

Abstract
During the development of neuronal circuits, axonal growth cones can contact many inappropriate targets before they reach an appropriate postsynaptic partner. Although it is well known that the contact with synaptic partners upregulates the secretory machinery of the presynaptic neuron, little is known about the signaling mechanisms involved in preventing the formation of connections with inappropriate target cells. Here, we show that the contact with a nonphysiological postsynaptic target inhibits neurotransmitter release from axonal terminals of the Helix serotonergic neuron C1 by means of an active mechanism requiring ongoing protein synthesis and leading to the inhibition of cAMP-dependent protein kinase (PKA) and mitogen-activated protein kinase (MAPK)-extracellular signal-related kinase (Erk) pathways. The reversal of the inhibitory effect of the nonphysiological target by blockade of protein synthesis was prevented by cAMP-PKA or MAPK-Erk inhibitors, whereas disinhibition of neurotransmitter release promoted by cAMP-PKA activation was not affected by MAPK-Erk inhibitors. The data indicate that the inhibitory effect of the nonphysiological target on neurotransmitter release is an active process that requires protein synthesis and involves the downregulation of the MAPK-Erk and cAMP-PKA pathways, the same protein kinases that are activated after contact with a physiological target neuron. These mechanisms could play a relevant role in the prevention of synapse formation between inappropriate partners by modulating the neurotransmitter release capability of growing nerve terminals according to the nature of the targets contacted during their development.
Keywords: target selection, exocytosis, protein synthesis, protein kinases, invertebrate neurons, Helix
Introduction
During axon elongation, growth cones can contact inappropriate targets before reaching their physiological postsynaptic partner(s) and establishing synaptic connections. It is well known that contact with appropriate targets induces the morphological and functional maturation of presynaptic terminals by means of retrograde signals, originating from contact-mediated interactions or from the release of diffusible molecules such as trophic factors and neurotransmitters (Hall and Sanes, 1993; Dan and Poo, 1994; Fitzsimonds and Poo, 1998; Munno and Syed, 2003), activating specific signal transduction pathways in the presynaptic neurons including cAMP-dependent protein kinase (PKA), mitogen-activated protein kinases (MAPKs), and tyrosine kinases (Dai and Peng, 1993; Funte and Haydon, 1993; Zoran et al., 1993; Hamakawa et al., 1999).
We have previously studied the modulation exerted by the target neuron on the development of the presynaptic cell by using as an experimental model the serotonergic neuron C1 of the land snail Helix cocultured in contact with a physiological or nonphysiological target (Cibelli et al., 1996; Ghirardi et al., 2000, 2001). In the presence of its physiological target B2, the neuron C1 presented a large number of varicosities containing neurotransmitter and synaptic vesicle proteins and formed chemical synaptic connections. On the contrary, the contact with a nonphysiological target such as the motoneuron C3 induced a dramatic reduction in the number of varicosities, an alteration in the distribution of synaptic vesicle proteins, and a decrease in evoked neurotransmitter release even with respect to a C1 neuron cultured in isolation. The inhibitory effect of the nonphysiological target on neurotransmitter release was reversed by a few minutes of contact with the physiological target B2, which induced an increase of neurotransmitter release associated with the formation of new varicosities. This buildup of transmitter release triggered by the physiological target was prevented by preincubation of the target neuron with an antibody against the Aplysia cell adhesion molecule (apCAM) (Keller and Schacher, 1990).
Although protein synthesis and the activation of several signal transduction pathways are known to be involved both in synaptogenesis (Feng et al., 1997; Hamakawa et al., 1999) and in many forms of long-term synaptic plasticity (Montarolo et al., 1986,1988; Frey et al., 1993), their role in preventing the formation of inappropriate synaptic connections during development and regeneration still has to be elucidated in detail.
To better understand the molecular mechanisms involved in the inhibition of synapse formation between inappropriate partners, we analyzed the role of protein synthesis as well as of the cAMP-PKA, MAPK-extracellular signal-related kinase (Erk), and tyrosine kinase pathways in the inhibitory effects exerted by the contact with a nonphysiological target on neurotransmitter release from the terminals of the Helix neuron C1 in culture. Here, we show that the contact with a nonphysiological target cell inhibits neurotransmitter release from developing terminals by means of an active mechanism requiring ongoing protein synthesis and leading to the inhibition of the same protein kinases that mediate the enhancement of neurotransmitter release triggered by contact with a physiological target.
Materials and Methods
Materials. Cell-labeling grade [35S]methionine (specific activity >1000 Ci/mmol) was from Amersham Biosciences (Milan, Italy). Nitrocellulose membranes (pore size 0.22 μm) were from Schleicher and Schuell (Milan, Italy). The following antibodies were used: rabbit polyclonal antibody against MAPK-Erk-2 (C-14) (Santa Cruz Biotechnology, Santa Cruz, CA); mouse monoclonal anti-phosphotyrosine antibody (Upstate Biotechnology, Lake Placid, NY); polyclonal antibody against c-Jun NH2-terminal kinase (Jnk-1/2) (New England Biolabs, Beverly, MA); phosphorylation state-specific polyclonal antibody against the active form (P-Thr183/P-Tyr185) of MAPK-Erk-1/2 and against the active form (P-Thr183/P-Tyr185) of Jnk-1/2 (Promega, Madison, WI); and horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies were (Bio-Rad, Milan, Italy). The chemiluminescence detection system (Renaissance Western Blot Chemiluminescence Reagent Plus) was from New England Nuclear (Brussels, Belgium). Culture reagents, protein synthesis inhibitors, adenosine-3′,5′-monophosphorothioate, Sp isomer (Sp-cAMPS), adenosine-3′5′-monophosphorothioate, Rp isomer (Rp-cAMPS), forskolin, 3-isobutyl-1-methylxanthine (IBMX), genistein, lavendustin, and U0126 were from Sigma (Milan, Italy). 2-(2-Amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98059) was obtained from Promega, and TGF-β1fromR&D Systems (Minneapolis, MN). All other chemicals, obtained from standard commercial suppliers, were of reagent grade.
Cell culture. All experiments were performed on identified cultured neurons of Helix pomatia. The cell culture technique of Helix neurons has been described previously by Ghirardi et al. (1996). Briefly, after protease treatment (10 mg/ml protease IX), neurons C1 and C3 from the cerebral ganglion and B2 from the buccal ganglion of juvenile animals were individually dissociated by using a thin glass micropipette and plated in dishes coated with poly-l-lysine (0.5 mg/ml in 0.1 m sodium tetraborate, pH 8.2) as a substrate and preincubated in Aplysia hemolymph for 24 hr. The cells were cultured for 3-4 d in L15 Leibovitz medium (with appropriate salt concentrations) at 18°C. Under this condition, neurons adhere to the substrate and extend new neurites. We prepared cultures containing isolated C1 neurons as well as cocultures of the C1 neuron in contact with either its physiological target B2 (C1B2) or the nonphysiological target C3 (C1C3) with which it does not form electrical or chemical connections in vivo.
Electrophysiology and detection of neurotransmitter release. The synaptic connection between C1 and B2 has been tested electrophysiologically by intracellular recording of the postsynaptic B2 neuron depolarization (hyperpolarized to -80 mV) after an intracellular stimulation of C1 to fire action potentials at 10 Hz for 10 sec by using glass micropipettes filled with 2.5 m KCl (10-15 MΩ) connected to an Axoclamp preamplifier in bridge mode. The resting membrane potential of the C1 and B2 neurons was between -45 and -50 mV, and neurons having membrane potential that changed >5 mV from the beginning to the end of the recording were not included in the study. All of the experiments were performed at room temperature. The secretory properties of the serotonergic C1 terminals were analyzed by adding a freshly dissociated sniffer neuron B2 to the 3- to 4-d-old C1C3 cocultures. The sniffer was impaled with a glass microelectrode for intracellular recording and micromanipulated in close contact with the neurites of C1 and left there throughout the experiment. Serotonin release was evoked by electrical stimulation of C1 (10 Hz for 10 sec) every 5 min and measured as the extent of depolarization recorded in the B2 sniffer. Because the basal release was very low when C1 was cocultured in the presence of the wrong target C3, the high-frequency stimulation was necessary to evoke a detectable depolarization in the B2 sniffer. Moreover, in the intact animal, C1 neurons are known to discharge at high frequencies in response to a chemical stimulation of the snail lips (Balaban, 2002).
Pharmacological treatments. To block protein synthesis, cultures were treated with either 10 μm anisomycin for 2 hr or 0.6 μm cycloheximide for 5 hr. Cells were treated with the cAMP competitive antagonist Rp-cAMPS (500 μm) for 30 min to inhibit the PKA pathway, with genistein (100 μm) or lavendustin (10 μm) for 2 hr to inhibit tyrosine kinases, or with U0126 or PD98059 (50 μm) for 2 hr to block the MAPK-Erk pathway. To stimulate the PKA pathway, cultured neurons were treated either with the cAMP analog Sp-cAMPS (500 μm) plus IBMX (500 μm) or with forskolin (50 μm) for 30 min. Stimulation of the MAPK-Erk pathway was performed by treating the cells with 10 ng/ml TGF-β1 for 5-15 min. The ability of genistein to inhibit tyrosine phosphorylation in Helix neurons was assessed by incubating dissociated neurons in the absence or presence of 100 μm genistein for 2 hr and then testing the tyrosine phosphorylation state of the extracted protein samples by immunoblotting with anti-phosphotyrosine antibodies (data not shown).
In vivo protein labeling of cultured Helix neurons with [35S]methionine. Neurons from the Helix cerebral ganglion were dissected out and plated at a cell density of 50-60 cells per dish in L15 medium 4 d before metabolic labeling. Dishes (six per experimental group) were subjected to the following sequential incubations: (1) complete L15 medium for 1 hr followed by methionine-free L15 medium containing 0.1 mCi per dish [35S]methionine for 2 hr (control group); (2) complete L15 medium containing 10 μm anisomycin for 1 hr followed by methionine-free L15 medium containing 10 μm anisomycin and 0.1 mCi per dish [35S]methionine for 2 hr (anisomycin-treated group); (3) complete L15 medium containing 10 μm anisomycin for 3 hr followed by complete L15 medium for 1 hr and by methionine-free L15 medium containing 0.1 mCi per dish [35S]methionine for 2 hr (anisomycin recovery group). All incubations were performed at room temperature. After metabolic labeling, cells were washed thoroughly with saline and solubilized in 1% SDS and boiled for 2 min. Samples containing equal amounts of total protein were subjected to SDS-PAGE (Laemmli, 1970) on 9% polyacrylamide resolving gels. Proteins separated on the gels were electrophoretically transferred to nitrocellulose membranes (Towbin et al., 1979) and autoradiographed on Kodak X-Omat films. Labeled protein bands in the various molecular weight ranges were excised from the membranes and counted by liquid scintillation spectrometry. The metabolic labeling experiments were repeated three times with different culture preparations and [35S]methionine batches.
Immunoblotting assays. Freshly dissected neurons from the Helix cerebral ganglia were treated with various drugs at room temperature according to the experimental protocol. After the treatment, neurons were washed in L15 medium and homogenized by sonication (XL-2020 sonifier, power 4, 10 pulses; Misonix Inc., Farmingdale, NY) in boiling 1% SDS/1 mm sodium orthovanadate/2 mm sodium fluoride/2 mm sodium pyrophosphate. Aliquots of the solubilized samples were taken for total protein determination, and the rest was boiled for 2 min after addition of concentrated Laemmli sample buffer. Samples containing equal amounts of total proteins (15-30 μg) were subjected to SDS-PAGE and transferred to nitrocellulose membranes as described above. Immunoblotting was performed as described previously (Tancredi et al., 2000). Membranes were blocked in Tris-buffered saline (50 mm Tris-Cl, 200 mm NaCl, pH 7.4)/5% (w/w) nonfat dry milk or 5% BSA (in the case of phospho-specific antibodies) and incubated overnight at 4°C with the following concentrations of primary antibodies: anti-total MAPK-Erk2, 0.05 μg/ml; anti-(P-Thr183/P-Tyr185)MAPK-Erk-1/2, 0.025 μg/ml; anti-total Jnk-1/2, 1:1000; anti-(P-Thr183/P-Tyr185) Jnk-1/2, 1:1000. After wash and incubation with horseradish peroxidase-conjugated goat anti-rabbit secondary antibodies (1:3000-1:10,000 dilution), immunoreactivity was detected using the chemiluminescence system and autoradiography. Before the immunoblots were performed with phosphorylation state-specific antibodies, the total kinase immunoreactivity was quantified using a brain homogenate standard curve as a reference system, and the samples were normalized to contain the same amounts of total kinases.
Other procedures. Protein concentrations were determined using the BCA assay (Pierce, Rockford, IL) with bovine serum albumin as standard. The results are expressed as means ± SEM. Statistic significance was determined using the one-way or two-way ANOVA followed by the Newman-Keuls or the Dunnett multiple comparison test.
Results
Anisomycin blocks protein synthesis in Helix neurons
In other invertebrates such as Aplysia and Lymnaea, anisomycin has been effectively used to reversibly block protein synthesis (Montarolo et al., 1986; Feng et al., 1997). To test whether protein synthesis is required for the target-dependent effects on the presynaptic neuron, we first checked the ability of an anisomycin treatment to block protein synthesis in Helix neurons (Fig. 1A,B). Cultured Helix neurons were metabolically labeled with [35S]methionine in the absence or presence of anisomycin and after washout of the inhibitor (recovery). An almost total blockade of [35S]methionine incorporation was obtained with 2 hr incubation in 10 μm anisomycin (3.3 ± 0.5% with respect to control; p < 0.01) for all molecular weight classes of proteins as compared with control. The inhibition was promptly reversible, and a significant recovery in [35S]methionine incorporation, ranging from approximately +60 to +300% in the various molecular weight classes of proteins, was observed after anisomycin withdrawal (mean total value 177 ± 12% with respect to control; p < 0.01). Treatment with high doses of anisomycin has been reported to induce cellular stress and affect cellular MAPK pathways by activating the stress-activated protein kinase Jnk-1/2 (Torocsik and Szeberenyi, 2000). We then tested, at the dose used to block protein synthesis in cultured Helix neurons, whether anisomycin affected the phosphorylation state-activation of MAPK-Erk and Jnk. Immunoblotting of control and anisomycin-treated Helix ganglion neurons with phosphorylation state-specific antibodies (Fig. 1C) revealed that the treatment with anisomycin was not associated with changes in the activation of either MAPK-Erk or Jnk.
Figure1.

Anisomycin completely blocks protein synthesis in Helix neurons without affecting MAPK-Erk and Jnk pathways. A, Metabolic labeling of cultured neurons with [35S]methionine was performed under control conditions (C), after 2 hr of anisomycin treatment (AM), or 1 hr after AM withdrawal [recovery (R)]. Labeled proteins were separated by SDS-PAGE and detected by autoradiography. Molecular mass markers are shown on the left in kilodaltons. B, The total amount of 35S radioactivity incorporated into SDS-PAGE-resolved proteins is reported as percentage of the respective value of the control group;*p<0.01 versus control group (n=6); Dunnett's multiple comparison test. C, The immunoblotting of extracts of Helix ganglion neurons incubated for 2 hr in the absence (C) or presence (AM) of anisomycin with phosphorylation state-specific antibodies against MAPK-Erk-1/2 and Jnk-1/2 reveals that the low dose of anisomycin used in the experimental protocol does not affect the state of activation of the Erk and Jnk pathways.
Protein synthesis is required for the inhibitory effect of the nonphysiological target
We have shown previously that the presence of the nonphysiological target C3 exerts a significant inhibitory effect on neurotransmitter release from the terminals of the Helix neuron C1 (Ghirardi et al., 2000, 2001). Immediately after the positioning of the B2 sniffer neuron in contact with the neurites of C1 (Fig. 2A), the mean depolarization recorded in the sniffer B2 evoked by C1 stimulation was ∼12.2 ± 1.55 mV (n = 15) in the case of stimulation of isolated C1 neurons and ∼14.1 ± 2.3 mV (n = 12) in the C1B2 cocultures (Fig. 2B,C). Such depolarization was dramatically reduced when C1 was cocultured with the nonphysiological target C3 to 1.82 ± 0.2 mV (n = 19; p < 0.001 vs the two former conditions). To investigate whether the inhibition of transmitter release attributable to the nonphysiological target was dependent on ongoing protein synthesis, the C1C3 cocultures were preincubated in 10 μm anisomycin for 2 hr, a treatment that had no significant effect on the neurosecretory properties of the C1 neuron cultured in isolation or in contact with the physiological target B2 (data not shown). Interestingly, we found that the depolarization evoked in the B2 sniffer by stimulation of C1 neurons contacting the nonphysiological target C3 was significantly larger after anisomycin treatment (14.9 mV ± 2.4; n = 15; p < 0.001) as compared with the untreated C1C3 condition. Similar results were obtained after 5 hr of incubation with cycloheximide, another protein synthesis inhibitor (12.8 ± 0.7 mV; n = 6; p < 0.001) (Fig. 2C). These values were similar to those recorded in the isolated C1 neurons or in the C1B2 cocultures in either the presence or absence of protein synthesis.
Figure 2.
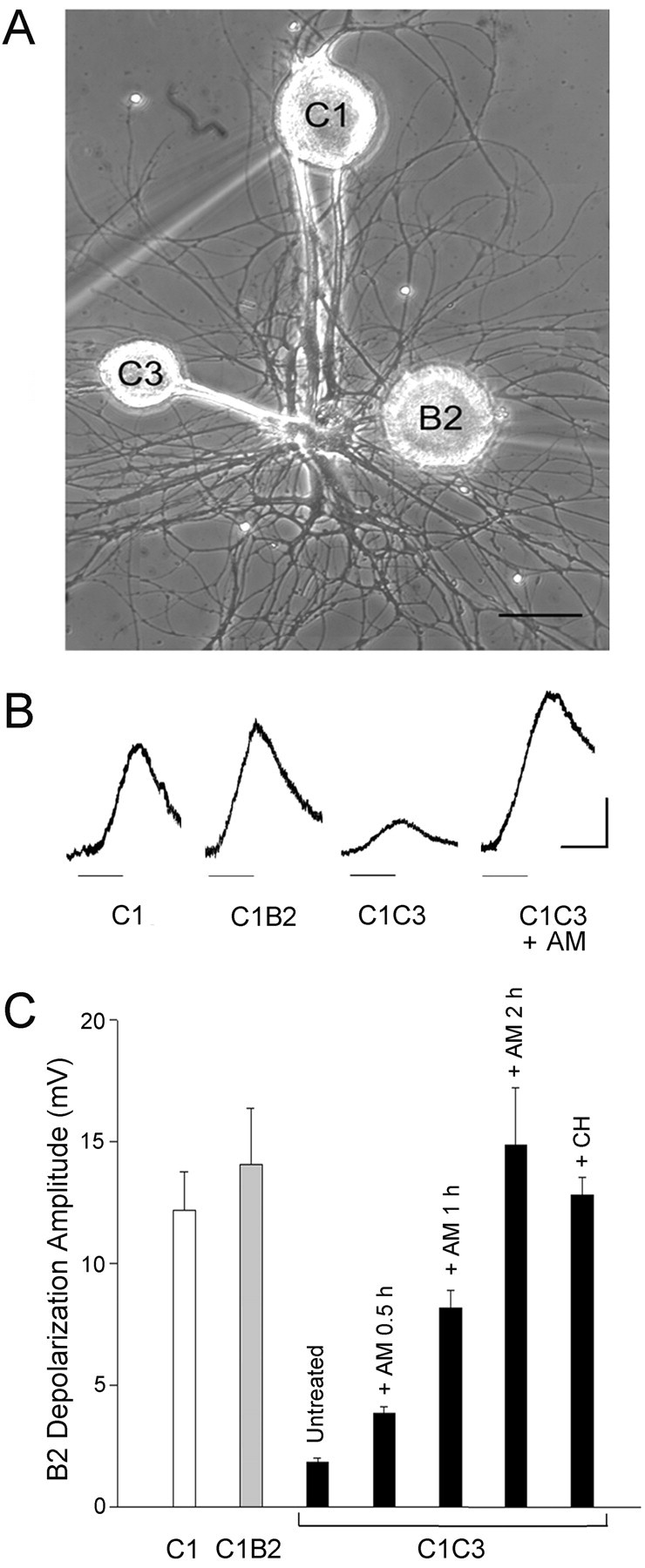
The inhibitory effect of the nonphysiological target requires protein synthesis. A, Phase-contrast micrograph of C1 and C3 Helix neurons in culture with the freshly dissociated sniffer neuron B2. The neurons C1 and B2 were impaled with microelectrodes for intracellular current-clamp recording, and the sniffer cell B2 was micromanipulated in contact with the axonal terminals of C1 at the time of recording. Scale bar, 100 μm. B, Representative traces of the depolarization recorded intracellularly in the sniffer B2 after C1 stimulation (10 sec at 10 Hz; horizontal bar) in C1, C1B2, and C1C3 cultures incubated in the absence or presence of anisomycin for 2 hr (AM). The B2 neuron was hyperpolarized to -80 mV. Calibration bars: 5 mV, 10 sec. C, Anisomycin and cycloheximide increased the depolarization recorded in the sniffer B2 after C1 stimulation in the presence of the nonphysiological target. The height of each bar represents the mean depolarization amplitude (±SEM) evoked by C1 stimulation and recorded in the B2 sniffer immediately after its contact with the neurites of the C1 neuron. The cultures containing the C1 neuron cultured alone (C1) or in the presence of the physiological target B2 (C1B2) or the nonphysiological target C3 (C1C3) were incubated in the absence or presence of anisomycin (AM) (10 μm for 0.5, 1, or 2 hr) or cycloheximide (CH) (0.6 μm for 5 hr). Statistical analysis indicates significant differences between the untreated C1C3 group and the other conditions (ANOVA: F(6,70) = 10.19, p < 0.0001; Newman-Keuls multiple comparison test: p < 0.01).
A significant, although less marked increase of neurotransmitter release after protein synthesis inhibition was also observed with shorter incubation times with inhibitors. The mean sniffer depolarization recorded after 30 and 60 min incubation with 10 μm anisomycin was 4.08 ± 0.49 mV (n = 6) and 8.25 ± 1.65 mV (n = 4), respectively. These increases represent ∼27 and 55% of the depolarization recorded in the sniffer B2 after C1 stimulation in the C1C3 coculture incubated with anisomycin for 2 hr (Fig. 2C). These data suggest that the inhibitory effect of the nonphysiological target on neurotransmitter release is rapidly reversible and is attributable primarily to mechanisms that require ongoing protein synthesis.
Role of the cAMP-PKA system in the modulation of neurotransmitter release in the presence of a nonphysiological target
Because several signal transduction pathways linked to PKA, MAPK, and tyrosine kinase activation have been implicated in the regulation of neuronal growth and synaptic plasticity (Bixby and Harris, 1991; Schmid et al., 1999; Purcell et al., 2003), we next analyzed whether these pathways are also implicated in the inhibition of neurotransmitter release induced by contact with a nonphysiological target. Incubation of C1C3 cocultures with the cAMP analog Sp-cAMPS (500 μm) in the presence of the phosphodiesterase inhibitor IBMX (500 μm) induced an approximately ninefold increase of the sniffer depolarization evoked by C1 stimulation (18.2 ± 2.5 mV; n = 17) as compared with untreated C1C3 cocultures (2.0 ± 0.2 mV; n = 20; p < 0.001) (Fig. 3A). A similar enhancement was observed after incubation with the adenylate cyclase activator forskolin (50 μm) (12.6 ± 1.1 mV; n = 11). Such values were similar to those measured after stimulation of C1 neurons either isolated (12.19 ± 1.11 mV; n = 26) or in C1C3 cocultures treated with anisomycin (10.65 ± 0.56 mV; n = 10). This indicates that the wrong target-dependent inhibition of neurotransmitter release results from a downregulation of the cAMP-PKA cascade that requires ongoing protein synthesis. This hypothesis was further confirmed by the observation that application of the cAMP competitive antagonist Rp-cAMPS (500 μm) to the C1C3 coculture did not affect neurotransmitter release of the C1 neuron in the presence of ongoing protein synthesis (mean sniffer depolarization 1.83 ± 0.22 mV; n = 9) but was able to completely abolish the stimulatory effect of anisomycin (2.3 ± 0.3 mV; n = 13) (Fig. 3A).
Figure 3.
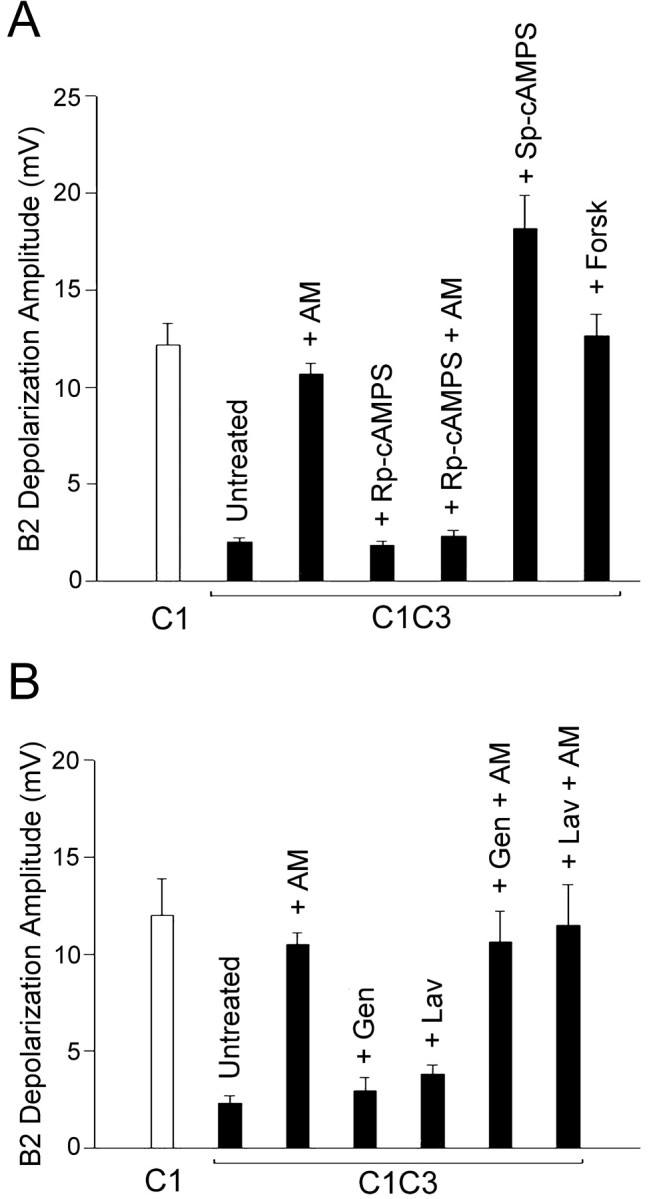
Involvement of the cAMP-PKA pathway, but not of tyrosine kinases, in the nonphysiological target-dependent inhibition of neurotransmitter release. A, Effects of application of the cAMP-PKA activators Sp-cAMPS and forskolin (Forsk) and of the PKA kinase inhibitor Rp-cAMPS on the evoked depolarization of the sniffer B2 after C1 stimulation in C1C3 cocultures incubated in the presence or absence of anisomycin (AM). The depolarization recorded in the B2 sniffer after the stimulation of the isolated, untreated C1 neuron is shown for comparison (open bar). Statistical analysis indicates a significant increase in neurotransmitter release after treatment with AM, Sp-cAMPS, or forskolin (ANOVA: F(6,99) = 23.08, p < 0.001; Newman-Keuls multiple comparison test: p < 0.05). B, C1C3 cocultures incubated in the absence or presence of anisomycin were treated with the tyrosine kinase inhibitors genistein (Gen) or lavendustin (Lav) before recording the C1 stimulation-induced depolarization in the sniffer B2. The data in the plot indicate that tyrosine kinase inhibition does not affect either the decrease of neurotransmitter release induced by the nonphysiological target or the rescuing effect of anisomycin (ANOVA: F(6,42) = 16.15, p < 0.001; Newman-Keuls multiple comparison test: p < 0.05) between the untreated C1C3 and C1C3 treated with AM, AM plus genistein or AM plus lavendustin.
Role of tyrosine kinases and MAPK-Erk systems in the modulation of neurotransmitter release in the presence of a nonphysiological target
To investigate the potential role of tyrosine kinases in the inhibition of neurotransmitter release induced by the wrong target, we incubated the C1C3 cocultures with the tyrosine kinase inhibitors genistein (100 μm) or lavendustin (10 μm), which have been previously demonstrated to be effective in Aplysia, Lymnaea, and Helix neurons (Hamakawa et al., 1999; Grinkevich et al., 2003; Purcell et al., 2003). We found that tyrosine kinase inhibition did not significantly affect neurotransmitter release from the C1 neuron cultured in contact with the wrong target C3. The depolarization recorded in the B2 sniffer after C1 stimulation was 2.96 ± 0.81 mV (n = 7) and 3.81 ± 0.47 mV (n = 13) after genistein and lavendustin treatment, respectively. These values were closely similar to those recorded in untreated C1C3 cocultures (2.33 ± 0.36 mV; n = 6) and significantly different from those obtained after stimulation of the isolated C1 neuron (12 ± 1.89 mV; n = 6; p < 0.05). Moreover, tyrosine kinase inhibition did not affect the disinhibition of neurotransmitter release performed by anisomycin in C1C3 cocultures. In the presence of anisomycin, the depolarization recorded in the sniffer B2 after C1 stimulation was 10.67 ± 1.59 mV (n = 6) and 11.6 ± 2.01 mV (n = 5) for genistein and lavendustin treatment, respectively. These values were similar to those recorded in control experiments in which C1C3 cocultures were treated with anisomycin in the absence of tyrosine kinase inhibitors (10.5 ± 1.52; n = 6) (Fig. 3B). These results suggest that tyrosine kinases are not directly involved in the inhibitory effect of the nonphysiological target on neurotransmitter release.
On the contrary, the stimulatory effect of anisomycin on neurotransmitter release from the C1 neurons contacting the nonphysiological target was virtually abolished when the MAPK-Erk pathway was inhibited by 50 μm of the MAP kinase kinase (MEK) inhibitor U0126 or PD98059. The depolarization of the sniffer B2 evoked by the stimulation of C1 in anisomycin-treated C1C3 cocultures was ∼12.14 ± 1.52 mV (n = 11; p < 0.01) compared with the value of 1.92 ± 0.27 mV (n = 20) recorded in the untreated C1C3 cocultures (Fig. 4A), whereas it was reduced to 2.3 ± 0.5 mV (n = 12; p < 0.01) after U0126 treatment and to 2.06 ± 0.3 mV (n = 8; p < 0.05) after PD98059 treatment. Such inhibitors were completely ineffective on the basal release of C1C3 cocultures in the absence of anisomycin (1.65 ± 0.28 mV, n = 6, and 1.91 ± 0.47 mV, n = 6, for U0126 and PD98059 treatments, respectively). Conversely, incubation of C1C3 cocultures with TGF-β1 (10 ng/ml), a MAPK-Erk activator, induced a fivefold increase in the depolarization recorded in the sniffer B2 after C1 stimulation (11.5 ± 1.5 mV; n = 17) with respect to untreated C1C3 cocultures (p < 0.001). The effect of TGF-β1 was blocked by treatment with the MAPK inhibitor U0126, which significantly reduced the depolarization of the sniffer B2 evoked by C1 stimulation to 3.3 ± 0.4 mV (n = 6; p < 0.05), a value similar to that recorded in untreated C1C3 cocultures (Fig. 4A).
Figure4.

Involvement of the MAPK-Erk pathway in the nonphysiological target-dependent inhibition of neurotransmitter release. A, Effects of the application of the MAPK-Erk activator TGF-β1 and the MEK inhibitors PD98059 and U0126 on the sniffer B2 depolarization after C1 stimulation in C1C3 cocultures incubated in the presence or absence of anisomycin (AM). The depolarization recorded in the sniffer B2 after the stimulation of the isolated, untreated C1 neuron is shown for comparison (open bar). Statistical analysis indicates a significant increase in neurotransmitter release after treatment with AM, U0126 plus Sp-cAMPS, or TGF-β1 (ANOVA: F(10,110) = 19.12, p < 0.001; Newman-Keuls multiple comparison test: p < 0.05). B, Representative immunoblots of extracts of Helix ganglion neurons incubated with TGF-β1 or with U0126 with phosphorylation state-specific antibodies against MAPK-Erk-1/2 and Jnk-1/2. The total amounts of the respective kinases in the blots have been normalized previously. The data show that MAPK-Erk is constitutively phosphorylated and its phosphorylation is markedly stimulated after treatment with TGF-β1 and inhibited by U0126 treatment, whereas the activation of MAPK-Jnk is not affected.
Immunoblotting analysis performed with phosphorylation state-specific antibodies in Helix ganglion neurons subjected to the same treatments revealed that MAPK-Erk is constitutively phosphorylated under basal conditions and its phosphorylation is stimulated markedly after treatment with TGF-β1 and inhibited after treatment with U0126 alone or in association with TGF-β1 (Fig. 4B). These treatments were specific for the MAPK-Erk pathway, because the phosphorylation state of Jnk-1/2 was not affected.
Interestingly, the inhibition of the MAPK-Erk cascade with U0126 did not affect the increase of neurotransmitter release from the C1 neuron in the presence of the wrong target induced by the cAMP-PKA pathway activator Sp-cAMPS (15.3 ± 1.7 mV; n = 7). Conversely, the increase in the C1 stimulation-evoked depolarization of the sniffer B2 induced after MAPK-Erk activation by TGF-β1 was suppressed by the inhibitor of the cAMP-PKA pathway, Rp-cAMPS (5.3 ± 0.5 mV; n = 8; p < 0.05) (Fig. 4A).
The induction of neurotransmitter release by contact with the physiological target requires PKA and MAPK-Erk activation
The presence of the physiological target B2 stimulates the growth of the presynaptic neuron C1 and is able to overcome the inhibition of neurotransmitter release exerted by a cocultured nonphysiological target within 30 min of contact (Ghirardi et al., 2000, 2001). In fact, a 30 min contact of the sniffer B2 with the C1 neurites in C1C3 cocultures induced a significant increase in the amount of C1 stimulation-evoked depolarization recorded in the sniffer B2 from 1.77 ± 0.17 mV immediately after contact up to 6.17 ± 0.94 mV after 30 min of contact (n = 6; p > 0.001) (Fig. 5). To investigate whether this induction of neurotransmitter release by the physiological target was dependent on the same pathways involved in the inhibitory effect of the nonphysiological target, we treated the C1C3 cocultures with PKA or MAPK-Erk or tyrosine kinase inhibitors and analyzed the time course of the sniffer B2 depolarization evoked by C1 stimulation during the 30 min of contact with the physiological target. Treatment of C1C3 cocultures with the cAMP antagonist Rp-cAMPS completely prevented the stimulatory effect of the physiological target on neurotransmitter release (with changes in B2 depolarization from 1.83 ± 0.22 mV immediately after the contact to 1.31 ± 0.23 mV after 30 min; n = 7). A similar prevention of the stimulatory effect of the physiological target on neurotransmitter release was observed with the MAPK-Erk inhibitors U0126 (with changes in the C1 stimulation-evoked depolarization of the B2 sniffer from 1.73 ± 0.4 mV immediately after contact to 1.5 ± 0.35 mV after 30 min; n = 4) and PD98059 (with changes in the C1 stimulation-evoked depolarization of the sniffer B2 from 1.2 ± 0.4 mV immediately after contact to 1.6 ± 0.35 mV after 30 min; n = 4) (Fig. 5). On the contrary, the tyrosine kinase inhibitor genistein did not significantly interfere with the upregulation of neurotransmitter release promoted by contact with the physiological target B2 in C1C3 cocultures. Similarly to untreated cultures, the depolarization of the sniffer B2 evoked by the stimulation of C1 in the presence of 100 μm genistein increased significantly after 30 min of contact (from 2.96 ± 0.7 mV immediately after contact to 8.14 ± 1.9 mV after 30 min; n = 7; p < 0.01) (Fig. 5). Similar results were obtained with 10 μm lavendustin (data not shown).
Figure 5.

PKA and MAPK cascades are required for the appropriate target-induced increase in neurotransmitter release from the C1 neuron in contact with the wrong target C3. Each point represents the mean ±SEM amplitude of the depolarization evoked in the sniffer B2 (appropriate target) by stimulation of the C1 neuron in C1C3 cocultures at the indicated time points after contact with B2 in the absence or presence of the PKA inhibitor Rp-cAMPS, the MEK inhibitors PD98059 or U0126, or the tyrosine kinase inhibitor genistein. Statistical analysis indicates significant differences in neurotransmitter release between untreated C1C3 cocultures and C1C3 cocultures treated with Rp-cAMPS, PD98059,or U0126 (2-way ANOVA: F(4,46) = 14.42, p < 0.001; Newman-Keuls multiple comparison test: p < 0.05 and 0.01 at 15 and 30 min after contact, respectively, for the various treatments). The treatment of the cocultures C1C3 with genistein did not induce any significant effect as compared with control untreated C1C3 cocultures.
These results suggest that, in the presence of an inappropriate target, the induction of neurotransmitter release by the physiological target retrogradely activates the same signal transduction pathways involved in the wrong target-mediated inhibition.
Discussion
The data reported here show that interaction with a nonphysiological target results in the inhibition of evoked neurotransmitter release from nerve terminals of the Helix C1 neuron, an active process requiring the synthesis of new proteins and involving a downregulation of the MAPK-Erk and cAMP-PKA pathways. In fact, such inhibitory action was blocked by incubation of C1C3 cocultures with the protein synthesis inhibitors anisomycin and cycloheximide. Moreover, the inhibitory effect of the nonphysiological target was overcome by stimulation of either the cAMP-PKA or MAPK-Erk cascades or by prolonged contact with the physiological target B2. The latter effect was also prevented by the inhibition of either the cAMP-PKA or the MAPK-Erk pathway, indicating that these pathways represent a final common pathway through which postsynaptic targets control the maturation and efficiency of presynaptic neurotransmitter release mechanisms.
In our experiments, the inhibitory effect of the wrong target on neurotransmitter release from the C1 neuron was strictly modulated by the presence of the appropriate target neuron B2. The physiological target is able to remove the wrong target-induced inhibition either after a 30 min contact with a preexisting C1C3 coculture, according to the ability of invertebrate neurons to rapidly form synaptic connections after contact between synaptic partners (Coulson and Klein 1997), or when it is plated in contact with C1 simultaneously with the wrong target (C3C1B2 cocultures). The presence of the neuron B2 in C3C1B2 cocultures overrides the inhibitory effect of the wrong target on the formation of varicosities containing 5HT (Ghirardi et al., 2000) and induces a significant increase in the amount of neurotransmitter release as compared with the C1C3 cocultures (M. Ghirardi, unpublished observations). On the other hand, the wrong target also fails to exert any inhibitory effect on neurotransmitter release when it is plated (for 0.5-2 hr) in contact with C1 neurons that have been grown in isolation or in contact with the appropriate target B2 for 3-4 d and have already developed an efficient releasing machinery.
Adhesion molecules such as apCAMs have been shown to play important roles in the formation and maturation of neuronal connections and in synaptic plasticity in Aplysia (Mayford et al. 1992; Zhu et al., 1994, 1995; Schacher et al., 2000) through pathways involving MAP kinase activation (Bailey et al., 1997). Because the blockade of the release-enhancing effect of the physiological target in Helix C1C3 cocultures by MAPK inhibitors is similar to the effect of incubation of the B2 neuron with anti-apCAM antibodies (Ghirardi et al., 2001), it is tempting to speculate that the signal transduction pathways activated by cell adhesion molecules could mediate the target-dependent effects on nerve terminal maturation. Possible targets of transmitter release modulation by apCAM-like molecules may be calcium channels, with changes in their functional properties and aggregation at release sites, or cytoskeletal proteins, because the cytoplasmic domains of N-CAM (neural cell adhesion molecule) and L1 are directly associated with ankyrins (Davis and Bennett, 1994). In Drosophila, the clustering of the adhesion molecule fasciclin II with Shaker K+ channels mediated by the DLG/PSD (discs large-postsynaptic density)-95 family of MAGUKs (membrane associated guanylate kinases) can be involved in the modulation of the level of activity and neurotransmitter release in the presynaptic motor neuron terminals (Thomas et al., 1997).
Several signal transduction pathways linked to PKA, PKC (protein kinase C), MAPK, and tyrosine kinase activation have been repeatedly implicated in the regulation of neuronal growth and synaptic plasticity in invertebrate as well as mammalian neurons with effects that are dependent, at least in part, on modulation of protein synthesis. Among them, the cAMP-PKA pathway appears to play a central role in several processes such as modulation of neuronal development and plastic changes underlying learning and memory (for review, see Silva et al., 1998; Kandel, 2001). In the presence of protein synthesis, we found that stimulation of the cAMP-PKA system is able to overcome the wrong target-dependent inhibitory effect on neurotransmitter release and that, conversely, its inhibition abolishes the release-enhancing effect of the protein synthesis blockade. These findings strongly indicate that the cAMP-PKA system, by acting downstream of protein synthesis, plays a key role in mediating the target-dependent effects on the development and maturation of the exocytotic machinery and that a tonic PKA activity is required for a correct nerve terminal maturation. These results are in agreement with previous findings showing that contact with the appropriate target muscle cells induces a PKA-dependent increase of intracellular calcium in Helisoma neurons (Funte and Haydon, 1993) and that the positive effects of the synaptic vesicle-associated phosphoprotein synapsin in Xenopus neural development are strongly dependent on PKA phosphorylation (Kao et al., 2002).
Although tyrosine kinases do not appear to participate in the target-dependent changes in our experimental model, a potential candidate in addition to PKA is the MAPK-Erk pathway. The MAPK pathway has been shown to regulate the formation of the Drosophila neuromuscular junction (Koh et al., 2002) and to phosphorylate proteins involved in transmitter release, such as synapsins (Angers et al., 2002). In addition, MAPK-Erk has a fundamental role in many forms of synaptic plasticity, both in invertebrates (Bailey et al., 1997; Martin et al., 1997; Sharma et al., 2003) and in mammals (Kawasaki et al., 1999; Roberson et al., 1999; Huang et al., 2000; Zhang et al., 2003). Similarly to what we observed for PKA, the stimulation of the MAPK-Erk in the presence of protein synthesis overcame the wrong target-induced inhibitory effect on neurotransmitter release, and conversely, its inhibition abolished the release-enhancing effect of the protein synthesis blockade. This suggests that the two pathways act synergistically downstream of protein synthesis. Interestingly, whereas MAPK-Erk activation is blocked by inhibition of the cAMP-PKA pathway, the effects of cAMP-PKA activation are not affected by MAPK-Erk blockade, indicating that the two pathways act on neurotransmitter release in series, rather than in parallel, with the cAMP-PKA system located downstream of the MAPK-Erk pathway.
Because a large body of experimental evidence suggests that novel protein synthesis is a requisite for the synapse to be formed (Montarolo et al., 1986; Feng et al., 1997; Martin et al., 2000; Schacher and Wu, 2002) and that contact with the physiological target is followed over time by an increase in transmitter release to a steady-state level (Grantyn et al., 1995; Cibelli et al., 1996; Verderio et al., 1999), we propose that signals from the physiological target activate a set of proteins that, through the MAPK-Erk and cAMP-PKA pathways, upregulate transmitter release from the presynaptic neuron. Recent data have shown the existence of protein synthesis and RNA translation not only in the cell body of neurons but also in neurites and synaptic sites (Kang and Schuman, 1996; Martin et al., 1998; Sotelo et al., 1999; Alvarez et al., 2000; Sigrist et al., 2000; Schacher and Wu, 2002). The presence of local protein synthesis in terminals could explain the rapidity of the anisomycin effect in increasing neurotransmitter release in C1C3 cocultures (Sherff and Carew, 2000; Wang et al., 2002; Zhang and Poo, 2002).
In conclusion, the present data indicate that contact with a nonphysiological target can inhibit neurotransmitter release from developing presynaptic terminals by means of an active mechanism requiring ongoing protein synthesis and leading to the inhibition of the same protein kinases, namely MAPK-Erk and PKA, that are activated after contact with a physiological target cell. The evidence that PKA- and MAPK-Erk-dependent events lie downstream of protein synthesis suggests that contact with an inappropriate target could trigger the translation of some molecules with inhibitory effects on PKA and MAPK activity. It is tempting to speculate that inhibition of the PKA pathway could be achieved by increasing the expression of the PKA catalytic inhibitor peptide (Demaille et al., 1977; Beavo and Mumby 1981), the PKA regulatory subunit RII, or some protein inhibiting the binding of RII to A kinase-anchoring proteins (AKAPs), a critical event for the correct compartmentalization of PKA within nerve terminals (Schwartz 2001). Inhibition of the latter binding prevented both short- and long-term facilitation in Aplysia sensorimotor synapses, processes in which an enhanced phosphorylation of presynaptic proteins is required (Liu et al., 2004). Another possibility is that the wrong target-dependent inhibition induces an increased expression of dual specificity phosphatases regulating MAPK activity in an inhibitory way (Clarke, 1994; Nebreda, 1994). Although the identity of the regulatory proteins involved remains to be elucidated, these mechanisms could represent a means for regulating the development of neuronal connectivity by preventing the formation of inappropriate connections.
Footnotes
This work was supported by Telethon-Italy Grants 1101 (P.G.M.) and 1131 (F.B.), the Italian Ministry of University (Cofin 2002 and Fondo per gli Investimenti della Ricerca di Base) (P.G.M., F.B.), and the Fisher Foundation for Alzheimer's Disease Research (F.B.).
Copyright © 2004 Society for Neuroscience 0270-6474/04/245054-09$15.00/0
Correspondence should be addressed to Dr. Mirella Ghirardi, Department of Neuroscience, University of Torino, Corso Raffaello, 30, 10125 Torino, Italy. E-mail: mirella.ghirardi@unito.it.
References
- Alvarez J, Giuditta A, Koenig E (2000) Protein synthesis in axons and terminals: significance for maintenance, plasticity and regulation of pheno-type. With a critique of slow transport theory. Prog Neurobiol 62: 1-62. [DOI] [PubMed] [Google Scholar]
- Angers A, Fioravante D, Chin J, Cleary LJ, Bean AJ, Byrne JH (2002) Serotonin stimulates phosphorylation of Aplysia synapsin and alters its sub-cellular distribution in sensory neurons. J Neurosci 22: 5412-5422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Kaang BK, Chen M, Martin KC, Lim CS, Casadio A, Kandel ER (1997) Mutation of the phosphorylation sites of MAP kinase blocks learning-related internalization of apCAM in Aplysia sensory neurons. Neuron 18: 913-924. [DOI] [PubMed] [Google Scholar]
- Balaban PM (2002) Cellular mechanisms of behavioral plasticity in terrestrial snail. Neurosci Biobehav Rev 26: 597-630. [DOI] [PubMed] [Google Scholar]
- Beavo JA, Mumby MC (1981) cAMP-dependent phosphorylation. In: Handbook of pharmacology, pp 363-389. Heidelberg: Springer
- Bixby JL, Harris WA (1991) Molecular mechanisms of axon growth and guidance. Annu Rev Cell Biol 7: 117-159. [DOI] [PubMed] [Google Scholar]
- Cibelli G, Ghirardi M, Onofri F, Casadio A, Benfenati F, Montarolo PG, Vitiello F (1996) Synapsin-like molecules in Aplysia punctata and Helix pomatia: identification and distribution in the nervous system and during the formation of synaptic contacts in vitro. Eur J Neurosci 8: 2530-2543. [DOI] [PubMed] [Google Scholar]
- Clarke PR (1994) Signal transduction. Switching off MAP kinases. Curr Biol 4: 647-650. [DOI] [PubMed] [Google Scholar]
- Coulson RL, Klein M (1997) Rapid development of synaptic connections and plasticity between sensory neurons and motor neurons of Aplysia in cell culture: implications for learning and regulation of synaptic strength. J Neurophysiol 77: 2316-2327. [DOI] [PubMed] [Google Scholar]
- Dai Z, Peng HB (1993) Elevation in presynaptic Ca2+ level accompanying initial nerve-muscle contact in tissue culture. Neuron 10: 827-837. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo M (1994) Evoked neuronal secretion of false transmitters. Neuron 13: 909-917. [DOI] [PubMed] [Google Scholar]
- Davis JQ, Bennett V (1994) Ankyrin binding activity shared by the neurofascin/L1/NrN-CAM family of nervous system cell adhesion molecules. J Biol Chem 269: 27163-27166. [PubMed] [Google Scholar]
- Demaille JG, Peters KA, Fischer EH (1977) Isolation and properties of the rabbit skeletal muscle protein inhibitor of adenosine 3′,5′-monophosphate dependent protein kinases. Biochemistry 12: 3080-3086. [DOI] [PubMed] [Google Scholar]
- Feng ZP, Klumperman J, Lukowiak K, Syed NI (1997) In vitro synaptogenesis between the somata of identified Lymnaea neurons requires protein synthesis but not extrinsic growth factors or substrate adhesion molecules. J Neurosci 17: 7839-7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzsimonds RM, Poo MM (1998) Retrograde signaling in the development and modifications of synapses. Physiol Rev 78: 143-170. [DOI] [PubMed] [Google Scholar]
- Frey U, Huang YY, Kandel ER (1993) Effects of cAMP simulates a late stage of LTP in hippocampal CA1 neurons. Science 260: 1661-1664. [DOI] [PubMed] [Google Scholar]
- Funte LR, Haydon PG (1993) Synaptic target contact enhances presynaptic calcium influx by activating cAMP-dependent protein kinase during synaptogenesis. Neuron 10: 1069-1078. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Casadio A, Santarelli L, Montarolo PG (1996) Aplysia hemolymph promotes neurite outgrowth and synaptogenesis of identified Helix neurons in cell culture. Invert Neurosci 2: 41-49. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Casadio A, Naretto G, Levi R, Montarolo PG (2000) Influence of the target on distribution and functioning of the varicosities of Helix pomatia metacerebral cell C1 in dissociated cell culture. Neuroscience 96: 843-853. [DOI] [PubMed] [Google Scholar]
- Ghirardi M, Naretto G, Fiumara F, Vitiello F, Montarolo PG (2001) Target-dependent modulation of neurotransmitter release in cultured Helix neurons involves adhesion molecules. J Neurosci Res 65: 111-120. [DOI] [PubMed] [Google Scholar]
- Grantyn R, Kraszewski K, Melnick I, Taschenberger H, Warton S (1995) In vitro development of vertebrate central synapses. Perspect Dev Neurobiol 2: 387-397. [PubMed] [Google Scholar]
- Grinkevich LN, Lisachev PD, Merkulova TI (2003) Formation of AP-1 transcription factors during learning in Helix Neurosci Behav Physiol 33: 39-47. [DOI] [PubMed] [Google Scholar]
- Hall ZW, Sanes JR (1993) Synaptic structure and development: the neuromuscular junction. Cell [Suppl] 72: 99-121. [DOI] [PubMed] [Google Scholar]
- Hamakawa T, Woodin MA, Bjorgum MC, Painter SD, Takasaki M, Lukowiak K, Nagle GT, Syed NI (1999) Excitatory synaptogenesis between identified Lymnaea neurons requires trophic factors and is mediated by receptor tyrosine kinases. J Neurosci 19: 9306-9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Martin K, Kandel ER (2000) Both protein kinase A and mitogen-activated protein kinase are required in the amygdala for the macromolecular synthesis-dependent late phase of long-term potentiation. J Neurosci 20: 6317-6325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294: 1030-1038. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM (1996) A requirement for local protein synthesis in neurotrophin-induced hippocampal synaptic plasticity. Science 273: 1402-1406. [DOI] [PubMed] [Google Scholar]
- Kao HT, Song HJ, Porton B, Ming GL, Hoh J, Abraham M, Czernik AJ, Pieribone VA, Poo MM, Greengard P (2002) A protein kinase A-dependent molecular switch in synapsins regulates neurite outgrowth. Nat Neurosci 5: 431-437. [DOI] [PubMed] [Google Scholar]
- Kawasaki H, Fujii H, Gotoh Y, Morooka T, Shimohama S, Nishida E, Hirano T (1999) Requirement for mitogen-activated protein kinase in cerebellar long term depression. J Biol Chem 274: 13498-13502. [DOI] [PubMed] [Google Scholar]
- Keller F, Schacher S (1990) Neuron-specific membrane glycoproteins promoting neurite fasciculation in Aplysia californica J Cell Biol 111: 2637-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh YH, Ruiz-Canada C, Gorczyca M, Budnik V (2002) The Ras1-mitogen-activated protein kinase signal transduction pathway regulates synaptic plasticity through fasciclin II-mediated cell adhesion. J Neurosci 22: 2496-2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680-685. [DOI] [PubMed] [Google Scholar]
- Liu J, Hu JY, Schacher S, Schwartz JH (2004) The two regulatory subunits of Aplysia cAMP-dependent protein kinase mediate distinct functions in producing synaptic plasticity. J Neurosci 24: 2465-2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin K, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER (1997) MAP kinase translocates into the nucleus of the presynaptic cell and is required for long term facilitation in Aplysia Neuron 18: 899-912. [DOI] [PubMed] [Google Scholar]
- Martin K, Barad M, Kandel ER (2000) Local protein synthesis and its role in synapse-specific plasticity. Curr Opin Neurobiol 10: 587-592. [DOI] [PubMed] [Google Scholar]
- Martin R, Vaida B, Bleher R, Crispino M, Giuditta A (1998) Protein synthesizing units in presynaptic and postsynaptic domains of squid neurons. J Cell Sci 111: 3157-3166. [DOI] [PubMed] [Google Scholar]
- Mayford M, Barzilai A, Keller F, Schacher S, Kandel ER (1992) Modulation of an NCAM-related adhesion molecule with long term synaptic plasticity in Aplysia Science 256: 638-644. [DOI] [PubMed] [Google Scholar]
- Montarolo PG, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S (1986) A critical period for macromolecular synthesis in long-term heterosynaptic facilitation in Aplysia Science 234: 1249-1254. [DOI] [PubMed] [Google Scholar]
- Montarolo PG, Kandel Er, Schacher S (1988) Long term heterosynaptic inhibition in Aplysia Nature 333: 171-174. [DOI] [PubMed] [Google Scholar]
- Munno DW, Syed NI (2003) Synaptogenesis in the CNS: an odyssey from wiring together to firing together. J Physiol (Lond) 552: 1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebreda AR (1994) Inactivation of MAP kinases. Trends Biochem Sci 19: 1-2. [DOI] [PubMed] [Google Scholar]
- Purcell AL, Sharma SK, Bagnall MW, Sutton MA, Carew TJ (2003) Activation of a tyrosine kinase-MAPK cascade enhances the induction of long-term synaptic facilitation and long-term memory in Aplysia Neuron 37: 473-484. [DOI] [PubMed] [Google Scholar]
- Roberson ED, English JD, Adams JP, Selcher JC, Kondratick C, Sweatt JD (1999) The mitogen-activated protein kinase cascade couples PKA and PKC to cAMP response element-binding protein phosphorylation in area CA1 of hippocampus. J Neurosci 19: 4337-4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacher S, Wu F (2002) Synapse formation in the absence of cell bodies requires protein synthesis. J Neurosci 22: 1831-1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schacher S, Wu F, Sun ZY, Wang D (2000) Cell-specific changes in expression of mRNAs encoding splice variants of Aplysia cell adhesion molecule accompany long-term synaptic plasticity. J Neurobiol 45: 152-161. [DOI] [PubMed] [Google Scholar]
- Schmid RS, Graff RD, Schaller MD, Chen S, Schachner M, Hemperly JJ, Maness P (1999) NCAM stimulates the Ras-MAPK pathway and CREB phosphorylation in neuronal cells. J Neurobiol 38: 542-558. [PubMed] [Google Scholar]
- Schwartz JH (2001) The many dimensions of cAMP signaling. Proc Natl Acad Sci USA 98: 13482-13484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma SK, Sherff CM, Shobe J, Bagnall MW, Sutton MA, Carew TJ (2003) Differential role of mitogen-activated protein kinase in three distinct phases of memory for sensitization in Aplysia J Neurosci 23: 3899-3907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherff CM, Carew TJ (2000) Induction of long-term facilitation at Aplysia sensorimotor synapses by coincident 5HT exposure requires postsynaptic protein synthesis. Soc Neurosci Abstr 26: 1523. [Google Scholar]
- Sigrist SJ, Thiel PR, Reiff DF, Lachance PE, Lasko P, Schuster CM (2000) Postsynaptic translation affects the efficacy and morphology of neuromuscular junctions. Nature 405: 1062-1065. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S (1998) CREB and memory. Annu Rev Neurosci 21: 127-148. [DOI] [PubMed] [Google Scholar]
- Sotelo JR, Kun A, Benech JC, Giuditta A, Morillas J, Benech CR (1999) Ribosomes and polyribosomes are present in the squid giant axon: an immunocytochemical study. Neuroscience 90: 705-715. [DOI] [PubMed] [Google Scholar]
- Tancredi V, D'Antuono M, Café C, Giovedì S, Bue MC, D'Arcangelo G, Onofri F, Benfenati F (2000) The inhibitory effects of interleukin-6 on synaptic plasticity in the rat hippocampus are associated with an inhibition of mitogen-activated protein kinase Erk. J Neurochem 75: 634-643. [DOI] [PubMed] [Google Scholar]
- Thomas U, Kim E, Kuhlendahl S, Koh YH, Gundelfinger ED, Sheng M, Garner CC, Budnik V (1997) Synaptic clustering of the cell adhesion molecule fasciclin II by discs-large and its role in the regulation of presynaptic structures. Neuron 19: 787-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torocsik B, Szeberenyi J (2000) Anisomycin uses multiple mechanisms to stimulate mitogen-activated protein kinase and gene expression and to inhibit neuronal differentiation in PC12 phaeochromocytoma cells. Eur J Neurosci 12: 527-532. [DOI] [PubMed] [Google Scholar]
- Towbin L, Staehelin T, Gordon J (1979) Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA 76: 4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Coco S, Pravettoni E, Bacci A, Matteoli M (1999) Synaptogenesis in hippocampal cultures. Cell Mol Life Sci 55: 1448-1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Iacoangeli A, Popp S, Muslimov IA, Imataka H, Sonenberg N, Lomakin IB, Tiedge H (2002) Dendritic BC1 RNA: functional role in regulation of translation initiation. J Neurosci 22: 10232-10241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JJ, Okutani F, Inoue S, Kaba H (2003) Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase signaling pathway leading to cyclic AMP response element-binding protein phosphorylation is required for the long term facilitation process of aversive olfactory learning in young rats. Neuroscience 121: 9-16. [DOI] [PubMed] [Google Scholar]
- Zhang X, Poo MM (2002) Localized synaptic potentiation by BDNF requires local protein synthesis in the developing axon. Neuron 36: 675-688. [DOI] [PubMed] [Google Scholar]
- Zhu H, Wu F, Schacher S (1994) Aplysia cell adhesion molecules and serotonin regulate sensory cell-motor cell interactions during early stages of synapse formation in vitro J Neurosci 14: 6886-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Wu F, Schacher S (1995) Changes in expression and distribution of Aplysia cell adhesion molecules can influence synapse formation and elimination in vitro J Neurosci 15: 4173-4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoran MJ, Funte LR, Kater SB, Haydon PG (1993) Neuron-muscle contact changes presynaptic resting calcium set-point. Dev Biol 158: 163-171. [DOI] [PubMed] [Google Scholar]