

Abstract
Previous studies of the lurcher mutation in GluR1 channels concluded that its main effect is to create constitutively active channels (Kohda et al., 2000; Taverna et al., 2000). GluR1Lc channels also exhibit slowed kinetics and a shift in their apparent affinity for glutamate (Kohda et al., 2000; Taverna et al., 2000). Here, we have undertaken a kinetic analysis of GluR1Lc channels to quantify the effects of lurcher and to determine the relative contribution of these effects to the lurcher phenotype. Analysis of GluR1Lc leak current demonstrated that the 2,3-dioxo-6-nitro-1,2,3,4-tetrahydro[f]quinoxaline-7-sulfonamide (NBQX)-sensitive portion of the leak current corresponded to a current generated by glutamate concentrations similar to the levels of contaminating glutamate measured in our normal external solutions. This result, and the small size of the leak current relative to the currents evoked by saturating glutamate, indicates that GluR1Lc channels exhibit little or no constitutive activity. Our results indicate that the primary effect of the lurcher mutation is to increase the affinity of GluR1 for glutamate and reduce the desensitization of GluR1 at nanomolar concentrations. We also found that the mutation makes both the rate and extent of GluR1Lc channel desensitization depend strongly on subunit occupancy. We conclude that the poor survival of GluR1Lc-transfected cells, and presumably cerebellar neurons in lurcher mice, results because channels carrying the lurcher mutation open and do not desensitize at ambient levels of glutamate.
Keywords: glutamate, AMPA receptor, desensitization, lurcher, kinetic modeling, gating
Introduction
Ionotropic AMPA-type glutamate receptors (GluRs) mediate a major portion of the fast excitatory transmission in the CNS, and mutations in GluRs can have extensive effects. The lurcher mutation in the GluR subunit GluRδ2 causes widespread Purkinje cell death in homozygous mice and was proposed to render this orphan receptor constitutively active (Phillips, 1960; Zuo et al., 1997). Lurcher is an alanine to threonine change (Ala636Thr) in the SYTANLAAF motif, which resides in the extracellular half of the second true transmembrane segment in all eukaryotic GluRs. When the same Ala-Thr mutation is made in GluR1 or GluR6, receptor desensitization and deactivation kinetics are slowed (Kohda et al., 2000; Taverna et al., 2000). Large leftward shifts were also reported for glutamate and kainate concentration-response curves (Taverna et al., 2000).
Although several groups have reported that the lurcher mutation results in channels that can open in the absence of glutamate (Kohda et al., 2000; Taverna et al., 2000; Schwarz et al., 2001), one group has reported that the same mutation in δ1 and NR1 does not result in constitutively active channels (Williams et al., 2003). In addition, the shifts in EC50 values reported by others raise the possibility that what appears to be constitutive activity is really channel activation by contaminating glutamate. Given these conflicting results, we sought to reexamine the effects of the lurcher mutation in GluR1 in the context of the available structural data and a kinetic model for AMPA-type channels.
Materials and Methods
Cell culture and patch-clamp recording. tsA201 cells were plated onto 12 mm glass coverslips that had been coated with poly-l-lysine (4.5 μg/ml). The culture medium was DMEM (Invitrogen, Gaithersburg, MD) containing 10% FBS. The cells were transiently transfected using Lipofectamine 2000 (Invitrogen) with 0.5-0.7 μg of total cDNA per coverslip. The solution used for transfection consisted of 200 μl of Opti-MEM (Invitrogen), 1.5 μl of Lipofectamine 2000, 1.5 μg of a reporter cDNA encoding green fluorescent protein in pRK5, and 4-5 μg of wild-type GluR1 (GluR1wt) or GluR1 with the lurcher mutation (GluR1Lc), both in a cytomegalovirus-driven mammalian expression vector. The GluR1 plasmid was kindly provided by Derek Bowie (Emory University, Atlanta, GA). The Ala-Thr mutation was inserted into the GluR1wt plasmid using PCR mutagenesis and primers described previously (Taverna et al., 2000). Both GluR1wt and GluR1Lc were the flip versions. Two hours after transfection, the cell culture media were changed to either DMEM-Glutamax (Invitrogen) or Neurobasal (Invitrogen) media supplemented with B-27. Some experiments were also performed on transfected cells, in which the glutamate scavenger glutamic pyruvic transaminase (United States Biochemicals, Cleveland, OH) and pyruvate (10 mm) were added to the culture media before recording.
Patch-clamp recordings were performed at room temperature with an EPC-9 amplifier (HEKA, Lambrecht/Pfalz, Germany) 12-36 hr after transfection. Whole-cell recordings of GluR1Lc currents were performed to measure the steady-state current evoked with CNQX and DNQX during prolonged applications. All other recordings were from excised outside-out patches. The holding potential was always set to -80 mV, and the seal resistance before patch excision was typically 3-10 GΩ. The only exception was the leak current analysis in which only patches with on-cell seal resistances of ≥25 GΩ were analyzed. The normal external solution was (in mm): 150 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, and 5 glucose, buffered with 10 HEPES (pH adjusted to 7.4 with NaOH). The N-methyl-d-glucamine (NMDG) external solution had all of the monovalent cations replaced with 153 mm NMDG (pH adjusted with HCl). The CsCl external solution had the same composition as the normal solution, but with 150 mm CsCl replacing NaCl. Patch pipettes (open tip resistance, 3-6 MΩ) were filled with a solution containing (in mm) 120 CsF, 33 CsOH, 2 MgCl2, 1 CaCl2, and 11 EGTA (pH adjusted to 7.4 with CsOH). Glutamate, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydro[f]quinoxaline-7-sulfonamide (NBQX), CNQX, DNQX, and cyclothiazide (CTZ) were added to the external solution.
Fast perfusion. All of the agonist-evoked responses in these experiments were obtained with a rapid perfusion system consisting of a theta glass pipette mounted on a piezoelectric bimorph (part number 62003/5H-144D; Morgan Matroc). The tips of the pipettes were broken to ∼300 μm, and the width of the septa separating the barrels of the theta glass was reduced by etching with hydrofluoric acid. Both barrels of the theta glass had multiple lines fed into them: three on one side, six on the other. Patches were positioned near the interface of the solutions flowing from the adjacent barrels, and the interface was moved by applying voltage across the bimorph with a constant voltage source (HVA-100; ALA Scientific). Voltage pulses were triggered with one of the analog-to-digital outputs on the EPC-9 amplifier and were analog low-pass filtered (200 Hz, -3 dB, four-pole Bessel type) to reduce mechanical oscillations of the piezoelectric device. The rate of solution exchange estimated from open-tip responses was 100-200 μsec. The recording chamber was superfused constantly with normal external solution flowing at a rate of 1 ml/min.
Data acquisition and analysis. Glutamate-evoked currents were analog low-pass filtered at 3 kHz (four-pole Bessel type, -3 dB) and were written directly to the hard drive of the computer at sampling rates of 5-125 kHz. The digital records were analyzed using Igor software (Wavemetrics, Lake Oswego, OR). Exponential functions were fitted to the decays of the currents as described previously (Robert et al., 2001). Concentration-response data from individual patches were normalized (see Results), and the mean normalized results were fitted with Hill-type functions to obtain EC50 and IC50 values and values for the Hill coefficient (nH). Glutamate preconditioning experiments were performed by first incubating the patch in varying concentrations of glutamate and then pulsing the patch with 5 mm glutamate. The amount of inhibition produced by the preincubation was calculated as the total peak current seen in the test glutamate pulse, normalized to the total peak current seen in the control glutamate response. Recovery data were obtained from two-pulse protocols. In experiments in which either 5 mm or 500 μm glutamate was applied during the pulse of each pair, the peak amplitude of the second pulse was expressed as a fraction of the peak amplitude of the paired first pulse. Results were pooled from several patches, and the mean data were fitted with a bi-exponential equation.
The dependence of GluR1Lc kinetics on receptor occupancy was determined by comparing parameters measured at different glutamate concentrations or by preincubating patches with the competitive antagonist NBQX. NBQX preincubation experiments were performed by first exposing the patch to varying concentrations of NBQX and making a 30 msec application of 500 μm glutamate. Inhibition by NBQX of glutamate-evoked currents was long-lasting and decayed exponentially with a time constant of 155 msec, indicating that NBQX dissociation was minimal during the 30 msec application of glutamate. The amount of inhibition produced by NBQX was calculated as the amount of current seen at 10 msec into the glutamate application, normalized to the current at 10 msec in the absence of any preincubating NBQX (see Fig. 5D). Concentration-response experiments gave an IC50 value of 121 nm for NBQX. The number of subunits available for glutamate binding after preexposure to NBQX was estimated as follows. We first assumed that the current generated by 500 μm glutamate is carried almost completely by fully occupied receptors. We then iteratively found a KD value for NBQX that gave an IC50 value of 120 nm (assuming binding is subunit independent) by using occupancy and binomial equations to calculate the fraction of channels with 0-4 subunits occupied by NBQX, multiplying (1 minus these fractions) by the relative conductance values we used, and summing the results. This analysis indicated that the KD for NBQX binding to individual subunits is ∼150 nm and that at 100 nm NBQX (the concentration used for the kinetic analysis), 53% of the receptors have two or fewer subunits available for glutamate.
Figure 5.

CNQX and DNQX are agonists at GluR1Lc channels. A, B, Currents through GluR1Lc channels evoked by applications (bar) of 500 μm glutamate (left) or 0.5, 1, and 50 μm CNQX or DNQX (right; A and B, respectively). The currents were recorded from a whole cell that was lifted off the coverslip. C, Mean concentration-response data for CNQX activation of GluR1Lc channels (n = 3-5 cells). The Hill-type fit to the data gave EC50 and nH values of 496 nm and 1.07. Error bars are SEM. D, Currents evoked by 30 msec applications (bar) of 500 μm glutamate in control solution and after preexposing the patch to the indicated concentrations of NBQX.
Values are reported as means ± SEM, unless indicated otherwise. Error estimates obtained from fits to mean data are the SDs calculated from the residuals of the fit.
Kinetic modeling. Kinetic modeling was done using Monte Carlo simulations with the software package ChannelLab (Synaptosoft, Atlanta, GA). All the simulations started in zero glutamate and included the effect of any conditioning pulses. The simulated pulses were filtered so that the rise time of the application matched the rise time of the junction potential recorded experimentally. Including the effect of contaminating glutamate (50 nm) had no significant effect on the results of the simulations.
Simulated recovery time courses were calculated as 1 minus probability of finding the channel in one of the desensitized states (for details, see Robert and Howe, 2003). The conductance values for the four open states were set to 5, 8, 15, and 25 pS (Derkach et al., 1999; Banke et al., 2000; Irizarry, 2001). The conductance of all other states was set to zero. All final simulations were performed with the rate constants in Table 1 using 5000-50,000 channels.
Table 1.
Rate constants for GluR1Lc and fold changes from GluR1wt rate constants
|
Rate constant |
GluR1Lc |
Fold change from GluR1wt |
|---|---|---|
| αa,b | 250 sec−1 | −15, −4.1, −1.2, +2 |
| −5.7, −3.8, −2.8, −1.9 | ||
| βa,b,c,d,e | 1400 sec−1 | |
| k1c | 2 × 107 m−1/sec−1 | |
| k−1c,d,e,f | 60 sec−1 | −150 |
| k−2f,g | 1 sec−1 | + 2.4 |
| δ0g,h | 0.069 s−1 | + 20 |
| γ0f,h,i | 5 sec−1 | + 5 |
| δ1b,h | 10 sec−1 | −180, −36, −27, −24 |
| γ1d,f,g | 16 sec−1 | + 2 |
| γ2d,f,j | 80 sec−1 | +10 |
| γ3d,f,j | 106.7 sec−1 | + 14 |
| γ4d,f,j | 120 sec−1 | + 15 |
| δ′d,f,j | 10 sec−1 | −20 |
| γ′b,f,h
|
1000 sec−1
|
+ 29 |
The rate constants refer to the kinetic model in Figure 7A. The right column gives the fold changes from the GluR1wt rate constants (Robert and Howe, 2003); when more than one value is given, each represents the fold change for each receptor occupancy (1-4). The experimental measurements or considerations that were the main constraints on the value of each rate constant are indicated below.
Deactivation time constants (brief and long applications).
Desensitization time constants.
Time course of activation.
Concentration-response data for peak currents.
Concentration-response data for plateau currents.
Time course of recovery.
Maximal recovery from bi-exponential fits.
Relative amplitude of plateau and peak currents.
Microscopic reversibility.
Inhibition-response data for glutamate preincubation.
Our goal was to determine how the values for the rate constants governing ligand binding, channel opening, and desensitization had been changed by the lurcher mutation. Because AMPA receptors are tetramers and each subunit contains a binding site for glutamate, any physically plausible model must include a large number of physically discrete states, which in principle could be connected in a very large number of ways. The models that we explored were based on the model proposed for GluR1 and GluR4 receptors (Robert and Howe, 2003), which built on previous work on AMPA receptor desensitization (Vyklicky et al., 1991; Raman and Trussell, 1995) and activation (Rosenmund et al., 1998; Smith and Howe, 2000; Smith et al., 2000). Accordingly, the models allowed desensitization from closed states, not from open states, and contained multiple open states with different unitary conductances. We also tested models that were similar to the GluR1wt model but conformed to the requirements of classical Monod-Wyman-Changeux (MWC) models (Monod et al., 1965). For either class of model, the inclusion of a constitutive open state (an unoccupied conducting state) had little effect on overall channel kinetics and mostly just increased the size of the leak current.
As discussed in detail previously (Robert and Howe, 2003), the large number of free parameters in the models tested made a statistical approach to obtaining values for the various rate constants unrealistic. Previous studies from our laboratory (Smith et al., 2000) and others (Colquhoun et al., 2003) have attempted maximum likelihood approaches (for models with fewer states than those tested here) and have failed to obtain convergent values for individual rate constants. The approach we took was similar to that described by Robert and Howe (2003), and their values for GluR1wt channel were used as a starting point. The data sets that primarily constrained values for individual rate constants are given in the footnotes to Table 1 and in the last paragraph of Results. Although alterations to multiple sets of rate constants were required to reproduce the entire data set, our requirement that there must be good agreement between the experimental and simulated data for several protocols ensured that the final set of values were well defined within the constraints imposed by any particular kinetic scheme.
Results
Comparison between GluR1wt and GluR1Lc
Figure 1 shows GluR1wt and GluR1Lc responses to a saturating concentration of glutamate. In contrast to GluR1wt receptors, which exhibit rapid and virtually complete desensitization (τdes = 2.2 ± 0.1 msec; plateau/peak current ratio, 0.007 ± 0.001; n = 4), GluR1Lc channels desensitize more slowly and incompletely (τdes = 8.0 ± 0.5 msec; plateau/peak current ratio, 0.65 ± 0.04; n = 6). CTZ, a drug known to remove desensitization for GluR1wt and native channels (Trussell et al., 1993; Wong and Mayer, 1993; Yamada and Tang, 1993), also removes desensitization for GluR1Lc channels (Fig. 1C,D). In addition, GluR1Lc deactivation kinetics are multi-exponential and much slower than GluR1wt. In response to a 1 msec application of glutamate, GluR1wt deactivation is rapid and follows a single exponential time course (τdeac = 0.92 ± 0.02 msec; n = 3) (Fig. 1E), whereas the decay of the GluR1Lc current is bi-exponential (τdeac1 = 1.9 ± 0.2 msec and τdeac2 = 16.3 ± 1.1 msec; n = 6) (Fig. 1F). The decay of the steady-state current on termination of a longer pulse of glutamate also displays multi-exponential components (Fig. 1B; Fig. 4).
Figure 1.

GluR1Lc kinetics are slower than GluR1wt kinetics. A, B, Currents evoked by 5 mm glutamate (100 msec) in outside-out patches containing GluR1wt (A) or GluR1Lc (B) channels. Desensitization of GluR1Lc channels is slower and much less complete than GluR1wt. C, D, Currents evoked in the same patches as in A and B by 5mm glutamate with or without CTZ (100 μm) for GluR1wt (C) and GluR1Lc (D). E, F, Currents evoked by a brief (1 msec) application of 5 mm glutamate to outside-out patches containing GluR1wt (E) or GluR1Lc (F). Deactivation of GluR1wt channels follows a simple exponential time course, whereas the decay of the currents through GluR1Lc channels shows multiple components. Single or double exponential fits to the various decays are superposed on the current traces (solid lines). The fit to the GluR1wt decay gave a time constant of 0.95 msec. The time constants (and relative amplitudes) of the fast and slow components detected in the GluR1Lc decay were 1.2 msec (0.26) and 11.5 msec (0.55). In this and subsequent figures, the current traces are averages of 3-10 consecutive records (unless stated otherwise).
Figure 4.
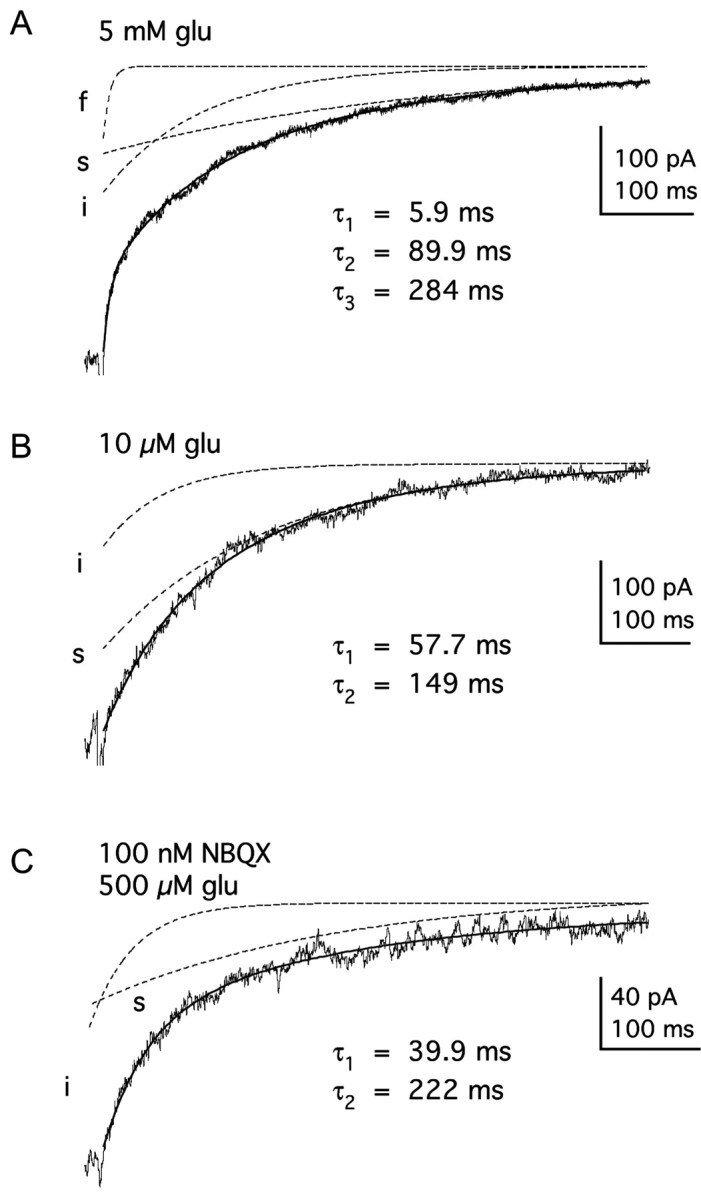
The decay of currents through GluR1Lc channels varies with receptor occupancy. A, Decay of the steady-state plateau current after removal of 5 mm glutamate has three exponential components. The tri-exponential fit to the results (solidline) is superposed on the data trace. The three individual components (fast, intermediate, and slow) had the indicated time constants and are shown separately (dashed lines). B, The decay of the plateau current after the removal of 10 μm glutamate has only two components (intermediate and slow). C, A saturating concentration of glutamate (0.5 mm) was applied after preincubation of the patch in 100 nm NBQX. The decay of the current contained two exponential components (intermediate and slow) that had time constants similar to the two slowest components seen in the absence of NBQX. The bi-exponential fits to the decays in B and C are superposed (solidlines) on the records. The individual components with the indicated time constants are shown are shown separately (dashed lines).
Previous studies have reported large shifts in the apparent affinity of GluR1Lc for glutamate and kainate, based on measurements of steady-state currents in oocytes or in whole-cell recordings (Kohda et al., 2000; Taverna et al., 2000). Because there is an obvious peak current seen with GluR1Lc, we used a rapid application system to measure the glutamate EC50 value for both the peak and plateau currents (Fig. 2A,D). Figure 2, A and B, shows representative examples of the current responses obtained over a range of glutamate concentrations with and without CTZ. In Figure 2D, the mean data from several patches are presented. Hill-type fits to the results for peak and plateau currents gave respective EC50 values of 110 ± 16 and 2.5 ± 0.3 μM (n = 4-9 values per concentration), values that are 6.5-fold and fivefold smaller than the corresponding values for GluR1wt (717 and 12.5 μM) (Robert and Howe, 2003). The glutamate EC50 value in the presence of CTZ (1.96 ± 0.43 μM; n = 3-5 per concentration) was close to the EC50 value for the plateau current with desensitization intact (Fig. 2D). CTZ also had little apparent effect on deactivation. The current decays in the presence of CTZ were multi-exponential (Fig. 2B), and fits to these decays gave time constants similar to those in the absence of CTZ.
Figure 2.
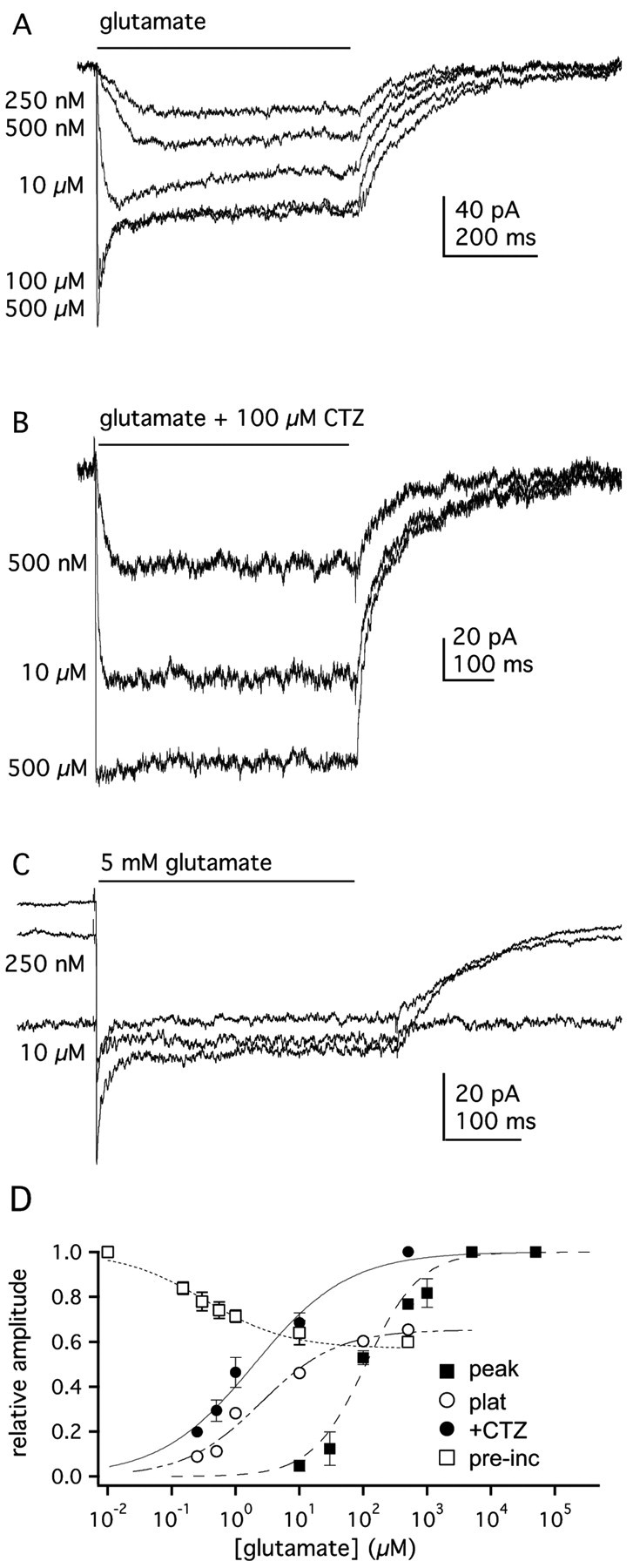
Concentration-response relationships for GluR1Lc channels. A, Currents evoked by applications (bar) of the indicated concentrations of glutamate to an outside-out patch containing GluR1Lc channels. B, GluR1Lc currents evoked in another patch by the indicated concentrations of glutamate in the presence of 100 μm CTZ. C, Responses of GluR1Lc channels in an outside-out patch evoked by preincubation of the patch in the indicated concentration of glutamate, followed by the application of 5 mm glutamate for 500 msec. D, Mean concentration-response data for glutamate-evoked peak (▪) and plateau (○) currents (5-9 patches per concentration). Also shown are concentration-response data for glutamate-evoked currents in the presence of CTZ (•; n = 3 patches) and data for the inhibition of responses to 5 mm glutamate by preexposure of the patches to low concentrations of glutamate (□; n = 4 patches). The EC50 (or IC50) and nH values obtained from Hill-type fits to each data set were: 110 μm and 0.99, peak currents; 2.46 μm and 0.69, plateau currents; 1.96 μm and 0.59, currents in CTZ; 0.33 μm and 0.83, preincubation inhibition. Errors bars represent SEM; some bars in this and subsequent figures are less than one-half the symbol size.
Wild-type and native AMPA-type channels desensitize at concentrations of glutamate that produce little detectable channel activation (Kiskin et al., 1986; Trussell and Fischbach, 1989; Patneau and Mayer, 1991; Colquhoun et al., 1992; Raman and Trussell, 1992). Although CTZ produced a modest potentiation of the responses of GluR1Lc at all glutamate concentrations, the increase was less at low glutamate concentrations. For example, for glutamate concentrations of 250 nm, 10 μM, and 500 μM, the CTZ-associated increases in current (expressed as a fraction of the peak current at saturating glutamate) were 0.11, 0.22, and 0.35, respectively. This result suggests that GluR1Lc channels exhibit little desensitization at low receptor occupancy. To test this more directly, we performed prepulse inhibition experiments. Patches were preexposed to various concentrations of glutamate for 500 msec and then challenged with 5 mm glutamate. As can be seen in Figure 2C, submicromolar concentrations of glutamate increased the holding current and suppressed the peak current in response to saturating glutamate. These experiments gave an IC50 value for glutamate of 0.33 ± 0.06 μM and a maximal inhibition of 40% (n = 4) (Fig. 2D). Thus, in contrast to GluR1wt channels (Robert and Howe, 2003), there are no concentrations of glutamate that produce substantial desensitization without producing substantial activation.
Contribution of GluR1Lc channels to the leak current
Given the conflicting conclusions about the constitutive nature of the lurcher mutation, we sought to quantify the contribution of GluR1Lc channels to the leak current. We measured the amount of leak current at -80 mV under a variety of conditions and normalized those results to the leak current present in our external solution (NaCl). Only patches that gave a discernable response to glutamate and that had on-cell seal resistances of at least 25 GΩ were included in this analysis. Replacement of all the monovalent cations with the impermeant cation NMDG blocked 80% of the leak current (±3.3%; n = 11) (Fig. 3A), confirming that the seal resistance of our patches did not contribute greatly to the total leak current.
Figure 3.
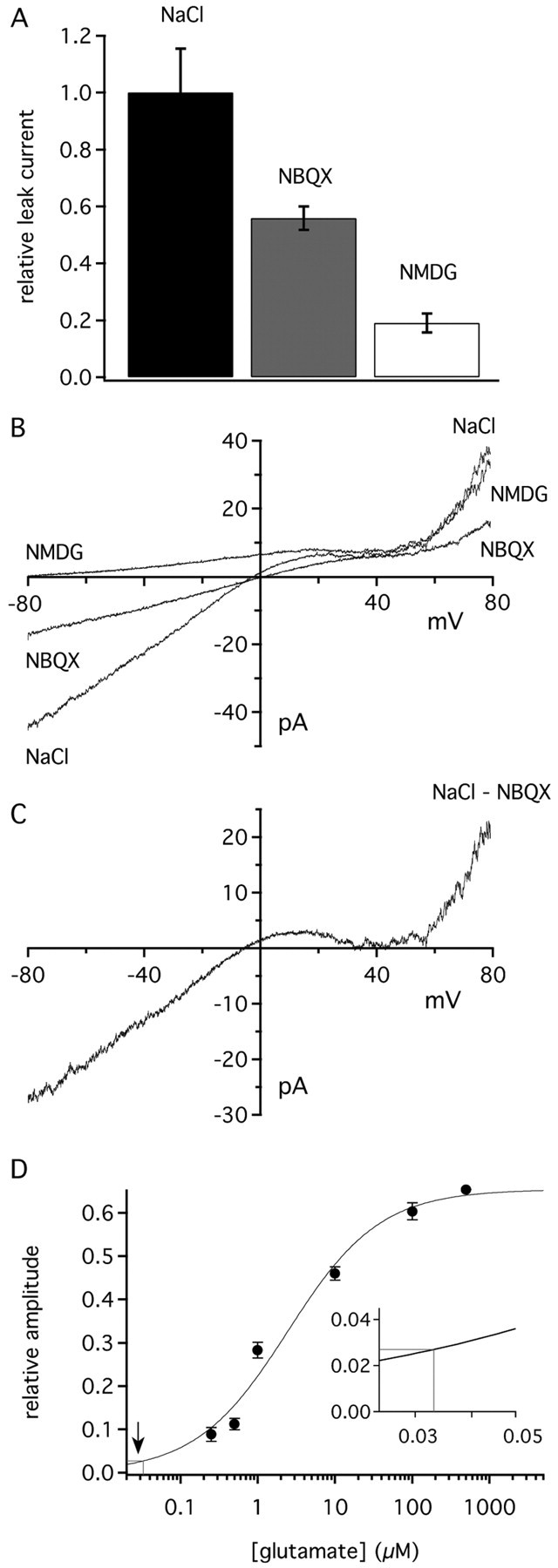
GluR1Lc leak current is reduced by NBQX and NMDG. A, Bar graph of GluR1Lc leak current amplitudes in NMDG and NBQX normalized to the amplitude of the leak current recorded in normal NaCl external solution. Error bars show SEM value (n = 6-11 patches). B, Current-voltage curve for the leak current in a typical GluR1Lc patch measured in normal external solution (NaCl), NaCl external containing 50 μm NBQX, or after replacing NaCl and KCl with NMDG. The curves were obtained from 510 msec voltage ramps. C, Current-voltage relationship for the NBQX-sensitive portion of the leak current obtained by subtracting the NBQX curve in B from the corresponding curve obtained in normal NaCl external. D, Average NBQX-sensitive leak current (solidline) plotted on the glutamate plateau concentration-response curve from Figure 2 D (solid curve). The inset shows an enlargement of the area (arrow) where the amplitude of the NBQX-sensitive leak current maps to the concentration-response curve.
We next measured how much of the leak current was sensitive to the competitive AMPA receptor antagonist NBQX. If the leak current is attributable to activation of GluR1Lc, then NBQX should effectively prevent the channels from gating. Indeed, previous work found that 50 μM NBQX blocked 55% of the leak current in GluR1Lc-transfected cells (Kohda et al., 2000). As shown in Figure 3A, 50 μM NBQX reduced the leak current in GluR1Lc patches by 45% (±4.1%; n = 6). The NBQX-sensitive portion of the leak current (Fig. 3B,C) exhibited the inward rectification typical of unedited GluR1 channels that results from block of the channels at positive potentials by internal polyamines (Bowie et al., 1999). In separate coapplication experiments, in which NBQX and glutamate were simultaneously applied, we confirmed that 50 μM NBQX was a sufficient concentration to inhibit glutamate-evoked responses up to 500 nm glutamate (data not shown). NBQX also had no effect on the leak current of untransfected cells.
For each patch, we also expressed the NBQX-sensitive leak current as a percentage of the plateau current evoked in the same patch by a saturating concentration of glutamate. Comparisons of these percentages with the concentration-response curve for the plateau current (Fig. 2C) indicated that the NBQX-sensitive portion of the leak current corresponded to the current expected, on average, in response to 33 nm glutamate (Fig. 3D). We measured the contaminating glutamate present in our normal external solution with both HPLC and an enzymatic cycling assay. The HPLC measurements gave values of 30-50 nm. The enzymatic assay gave a mean ± SD value of 28 ± 9 nm (n = 12). Taken together, the results are consistent with the idea that the NBQX-sensitive portion of the leak arose primarily from glutamate activation of GluR1Lc rather than constitutive channel activity.
Lurcher kinetics depend on receptor occupancy
For GluR1Lc channels, our CTZ results suggested that desensitization is minimal at low receptor occupancy. In addition, steady-state current deactivation decays contained multiple exponential components. These components might reflect the gating kinetics of channels with different numbers of glutamate molecules bound (Rosenmund et al., 1998; Smith and Howe, 2000). We, therefore, examined further the extent to which desensitization and deactivation of GluR1Lc channels depend on receptor occupancy.
Unlike GluR1wt (Robert and Howe, 2003), the rate of GluR1Lc desensitization was concentration dependent and became faster as the concentration of glutamate was increased from 100 μM to 5 mm (τdes = 12.8 ± 1.1, 10.4 ± 0.6, 9.1 ± 1.6, and 8.1 ± 0.5 msec at 0.1, 0.5, 1, and 5 mm glutamate, respectively; n = 6-15). The desensitization decays for 500 and 100 μM were significantly different from the decays for 5 mm (p < 0.025 and p < 0.005, respectively).
The decay of the steady-state current through GluR1Lc channels also depended on glutamate concentration. At saturating glutamate, three exponential components were consistently distinguished in these decays. Figure 4A shows an example of results obtained with 5 mm glutamate, in which the current decay has been fitted with three exponential components. (τfast = 4.6 ± 0.9 msec, τinter = 46 ± 10 msec, and τslow = 202 ± 25 msec, with the respective relative amplitudes of 0.19 ± 0.04, 0.34 ± 0.03, and 0.45 ± 0.05; n = 6). The fast component exhibited the greatest concentration dependence. The mean time constant of this component increased (from 4.5 to 9.2 msec) as the glutamate concentration was lowered from 5 mm to 10 μM. The fast time constants for 0.01, 0.03, and 0.1 mm were significantly different from the τfast measured for 5 mm glutamate (p < 0.05, 0.025, and 0.01, respectively). At low and intermediate glutamate concentrations (0.1-10 μM), the fast component was often missing (Fig. 4B).
The results of experiments in which patches were preincubated with NBQX also indicated that the decay of the plateau current depended on receptor occupancy. Outside-out patches were preincubated with various concentrations of NBQX and then challenged with 500 μM glutamate for 30 msec (as in Fig. 5D). We scaled the traces to the NBQX-free response and then fit the decays with multi-exponential equations. As we found with glutamate, the number of components, as well as their time constants and relative amplitudes, varied with NBQX concentration. Concentrations of NBQX that reduced the peak current also slowed the decay of the plateau current (Fig. 4C). Preincubation with 100 nm NBQX reduced the currents evoked by saturating glutamate 46 ± 3% (mean ± SD; n = 7), suggesting that at this concentration more than half the channels have two or fewer subunits available for glutamate (see Materials and Methods for details). The steady-state glutamate-evoked currents measured after preincubation with 100 nm NBQX decayed with only two components (τinter = 41 ± 8 msec and τslow = 192 ± 31 msec; n = 6) (Fig. 4C).
DNQX is an agonist for GluR1Lc
The series of quinoxalinediones, CNQX, DNQX, and NBQX, are all competitive antagonists of native and wild-type AMPA receptors (Honore et al., 1988). The structure of the binding domain in complex with DNQX shows that DNQX induces only a small amount of binding domain closure (2.5°, on average), in part because it braces the binding cleft open via interactions between the nitro group on the quinoxaline ring and a threonine residue (T686) that forms a cross-cleft hydrogen bond in agonist-bound structures (Armstrong and Gouaux, 2000). Although structures for CNQX and NBQX are not available, the quinoxalinediones only differ in the size of the substituent at what is the sixth position in CNQX [cyano (CNQX) < nitro (DNQX) < phenylsulphonamide (NBQX)], suggesting that the amount of domain closure might be greatest with CNQX and least with NBQX. This might explain why the lurcher mutation renders CNQX an agonist (Taverna et al., 2000), whereas NBQX remains an antagonist. Previous work did not compare the efficacies of CNQX and glutamate, but the extent of binding domain closure is known to correlate with agonist efficacy (Jin et al., 2003). We, therefore, compared CNQX-, DNQX-, and glutamate-evoked currents.
Both CNQX and DNQX produced detectable currents through GluR1Lc channels (Fig. 5A, B), and concentration-response data gave an EC50 value of 496 nm for CNQX (Fig. 5C). The small size of DNQX-evoked currents limited concentration-response analysis, but its EC50 value was ∼600 nm. When saturating concentrations were compared in the same patches (Fig. 5A,B), the maximum currents evoked by CNQX and DNQX were 24 ± 5.6 and 13 ± 4.2% (n = 3-5), respectively, of the glutamate-evoked responses. NBQX applications never evoked currents from GluR1Lc channels (Fig. 5D), and an IC50 value of 121 ± 0.06 nm was obtained from preincubation experiments (nH = 0.92; n = 9 patches). Thus, the relative order of efficacy of the three quinoxalinediones (CNQX > DNQX > NBQX) does indeed inversely correlate with the bulkiness of the different substituents, a result consistent with other findings that the extent of binding domain closure is directly related to agonist efficacy (Jin et al., 2003).
Unlike glutamate, both CNQX and DNQX produced only plateau currents, with no sign of a peak even at high concentrations. However, the deactivation decays of CNQX-evoked currents were multi-exponential, suggesting that this feature of GluR1Lc channels is agonist independent.
GluR1Lc recovery from desensitization
One clear difference between GluR1wt and GluR1Lc channels is that for GluRLc the steady-state current is a much larger fraction of the peak current evoked at saturating glutamate. Because GluR1wt and GluR1Lc enter desensitization at similar rates, this suggests that the lurcher mutation increases substantially the rate constant for leaving desensitization, which for GluR1wt channels is the primary determinant of the rate at which channels recover from desensitization (Robert and Howe, 2003). If this is also true for GluR1Lc channels, then they should recover from desensitization more rapidly than GluR1wt.
To test whether GluR1Lc channels show accelerated recovery from desensitization, we measured recovery using a two-pulse protocol. A 50 or 100 msec application of glutamate was first made to desensitize the channels, and a paired application was made at increasing time intervals to determine what fraction of the channels had recovered (Fig. 6A). Only patches in which the difference between the peak and plateau currents was at least 50 pA were used in this analysis. Mean data from several patches are presented in Figure 6B. In contrast to GluR1wt, the recovery time course for GluRLc is not sigmoid and is best described by a bi-exponential equation. The bi-exponential fit to the mean data in Figure 6B gave τrecov1 and τrecov2 values of 16.4 and 166 msec, with relative amplitudes for the fast and slow components of 0.21 and 0.73. For comparison, the recovery time course for GluR1wt channels is shown as a dotted line in Figure 6B. As can be seen, at short intervals, the fraction of GluR1Lc channels that have recovered from desensitization is larger than GluR1wt (5 msec, 13 vs 0.5%; 20 msec, 26 vs 5%; 100 msec, 57 vs 41%). Recovery is indeed faster for GluR1Lc channels, especially at short recovery intervals.
Figure 6.

GluR1Lc channels recover faster from desensitization than GluR1wt. A, Selected records from a typical two-pulse protocol to measure recovery from desensitization. After an initial application of 500 μm glutamate (first black bar), a second application was given (second black bar) at varying time intervals (dashed lines). Fractional recovery was measured as the difference between the peak current during the second application and the steady-state current at the end of first application of the pair. B, Mean recovery data from seven patches were fitted with abi-exponential equation (solidline) that gave time constants of 16.4 and 166 msec for the two components. The relative amplitudes of the two components were 0.21 ± 0.07 and 0.73 ± 0.05, respectively. Bars indicate SEM. The dotted curve is the recovery time course for GluR1wt channels measured with 5 mm glutamate (Robert and Howe, 2003).
Modeling of GluR1Lc kinetics
To further characterize the effects of the lurcher mutation in GluR1, we sought to interpret our results in the context of a kinetic model. Although we considered many types of models, they all incorporated the basic scheme depicted in Figure 7. All models tested had five closed states (zero to four glutamates bound) and at least four open states (four concentration-dependent open levels). Channel activation and desensitization were assumed to proceed in parallel from the same closed states (Vyklicky et al., 1991; Raman and Trussell, 1995). Our goal was to find a single set of rate constants that could reproduce the GluR1Lc current at high agonist concentrations, the concentration-response curves for glutamate, the time courses of entry into and recovery from desensitization, the concentration dependence of desensitization, and the multi-exponential decay of the steady-state current. Here, we compare two types of kinetic models that have been proposed for AMPA-type GluRs: a classical MWC model and the model proposed by Robert and Howe (2003).
Figure 7.

Modeling GluR1Lc kinetics. A, Kinetic model used to simulate the results obtained with GluR1Lc. The rate constants used are reported in Table 1. B-D, Simulated current response to 5 mm glutamate (B), concentration- and inhibition-response curves for glutamate (C), and channel recovery from desensitization (D). For C and D, the points represent the mean experimental data, whereas the fits are the simulated data. Fits to the simulated data gave peak, plateau, and +CTZ EC50 values of 159 ± 14.2, 1.5 ± 0.09, and 3.3 ± 0.13 μm, respectively, and Hill coefficients of 0.98, 0.72, and 0.69. The fit to the inhibition data gave an IC50 value of 1.61 ± 0.19 μm with nH = 0.61. The simulated recovery time course was fitted with a bi-exponential equation and gave values ofτ1 = 20 msec,τ2 = 166 msec with relative amplitudes of 0.26 and 0.80. E, Simulated deactivation decay after a 1 msec pulse of 5 mm glutamate. The fits to the decay are indicated on the figure and gave values ofτ1 = 3.0 msec andτ2 = 24 msec. F, Open-state probabilities for the trace shown in E. During the 1 msec pulse, which begins at 10 msec, most channels quickly reach O4. When glutamate is removed, the channels visit O3 to O1 as glutamate sequentially unbinds from individual subunits. The repeated openings to successively smaller conductance levels result in the longer decay component seen in E. G, Cartoon representing three possible conformations of the ligand-binding domains of AMPARs. Glutamate is shown as a grayball. The transitions between the open-bound and closed-bound states are represented by the rate constants ρ and ϵ. H, Energy-state diagrams for GluR1Lc with a single subunit occupied (black dashed trace) and all four subunits occupied (solid black trace). As subunit occupancy increases (arrows), the stability of the open state decreases and the stability of the desensitized state increases. Also shown is an energy-state diagram for the fully occupied GluR1wt channel (solid gray trace). The relative depths of the wells were calculated from the rate constants in Table 1 and Robert and Howe (2003).
Classical MWC models have two distinguishing features: (1) the transition from one set of states to another is concerted (i.e., all receptor subunits adopt identical conformations); and (2) ligand affinity differs for different sets of states. In the kinetic model depicted in Figure 7, an MWC model would require that glutamate binding to open states be allowed and that glutamate bound with higher affinity to at least one set of states (open, closed, or desensitized) than to the others. To maintain microscopic reversibility, substantial differences in affinity between closed and open (or closed and desensitized) states also require correspondingly substantial differences in the ratios of the rate constants for channel opening and closing (β/α) or channel entry into and exit from desensitization (δ/γ) for states with different occupancies.
Invariably, MWC models reproduced the experimental results poorly. For example, models in which the affinity of glutamate binding was highest to open states paradoxically predicted that at low glutamate concentrations most channels are desensitized (a prediction at odds with our data). This is a consequence of the strong dependence of β and α on receptor occupancy that is necessary to satisfy microscopic reversibility, which led to desensitization being much more likely than channel opening (δ >> β) with one or two glutamates bound. If δ and γ were altered to decrease the fraction of desensitized channels, the rate of entry into desensitization became too slow for fully occupied channels. Models in which the affinity of desensitized channels was highest gave open probabilities that were too low. Increases in β that corrected this deficiency, and came close to reproducing the peak/plateau current ratio, gave τdes values that were much larger than those observed. In addition, all MWC models explored predicted that recovery from desensitization was far too slow and incomplete.
We also considered a model proposed recently for wild-type GluR1 and GluR4 receptors (Robert and Howe, 2003). This model is identical to the one shown in Figure 7A (except that the O1 state was not included) and was based on available kinetic and structural data for AMPA-type GluRs. Briefly, AMPA-type receptors have four binding sites per receptor, one per subunit. Channel activation is triggered by closure of the binding domains once ligand is bound and desensitization occurs because of rearrangement of the dimer interface (Sun et al., 2002). For fully occupied desensitized receptors, it was found that two glutamates dissociated rapidly and two slowly. It was proposed that this arose because the binding domain of one subunit in each dimer was stabilized in a closed conformation. In the wild-type model, the time course of recovery is determined by the rate at which subunits with closed binding domains resensitize (γ) and not the rate at which glutamate unbinds (k-1). Another key feature of this model is that the ligand affinity is the same for all states of the receptor, with the exception that unbinding from the D22 and D1 states is presumed to be slow, because it represents glutamate dissociation from a closed binding domain (for further discussion, see Robert and Howe, 2003).
We began the kinetic modeling of GluR1Lc by decreasing wild-type values for k-1 and δ, and increasing γ, which led to a set of rate constants that reproduced well the slower and incomplete desensitization seen with GluR1Lc, as well as the concentration-response curve for steady-state currents. These simple changes, however, failed to reproduce the recovery time course, the decay of the steady-state current, or the concentration dependence of desensitization and the peak current. For GluR1wt, there is little dependence of any of the rate constants on receptor occupancy (Robert and Howe, 2003). However, it soon became clear that the remaining results for GluR1Lc channels could only be reproduced if some of the rate constants varied substantially with occupancy. Introducing negative cooperativity of binding gave multi-exponential steady-state current decays but failed to resolve the other deficiencies, and recovery from desensitization was much too slow. By allowing δ and γ to vary with receptor occupancy, we were able to reproduce both the time course of recovery and the concentration dependence of desensitization. However, changes in k-1 that then gave the correct multi-component decays of the plateau currents resulted in EC50 values for these same currents that were smaller than those measured. In contrast, by letting β and α vary with occupancy, it was possible to reproduce both the decays and all three concentration-response curves (Fig. 7B,C).
The final model for GluR1Lc kinetics is shown in Figure 7A. Simulated results using this model and the rate constants in Table 1 are presented in Figure 7. The simulated response to saturating concentrations of glutamate (Fig. 7B) mimics several characteristics of the GluR1Lc current response. Both the peak and plateau current concentration-response curves are reproduced well by this model, with EC50 values within 50% of the experimental values (Fig. 7C). The recovery predicted by the model also matches the experimental time course (Fig. 7D). Our model also reproduces GluR1Lc deactivation in response to a brief application of glutamate, an experimental result that was not used to define the final model or rate constants (Fig. 7E). The slow deactivation component arises from continued channel opening after glutamate is removed and results from the slow rate of glutamate dissociation for GluR1Lc. During the brief glutamate application, the channels rapidly fill the O4 state; after the removal of glutamate, the channels transit through the other open states (O3-O1) as glutamate sequentially dissociates (Fig. 7F). The model also gave activation rise times over a range of glutamate concentrations that were similar to those recorded experimentally (data not shown). The only experimental data that our model did not reproduce well was the glutamate inhibition-response curve. The simulated IC50 value (1.6 μM) is fivefold larger than our experimental value of 0.33 μM. One possible reason for this discrepancy is that the glutamate IC50 is especially sensitive to the presence of contaminating glutamate in the external solution.
Table 1 gives the fold changes from GluR1wt values for GluR1Lc for each receptor occupancy. The largest differences between the rate constants used for GluR1Lc and those found for GluR1wt are in the rate constants for glutamate dissociation, k-1, and channel desensitization, δ. GluR1Lc kinetics requires a 150-fold decrease in the value for k-1 and 180- to 24-fold decreases in δ. Another rate constant that has changed substantially in our model is γ. For the fully occupied receptor, the value of γ is 15-fold larger than the GluR1wt value.
Discussion
Compared with GluR1wt, GluR1Lc channels have a decreased rate and extent of desensitization. Our kinetic modeling indicates that GluR1Lc channels also exhibit a marked increase in their affinity for glutamate. Although our results do not rule out the possibility that lurcher increases the frequency of unliganded openings, the NBQX-sensitive leak current is a small fraction of the activity evoked by saturating glutamate, and the size of the NBQX-sensitive current suggests it likely arises predominantly from activation of GluR1Lc channels by contaminating glutamate present in the external solution.
GluR1Lc shows reduced desensitization at ambient levels of glutamate
Our results indicate that one main effect of lurcher is to substantially reduce the likelihood that GluR1 channels are desensitized at low receptor occupancies. At levels of glutamate found in vivo (Meldrum, 2000), little activation of native GluRs occurs (Colquhoun et al., 1992; Hausser and Roth, 1997). In contrast, GluR1Lc channels would generate a small, but sustained, current. The resulting influx of cations could trigger apoptotic pathways, some of which have been shown to be activated in lurcher mice (Doughty et al., 2000; Selimi et al., 2000). We conclude that decreased desensitization is the main effect of the lurcher mutation, not the creation of a constitutively active channel as postulated before (Kohda et al., 2000; Taverna et al., 2000; Wollmuth et al., 2000; Schwarz et al., 2001).
Lurcher changes the route of channel recovery
In addition to the large reduction in the desensitization rate constant at low receptor occupancies, changes in the resensitization rate constant at high occupancies were required to reproduce the altered peak/plateau current ratio and the recovery time course seen with lurcher. GluR1wt channels follow a stepwise recovery composed of fast unbinding and slow resensitization steps (Robert and Howe, 2003), and because GluR1wt resensitization is much slower than glutamate dissociation, almost all GluR1wt channels escape desensitization from the singly liganded desensitized state, D1. For GluR1Lc channels, the combination of large values for the resensitization rate constants, together with the much smaller unbinding rate constants, greatly increases the likelihood that channels will escape from desensitization with two to four subunits occupied. In addition, channels that do escape are more likely to reenter desensitization than lose glutamate. As a result, the time course of recovery is a complex function of several rate constants and no longer simply reflects the rate of re-sensitization.
A pore mutation that alters ligand affinity
The modest shifts in EC50 values we find appear at odds with the large shift in the glutamate EC50 value reported previously (Taverna et al., 2000). Some of this discrepancy may be attributable to differences in the two types of expression systems used (outsideout patches from mammalian cells vs oocytes). It is also notable, however, that the EC50 value reported by Taverna et al. (2000) is more than an order of magnitude above other reported values for GluR1wt and related subunits (20-25 μM) (Patneau and Mayer, 1990; Armstrong et al., 2003; Leever et al., 2003).
Our kinetic modeling does indicate, however, that lurcher markedly increases the affinity of the receptors for glutamate. This large change in affinity, absent large changes in apparent potency, is primarily explained by the different desensitization behavior of wild-type and lurcher channels. Because GluR1Lc desensitizes less than GluR1wt, the half-maximal plateau current, and especially the half-maximal peak current, occur at receptor occupancies substantially greater than the corresponding occupancies for wild-type channels. This tends to blunt true differences in affinity when EC50 values are compared. Indeed, when we measure EC50 values for lurcher and wild type in the presence of CTZ, the difference (∼80-fold) more closely parallels the difference in affinity that we estimate from the kinetic simulations.
Why does a mutation in the pore produce such a large difference in the affinity of binding? Although binding domain closure is the conformational change that triggers gating (Armstrong and Gouaux, 2000; Armstrong et al., 2003; Jin et al., 2003; Jin and Gouaux, 2003), in our kinetic scheme (Fig. 7A), cleft closure and opening are ignored. If cleft closure is included (as in Fig. 7G), then for closed states the binding domain of an individual subunit would exist in at least three conformations: open, open-bound, and closed-bound. The transition between the first two conformations corresponds to glutamate binding to an open cleft, and the next transition represents the cleft closure that precedes channel opening or desensitization. Because glutamate can only dissociate at an appreciable rate from an open binding cleft (Armstrong and Gouaux, 2000), the more time spent in the closed-bound conformation, the slower the rate of glutamate dissociation. Given the location of the lurcher mutation, we suggest that what appears in our modeling as a large decrease in k-1 is, in fact, the effect of the mutation to stabilize a state that is inherently unstable in wild-type channels, namely the closed-bound transition state that precedes channel gating.
A comparison of the crystal structure of KcsA (Doyle et al., 1998) with scanning mutagenesis studies indicates that the structure of KcsA is a good model for the GluR pore (Panchenko et al., 2001; Sobolevsky et al., 2003). When mapped directly onto the KcsA structure, the side chain of the lurcher alanine points directly at conserved residues within TM1, and SCAM (substituted cysteine accessibility methods) studies of GluR1 indicate that the lurcher alanine was the only residue in the SYTANLAAF motif inaccessible to modification, suggesting that it never faces the channel vestibule (Sobolevsky et al., 2003). Mutation of the lurcher alanine to threonine may promote formation of a hydrogen bond between the hydroxyl group of threonine and residues within TM1 of the same subunit, thereby strengthening helix-helix interactions. If rearrangement of the helices is required for gating, as recent work suggests (Sobolevsky et al., 2004), this might explain why lurcher decreases the rate constants for channel opening and entry into desensitization.
The effect of lurcher varies with receptor occupancy
For GluR1wt, the steady-state distribution of channels lies toward the desensitized conformation and shows little dependence on receptor occupancy. In contrast, the GluR1Lc distribution lies toward the open state, presumably because lurcher opposes rearrangement of the dimer interface (Sun et al., 2002). In addition, deactivation and desensitization for GluR1Lc resemble wild-type channels more as occupancy increases. As depicted in Figure 7H, the free energy of the desensitized state decreases as receptor occupancy increases (compare solid and dashed black lines), whereas the opposite is true for the open state.
A possible explanation for the concentration dependence of GluR1Lc deactivation and desensitization kinetics may involve interactions within and between receptor dimers that are thought to govern channel activation and desensitization (Mansour et al., 2001; Robert et al., 2001; Bowie and Lange, 2002; Sun et al., 2002; Robert and Howe, 2003). It has been proposed that when glutamate is bound and the channel is closed, there is large strain on the dimer interface, which is relieved either by channel opening or rearrangement of the interface and desensitization (Sun et al., 2002). At low concentrations of glutamate, only one or two subunits are occupied by ligand, and the likelihood that both subunits within a dimer are occupied is low. The strain on the dimer interface may be less for singly than doubly occupied dimers and may be more easily countered by any interactions between the transmembrane helices. At higher glutamate concentrations, doubly occupied dimers are common, and the closed-bound transition state may be inherently less stable.
Our proposed structural explanation for the effect of the lurcher mutation predicts that other residues in TM1, and perhaps TM3, would influence channel activation and desensitization. A systematic mutagenesis study of these regions may yield more information regarding the role of the transmembrane domains in channel function. Although the structural explanation for the effects of lurcher remains to be proven, our results demonstrate that the main effects of lurcher are to greatly reduce desensitization at low glutamate concentrations and greatly slow glutamate dissociation by stabilizing closure of the binding cleft.
Footnotes
This work was supported by a National Science Foundation predoctoral fellowship (R.M.K.) and National Institutes of Health Grant GM58926. We thank Antoine Robert, Fred Sigworth, Reiko Fitzsimonds, and Lise Heginbotham for useful discussions.
Correspondence should be addressed to Dr. J. R. Howe, Department of Pharmacology, Yale University School of Medicine, 333 Cedar Street, New Haven, CT 06520. E-mail: james.howe@yale.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244941-11$15.00/0
References
- Armstrong N, Gouaux E (2000) Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron 28: 165-181. [DOI] [PubMed] [Google Scholar]
- Armstrong N, Mayer M, Gouaux E (2003) Tuning activation of the AMPA-sensitive GluR2 ion channel by genetic adjustment of agonist-induced conformational changes. Proc Natl Acad Sci USA 100: 5736-5741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banke TG, Bowie D, Lee H, Huganir RL, Schousboe A, Traynelis SF (2000) Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J Neurosci 20: 89-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Lange GD (2002) Functional stoichiometry of glutamate receptor desensitization. J Neurosci 22: 3392-3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Bahring R, Mayer ML (1999) Block of AMPA and kainate receptors by polyamines and arthopod toxins. In: Ionotropic glutamate receptors in the CNS (Jonas P, Monyer H, Adelmann G, eds), pp 251-274. Berlin: Springer.
- Colquhoun D, Jonas P, Sakmann B (1992) Action of brief pulses of glutamate on AMPA/kainate receptors in patches from different neurones of rat hippocampal slices. J Physiol (Lond) 458: 261-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Hatton CJ, Hawkes AG (2003) The quality of maximum likelihood estimates of ion channel rate constants. J Physiol (Lond) 547: 699-728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach V, Barria A, Soderling TR (1999) Ca2+/calmodulin-kinase II enhances channel conductance of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc Natl Acad Sci USA 96: 3269-3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty ML, De Jager PL, Korsmeyer SJ, Heintz N (2000) Neurodegeneration in lurcher mice occurs via multiple cell death pathways. J Neurosci 20: 3687-3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R (1998) The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science 280: 69-77. [DOI] [PubMed] [Google Scholar]
- Hausser M, Roth A (1997) Dendritic and somatic glutamate receptor channels in rat cerebellar Purkinje cells. J Physiol 501: 77-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honore T, Davies SN, Drejer J, Fletcher EJ, Jacobsen P, Lodge D, Nielsen FE (1988) Quinoxalinediones: potent comeptitive non-NMDA glutamate receptor antagonists. Science 241: 701-703. [DOI] [PubMed] [Google Scholar]
- Irizarry SN (2001) Agonist-dependent activation and desensitization of recombinant AMPA receptors. PhD thesis, Yale University.
- Jin R, Gouaux E (2003) Probing the function, conformational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: studies of 5-substituted Willardiines and GluR2 S1S2 in the crystal. Biochemistry 42: 5201-5213. [DOI] [PubMed] [Google Scholar]
- Jin R, Banke TG, Mayer ML, Traynelis SF, Gouaux E (2003) Structural basis for partial agonist action at ionotropic glutamate receptors. Nat Neurosci 6: 803-810. [DOI] [PubMed] [Google Scholar]
- Kiskin NI, Krishtal OA, Tsyndrenko A (1986) Excitatory amino acid receptors in hippocampal neurons: kainate fails to desensitize them. Neurosci Lett 63: 225-230. [DOI] [PubMed] [Google Scholar]
- Kohda K, Wang Y, Yuzaki M (2000) Mutation of a glutamate receptor motif reveals its role in gating and delta2 receptor channel properties. Nat Neurosci 3: 315-322. [DOI] [PubMed] [Google Scholar]
- Leever JD, Clark S, Weeks AM, Partin KM (2003) Identification of a site in GluR1 and GluR2 that is important for modulation of deactivation and desensitization. Mol Pharmacol 64: 5-10. [DOI] [PubMed] [Google Scholar]
- Mansour M, Nagarajan N, Nehring RB, Clements JD, Rosenmund C (2001) Heteromeric AMPA receptors assemble with a preferred subunit stoichiometry and spatial arrangement. Neuron 32: 841-853. [DOI] [PubMed] [Google Scholar]
- Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130: 1007S-1015S. [DOI] [PubMed] [Google Scholar]
- Monod J, Wyman J, Changeux J-P (1965) On the nature of allosteric transitions: a plausible model. J Mol Biol 12: 88-118. [DOI] [PubMed] [Google Scholar]
- Panchenko VA, Glasser CR, Mayer ML (2001) Structural similarities between glutamate receptor channels and K(+) channels examined by scanning mutagenesis. J Gen Physiol 117: 345-360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML (1990) Structure-activity relationships for amino acid transmitter candidates acting at N-methyl-D-aspartate and quisqualate receptors. J Neurosci 10: 2385-2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patneau DK, Mayer ML (1991) Kinetic analysis of interactions between kainate and AMPA: evidence for activation of a single receptor in mouse hippocampal neurons. Neuron 6: 785-798. [DOI] [PubMed] [Google Scholar]
- Phillips R (1960) “Lurcher,” new gene in linkage group XI of the house mouse. J Genet 57: 35-42. [Google Scholar]
- Raman IM, Trussell LO (1992) The kinetics of the response to glutamate and kainate in neurons of the avian cochlear nucleus. Neuron 9: 173-186. [DOI] [PubMed] [Google Scholar]
- Raman IM, Trussell LO (1995) The mechanism of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor desensitization after removal of glutamate. Biophys J 68: 137-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Howe JR (2003) How AMPA receptor desensitization depends on receptor occupancy. J Neurosci 23: 847-858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A, Irizarry SN, Hughes TE, Howe JR (2001) Subunit interactions and AMPA receptor desensitization. J Neurosci 21: 5574-5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stern-Bach Y, Stevens CF (1998) The tetrameric structure of a glutamate receptor channel. Science 280: 1596-1599. [DOI] [PubMed] [Google Scholar]
- Schwarz MK, Pawlak V, Osten P, Mack V, Seeburg PH, Kohr G (2001) Dominance of the lurcher mutation in heteromeric kainate and AMPA receptor channels. Eur J Neurosci 14: 861-868. [DOI] [PubMed] [Google Scholar]
- Selimi F, Doughty M, Delhaye-Bouchaud N, Mariani J (2000) Target-related and intrinsic neuronal death in lurcher mutant mice are both mediated by caspase-3 activation. J Neurosci 20: 992-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TC, Howe JR (2000) Concentration-dependent substate behavior of native AMPA receptors. Nat Neurosci 3: 992-997. [DOI] [PubMed] [Google Scholar]
- Smith TC, Wang LY, Howe JR (2000) Heterogeneous conductance levels of native AMPA receptors. J Neurosci 20: 2073-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Yelshansky MV, Wollmuth LP (2003) Different gating mechanisms in glutamate receptor and K+ channels. J Neurosci 23: 7559-7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky AI, Yelshansky MV, Wollmuth LP (2004) The outer pore of the glutamate receptor channel has 2-fold rotational symmetry. Neuron 41: 367-378. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E (2002) Mechanism of glutamate receptor desensitization. Nature 417: 245-253. [DOI] [PubMed] [Google Scholar]
- Taverna F, Xiong ZG, Brandes L, Roder JC, Salter MW, MacDonald JF (2000) The lurcher mutation of an alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit enhances potency of glutamate and converts an antagonist to an agonist. J Biol Chem 275: 8475-8479. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Fischbach GD (1989) Glutamate receptor desensitization and its role in synaptic transmission. Neuron 3: 209-218. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM (1993) Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron 10: 1185-1196. [DOI] [PubMed] [Google Scholar]
- Vyklicky Jr L, Patneau DK, Mayer ML (1991) Modulation of excitatory synaptic transmission by drugs that reduce desensitization at AMPA/kainate receptors. Neuron 7: 971-984. [DOI] [PubMed] [Google Scholar]
- Williams K, Dattilo M, Sabado TN, Kashiwagi K, Igarashi K (2003) Pharmacology of delta2 glutamate receptors: effects of pentamidine and protons. J Pharmacol Exp Ther 305: 740-748. [DOI] [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Jatzke C, Seeburg PH, Heintz N, Zuo J (2000) The lurcher mutation identifies delta 2 as an AMPA/kainate receptor-like channel that is potentiated by Ca(2+). J Neurosci 20: 5973-5980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LA, Mayer ML (1993) Differential modulation by cyclothiazide and concanavalin A of desensitization at native alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid- and kainate-preferring glutamate receptors. Mol Pharmacol 44: 504-510. [PubMed] [Google Scholar]
- Yamada KA, Tang CM (1993) Benzothiadiazides inhibit rapid glutamate receptor desensitization and enhance glutamatergic synaptic currents. J Neurosci 13: 3904-3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N (1997) Neurodegeneration in lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature 388: 769-773. [DOI] [PubMed] [Google Scholar]