatively fast (maximal inhibition at ∼1 min after agonist application) and partially reversible inhibition (Fig. 6B). The inhibition was characterized by a dramatic decrease in both the steady-state outward current (measured before hyperpolarizing pulse) and the time-dependent current relaxation measured during the step to -60 mV (Fig. 6B). IM inhibition was determined by normalizing IM amplitude after 1 min of agonist application to the pre-drug amplitude. BAM8-22-mediated IM inhibitions in uninjected and hSNSR4-injected neurons were -3.9 ± 1.8% (n = 5) and 87.3 ± 2.7% (n = 6), respectively (Fig. 6C). PTX pretreatment (0.5 μg/ml; 24-26 hr) did not significantly attenuate BAM8-22-mediated IM inhibition (p = 0.35) despite the fact that GPCRs linked to PTX-sensitive Gi/o-proteins can activate some PLC-β isoforms via Gβγ (Rhee, 2001). The inhibition of IM amplitude in hSNSR4-injected neurons pretreated with PTX was 80.9 ± 6.0% (n = 7). These data demonstrate that hSNSR4, when heterologously expressed in SCG neurons, couples to M-type K+ channels via PTX-insensitive G-proteins.
Figure 6.
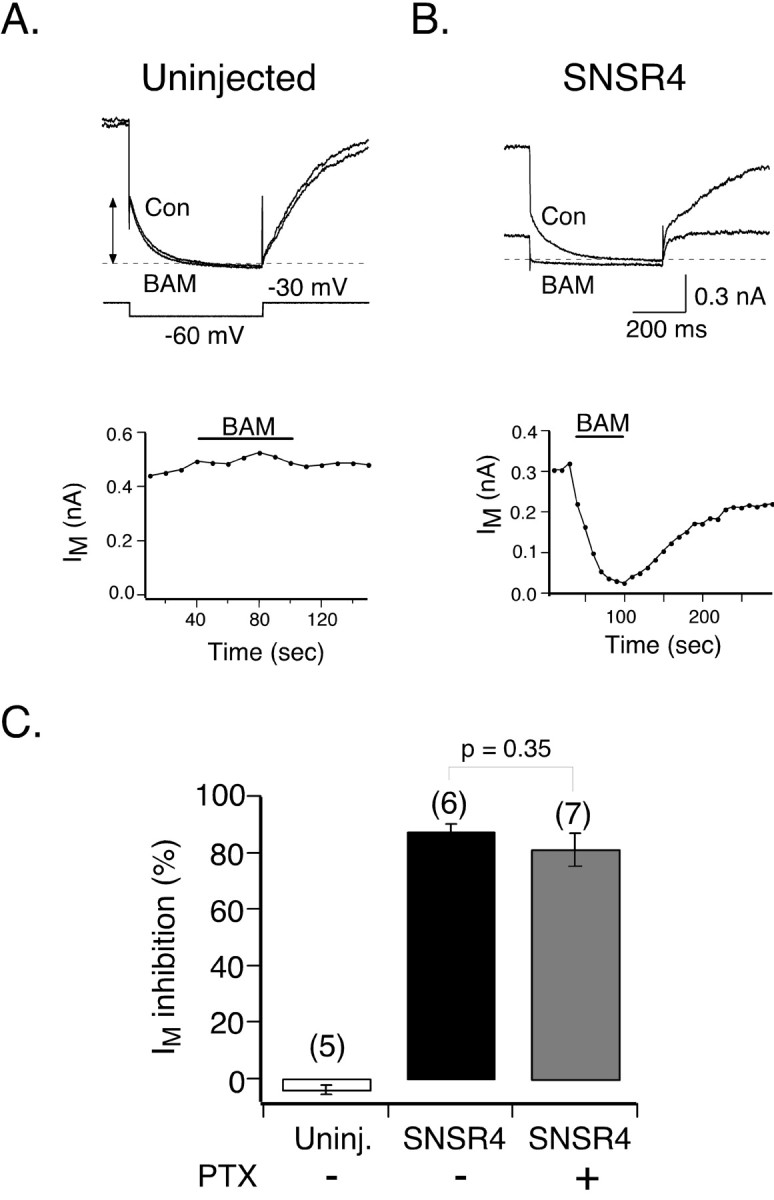
Activation of hSNSR4 inhibits M-type K+ channels in rat SCG neurons. A, B, Comparison of BAM8-22 (BAM)-induced inhibition of M-type K+ current (IM) in control (Con) and hSNSR4-expressing neurons. Representative IM traces (top) recorded from an uninjected (A) and hSNSR4-expressing (B) SCG neurons in the absence (control) or presence of BAM8-22 (3 μm). IM was evoked from a holding potential of -30 mV with the voltage protocol illustrated (A). The dashed line represents the zero current level. Time courses of IM amplitude (below) from the same neurons in the absence or presence of BAM8-22. IM amplitude was determined as the current component deactivating during a 500 msec voltage step to -60 mV (arrows in A, top). C, hSNSR4-mediated inhibition of IM was insensitive to overnight pretreatment with PTX (0.5 μg/ml). Mean (±SEM) percentage IM inhibition was produced by application of BAM8-22 under the conditions indicated. IM inhibition was determined from traces acquired just before and 1 min after the start of agonist application. The number of neurons tested is indicated in parentheses. Uninj., Uninjected.
Heterologously expressed hSNSR4 modulates synaptic transmission in rat hippocampal neurons
To test whether expression of hSNSR4 results in modulation of synaptic transmission, we used recombinant adenoviruses to coexpress the receptor and EGFP in hippocampal neurons grown in microisland culture (Fig. 7A). This preparation has some favorable features for the study of synaptic transmission. Single neurons grown on isolated permissive microislands are forced to develop a large number of recurrent synaptic contacts (i.e., autapses), resulting in a large amplitude synaptic response and, consequently, a favorable signal-to-noise ratio and low variability. Additionally, autaptic responses are unambiguously mono-synaptic. Conventional whole-cell patch-clamp techniques were used to record EPSCs from neurons that had been infected for 30-60 hr. To evoke autaptic responses, hippocampal neurons were held at -60 mV (or in some cases at -40 mV to reduce driving force) and were stimulated every 10 sec with a pair of square depolarizing pulses (2 msec pulse, 60 mV, 50 msec interval between prepulse and postpulse). As shown in Figure 7, B and D, application of BAM8-22 (3 μm) robustly inhibited prepulse-evoked EPSCs in neurons coexpressing hSNSR4 and EGFP (55.9 ± 4.9%; n = 21), whereas the peptide was ineffective in neurons expressing EGFP alone (3.2 ± 0.9%; n = 6). The inhibition was relatively rapid, reaching nearly maximal inhibition within 30 sec and partially reversible after washout of the agonist (Fig. 7B).
Figure 7.

Activation of hSNSR4 induces presynaptic inhibition in rat hippocampal neurons. A, Recombinant adenoviruses-mediated coexpression of hSNSR4 and EGFP in a hippocampal neuron cultured on a microisland of permissive substrate for 2 weeks. The phase-contrast (left) and fluorescent (right) micrographs were taken from the same cell. con, Control. B, Top, Representative whole-cell current traces obtained from a neuron expressing hSNSR4 showing depression of the EPSCs during application of BAM8-22 (BAM; 3 μm). EPSCs were evoked by pairs of 2 msec depolarizing commands (50 msec interval between prepulse and postpulse) applied at 0.1 Hz. B, Middle, Time course of BAM8-22-induced inhibition of EPSCs evoked by prepulse (filled circles) and postpulse (open circles) depolarizations. B, Bottom, PPR increased during application of BAM8-22, indicating presynaptic locus of inhibition. C, Bar graph depicting mean (±SEM) PPR in the absence (open) or presence (filled) of BAM8-22 in neurons expressing hSNSR4. D, Bar graph comparing BAM8-22-induced mean (±SEM) percentage inhibition of EPSCs (evoked by prepulse) in neurons expressing EGFP (open, left bar) or hSNSR4 either untreated (filled, middle bar) or after overnight treatment with PTX (filled, right bar). The number of neurons tested is indicated in parentheses.
As shown in Figure 7B (top), in comparison with postpulse-evoked EPSC, EPSCs evoked by the prepulse were more susceptible to BAM8-22-mediated inhibition. This finding is more clearly illustrated in the plot of paired pulse facilitation ratio (PPR; the amplitude ratio of postpulse-over prepulse-evoked EPSCs) versus time (Fig. 7B, bottom). BAM8-22 induced a reversible increase of PPR that coincided with the peptide-induced inhibition of synaptic response. PPR is an indicator that has often been used to indirectly evaluate the release probability of synaptic vesicles (Manabe et al., 1993; Zucker and Regehr, 2002). As summarized in Figure 7C, PPR in neurons expressing hSNSR4 was 1.24 ± 0.07 (n = 21) in the absence of agonist and markedly increased after application of BAM8-22 (1.59 ± 0.10; n = 21; p < 0.001; paired t test). These data suggested that BAM8-22-mediated inhibition of EPSCs in neurons expressing hSNSR4 was caused, at least in part, by a reduction in release probability of vesicles at presynaptic terminals.
The most common mechanism of GPCR-induced presynaptic inhibition is the inhibition of Ca2+ channels by PTX-sensitive Gi/o-proteins (Wu and Saggau, 1997). Additionally, intracellular Ca2+ stores may contribute to short-term synaptic plasticity in some cases (Galante and Marty, 2003; but see Carter et al., 2002). As shown above, when expressed in SCG or DRG neurons, hSNSR4 can couple to both PTX-sensitive Gi/o and PTX-insensitive G-proteins. We thus tested the effect of pretreatment with PTX (100 ng/ml; >24 hr) on EPSC inhibition. As summarized in Figure 7D, PTX treatment abolished BAM8-22-induced inhibition of prepulse-evoked EPSCs in neurons expressing hSNSR4 (-6.7 ± 3.2%; n = 11; p = 0.29; in comparison with control EGFP-expressing neurons) after adenovirus infection. Interestingly, application of BAM8-22 to one neuron expressing hSNSR4 resulted in an increase of prepulse-evoked EPSC amplitude (27.8%) with accompanying reduction of PPR from 1.63 to 1.34, indicating an increased release probability. Together, these data suggest activation of hSNSR4 after adenoviral infection can induce inhibition of transmitter release mediated by PTX-sensitive Gi/o-proteins.
Discussion
SNSRs/mgrXs are a new family of human GPCRs with an expression profile restricted to small nociceptors of primary sensory ganglia. Their unique expression pattern raises the exciting possibility that SNSRs/mgrXs may provide novel targets for the development of improved analgesic agents, although at present, their neuronal function is unknown. Unlike the great majority of human GPCRs, orthologs of SNSRs/mgrXs probably do not exist in rodents, and the gene family appears to have undergone rapid and divergent evolution (Choi and Lahn, 2003; Zylka et al., 2003). This divergence makes it difficult to assess physiological functions by studying natively expressed receptors in rodent sensory ganglia or using murine gene knock-out technology. As a first step toward understanding the neuronal function of this family of receptors, we used recombinant adenoviral infection and nuclear injection techniques to express one of the human SNSRs/mrgXs, hSNSR4, in cultured rat neurons and found that the receptor can modulate neuronal ion channels as well as synaptic transmission.
Consistent with molecular screening efforts (Dong et al., 2001; Lembo et al., 2002) and bioinformatics searches (Zylka et al., 2003) that failed to identify rodent orthologs of human SNSRs, application of BAM8-22, a potent peptide agonist of human SNSR3/4, did not alter the amplitude of HVA and LVA ICa in a number (n = 41) of control rat and mouse DRG neurons. In contrast, the peptide was highly effective in modulating HVA ICa but had little or no effect on ICa arising from LVA (T-type) Ca2+ channels in acutely dissociated rat DRG neurons that had been infected with adenoviruses coexpressing both EGFP (as a marker) and hSNSR4. These data establish that hSNSR4 efficiently couples to pre-existing Ca2+ channels and signaling components after heterologous expression. Our data suggest that orthologs of human SNSR3/4 do not exist in rodent primary sensory neurons as previously predicted. However, it should be noted that BAM8-22 [termed BAM-15 by Han et al. (2002)] is a fairly potent agonist for (murine) mrgC11, and a rat ortholog of this receptor has been predicted. Thus, GPCRs responsive to BAM8-22 may exist in rodent neurons but expressed in an extremely small subset of sensory neurons or targeted to a cellular location such as peripheral or central terminals not accessible in our preparation. However, our results demonstrate unequivocally that heterologously expressed hSNSR4s couple to native Ca2+ channels using endogenous signaling pathways. Ligand-binding assays using radioactive BAM8-22 or immunochemistry using antibodies generated against human SNSRs should help resolve this issue.
Primary sensory neurons express multiple types of Ca2+ channels, including T-, N-, P/Q-, R-, and L-types (Fedulova et al., 1985; Fox et al., 1987; Kostyuk et al., 1988; Regan et al., 1991; Mintz et al., 1992). From our data, it is not clear which type(s) of HVA Ca2+ channels is subjected to the inhibition after activation of hSNSR4, although one assumes that N- and P/Q-type Ca2+ channels are involved because these channel types are highly modulated by GPCRs. LVA Ca2+ channels apparently were not affected by hSNSR4 activation. This result is interesting within the context of a recent study (Wolfe et al., 2003) demonstrating that T-type Ca2+ channels are modulated by a specific Gβγ combination, namely Gβ2γ2. To study hSNSR4-mediated modulation on a defined HVA Ca2+ channel subtype, receptors were expressed in rat SCG neurons. Ca2+ channels in this system are homogeneous and consist of current arising primarily from N-type Ca2+ channels (Ikeda, 1991). Application of BAM8-22 induced robust inhibition (66.6%) of ICa in SCG neurons previously injected with expression vectors encoding hSNSR4, whereas the peptide was ineffective in control neurons. The finding that hSNSR4 modulates N-type Ca2+ channels might be physiologically relevant, because evoked transmitter release at excitatory synapses formed between small-diameter nociceptors and the neurons located in layers I and II of the spinal dorsal horn are controlled primarily by Ca2+ influx via N-type Ca2+ channels (Bao et al., 1998; Craig, 2003).
We also found that BAM8-22-induced ICa inhibition in SCG neurons was comprised of approximately two equal components: VD inhibition mediated by PTX-sensitive Gi/o-proteins and VI inhibition mediated by PTX-insensitive G-proteins. VI inhibition in SCG neurons is usually mediated by Gq/11-proteins (Kammermeier et al., 2000; Dolphin, 2003). Additionally, application of the peptide nearly abolished the current derived from M-type K+ channels in SCG neurons heterologously expressing hSNSR4, whereas it was ineffective in control SCG neurons. IM is a noninactivating, voltage-dependent K+ conductance that has been found to play an important role in controlling neuronal excitability (Brown and Adams, 1980). Thus far, only Gq/11-coupled receptors (under native conditions) have been found to inhibit IM. Consistent with this, we found that pretreatment with PTX did not alter BAM8-22-induced inhibition of IM. IM has been recorded from many types of central and peripheral neurons, but it was not until recently that the presence of IM in mammalian primary sensory neurons was documented and shown to be involved in chronic pain transmission (Passmore et al., 2003). If sensory neurons in humans also express IM channels, then the coupling of hSNSR4 to IM, as shown here, might have relevance to human nociceptive pathways. These data indicate that hSNSR4 can couple to both Gi/o-proteins and PTX-insensitive G-proteins, most likely Gq/11. Because Gq/11-coupled receptors such as bradykinin receptors can sensitize the heat-responsive vanilloid receptor VR1 (Chuang et al., 2001), it would be interesting to test whether hSNSR4 modulates the function of VR1.
Thus far, ∼40 mrg gene family members with intact coding sequences have been reported. Potent agonists have been discovered for seven members (including hSNSR4), thereby facilitating studies of how receptor activation influences intracellular [Ca2+] and cAMP levels in mammalian cell lines. Results from these experiments indicate that human SNSR3, hSNSR4 (mrgX1), and mrgX2, and murine mrgA1, mrgA10, and mrgC11 signal via Gq/11- but probably not Gi/o- or Gs-proteins (Dong et al., 2001; Han et al., 2002; Lembo et al., 2002; Robas et al., 2003), whereas the rat adenine receptor (a likely ortholog of murine mrgA10) can couple to Gi/o-proteins (Bender et al., 2002). Here, we demonstrated that hSNSR4, when expressed in rat neurons, couples to both PTX-insensitive (most likely Gq/11) and PTX-sensitive (Gi/o family) G-proteins. This is the first demonstration, to our knowledge, of the pleiotropic G-protein-coupling potential of mrg genes.
In a subset of SCG neurons heterologously expressing hSNSR4, the receptor appeared in the absence of overt agonist to inhibit Ca2+ channels in a voltage-dependent manner. This “constitutive activity” was not seen in PTX-pretreated neurons, indicating a requirement for the activation of Gi/o-proteins and ruling out a simple upregulation of basal Gβγ expression as a source of the activity. Because our assay relies on strong depolarization-induced relief of inhibition, constitutive VI Ca2+ channel inhibition, if present, would not be detected. Therefore, we cannot rule out the possibility that hSNSR4 also constitutively activates Gq/11-proteins, although the basal IM amplitude in SCG neurons was not significantly altered by expression of the receptor (p = 0.14; data not shown). Constitutive activity has been found in >60 wild-type or mutant GPCRs and can cause diseases in some cases (Seifert and Wenzel-Seifert, 2002). The significance of the constitutive activity of hSNSR4 remains unknown. Interestingly, it was reported recently that nociceptin receptors in rat DRG neurons constitutively inhibit the Ca2+ channel function (perhaps via a direct physical interaction) and that this activity may contribute to the mechanisms of nociceptive signaling (Beedle et al., 2004).
Finally, we also examined the potential role of hSNSR4 in synaptic neurotransmission. Activation of hSNSR4 heterologously expressed in hippocampal neurons induced marked inhibition of synaptic responses. The inhibition was abolished by PTX pretreatment, indicating that it was mediated by Gi/o-proteins. Additionally, the inhibition was associated with an increase in paired-pulse facilitation, thus suggesting a presynaptic locus of action. From these results, it is clear that hSNSR4 can traffic to presynaptic nerve terminals, couple to native signaling proteins, and interact with the release apparatus to alter synaptic transmission. If analogous processes occur in human DRG neurons, SNSRs could play a role in pain message signaling in vivo.
Footnotes
We thank Dr. T.-C. He (Johns Hopkins University, Baltimore, MD) for providing the AdEasy system and Dr. Christopher Thaler (National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Bethesda, MD) for critiquing a previous version of this manuscript.
Correspondence should be addressed to Dr. Stephen R. Ikeda, Laboratory of Molecular Physiology, National Institute on Alcohol Abuse and Alcoholism, National Institutes of Health, Park Building, Room 150, 12420 Parklawn Drive, MSC 8115, Bethesda, MD 20892-8115. E-mail:sikeda@mail.nih.gov.
Copyright © 2004 Society for Neuroscience 0270-6474/04/245044-10$15.00/0
References
- Bao J, Li JJ, Perl ER (1998) Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci 18: 8740-8750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP (1989) Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 240: 153-156. [DOI] [PubMed] [Google Scholar]
- Beedle AM, McRory JE, Poirot O, Doering CJ, Altier C, Barrere C, Hamid J, Nargeot J, Bourinet E, Zamponi GW (2004) Agonist-independent modulation of N-type calcium channels by ORL1 receptors. Nat Neurosci 7: 118-125. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF (1991) Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proc Natl Acad Sci USA 88: 7834-7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender E, Buist A, Jurzak M, Langlois X, Baggerman G, Verhasselt P, Ercken M, Guo HQ, Wintmolders C, Van den Wyngaert I, Van Oers I, Schoofs L, Luyten W (2002) Characterization of an orphan G protein-coupled receptor localized in the dorsal root ganglia reveals adenine as a signaling molecule. Proc Natl Acad Sci USA 99: 8573-8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Adams PR (1980) Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature 283: 673-676. [DOI] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA, Regehr WG (2002) Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci 22: 21-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Lambert NA (2000) Endogenous regulators of G protein signaling proteins regulate presynaptic inhibition at rat hippocampal synapses. Proc Natl Acad Sci USA 97: 12810-12815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SS, Lahn BT (2003) Adaptive evolution of MRG, a neuron-specific gene family implicated in nociception. Genome Res 13: 22-2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D (2001) Bradykinin and nerve growth factor release the capsaicin receptor from PtdIns(4,5)P2-mediated inhibition. Nature 411: 957-962. [DOI] [PubMed] [Google Scholar]
- Craig AD (2003) Pain mechanisms: labeled lines versus convergence in central processing. Annu Rev Neurosci 26: 1-30. [DOI] [PubMed] [Google Scholar]
- Dolphin AC (2003) G protein modulation of voltage-gated calcium channels. Pharmacol Rev 55: 607-627. [DOI] [PubMed] [Google Scholar]
- Dong X, Han S, Zylka MJ, Simon MI, Anderson DJ (2001) A diverse family of GPCRs expressed in specific subsets of nociceptive sensory neurons. Cell 106: 619-632. [DOI] [PubMed] [Google Scholar]
- Elmslie KS (2003) Neurotransmitter modulation of neuronal calcium channels. J Bioenerg Biomembr 35: 477-489. [DOI] [PubMed] [Google Scholar]
- Elmslie KS, Zhou W, Jones SW (1990) LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron 5: 75-80. [DOI] [PubMed] [Google Scholar]
- Fedulova SA, Kostyuk PG, Veselovsky NS (1985) Two types of calcium channels in the somatic membrane of new-born rat dorsal root ganglion neurones. J Physiol (Lond) 359: 431-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW (1987) Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J Physiol (Lond) 394: 149-172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galante M, Marty A (2003) Presynaptic ryanodine-sensitive calcium stores contribute to evoked neurotransmitter release at the basket cell-Purkinje cell synapse. J Neurosci 23: 11229-11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ (1981) Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch 391: 85-100. [DOI] [PubMed] [Google Scholar]
- Han SK, Dong X, Hwang JI, Zylka MJ, Anderson DJ, Simon MI (2002) Orphan G protein-coupled receptors MrgA1 and MrgC11 are distinctively activated by RF-amide-related peptides through the Galpha q/11 pathway. Proc Natl Acad Sci USA 99: 14740-14745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509-2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Garcia DE, Mackie K, Hille B, Scheuer T, Catterall WA (1996) Modulation of Ca2+ channels by G-protein βγ subunits. Nature 380: 258-262. [DOI] [PubMed] [Google Scholar]
- Ikeda SR (1991) Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. J Physiol (Lond) 439: 181-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR (1996) Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature 380: 255-258. [DOI] [PubMed] [Google Scholar]
- Ikeda SR (2004) Expression of G-protein signaling components in adult mammalian neurons by microinjection. Methods Mol Biol 259: 167-182. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Dunlap K (1999) Voltage-dependent modulation of N-type calcium channels: role of G protein subunits. Adv Second Messenger Phosphoprotein Res 33: 131-151. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI (2001) Molecular mechanisms of nociception. Nature 413: 203-210. [DOI] [PubMed] [Google Scholar]
- Kammermeier PJ, Ruiz-Velasco V, Ikeda SR (2000) A voltage-independent calcium current inhibitory pathway activated by muscarinic agonists in rat sympathetic neurons requires both Gαq/11 and Gβγ. J Neurosci 20: 5623-5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H (1991) Tonic inhibition and rebound facilitation of a neuronal calcium channel by a GTP-binding protein. Proc Natl Acad Sci USA 88: 8855-8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostyuk PG, Shuba YM, Savchenko AN (1988) Three types of calcium channels in the membrane of mouse sensory neurons. Pflügers Arch 411: 661-669. [DOI] [PubMed] [Google Scholar]
- Kozak M (1999) Initiation of translation in prokaryotes and eukaryotes. Gene 234: 187-208. [DOI] [PubMed] [Google Scholar]
- Lembo PM, Grazzini E, Groblewski T, O'Donnell D, Roy MO, Zhang J, Hoffert C, Cao J, Schmidt R, Pelletier M, Labarre M, Gosselin M, Fortin Y, Banville D, Shen SH, Strom P, Payza K, Dray A, Walker P, Ahmad S (2002) Proenkephalin A gene products activate a new family of sensory neuron-specific GPCRs. Nat Neurosci 5: 201-209. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA (1993) Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol 70: 1451-1459. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP (1992) P-type calcium channels in rat central and peripheral neurons. Neuron 9: 85-95. [DOI] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA (2003) KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci 23: 7227-7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regan LJ, Sah DWY, Bean BP (1991) Ca2+ channels in rat central and peripheral neurons: high-threshold current resistant to dihydropyridine blockers and ω-conotoxin. Neuron 6: 269-280. [DOI] [PubMed] [Google Scholar]
- Rhee SG (2001) Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70: 281-312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robas N, Mead E, Fidock M (2003) MrgX2 is a high potency cortistatin receptor expressed in dorsal root ganglion. J Biol Chem 278: 44400-44404. [DOI] [PubMed] [Google Scholar]
- Seifert R, Wenzel-Seifert K (2002) Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn Schmiedebergs Arch Pharmacol 366: 381-416. [DOI] [PubMed] [Google Scholar]
- Suh BC, Hille B (2002) Recovery from muscarinic modulation of M current channels requires phosphatidylinositol 4,5-bisphosphate synthesis. Neuron 35: 507-520. [DOI] [PubMed] [Google Scholar]
- Wolfe JT, Wang H, Howard J, Garrison JC, Barrett PQ (2003) T-type calcium channel regulation by specific G-protein βγ subunits. Nature 424: 209-213. [DOI] [PubMed] [Google Scholar]
- Wu LG, Saggau P (1997) Presynaptic inhibition of elicited neurotransmitter release. Trends Neurosci 20: 204-212. [DOI] [PubMed] [Google Scholar]
- Young D, Waitches G, Birchmeier C, Fasano O, Wigler M (1986) Isolation and characterization of a new cellular oncogene encoding a protein with multiple potential transmembrane domains. Cell 45: 711-719. [DOI] [PubMed] [Google Scholar]
- Yusaf SP, Goodman J, Pinnock RD, Dixon AK, Lee K (2001) Expression of voltage-gated calcium channel subunits in rat dorsal root ganglion neurons. Neurosci Lett 311: 137-141. [DOI] [PubMed] [Google Scholar]
- Zhang H, Craciun LC, Mirshahi T, Rohacs T, Lopes CM, Jin T, Logothetis DE (2003) PIP2 activates KCNQ channels, and its hydrolysis underlies receptor-mediated inhibition of M currents. Neuron 37: 963-975. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR (1994) VIP inhibits N-type Ca2+ channels of sympathetic neurons via a pertussis toxin-insensitive but cholera toxin-sensitive pathway. Neuron 13: 657-669. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]
- Zylka MJ, Dong X, Southwell AL, Anderson DJ (2003) Atypical expansion in mice of the sensory neuron-specific Mrg G protein-coupled receptor family. Proc Natl Acad Sci USA 100: 10043-10048. [DOI] [PMC free article] [PubMed] [Google Scholar]