

Abstract
Intracellular accumulations of filamentous tau inclusions are neuropathological hallmarks of neurodegenerative diseases known as tauopathies. The discovery of multiple pathogenic tau gene mutations in many kindreds with familial frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17) unequivocally confirmed the central role of tau abnormalities in the etiology of neurodegenerative disorders. To examine the effects of tau gene mutations and the role of tau abnormalities in neurodegenerative tauopathies, transgenic (Tg) mice were engineered to express the longest human tau isoform (T40) with or without the R406W mutation (RW and hWT Tg mice, respectively) that is pathogenic for FTDP-17 in several kindreds. RW but not hWT tau Tg mice developed an age-dependent accumulation of insoluble filamentous tau aggregates in neuronal perikarya of the cerebral cortex, hippocampus, cerebellum, and spinal cord. Significantly, CNS axons in RW mice contained reduced levels of tau when compared with hWT mice, and this was linked to retarded axonal transport and increased accumulation of an insoluble pool of RW but not hWT tau. Furthermore, RW but not hWT mice demonstrated neurodegeneration and a reduced lifespan. These data indicate that the R406W mutation causes reduced binding of this mutant tau to microtubules, resulting in slower axonal transport. This altered tau function caused by the RW mutation leads to increased accumulation and reduced solubility of RW tau in an age-dependent manner, culminating in the formation of filamentous intraneuronal tau aggregates similar to that observed in tauopathy patients.
Keywords: tau R406W mutation, transgenic mice, tauopathy, FTDP-17, axonal transport, tau proteins
Introduction
Filamentous tau pathologies in the CNS are hallmark lesions of a class of neurodegenerative diseases collectively known as tauopathies (Lee et al., 2001). Although Alzheimer's disease (AD) is a mixed tauopathy with associated amyloid β deposits, examples of primary tauopathies include progressive supranuclear palsy, Pick's disease, corticobasal degeneration, amyotrophic lateral sclerosis/parkinsonism-dementia complex, and autosomal dominantly inherited frontotemporal dementia with parkinsonism linked to chromosome 17 (FTDP-17). Indeed, discovery of numerous pathogenic tau mutations in multiple FTDP-17 kindreds provided unequivocal proof that tau abnormalities alone are sufficient to cause neurodegeneration. To date, >30 different intronic and exonic pathogenic mutations have been identified in the tau gene of >50 distinct FTDP-17 kindreds (Clark et al., 1998; Dumanchin et al., 1998; Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998).
Several mechanisms have been proposed to explain how FTDP-17 mutations alter tau and cause neurodegeneration. For example, intronic and some exonic tau gene mutations affect alternative splicing of exon 10 and consequently change the relative proportion of tau isoforms with four or three microtubule (MT) binding repeats (Hong et al., 1998; Hutton et al., 1998; D'Souza et al., 1999; Delisle et al., 1999; Grover et al., 1999; Hasegawa et al., 1999; Varani et al., 1999; Yasuda et al., 1999, 2000; Spillantini et al., 2000; Stanford et al., 2000). Other exonic tau gene mutations (e.g., P301L, R406W) impair the ability of tau to bind to and stabilize MTs as well as to promote the assembly of MTs (Hasegawa et al., 1998; Hong et al., 1998). Some of these mutations (e.g., P301L) also facilitate the pathological fibrillogenesis of tau (Hasegawa et al., 1998; Hong et al., 1998; Bugiani et al., 1999; D'Souza et al., 1999; Murrell et al., 1999; Rizzu et al., 1999; Barghorn et al., 2000; Pickering-Brown et al., 2000; Rizzini et al., 2000). Finally, several tau gene mutations, most notably R406W, alter the phosphorylation state and solubility of tau, thereby contributing to the formation of fibrillary tau aggregates (Lee et al., 1991; Crowther and Goedert, 2000; Perez et al., 2000; Sahara et al., 2000; Vogelsberg-Ragaglia et al., 2000; Connell et al., 2001; Delobel et al., 2002).
Transgenic (Tg) mouse models of tauopathies have provided insights into the pathogenesis of tau lesions and their contributions to the onset and progression of neurodegenerative disease. Overexpression of human wild-type (hWT) tau in Tg mice can result in somato-dendritic and/or axonal tau aggregates associated with an early onset neurodegenerative phenotype (Ishihara et al., 1999, 2001a,b; Spittaels et al., 1999; Probst et al., 2000; Lee et al., 2001). Some of these Tg mice also develop AD-like neurofibrillary tangles (NFTs) in neurons with advancing age (Ishihara et al., 2001b). Furthermore, there is accumulating evidence that FTDP-17 mutations, such as P301L (Lewis et al., 2000; Götz et al., 2001), V337M (Tanemura et al., 2002) and R406W (Lim et al., 2001; Tatebayashi et al., 2002), accelerate the formation of NFT-like intraneuronal inclusions by mutant tau proteins and induce a more severe neurodegenerative phenotype in Tg mice that express mutant tau compared with hWT tau mice. Nonetheless, the mechanisms whereby tau mutations induce more profound accumulations of fibrillary tau inclusions in neuronal perikarya than hWT tau in Tg mice remains poorly understood.
To address this question, we generated lines of Tg mice engineered to overexpress either hWT tau or R406W (RW) mutant tau. The RW mutation, located on exon 13 of the tau gene, was identified in three distinct FTDP-17 kindreds (Hutton et al., 1998; Rizzu et al., 1999; Van Swieten et al., 1999). The RW mutation reduces the ability of tau to bind MTs (Hong et al., 1998) and impairs the assembly as well as the stability of MTs in vitro and in cell culture systems (Rizzu et al., 1999; Barghorn et al., 2000; Vogelsberg-Ragaglia et al., 2000). Because RW tau Tg mice express tau much more prominently in the somato-dendritic compartment than in axons compared with hWT tau Tg mice, we speculated that this may result from a mutation-induced reduced binding of tau to MTs and therefore retardation of the slow axonal transport of RW tau. The data presented here confirm this hypothesis and suggest that RW tau may cause FTDP-17 by a novel mechanism that impairs the axonal transport of this tau mutant attributable to reduced binding to MTs.
Materials and Methods
Generation of hWT and RW tau Tg mice. Two cDNA constructs containing the longest human brain tau isoform (T40) with (RW tau) or without (hWT tau) the R406W tau mutation were cloned into the mouse prion promoter (MoPrP.Xho) expression vector at the XhoI site. A 15.3 kb NotI fragment containing T40, the MoPrP promoter, together with its 3′ and 5′ untranslated sequences, was microinjected into fertilized C57BL6 × C3H (B6C3/F1) mouse eggs and then implanted into pseudopregnant females. Genomic DNA samples were isolated from the tails of Tg and non-Tg mice using the Puregene DNA Isolation kit (Gentra Systems, Minneapolis, MN). Potential founders and Tg offspring were identified by slot blots with a 32P-labeled T40 cDNA probe, and homozygous Tg mouse lines were generated. All of the mice used in this study were maintained in the B6C3 background.
Determination of expression levels and phosphorylation state of tau. tau expression in the brain and the spinal cord of Tg mice were determined by Western blot analysis as described previously (Ishihara et al., 1999). Briefly, mice were deeply anesthetized, and the brain and spinal cord were removed. To determine the total tau protein expressed in Tg mice, tissue was directly homogenized in 2 ml/gm 2% of SDS buffer (15% sucrose, 50 mm Tris, 1 mm PMSF, 0.1% protease inhibitor mixtures containing 1 μg/ml each of pepstatin A, leupeptin, N-tosyl-l-phenylalanine chloromethyl ketone, N-tosyl-l-lysine chloromethyl ketone, and soybean trypsin inhibitor, and 100 mm EDTA, pH 7.6-8.0), sonicated, and centrifuged at 50,000 × g for 60 min at 22°C in a TL-100 ultracentrifuge (Beckman Instruments, Fullerton, CA). Protein concentrations were then determined using the BCA assay kit (Pierce, Rockford, IL). Equal amounts (60 μg) of samples were subsequently resolved on 7.5% SDS-PAGE slab gels and transferred onto nitrocellulose membranes. The membranes were probed with the antibodies described below and detected quantitatively using either [125I]-labeled goat anti-mouse IgG or [125I]-labeled Protein A (NEN, Boston, MA) as secondary antibodies.
To generate heat-stable fractions, tissue was homogenized in 2 ml/gm ice-cold high-salt reassembly buffer (hi-salt buffer) (0.1 m MES, 1 mm EGTA, 0.5 mm MgSO4, 0.75 m NaCl, 0.02 m NaF, 1 mm PMSF, and 0.1% protease inhibitor mixtures) and centrifuged at 50,000 × g for 40 min at 4°C in a TL-100 ultracentrifuge (Beckman Instruments). The supernatants were boiled for 5 min, chilled on ice for 5 min, and recentrifuged at 10,000 × g for 20 min at 4°C. Protein concentrations were then determined for the second supernatants using the BCA assay kit (Pierce). Equal amounts (15 μg) of samples were subsequently resolved on 7.5% SDS-PAGE slab gels and transferred onto nitrocellulose membranes. The membranes were probed with primary antibodies and the appropriate [125I]-labeled secondary antibodies.
tau protein solubility in the CNS of Tg mice. The solubility of tau proteins in the Tg mouse CNS was analyzed by immunoblot using serially extracted CNS samples. Soluble protein was extracted from cerebral cortex and spinal cord from 1-, 6-, and 12-month-old RW and hWT tau Tg and non-Tg mice (n = 3 per group) with RAB hi-salt buffer (0.1 m MES, 1 mm EGTA, and 0.5 mm MgSO4, pH 7.0). The pellets were rehomogenized with 1 m sucrose-RAB buffer and centrifuged at 50,000 × g for 20 min at 4°C to remove myelin and related lipids. The resulting pellets were reextracted with 1 ml/gm radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris, 150 mm NaCl, 1% NP-40, 5 mm EDTA, 0.5% sodium deoxycholate, and 0.1% SDS, pH 8.0) and centrifuged at 50,000 × g for 20 min at 4°C to generate RIPA-soluble samples. Finally, the RIPA-insoluble pellets were sonicated with 70% formic acid (FA) to recover the most insoluble aggregates. Quantitative Western blot analyses were used to determine tau levels in each fraction as described previously (Ishihara et al., 1999).
Microtubule binding assay. Brains from 12-month-old hWT and RW Tg mice (n = 3 per group) were homogenized in 1 ml/gm PIPES buffer (PB) (80 mm PIPES, 1 mm MgCl2·6H2O, 1 m EGTA, 0.1% protease inhibitor mixture, and 1 mm PMSF, pH 6.8), put on ice for 20 min, and centrifuged at 12,000 × g for 20 min at 4°C. The supernatants containing 100 mm GTP were recentrifuged at 50,000 × g for 30 min at 4°C in a TL-100 ultracentrifuge (Beckman Instruments). To assemble MTs, 100 mm GTP and equal amount of PB-glycerol (66% glycerol with density 1.25 mg/ml) were added to the second supernatants, and the mixture was warmed to 37°C for 20 min in a water bath. The samples were then centrifuged at 50,000 × g for 30 min at 30°C in a TL-100 ultracentrifuge. The supernatants (containing unassembled tubulin and free tau) and pellets (containing MTs and bound tau) were harvested, and PB equivalent to those in the supernatant was used to elute tau bound to MTs. Forty microliter samples from supernatants and 20 μl samples from pellet were resolved on 7.5% SDS-PAGE slab gels and transferred onto nitrocellulose membranes. The membranes were probed with primary rabbit anti-tau antibody 17026, followed by [125I]-labeled Protein A as described above. A statistical analysis using Student's t tests was then performed on the quantitative Western blot studies.
Immunohistochemical analyses. RW and hWT tau Tg mice and non-Tg mice (1-, 6-, and 12-month-old; n = 3 per age group) were perfused with 4% paraformaldehyde in PBS or 70% ethanol in isotonic saline after being lethally anesthetized by an intraperitoneal injection of ketamine hydrochloride (1 mg/10 gm) and xylazine (0.1 mg/10 gm), in accordance with protocols approved by the University of Pennsylvania. The brains and spinal cords of mice were removed and processed as described previously (Zhang et al., 1997). Paraffin-embedded tissue was cut into 6-μm-thick sections, and every fifth section was examined by immunohistochemistry (Tu et al., 1996; Ishihara et al., 1999).
Analysis of slow axonal transport in Tg mice. Sixty RW (from three different lines) and age-matched hWT tau Tg mice at 2, 6, and 12 months of age (n = 6 per age group) were deeply anesthetized, a laminectomy was performed to expose the T13 to L1 segments of spinal cord, and 200 μCi [35S]-labeled methionine in 0.7 μl of saline were microinjected into three anterior horn sites (L4, L5, and L6) over a period of 10 min/injection using a stereotactic apparatus as described previously (Zhang et al., 1997). Mice from RW line 37 and hWT line 23 at 2, 6, and 12 months of age and mice from RW line 60 and 65 at 6 months old were used in this study. Mice were killed 7 d after microinjection, and the L4-L5 ventral roots and the contiguous spinal nerves were removed and cut into five to seven consecutive 2 mm segments on a steel ruler. The spinal cords at L4-L5 were also dissected as a positive control for metabolic labeling. The corresponding ventral root segments from three mice were pooled and extracted in 200 μl of RIPA buffer, sonicated for 20 sec, and centrifuged at 14,000 × g for 10 min at 4°C. tau or low molecular weight neurofilament subunit (NFL) was immunoprecipitated from the supernatant with 17026 or anti-NFL coupled to Protein A/G plus agarose (beads; Santa Cruz Biotechnology, Santa Cruz, CA) overnight at 4°C. Bound tau or NFL proteins were eluted by boiling in sample buffer for 5 min, resolved by SDS-PAGE, and immunoblotted in triplicate gels as described previously (Zhang et al., 1997). Each axonal transport experiment for each age group was repeated twice using three hWT and three R406W mutant Tg mice per experiment. The amount of radiolabeled tau or NFL transported per segment was expressed as a percentage of total radiolabeled transported tau or NFL protein within all five to seven segments of ventral roots. Quantification of transported [35S]methionine radiolabeled tau and NFL were the average of six mice from the two experiments.
Levels of RW and hWT tau as well as the middle molecular weight neurofilament subunit (NFM) in the ventral root axons of Tg mice were further analyzed after nerve ligation by Western blots as describe above. RW and hWT Tg and non-Tg mice at 2 and 12 months (n = 3 per age group) underwent left or right laminectomy to expose the L4-L6 spinal cord segments under deep anesthesia. The L5 dorsal root ganglia were exposed, and the L5 ventral root nerve proximal to the junction of motor and sensory nerves was ligated unilaterally with 9-0 nylon sutures in the nerves. One week after ligation, 12 mm segments of the L5 ventral root nerves were dissected from the ligated and nonligated sides and prepared for quantitative Western blot analysis, and statistical analysis was performed (Zhang et al., 1997).
To investigate the possibility that the retardation of tau transport resulted in increased tau accumulation and aggregation in an insoluble pool, we examined the solubility of the newly synthesized radiolabeled tau proteins in the spinal cord of RW and hWT Tg mice 7 d after injection of [35S]methionine into the spinal cords as described above. tau protein was sequentially extracted from spinal cords with RAB hi-salt buffer, followed by 2% SDS and finally with 70% FA from all three lines of RW and age-matched paired hWT tau Tg mice (n = 3 per group) as described above. Radiolabeled tau proteins from serial extractions were collected by immunoprecipitation with the anti-tau antibody 17026 and separated by SDS-PAGE. Statistical analyses were performed as described previously (Zhang et al., 1997; Ishihara et al., 1999).
Preembedding immunoelectron microscopy. Preembedding immunoelectron microscopy (immuno-EM) was performed for the RW and hWT tau Tg mice and non-Tg mice at 1, 6, and 12 months of age (n = 2 per group). The mice were deeply anesthetized and perfused intracardially with 10 ml of 0.05% glutaraldehyde and 0.5% paraformaldehyde in 0.1 m cacodylate buffer, pH 7.4, followed by 50 ml of 0.2% glutaraldehyde and 2% paraformaldehyde in 0.1 m cacodylate buffer. The brains and spinal cords were removed and postfixed with 4% paraformaldehyde, 0.2% glutaraldehyde, and 0.2% picric acid in 0.1 m cacodylate buffer overnight. The cerebral cortex, hippocampus, cerebellum, and L5 spinal cord segments were dissected and cut into 50-μm-thick sections with a vibratome. Sections were quenched in 0.1% sodium borohydride in Tris buffered saline for 10 min and subsequently in 20% ethanol for 10 min. The sections were blocked in 5% donor horse serum in PBS with 0.1% cold water fish skin gelatin and 1% chicken egg albumin for 60 min, incubated with 17026, a rabbit polyclonal antibody to recombinant tau protein in 0.1% bovine serum albumin, and PBS overnight at 4°C. Subsequently, sections were washed and incubated with either biotinylated goat anti-rabbit IgG for 2 hr at room temperature. Sections were visualized with diaminobenzidine, followed by silver-gold intensification using silver methenamine developer containing 3% methenamine, 5% silver nitrate, and 1% sodium tetraborate at 60°C for 10 min as described previously (Ishihara et al., 2001b). The reaction was stopped with 2% sodium acetate and then stabilized in 3% sodium thiosulphate for 5 min. Gold toning was performed by incubating the sections in 0.1% gold chloride for 5 min, followed by stabilizing with 3% sodium thiosulfate for 5 min. Sections were fixed with 2% glutaraldehyde in PBS buffer overnight. Sections with positive staining were postfixed in 0.5% osmium tetroxide for 20 min at 4°C, dehydrated with graded series of ethanols, and embedded in Epon-Araldite resin at 60°C for 48 hr. Ultrathin sections of these blocks were cut and mounted on 100 mesh EM grids and examined using a JEM1010 electron microscope (Jeol, Peabody, MA) at 80 kV.
Tail suspension test. To monitor motor behavior, we performed the tail suspension test on the RW and hWT tau Tg mice at 1, 6, 12, 15, and 18 months of age (n = 20 per age group) as described previously (Ishihara et al., 2001). Briefly, Tg and non-Tg mice were suspended by their tails for 15 sec, and abnormal motor signs were defined as dystonic movements of the hindlimbs, or a combination of hindlimbs, forelimbs, and trunk, with the limbs retracted into the body. Under these conditions, non-Tg control mice do not display dystonic movements.
Antibodies. The following tau antibodies were used in this study: (1) T14, a monoclonal antibody specific for amino acids 141-178 of human tau; (2) 17026, rabbit polyclonal antiserum raised to recombinant human tau and recognize both human and mouse tau; (3) NFL, a polyclonal rabbit antiserum specific for NFL (Tu et al., 1995; Zhang et al., 1997), and (4) β-tubulin, a monoclonal antibody (Sigma, St. Louis, MO).
Results
Generation of RW and hWT T40 tau Tg mice
Tg mice were generated to express the cDNA transgene encoding RW or hWT T40 tau under the control of the mouse prion protein promoter. Three lines of RW tau Tg mice (lines 37, 60, and 65) and two lines of hWT tau Tg mice (lines 1 and 23) that stably expressed the T40 human tau isoform were identified. Homozygous RW tau Tg mice were subsequently generated from heterozygous line 37 and 65 mice.
The expression level of total tau proteins in the hWT and RW Tg mice was determined by Western blot analysis of brain and spinal cord extracts directly solubilized in 2% SDS (Fig. 1A). We found that human tau expression levels in brain of heterozygous (+/-) hWT tau Tg line 23+/- and homozygous (+/+) RW tau Tg line 37+/+ were similar and are ∼8- to 10-fold over endogenous mouse tau. Heterozygous RW mice from tau Tg line 60+/- showed the highest tau expression level, whereas animals from the RW tau line 65+/+ expressed the lowest level of human tau among all lines at levels comparable with endogenous mouse tau. Although tau was not detected in line 65+/+ with 60 μg of brain extract (Fig. 1A), low tau protein levels were detected by overloading the gel with 10-fold more protein (600 μg) (Fig. 1B). Because the level of human tau in mice of the RW tau Tg line 37+/+ was similar to that of hWT line 23+/- mice (Fig. 1C), the majority of the studies described below were performed using these two lines of Tg mice. However, RW lines 65+/+ and 60+/- mice were also used in the axonal transport and solubility studies.
Figure 1.
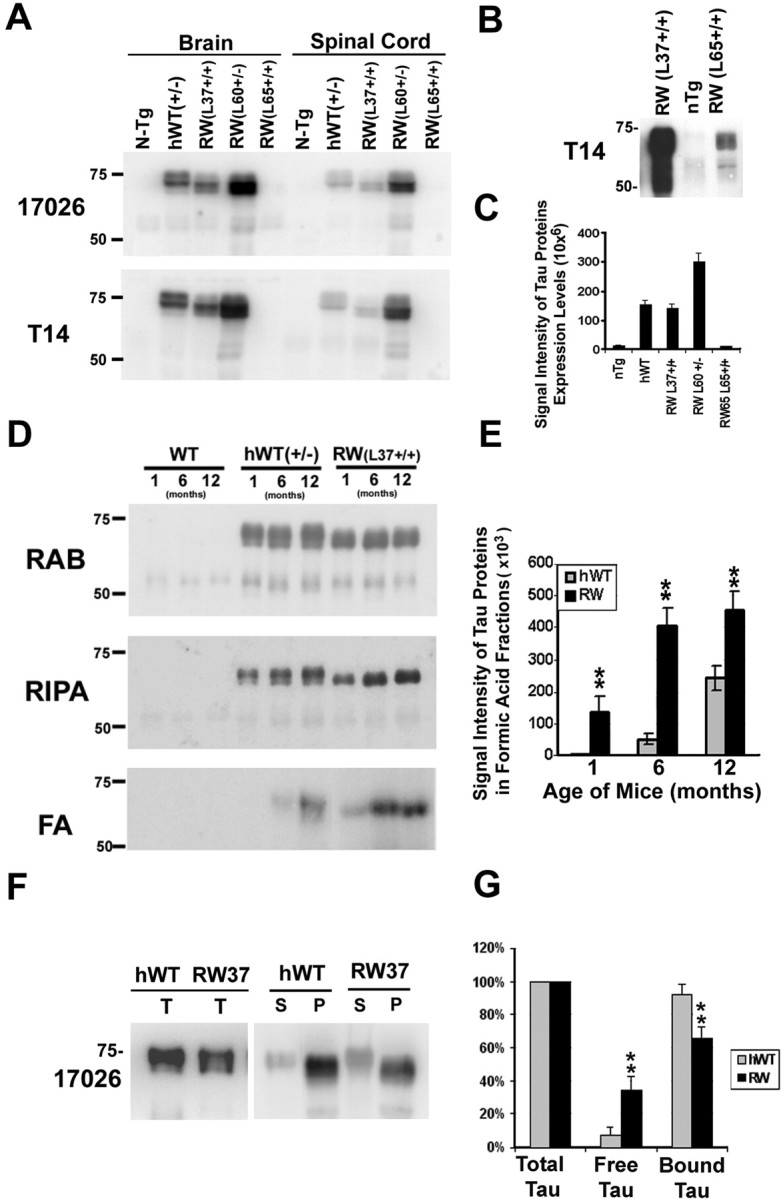
RW mutant tau in Tg mice accumulates as an insoluble pool with advancing age and shows reduced binding to MTs. A-C, To compare the levels of hWT and RW tau expression in several lines of Tg mice, Western blot analysis of transgenic human tau and total tau (human plus mouse) levels were estimated using T14, a human-specific anti-tau monoclonal antibody, and 17026, a polyclonal anti-tau antibody that recognizes both human and mouse tau. A, Total tau in brains from the different Tg mice were recovered after direct extraction with 2% SDS buffer and resolved on 7.5% SDS-PAGE gels. Equal amounts of total proteins (60 μg) were loaded for non-Tg (N-Tg) and heterozygous hWT (line 23+/-) and RW (line 60+/- and 65+/+) and homozygous RW tau Tg mice (line 37+/+). Heterozygous hWT and homozygous RW tau Tg mice (line 37) show similar expression levels of tau in the brain and the spinal cord at 6 month of age (A). Although no obvious tau was clearly detected in RW tau Tg line 65 when 60 μg of total CNS proteins were loaded (A), the loading of 10-fold more total proteins (600 μg) resulted in the visualization of a protein band that was recognized by the human tau-specific T14 monoclonal antibody (B). Quantification of the relative tau levels in hWT and all three lines of RW mice are shown in C. To demonstrate the presence of increase level of less soluble tau protein, brains from 1-, 6-, and 12-month-old non-Tg and hWT and RW Tg mice were sequentially extracted using RAB hi-salt buffer, RIPA buffers, and 70% FA (D). Tau protein levels were determined by quantitative Western blotting using the 17026 polyclonal antibody, followed by [125I]Protein A. Human transgenic T40 in the RIPA and FA fractions progressively accumulate in the brains of both RW and hWT tau Tg mice but not in the brains of non-Tg mice. However, the age-related increase of the FA-extractable tau species is larger in the RW mice than the hWT mice (E). **p < 0.01. To compare the binding of hWT and RW tau to endogenous mouse MTs, MT-binding assays were conducted (F). RW mutant tau showed reduced binding to MTs when compared with hWT tau from Tg mice (G). Total tau (T) from hWT and RW mouse brains were used for MT binding assays, and quantification of tau bound to MTs in the pellet (P) versus those that remained in the supernatant (S) is shown in G. n = 3. **p < 0.001.
RW tau showed an increased accumulation of insoluble tau and reduced binding to MTs when compared with hWT tau
Soluble and insoluble tau proteins from brain tissue of Tg mice were analyzed by sequential protein extraction as described previously (Ishihara et al., 1999) (Fig. 1D). The RAB (i.e., soluble) tau extracts showed consistent tau protein levels independent of age. In contrast, there was an increase in the amounts of both RIPA buffer and FA-extractable human RW tau relative to hWT tau in the Tg mice at each age examined here. For instance, the amount of RIPA-extractable tau in the RW and hWT mice at 1 month of age was 13 and 10% of total tau proteins, respectively, and the differences between the RW and hWT tau Tg mice increased to 21 and 16% of total tau proteins, respectively, at 12 months of age (Fig. 1D,E). As expected, the FA-extractable tau fraction in the RW mice was significantly higher than that in hWT mice at each of the three different ages examined here, although the actual amount of FA-extractable tau as a percentage of total tau remains low at only ∼0.5% for RW and 0.3% for hWT mice by 12 months of age (Fig. 1D,E). Non-Tg control mice showed no accumulation of insoluble tau in either the RIPA or FA fractions (Fig. 1D). Furthermore, RW tau is less phosphorylated than hWT tau as judged by the absence of more highly phosphorylated slower-migrating tau isoforms in RW when compared with hWT mice (Fig. 1D). Western blot analyses using a panel of phosphorylation-dependent anti-tau antibodies demonstrate a selective and dramatic reduction in the phosphorylation of Ser396 and Ser404, consistent with previously published studies (data not shown) (Crowther and Goedert, 2000; Perez et al., 2000; Sahara et al., 2000; Vogelsberg-Ragaglia et al., 2000, Delobel et al., 2002). Finally, human tau serially extracted from the spinal cord of RW and hWT mice also showed a similar age-related increase in insoluble tau and reduction in Ser396-Ser404 phosphorylation (data not shown).
We next examined the ability of RW and hWT tau expressed in Tg mice to bind endogenous mouse brain MTs in an in vitro MT-binding assay. Significantly higher amounts of hWT tau (92%) were recovered bound to MTs compared with RW tau (65%) (Fig. 1F, G). Interestingly, the total amount of soluble free tau in supernatant and MT-bound tau recovered in the pellet in hWT mice appeared higher than that in RW mice, and this is most likely attributable to the accumulation of some of the RW tau in an insoluble pool.
RW tau Tg mice develop somatodendritic tau pathology with aging
The distribution of transgenic tau proteins in the CNS of the Tg mice was examined immunohistochemically. tau expression in neurons was observed in multiple CNS regions, including the neocortex, amygdala, hippocampus, brainstem, and spinal cord. Notably, the somato-dendritic compartment of neurons in the CNS of the RW mice showed the most intense tau immunostaining (Fig. 2B,D,F) when compared with much weaker somato-dendritic tau staining in CNS neurons of the hWT mice (Fig. 2A,C,E). The RW tau Tg mice also showed an age-dependent increase in the intensity of the somato-dendritic tau staining, and NFT-like tau pathology was detected in the hippocampus of 12-month-old RW mice (Fig. 2G) that resembled NFTs in the hippocampus of a human FTDP-17 patient with the same R406W tau gene mutation (Fig. 2H). Furthermore, a subset of these tau inclusions also was stained with thioflavin S in the RW mice (Fig. 2I), similar to patients with this mutation (Fig. 2J). In contrast, thioflavin S-positive inclusions were not detected in the hWT mice. Thus, these findings demonstrate that the neuronal tau inclusions in the CNS of the RW mice and human FTDP-17 patients share common structural characteristics. Finally, in the Tg but not in the non-Tg mice, numerous reactive astrocytes were detected with a monoclonal antibody specific for glial fibrillary acidic protein in brain and spinal cord (data not shown). In particular, we observed profound gliosis associated with neuronal tau pathology in the hippocampus of the RW mice.
Figure 2.

RW tau Tg mice develop somato-dendritic NFT-like tau pathology. A-F, Immunostaining using the polyclonal anti-tau antibody 17026 in the spinal cord (A, B), neocortex (C, D), and cerebellum (E, F) of hWT and RW tau Tg mice at 12 months of age. Neurons in RW tau Tg mice progressively accumulated tau in their somato-dendritic compartment, where as neurons in the hWT tau Tg mice showed little or no perikaryal tau immunoreactivity. G, H, High-power photomicrographs demonstrate intense perikaryal tau immunoreactivity in hippocampal hilar neurons of an 18-month-old RW Tg mouse using the 17026 antibody (G), which resemble the filamentous neuronal tau inclusions in the hippocampus of a patient with FTDP-17 caused by a R406W tau gene mutation (H). Some of the tau-positive neurons in the RW tau Tg mice are thioflavin S positive (I), similar to the NFTs in an R406W FTDP-17 brain (J). Scale bars: A-F, 100 μm; G-J, insets, 10 μm.
Retardation of orthograde slow axonal transport of RW tau in the ventral roots of the RW tau Tg mice
The reduced binding of RW tau to MTs noted above and the increase in perikarya neuronal tau staining observed in the RW mice prompted us to hypothesize that the reduced binding of RW tau to MTs could lead to reduced transport of this mutant tau isoform, culminating in the aggregation of RW tau in the somatodendritic compartment of neurons. Tau proteins have been shown previously to move within the slow component of axonal transport (Mercken et al., 1995). Thus, the transport of tau was quantified by monitoring radiolabeled tau proteins in ventral roots 7 d after microinjection of [35S]-labeled methionine into the L4-L6 anterior horns of RW and age-matched hWT tau Tg mice at 2, 6, and 12 months of age. Newly synthesized radiolabeled tau proteins were detected by immunoprecipitation and autoradiography. As shown in Figure 3, there was significant reduction of radiolabeled tau proteins in the distal segments of the ventral roots in line 37+/+ of adult RW mice compared with hWT mice at all ages studied here. In fact, human tau was barely detectable in the most distal segment of the ventral roots of adult RW mice, although it was readily detectable in the same roots of the hWT mice. Furthermore, the retardation of tau axonal transport in RW Tg line 37+/+ mice was most pronounced at 6 months of age, in which the peak of radiolabeled hWT tau transported was 4-6 mm along the ventral root compared with 2 mm for radiolabeled RW tau (Fig. 3E,G). This observation was also supported in similar studies using RW line 60+/- at 6 month of age (Fig. 3M,O) and RW line 65+/+ at 6 month of age (data not shown). In contrast, there was no reduction or change in the transport of NFL in the same lines of RW mice at any age, and this is significant because NFL is one of the major cytoskeletal proteins translocated in the slow component of axonal transport (Fig. 3). Thus, the R406W tau gene mutation specifically impairs the transport of the mutant tau in the slow component of axonal transport.
Figure 3.
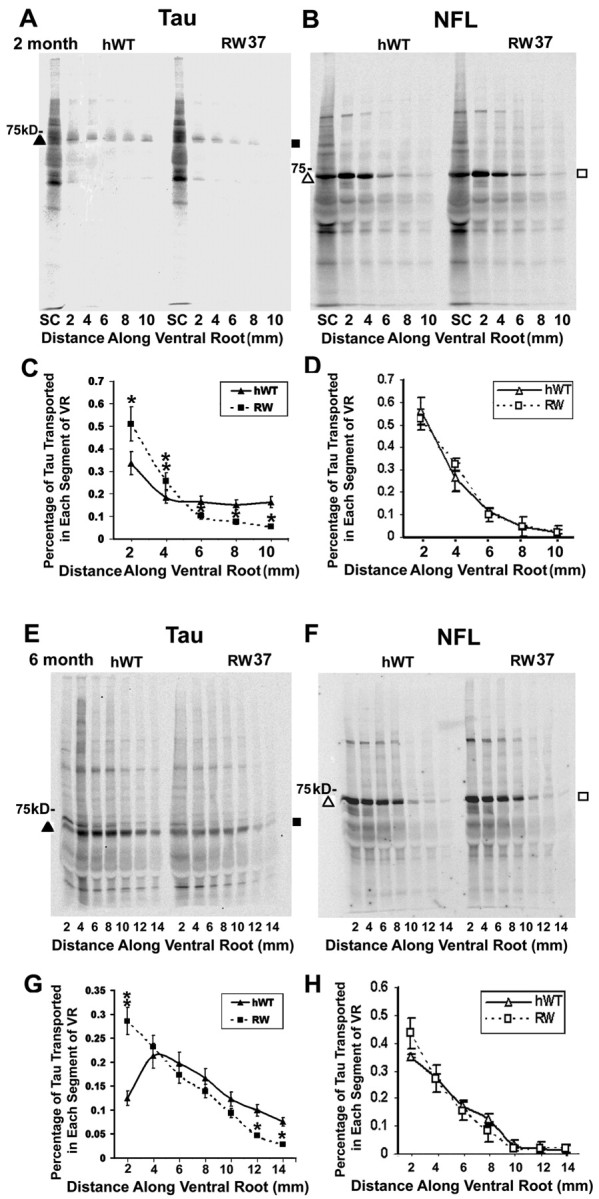

The slow axonal transport of RW mutant tau is retarded in the ventral roots of RW tau Tg mice. SDS-PAGE shows progressive retardation of the slow axonal transport for tau in the ventral roots (VR) and contiguous spinal nerves (SN) of RW line 37 tau Tg mice at 2 months of age (A, C), at 6 months of age (E, G), and at 12 months of age (I, K) compared with age-matched hWT tau Tg mice, but the slow axonal transport of NFL remains the same in the RW and hWT tau Tg mice at 2 months (B, D), 6 months (F, H), and 12 months of age (J, L). Similar retardation of RW tau transport was also detected in a second RW line, i.e., line 60 at 6 months of age (M, O), with no obvious differences in NFL transport (N, P). tau or NFL protein samples were collected by immunoprecipitation with the anti-tau antibody 17026 (A, C, E, G, I, K, M, O) or the anti-NFL antibody (B, D, F, H, J, L, N, P) 7 d after injection of [35S]methionine into spinal cord (SC). The graphs illustrate quantitative measurements of tau (C, G, K, O) and NFL (D, H, L, P) proteins conveyed by slow axonal transport in pairs of age-matched RW and hWT Tg mice (n = 6 per age group). At all ages studies, slow axonal transport of tau in both lines of RW Tg mice is significantly reduced compared with hWT, but no differences were found in the slow axonal transport of NFL between the RW lines and hWT tau Tg mice. *p < 0.05; **p < 0.01.
Nerve ligation studies further support reduced transport of RW tau
To further demonstrate retardation of axonal transport in the RW mice, we ligated ventral nerve roots to measure the levels of transported tau proteins that accumulated proximal to the ligatures. The amounts of human tau protein in the nonligated ventral roots were reduced in the RW mice compared with the hWT mice, and this is consistent with decreased steady-state levels of tau in these axons of RW mice (Fig. 4A-C). Moreover, RW mice accumulated less tau proximal to the ligation site than did the hWT mice, which is also consistent with the observations above demonstrating retarded tau transport in these animals (Fig. 4A-D). In sharp contrast, the steady-state and transported NFM in the ventral roots of the RW and hWT tau Tg mice did not differ significantly at any age examined here (Fig. 4A,B,E). Thus, our data suggest that the R406W tau gene mutation specifically retards the slow axonal transport of tau proteins in the RW mice, and we infer that the pathogenic effects of the R406W mutation in FTDP-17 patients are mediated at least in part by a novel mechanism involving alterations in the transport of R406W mutant tau.
Figure 4.

Nerve ligation of L5 ventral roots in RW mice showed retardation of slow axonal transport of RW mutant tau (A, B). Quantitative Western blot analysis of tau (bottom panels) and NFM (top panels) protein levels in the ligated L5 ventral roots (L) compared with nonligated L5 ventral roots (U) of non-Tg as well as the hWT and RW mice at 2 (A) and 12 (B) months of age. Samples were resolved on 7.5% SDS-PAGE and blotted with the 17026 antibody. Equal volumes of sample (10 μl) were loaded in all lanes. Non-Tg mice showed no detectable signals in the ventral roots (A, B). C, A histogram demonstrates the signal intensity of tau proteins from Western blots in the ligated and nonligated ventral roots of the non-Tg, hWT, and RW Tg mice. D, E, These histograms show the ratio of tau (D) or NFM (E) proteins in the ligated verse nonligated ventral roots. The ratio of tau in RW Tg mice was significantly lower than that in hWT Tg mice at both 2 and 12 months of age (D). In contrast, there was no significant difference in the ratio of NFM among non-Tg and hWT and RW Tg mice at 2 and 12 months of age (E). *p < 0.05; **p < 0.01.
Retardation of slow axonal transport of tau in the RW Tg mice leads to the accumulations of newly synthesized tau in a less soluble pool
To determine whether or not the reduced binding of RW tau to MTs and the reduced transport of this mutant tau isoform promote aggregation of RW tau in neuronal perikarya into a less soluble pool, we assessed the fate of newly synthesized RW and hWT tau in three lines of RW mice and one line of hWT mice. Seven days after [35S]methionine injection into the spinal ventral horns of RW and hWT mice, tau proteins in the spinal cords were sequentially extracted with RAB hi-salt buffer, 2% SDS, and 70% FA, and tau in each fraction was collected by immunoprecipitation with the anti-tau antibody 17026. Although the amount of newly synthesized radiolabeled tau in the RAB hi-salt buffer fractions from ventral horn of the spinal cord are comparable in RW lines 37+/+, RW 65+/+, and hWT mice, almost twice as much RW tau was recovered from line 60+/- when compared with hWT mice (Fig. 5A). However, significant increases in newly synthesized tau proteins were found in the SDS-extracted fractions from ventral horn of the spinal cord in all three lines of RW Tg mice compared with age-matched hWT mice (Fig. 5A,B). We did not detect any tau protein in the FA fractions from both RW and hWT mice because the majority of the insoluble tau was recovered after extraction with 2% SDS. These results suggest that reduced transport of newly synthesized RW tau proteins results in their accumulation in a less soluble pool in the spinal cord.
Figure 5.

Increased accumulation of newly synthesized tau as a less soluble pool in the spinal cords of the RW Tg mice. A, Sequential extraction of tau with RAB hi-salt buffer, followed by 2% SDS 7 d after [35S]methionine injection into ventral horns of the spinal cords of three different RW lines and age-matched hWT mice. Radiolabeled tau was collected by immunoprecipitation with the anti-tau antibody 17026. B, Bar graph shows significant increases in newly synthesized tau proteins in the SDS fractions of ventral horn of the spinal cord in all three lines of RW Tg mice compared with age matched hWT mice. n = 18. **p < 0.01.
RW Tg mice accumulate tau-immunoreactive straight filaments in neuronal perikarya
To further characterize the neuronal tau inclusions in the Tg mice, we performed ultrastructural studies using preembedding immuno-EM. In both hWT and RW tau Tg mice, there was only diffuse perikayal tau staining without filamentous aggregates in neurons of the cerebral cortex, hippocampus, cerebellum, and spinal cord at 1 (Fig. 6A, B) to 6 months of age (data not shown). In contrast, straight filaments with diameters of 10-15 nm were detected in the perikarya of neurons in the above-mentioned CNS regions of 12-month-old RW mice (Fig. 6D-H), but similar tau filaments were not identified in age-matched hWT tau Tg animals (Fig. 6C). However, we did observe occasional tau-immunolabeled filaments in the hWT tau Tg mice over 18 months of age, consistent with previous studies (Ishihara et al., 2001b), but they were far less abundant than in 12-month-old RW tau Tg mice (data not shown). Significantly, none of these filamentous aggregates were recognized by antibodies to β-tubulin or NFL (data not shown), thereby indicating that these tau-immunolabeled filaments were composed exclusively of assembled tau without any associated NFs or MTs.
Figure 6.
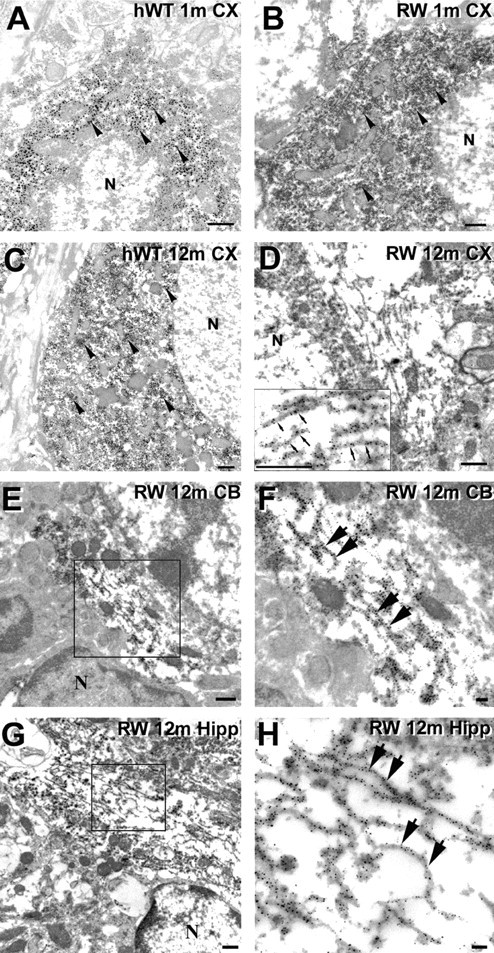
Intraneuronal tau inclusions in the RW tau Tg mice are composed of straight filaments. A-H, Preembedding immuno-EM using 17026 antibodies show tau immunostaining in neuronal perikarya of the hWT and RW tau Tg mice. Neocortical neurons of a 1-month-old hWT mouse (A), a 1-month-old RW mouse (B), and a 12-month-old hWT mouse (C) demonstrate diffuse tau staining (arrowheads) but no obvious tau filaments. D, Randomly oriented tau-positive straight filaments are demonstrated in the perikaryon of a neocortical neuron of a 12-month-old RW tau Tg mouse, and a higher-power view of these filaments (small arrows) is shown in the inset. E-H, Preembedding immuno-EM using the 17026 antibody shows tau-immunoreactive straight filaments in neurons from several CNS areas of a 12-month-old RW mouse. Higher-power views of the tau filaments (large arrows in F, H) are from the boxed areas in the adjacent panels showing lower-power views (E, G). The tau-immunoreactive filaments were found in the cerebellum (E, F) and hippocampus (G, H). N, Nucleus. Scale bars: A-E, G, 500 nm; F, H, 100 nm.
Lifespan is shorter in the RW versus hWT tau Tg mice
The RW 37+/+ mice had a shorter average lifespan than the hWT mice. Whereas 90% of hWT mice survived to 24 months of age, only 55% of RW 37+/- tau Tg mice survived this long, and only 10% of the RW 37+/+ tau Tg mice survived for 2 years, although they expressed tau levels that were comparable with those in the hWT tau Tg mice that survived 24 months (Fig. 7). In addition, the RW mice developed progressive motor weakness with advancing age, as demonstrated by dystonic movements of their hindlimbs when lifted by their tails (data not shown). One-third of RW+/+ tau Tg mice showed this phenotype by ∼12 months of age. Similar motor impairments were not observed in the non-Tg or hWT tau Tg mice, suggesting that there is gene dose-dependent neurotoxic effect of the R406W mutation, although the nature of this toxicity is unclear.
Figure 7.

RW tau Tg mice show reduced longevity. Longevity of hWT and RWL37 tau Tg mice was studied in cohorts of pups weaned at 1 month of age, and survival ratio of mice in each cohort was assessed at 6, 12, 18, and 24 months of age (n = 20 mice per age group). Approximately 90 and 55% of hWT+/- and RWL37+/- tau Tg mice survived up to 24 months of age, respectively, and 40% of RWL37+/+ tau Tg mice died by 12 months of age.
Discussion
The present study provides compelling data to implicate a distinct molecular mechanism underlying the pathogenesis of neuronal tau inclusions and the ensuing onset and progression of a neurodegenerative tauopathy in a Tg mouse model of human FTDP-17. Specifically, overexpression of RW tau in Tg mice resulted in a marked alteration in the translocation and distribution of this tau mutant in CNS neurons of these mice at the earliest ages examined here, and this was followed by an age-dependent progressive accumulation of filamentous tau inclusions in the perikarya of these CNS neurons. Similar accumulations of neuronal RW tau as filamentous inclusions also were detected in two previous studies overexpressing the RW mutation in Tg mice (Lim et al., 2001; Tatebayashi et al., 2002), but the mechanisms underlying this were not addressed. Thus, similar to FTDP-17 patients harboring the R406W tau gene mutation, the RW mice developed a neurodegenerative tauopathy with advancing age that was characterized neuropathologically by filamentous intraneuronal tau inclusions that resembled the NFTs observed in AD as well as in sporadic and hereditary human tauopathies, including FTDP-17. Although other tau Tg mice with a neurodegenerative tauopathy phenotype have been reported, we demonstrate for the first time here that the R406W tau gene mutation leads to a selective impairment in the axonal transport of this human tau mutant in the RW mice, and this observation suggests a novel mechanism to account for the pathogenesis of the tau inclusions in these mice.
Previous studies have shown that normal tau proteins move primarily at 0.2-0.4 mm/d in the slow component of axonal transport in vivo (Mercken et al., 1995). Our analysis of slow axonal transport in the tau Tg mice described here demonstrated that newly synthesized human RW and hWT tau proteins were translocated from neuronal perikarya into axons but that the slow axonal transport of RW tau was retarded compared with that of hWT tau. However, the RW mice did not show a generalized disturbance of slow axonal transport because the slow axonal transport of NFL and NFM were similar in both the RW and the hWT mice. In contrast to the RW mice, we reported previously that fast axonal transport was retarded in Tg mice that expressed very high levels of the shortest human tau isoform (more than sevenfold) and developed a neurodegenerative tauopathy phenotype (Ishihara et al., 1999). However, in these tau Tg mice, we showed that the impairment in fast axonal transport was caused by the formation of tau axonal spheroids and a reduction in MTs. Retardation of fast axonal transport also was described in Tg mice overexpressing four repeat tau isoforms (Künzi et al., 2002). Other studies reported recently showed that the overexpression of tau in cultured neuronal cells blocked the axonal transport of NFs and other organelles, but the authors of this study suggested that this is most likely attributable to the physical obstruction of axonal transport by the increased binding of the overexpressed tau to MTs (Stamer et al., 2002). Unlike both of the scenarios described above, the primary defect of the RW mutant tau in the RW mice is reduced binding of the mutant tau to MTs because this mutant tau preferentially accumulates in neuronal perikarya rather than in axons and the slow axonal transport of other proteins is not impaired. Our data presented here differ significantly from a recent slow axonal transport study of WT and mutant tau in cultured cortical neurons in which the authors did not find any differences in the rates of tau transport (Utton et al., 2002). The differences between the two studies are numerous and may be attributable to the following: (1) the different model systems used in the two studies, i.e., immature cultured neurons versus mature as well as aged Tg mice; (2) the transport of enhanced green fluorescent protein (EGFP)-tagged tau proteins in the previous study versus untagged tau protein in our study; (3) the time course of tau transport in cultured cortical neurons (in hours) versus transport in ventral roots of spinal cord neurons in intact animals (in days); and (4) expression of EGFP-tagged tau by transfection in a few neurons versus the stable and permanent expression of transgenic tau in mice. These and other factors may explain why Utton et al. (2002) did not detect differences in the transport rate of wild-type and mutant tau proteins.
Other insights into the consequences of FTDP-17 tau gene mutations emerged from studies on the RW mutant tau in cultured cells as well as in Tg mice. For example, we and others have shown that the R406W mutant tau, when overexpressed in cultured cells, in the RW mice and in FTDP-17 patients with the R406W tau mutation was less phosphorylated, especially at Ser396 and Ser404, phosphorylation sites that are in close proximity to this missense mutation (Perez et al., 2000; Vogelsberg-Ragaglia et al., 2000; Miyasaka et al., 2001). These data suggest that the R406W tau gene mutation may induce a conformational change at sites flanking this mutation because this structural and/or conformational change in tau probably affects the MT-binding domains and could lead to reduced MT binding and its ability to promote MT assembly. Indeed, our data demonstrate that RW mutant tau bind less well to endogenous mouse brain MTs than hWT tau is consistent with previous in vitro MT binding and assembly assays as well as cell culture studies in which the R406W tau gene mutation markedly reduces the ability of tau to bind MTs and promote MT assembly when compared with other tau mutants or wild-type tau proteins (Hong et al., 1998; Rizzu et al., 1999; Barghorn et al., 2000; Vogelsberg-Ragaglia et al., 2000). Thus, we suggest that a reduction in the MT-binding ability of R406W mutant tau attributable to the conformational change proposed here contributes to the selective retardation in the slow axonal transport of tau in the RW mice. The impaired axonal transport of RW tau causes its accumulation in the somato-dendritic domain of neurons, wherein it aggregates over time into filamentous inclusions that eventually lead to axonal degeneration.
This scenario may account for the fact that the RW tau Tg mice progressively develop fibrillary tau inclusions in the perikarya of CNS neurons with advancing age, and these tau lesions closely resemble authentic human NFTs observed in AD, FTDP-17, and other tauopathies. For example, in addition to being filamentous and tau immunoreactive, some tangle-like inclusions were stained with the amyloid binding dye thioflavin S, which indicates that the tau filaments in these inclusions form β-pleated sheet structures similar to those in the NFTs of human neurodegenerative diseases. Moreover, because the insoluble tau aggregates and NFT-like inclusions in the RW mice are composed solely of mutant human tau without any endogenous mouse tau, we conclude that R406W mutant tau forms NFT-like filamentous tau lesions by itself. However, at the ultrastructural level, the neuronal cytoplasmic tau lesions in the RW mice are composed primarily of straight filaments rather than the paired helical filaments as observed in the brains of patients with AD or with FTDP-17 caused by an R406W tau gene mutation (Reed et al., 1997). Thus, these results suggest that all six tau isoforms may be required for the formation of authentic paired helical filaments with the characteristic morphology.
Because other FTDP-17 tau gene mutations in addition to the R406W substitution also reduce the binding of tau to MTs (Hong et al., 1998), the data presented here may indicate that impaired transport of tau into axons is one of the primary molecular mechanisms that result in the formation of neuronal cytoplasmic tau lesions in FTDP-17. Moreover, because the phosphorylation of tau has been shown to reduce the ability of tau to bind to MTs (Lovestone et al., 1996; Hong and Lee, 1997), our data provide a plausible explanation for the abnormal redistribution of tau from axons to neuronal perikarya in AD and diverse neurodegenerative tauopathies that are characterized by large accumulations of hyperphosphorylated tau proteins. Future transport studies on Tg mice with other tau gene mutations should resolve this issue.
In summary, our findings in the RW tau Tg mice suggest that the R406W tau gene mutation promotes the formation of filamentous tau lesions with advancing age in neuronal perikarya by a novel mechanism of disease that may be initiated by a mutation-induced impairment of MT binding. The altered MT binding leads to retardation in the slow axonal transport of tau, followed by a cascade of events that include the perikaryal accumulation of insoluble tau to culminate in the fibrillization of tau and the formation of NFTs. Although this mechanistic scenario could be associated exclusively with the R406W tau gene mutation, we speculate that similar types of processes or variations of this scenario may underlie AD as well as other sporadic and hereditary neurodegenerative tauopathies.
Footnotes
These studies were supported by grants from the National Institute on Aging and the Oxford Foundation. V.M.-Y.L. is the John H. Ware III Professor for Alzheimer's Disease Research, and J.Q.T. is the William Maul Measy-Truman G. Schnabel Jr MD Professor of Geriatric Medicine and Gerontology. We appreciate technical assistance from personnel in the Biomedical Imaging Core Facility of The University of Pennsylvania.
Correspondence should be addressed to Dr. Virginia M.-Y. Lee, Department of Pathology and Laboratory Medicine, University of Pennsylvania School of Medicine, Maloney 3, HUP, 3600 Spruce Street, Philadelphia, PA 19104-4283. E-mail: vmylee@mail.med.upenn.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244657-11$15.00/0
References
- Barghorn S, Zheng-Fischhofer Q, Ackmann M, Biernat J, von Bergen M, Mandelkow E, Mandelkow E (2000) Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 39: 11714-11721. [DOI] [PubMed] [Google Scholar]
- Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo MG, Spillantini F, Tagliavini M, Goedert M, Ghetti B (1999) Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol 58: 667-677. [DOI] [PubMed] [Google Scholar]
- Clark LN, Poorkaj P, Wszolek Z, Geschwind DH, Nasreddine ZS, Miller B, Li D, Payami H, Awert F, Markopoulou K, Andreadis A, D'Souza I, Lee VM-Y, Reed L, Trojanowski JQ, Zhukareva V, Bird T, Schellenberg G, Wilhelmsen KC (1998) Pathogenic implications of mutations in the tau gene in pallidopontonigral degeneration and related neurodegenerative disorders linked to chromosome 17. Proc Natl Acad Sci USA 95: 13103-13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JW, Gibb GM, Betts JC, Blackstock JC, Gallo J, Lovestone S, Hutton M, Anderton BH (2001) Effects of FTDP-17 mutations on the in vitro phosphorylation of tau by glycogen synthase kinase 3β identified by mass spectrometry demonstrate certain mutations exert long-range conformational changes. FEBS Lett 493: 40-44. [DOI] [PubMed] [Google Scholar]
- Crowther RA, Goedert M (2000) Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol 130: 271-279. [DOI] [PubMed] [Google Scholar]
- D'Souza I, Poorkaj P, Hong M, Nochlin D, Lee VM-Y, Bird TD, Schellenberg GD (1999) Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc Natl Acad Sci USA 96: 5598-5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delisle MB, Murrell JR, Richardson R, Trofatter JA, Rascol O, Soulages X, Mohr M, Calvas P, Ghetti B (1999) A mutation at codon 279 (N279K) in exon 10 of the tau gene causes a tauopathy with dementia and supranuclear palsy. Acta Neuropathol (Berl) 98: 62-77. [DOI] [PubMed] [Google Scholar]
- Delobel P, Flament S, Hamdane M, Jakes R, Rousseau A, Delacourte A, Vilain JP, Goedert M, Buee L (2002) Functional characterization of FTDP-17 tau gene mutations through their effects on Xenopus oocyte maturation. J Biol Chem 277: 9199-9205. [DOI] [PubMed] [Google Scholar]
- Dumanchin C, Camuzat A, Campion D, Verpillat P, Hannequin D, Dubois B, Saugier-Veber P, Martin C, Penet C, Charbonnier F, Agid Y, Frebourg T, Brice A (1998) Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and Parkinsonism. Hum Mol Genet 7: 1825-1829. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, Barmettler R, Nitsch RM (2001) tau filament formation in transgenic mice expressing P301L tau. J Biol Chem 276: 529-534. [DOI] [PubMed] [Google Scholar]
- Grover A, Houlden H, Baker M, Adamson J, Lewis J, Prihar G, Pickering-Brown S, Duff K, Hutton M (1999) 5′ splice site mutations in tau associated with the inherited dementia FTDP-17 affect a stem-loop structure that regulates alternative splicing of exon 10. J Biol Chem 274: 15134-15143. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Goedert M (1998) Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett 437: 207-210. [DOI] [PubMed] [Google Scholar]
- Hasegawa M, Smith MJ, Iijima M, Tabira T, Goedert M (1999) FTDP-17 mutations N279K and S305N in tau produce increased splicing of exon 10. FEBS Lett 443: 93-96. [DOI] [PubMed] [Google Scholar]
- Hong M, Lee VM-Y (1997) Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem 272: 19547-19553. [DOI] [PubMed] [Google Scholar]
- Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BI, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VM-Y (1998) Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 282: 1914-1917. [DOI] [PubMed] [Google Scholar]
- Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Reed L, Trojanowski JQ, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J, Hillebrand M, Joosse M, Kwon JM, Nowotny P, Heutink P (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393: 702-705. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VM-Y (1999) Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron 24: 751-762. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Higuchi M, Zhang B, Yoshiyama Y, Hong M, Trojanowski JQ, Lee VM-Y (2001a) Attenuated neurodegenerative disease phenotype in tau transgenic mouse lacking neurofilaments 1. J Neurosci 21: 6026-6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Zhang B, Higuchi M, Yoshiyama Y, Trojanowski JQ, Lee VM-Y (2001b) Age-dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am J Pathol 158: 555-562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Künzi V, Glatzel M, Nakano MY, Greber UF, Leuven FV, Aguzzi A (2002) Unhampered prion neuroinvasion despite impaired fast axonal transport in transgenic mice overexpressing four-repeat tau. J Neurosci 22: 7471-7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM-Y, Balin BJ, Otvos Jr L, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal tau. Science 251: 675-678. [DOI] [PubMed] [Google Scholar]
- Lee VM-Y, Goedert M, Trojanowski JQ (2001) Neurodegenerative tauopathies. Annu Rev Neurosci 24: 1121-1159. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, van Slegtenhorst M, Gwinn-Hardy K, Paul MM, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: 402-405. [DOI] [PubMed] [Google Scholar]
- Lim F, Hernández F, Lucas JJ, Gómez-Ramos P, Morán MA, Ávila J (2001) FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and tau filaments in forebrain. Mol Cell Neurosci 18: 702-714. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Hartley CL, Pearce J, Anderton BH (1996) Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: the effects on the organization and stability of microtubules. Neuroscience 73: 1145-1157. [DOI] [PubMed] [Google Scholar]
- Mercken M, Fisher I, Kosik KS, Nixon RA (1995) Three distinct axonal transport rates for tau, tubulin, and other microtubule-associated proteins: evedence for dynamic interactions of tau with microtubules in vivo J Neurosci 15: 8259-8267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka T, Morishima-Kawashima M, Ravid R, Heutink P, van Swieten JC, Nagashima K, Ihara Y (2001) Molecular analysis of mutant and wild-type tau deposited in the brain affected by the FTDP-17 R406W mutation. Am J Pathol 158: 373-379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell JR, Spillantini MG, Zolo P, Guazzelli M, Smith MJ, Hasegawa M, Redi F, Crowther RA, Pietrini P, Ghetti B, Goedert M (1999) tau gene mutation G389R causes a tauopathy with abundant pick body-like inclusions and axonal deposits. J Neuropathol Exp Neurol 58: 1207-1226. [DOI] [PubMed] [Google Scholar]
- Perez M, Lim F, Arrasate M, Avila J (2000) The FTDP-17-linked mutation R406W abolishes the interaction of phosphorylated tau with microtubules. J Neurochem 74: 2583-2589. [DOI] [PubMed] [Google Scholar]
- Pickering-Brown S, Baker M, Yen SH, Liu WK, Hasegawa M, Cairns N, Lantos PL, Rossor M, Iwatsubo T, Davies Y, Allsop D, Furlong R, Owen F, Hardy J, Mann D, Hutton M (2000) Pick's disease is associated with mutations in the tau gene. Ann Neurol 48: 859-867. [PubMed] [Google Scholar]
- Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD (1998) tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 43: 815-825. [DOI] [PubMed] [Google Scholar]
- Probst A, Gotz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Lee VM-Y, Trojanowski JQ, Ishihara T, Hong M, Jakes R, Crowther RA, Spillantini MG, Burki K, Goedert M (2000) Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol 99: 469-481. [DOI] [PubMed] [Google Scholar]
- Reed LA, Grabowski TJ, Schmidt ML, Morris JC, Goate A, Solodkin A, Van Hoesen GW, Schelper RL, Talbot CJ, Wragg MA, Trojanowski JQ (1997) Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol 42: 564-572. [DOI] [PubMed] [Google Scholar]
- Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG (2000) tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol 59: 990-1001. [DOI] [PubMed] [Google Scholar]
- Rizzu P, Van Swieten JC, Joosse M, Hasegawa M, Stevens M, Tibben A, Niermeijer MF, Hillebrand M, Ravid R, Oostra BA, Goedert M, van Duijn CM, Heutink P (1999) High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in The Netherlands. Am J Hum Genet 64: 414-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahara N, Tomiyama T, Mori H (2000) Missense point mutations of tau to segregate with FTDP-17 exhibit site-specific effects on microtubule structure in COS cells: a novel action of R406W mutation. J Neurosci Res 60: 380-387. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B (1998) Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA 95: 7737-7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Yoshida H, Rizzini C, Lantos PL, Khan N, Rossor MN, Goedert M, Brown J (2000) A novel tau mutation (N296N) in familial dementia with swollen achromatic neurons and corticobasal inclusion bodies. Ann Neurol 48: 939-943. [DOI] [PubMed] [Google Scholar]
- Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F (1999) Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol 155: 2153-2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM (2002) tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156: 1051-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford PM, Halliday GM, Brooks WS, Kwok JB, Storey CE, Creasey H, Morris JG, Fulham MJ, Schofield PR (2000) Progressive supranuclear palsy pathology caused by a novel silent mutation in exon 10 of the tau gene: expansion of the disease phenotype caused by tau gene mutations. Brain 123: 880-893. [DOI] [PubMed] [Google Scholar]
- Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ichikawa M, Yamaguchi H, Takashima A (2002) Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci 22: 133-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatebayashi Y, Miyasaka T, Chui D-H, Akagi T, Mishima K-I, Iwasaki K, Fujiwara M, Tanemura K, Murayama M, Ishiguro K, Planel E, Sato S, Hashikawa T, Takashima A (2002) tau filament formation and associative memory deficit in aged mice expressing mutant (R406W) human tau. Proc Natl Acad Sci USA 99: 13896-13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VM-Y (1995) Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol 129: 1629-1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu PH, Raju P, Robinson KA, Gurney ME, Trojanowski JQ, Lee VM-Y (1996) Transgenic mice carrying a human mutant superoxide dismutase transgene develop neuronal cytoskeletal pathology resembling human amyotrophic lateral sclerosis lesions. Proc Natl Acad Sci USA 93: 3155-3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utton MA, Connell J, Asuni AA, Slegtenhorst MV, Hutton M, Silva RD, Lees AJ, Miller CCJ, Anderton BH (2002) The slow axonal transport of the microtubule-associated protein tau and the transport rates of different isoforms and mutants in cultured neurons. J Neurosci 22: 6394-6400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Swieten JC, Stevens M, Rosso SM, Rizzu P, Joosse M, de Koning I, Kamphorst W, Ravid R, Spillantini MG, Niermeijer MF, Heutink P (1999) Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol 46: 617-626. [DOI] [PubMed] [Google Scholar]
- Varani L, Hasegawa M, Spillantini MG, Smith MJ, Murrell JR, Ghetti B, Klug A, Goedert M, Varani G (1999) Structure of tau exon 10 splicing regulatory element RNA and destabilization by mutations of frontotemporal dementia and parkinsonism linked to chromosome 17. Proc Natl Acad Sci USA 96: 8229-8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelsberg-Ragaglia V, Bruce J, Richter-Landsberg C, Zhang B, Hong M, Trojanowski JQ, Lee VM-Y (2000) Distinct FTDP-17 missense mutations in tau produce tau aggregates and other pathological phenotypes in transfected CHO cells Mol Biol Cell 11: 4093-4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M, Kawamata T, Komure O, Kuno S, D'Souza I, Poorkaj P, Kawai J, Tanimukai S, Yamamoto Y, Hasegawa H, Sasahara M, Hazama F, Schellenberg GD, Tanaka C (1999) A mutation in the microtubule-associated protein tau in pallido-nigro-luysian degeneration. Neurology 53: 864-868. [DOI] [PubMed] [Google Scholar]
- Yasuda M, Takamatsu J, D'Souza I, Crowther RA, Kawamata T, Hasegawa M, Hasegawa H, Spillantini MG, Tanimukai S, Poorkaj P, Varani L, Varani G, Iwatsubo T, Goedert M, Schellenberg DG, Tanaka C (2000) A novel mutation at posititon +12 in the intron following exon 10 of the tau gene in familial frontotemporal dementia (FTD-Kumamoto). Ann Neurol 47: 422-429. [PubMed] [Google Scholar]
- Zhang B, Tu P, Abtahian F, Trojanowski JQ, Lee VM-Y (1997) Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J Cell Biol 139: 1307-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]