

Abstract
Alzheimer's disease (AD), the most common neurodegenerative disorder, results in progressive degeneration of synapses and aberrant sprouting of axon terminals. The mechanisms underlying these seemingly opposing cellular phenomena are unclear. We hypothesized that Fyn kinase may play a role in one or both of these processes because it is increased in AD brains and because it is involved in synaptic plasticity and axonal outgrowth. We investigated the effects of Fyn on AD-related synaptotoxicity and aberrant axonal sprouting by ablating or overexpressing Fyn in human amyloid precursor protein (hAPP) transgenic mice.
On the fyn+/+ background, hAPP/amyloid β peptide (Aβ) decreased hippocampal levels of synaptophysin-immunoreactive presynaptic terminals (SIPTs), consistent with previous findings. On the fyn-/- background, hAPP/Aβ did not affect SIPTs. SIPT reductions correlated with hippocampal Aβ levels in hAPP/fyn+/+, but not hAPP/fyn-/-, mice suggesting that Fyn provides a critical link between hAPP/Aβ and SIPTs. Furthermore, overexpression of Fyn exacerbated SIPT reductions in hAPP mice. We also found that the susceptibility of mice to hAPP/Aβ-induced premature mortality was decreased by Fyn ablation and increased by Fyn overexpression. In contrast, axonal sprouting in the hippocampus of hAPP mice was unaffected. We conclude that Fyn-dependent pathways are critical in AD-related synaptotoxicity and that the pathogenesis of hAPP/Aβ-induced neuronal alterations may be mechanistically heterogenous.
Keywords: Alzheimer's disease, amyloid β, Fyn kinase, synaptic deficits, signaling, sprouting, GAP-43, neurodegeneration
Introduction
The amyloid precursor protein (APP) is expressed in many cell types and is particularly abundant in synapses. One of its metabolites, amyloid β peptide (Aβ), suppresses synaptic transmission and may participate in the regulation of neuronal activity (Hsia et al., 1999; Kamenetz et al., 2003). Mutations in the human APP (hAPP) gene that lead to increased levels of Aβ cause autosomal dominant familial Alzheimer's disease (FAD) (Selkoe and Schenk, 2003). Of several species of Aβ produced, the 42-residue form (Aβ1-42) is particularly susceptible to aggregation and is a primary constituent of neuritic amyloid plaques, pathological hallmarks of the disease (Selkoe and Schenk, 2003). Both FAD and sporadic AD are associated with a progressive degeneration of neurons and synapses (DeKosky and Scheff, 1990; Terry et al., 1999) and an aberrant sprouting of axon terminals (Geddes et al., 1985; Masliah et al., 1991; Arendt, 2001). The precise relationship between hAPP, Aβ, plaques, synaptic loss, axonal sprouting, and cognitive decline in AD is unknown.
Although amyloid plaques are a diagnostic feature of AD, growing evidence suggests that plaques may not be the primary cause of AD-related synaptic alterations and cognitive decline. In transgenic (TG) mice overexpressing hAPP/Aβ in neurons, synaptic and behavioral deficits are detectable well before plaque formation (Holcomb et al., 1999; Hsia et al., 1999; Mucke et al., 2000; Raber et al., 2000; Buttini et al., 2002; Westerman et al., 2002; Palop et al., 2003). Aβ1-42 can form small neurotoxic assemblies that act intracellularly or extracellularly and could impair neuronal and synaptic functions independent of plaques (Lambert et al., 1998; Caughey and Lansbury, 2003). Aβ interacts with a number of cell surface receptors on neurons, including integrins (Sabo et al., 1995; Bi et al., 2002),α7 nicotinic ACh receptors (Dineley et al., 2001), and the p75 neurotrophin receptor (Yaar et al., 1997). Additionally, several intracellular molecules have been implicated in Aβ-induced alterations of signaling cascades that could result in the dysfunction or degeneration of neurons (Klein, 2000; Small et al., 2001; Williamson et al., 2002; Grace and Busciglio, 2003).
One of these intracellular molecules is Fyn kinase, a member of the src family of nonreceptor tyrosine kinases. Fyn immunoreactivity (IR) is increased in AD brains (Shirazi and Wood, 1993), and genetic ablation of Fyn rendered hippocampal slices resistant to the neurotoxic effects of Aβ oligomers (Lambert et al., 1998). Here, we used genetic modulations of Fyn (overexpression vs ablation) to test the hypotheses that Fyn is involved in degenerative (synaptic deficits) and trophic (aberrant sprouting) effects of hAPP/Aβ.
Materials and Methods
TG mice. hAPPFAD TG lines J20 and J9 produce hAPP carrying Swedish (K670N, M671L) and Indiana (V717F) FAD mutations (hAPP770 numbering) (Rockenstein et al., 1995; Mucke et al., 2000). Lines were crossed for 6-10 generations onto a C57BL/6 background using mice from The Jackson Laboratory (Bar Harbor, ME). Heterozygous FYN TG mice (line N8) overexpress wild-type mouse Fyn directed by the calcium/calmodulin-dependent protein kinase IIα promoter on a C57BL/6 background (Kojima et al., 1997, 1998). Fyn-/- mice (Grant et al., 1992) on a 129/SVJ background were obtained from The Jackson Laboratory. Measurements were performed on gender-balanced groups. Mice were anesthetized and flush-perfused transcardially with PBS. Hemibrains were fixed in 4% phosphate-buffered paraformaldehyde or stored at -70°C.
Growth-associated protein 43 analysis. Vibratome (50 μm) or sliding microtome (30 μm) sections were avidin-biotin/immunoperoxidase stained using anti-growth-associated protein 43 (GAP-43) (1:400; Sigma, St. Louis, MO), biotinylated donkey anti-mouse (1:500; Jackson ImmunoResearch, West Grove, PA), and DAB. For quantitation of GAP-43 IR, two sections per mouse were used. In each section, the integrated optical density of staining in two areas of the outer molecular layer (OML) and two areas of the middle molecular layer (MML) of the dentate gyrus was determined by BioQuant Image Analysis (R&M Biometrics, Nashville, TN). Measurements for each area were averaged and used to calculate group means.
Synaptophysin-immunoreactive presynaptic terminal analysis. Vibratome sections (50 μm) were labeled with anti-synaptophysin (1 μg/ml; Boehringer Mannheim, Indianapolis, IN) and FITC-conjugated horse anti-mouse IgG (1:75; Vector Laboratories, Burlingame, CA) and imaged by confocal microscopy (MRC1024; Bio-Rad, Hercules, CA). For each experiment, we first determined the linear range of the fluorescence intensity of synaptophysin-immunoreactive presynaptic terminals (SIPTs) in control sections from nontransgenic (NTG) wild-type mice. This setting was then used to collect all images analyzed in the same experiment. Sections were blind coded and processed in parallel. Twelve confocal images of the molecular layer of the dentate gyrus were obtained from three sections per mouse and analyzed with NIH Image software. The area occupied by SIPTs of defined signal intensity was quantified and expressed as a percentage of the total image area, as described (Masliah et al., 1992; Buttini et al., 1999). Previous studies indicated that SIPT measurements are not significantly influenced by genetic backgrounds (Mucke et al., 1994).
Aβ ELISAs. Snap-frozen hippocampi were homogenized in guanidine buffer, and Aβ peptides were quantitated by ELISA, as described (Johnson-Wood et al., 1997).
Plaque quantitation. Vibratome sections were immunoperoxidase labeled with an Elite kit (Vector Laboratories), using biotinylated anti-Aβ1-5 (5 μg/ml, 3D6; Elan Pharmaceuticals, South San Francisco, CA) (Johnson-Wood et al., 1997) and DAB, and counterstained with 1% hematoxylin. The percentage of area of the hippocampus covered by 3D6 IR was determined with a Quantimet 570C (Leica, Deerfield, IL). Three sections were analyzed per mouse, and the average was used to calculate group means.
Statistical analyses. Statistical analyses were performed with Statview 5.0 (SAS Institute, Cary, NC). Differences between means were assessed by Student's t test or two-factor ANOVA, followed by the Tukey-Kramer post hoc test. Correlations were assessed by simple regression analysis.
Results
Modulating Fyn levels alters hAPPFAD/Aβ-induced synaptotoxicity
We examined SIPT levels in hAPPFAD mice in the context of wild-type, increased, or absent expression of Fyn. First, we crossed heterozygous FYN TG mice in which wild-type mouse Fyn is overexpressed in the forebrain (Kojima et al., 1997, 1998) with heterozygous TG mice from the low hAPPFAD/Aβ expresser line J9 (Hsia et al., 1999; Mucke et al., 2000), resulting in fyn+/+ littermates of four genotypes: hAPPlow/FYN, hAPPlow, FYN, and NTG.
Overexpression of Fyn worsened SIPT reductions in hAPPlow mice (Fig. 1A). Both hAPPlow (p < 0.0001) and FYN (p < 0.001) had a clear effect on SIPT levels. Post hoc analysis revealed that SIPT levels were significantly lower in doubly TG hAPPlow/FYN mice than in singly TG hAPPlow (p < 0.05) or FYN (p < 0.001) mice (Fig. 1B). Although the hAPPlow-induced SIPT reductions were relatively modest, we have previously shown that such SIPT reductions are accompanied by major reductions in synaptic transmission strength, which underscores their pathophysiological significance (Hsia et al., 1999).
Figure 1.
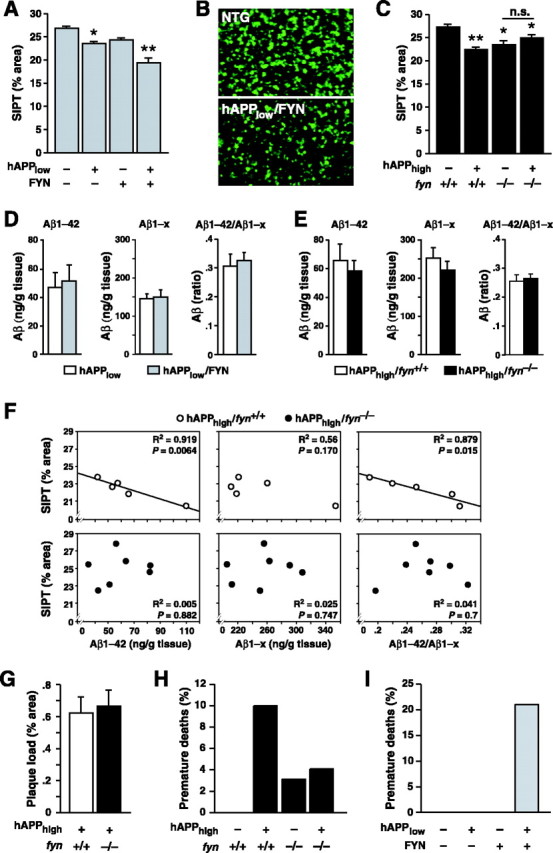
Genetic modulation of mouse Fyn alters the extent of hAPPFAD/Aβ-induced reductions in SIPT and premature mortality. FYN refers to the Fyn transgene, and fyn refers to the endogenous gene. n.s., Not significant. *p = 0.001; **p < 0.0005 versus NTG wild-type littermate controls. A, Overexpressing Fyn exacerbated SIPT reductions in hAPPlow mice (n = 5-9 mice/genotype; age, 6-8 months). B, Confocal microscopic images illustrating SIPT in the OML of an NTG mouse (top) and an hAPPlow/FYN doubly TG mouse (bottom). C, Ablating Fyn reduced SIPT levels but also prevented additional SIPT reductions in hAPPhigh mice (n = 5-8 mice/genotype; age, 4-5 months). D, Overexpressing Fyn did not alter Aβ levels in hAPPlow mice (n = 5-9 mice/genotype; age, 6-8 months). E, Ablating Fyn did not alter Aβ levels in hAPPhigh mice (n = 5-8 mice/genotype; age, 4-5 months). F, SIPT levels correlated inversely with Aβ1-42 levels and Aβ1-42/Aβ1-x ratios in hAPPhigh mice on the fyn+/+, but not fyn-/-, background (age, 4-5 months). G, Comparable plaque load in hAPPhigh mice on fyn+/+ and fyn-/- backgrounds (n = 7-8 mice/genotype; age, 8-10 months). H, I, Proportion of mice that died prematurely in each of the groups indicated during the first 6 months postnatally (percentage of 38-64 mice/genotype). Husbandry conditions were similar for all groups.
Next, we crossed Fyn-deficient (fyn-/-) mice (Grant et al., 1992) for two generations with heterozygous TG mice from the high hAPPFAD/Aβ expresser line J20 (Mucke et al., 2000; Palop et al., 2003), yielding hAPPhigh/fyn+/+, hAPPhigh/fyn-/-, fyn+/+, and fyn-/- mice. Although ablation of Fyn per se decreased SIPT levels compared with fyn+/+ littermates (Fig. 1C), there was a significant interaction between the hAPPhigh and fyn genotype (p < 0.0001), indicating that hAPPhigh differentially affects pre-synaptic terminals in the presence or absence of wild-type Fyn. Indeed, overexpression of hAPPhigh significantly reduced SIPT levels in fyn+/+, but not fyn-/-, mice (Fig. 1C).
Modulating Fyn levels does not alter Aβ levels but changes the relationship between Aβ and SIPT
To determine whether the genetic modulation of Fyn affects the production or metabolism of Aβ, we examined hippocampal Aβ levels by ELISA. The levels of Aβ1-42 and Aβ1-x (approximates total Aβ), and Aβ1-42/Aβ1-x ratios, were comparable in 6- to 8-month-old doubly TG hAPPlow/FYN mice and singly TG hAPPlow mice (Fig. 1D), suggesting that increased levels of Fyn do not alter the production or degradation of Aβ.
Aβ levels in 4- to 5-month-old hAPPhigh mice were also comparable on the fyn+/+ and fyn-/- backgrounds (Fig. 1E). Interestingly, SIPT levels at this age correlated inversely with Aβ1-42 levels and Aβ1-42/Aβ1-x ratios in hAPPhigh/fyn+/+ mice but not in hAPPhigh/fyn-/- mice (Fig. 1E), suggesting that Fyn is critical in linking Aβ with SIPT reductions.
At 4-5 months, hAPPhigh mice had no or only few plaques on the fyn+/+ or fyn-/- backgrounds (data not shown). We therefore compared plaque loads in hAPPhigh mice at 8-10 months of age, when all of them had plaques. Their hippocampal plaque loads were comparable on the fyn+/+ and fyn-/- backgrounds (Fig. 1G), suggesting that the differential effects of hAPPFAD/Aβ on these backgrounds were not attributable to modulation of Aβ deposition.
Premature death in hAPPFAD mice is modulated by Fyn
Some lines of hAPP mice exhibit premature mortality (Hsiao et al., 1995; Carlson et al., 1997), but the mechanisms remain unknown. Approximately 10% of our hAPPhigh/fyn+/+ mice died before 6 months of age (Fig. 1H), compared with 0% of NTG fyn+/+ controls. Although fyn-/- mice were slightly more susceptible to premature death than fyn+/+ mice, ablation of Fyn improved survival in hAPPhigh mice, and hAPPhigh did not increase premature mortality on the fyn-/- background (Fig. 1H). Notably, NTG and hAPPlow or FYN singly TG mice did not die prematurely, whereas 20% of hAPPlow/FYN doubly TG mice died during the first 6 months after birth (Fig. 1I).
Aberrant axonal sprouting in hAPPFAD mice depends on levels of hAPPFAD/Aβ expression
In AD, the loss of presynaptic terminals is associated with aberrant axonal sprouting (Geddes et al., 1985; Masliah et al., 1991; Arendt, 2001). To determine whether these processes are linked mechanistically, we analyzed axonal sprouting in our TG models with antibodies against GAP-43, which is expressed by many types of growing axons (Benowitz and Perrone-Bizzozero, 1991). hAPPhigh mice showed prominent age-dependent axonal sprouting in the molecular layer of the dentate gyrus (Fig. 2A,B). This layer contains the dendritic arbors of the dentate granule cells and pre-synaptic terminals of afferent projections from other regions such as the entorhinal cortex (glutamatergic) and the basal fore-brain and medial septum (cholinergic). At 4 months of age, hAPPhigh mice showed aberrant sprouting primarily in the OML (Fig. 2B). By 6 months, significant sprouting was observed in both the OML and the MML (Fig. 2A,B,D). In contrast, hAPPlow mice had no significant increase in axonal sprouting at 6 (Fig. 2C,E) or 20 (data not shown) months, suggesting that a threshold level of hAPPFAD/Aβ expression is required to elicit aberrant sprouting.
Figure 2.
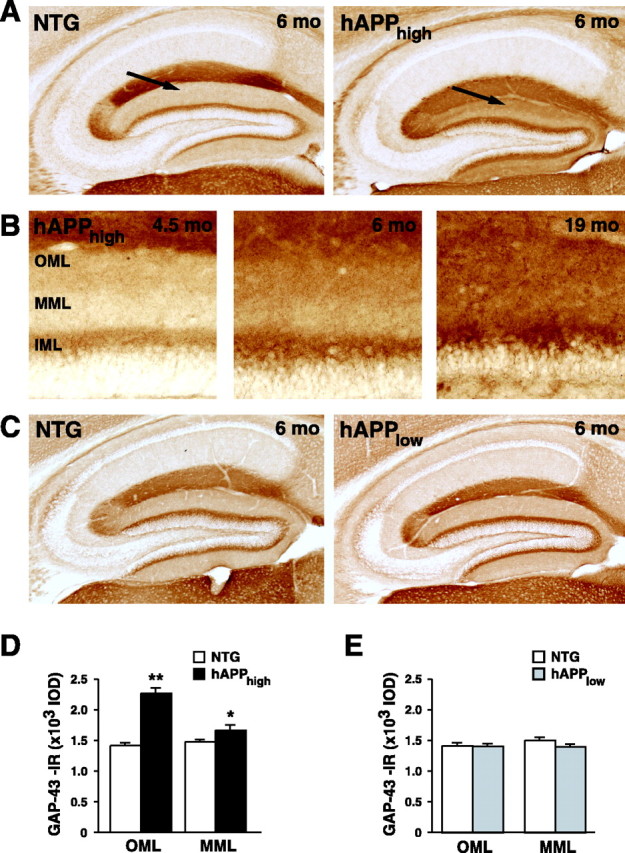
Aberrant axonal sprouting in hAPPFAD mice depends on age and hAPPFAD expression levels. A, Photomicrographs of GAP-43-IR in the hippocampus of a 6-month-old NTG mouse and an hAPPhigh TG littermate. Prominent aberrant sprouting is evident in the molecular layer of the hAPPhigh mouse (arrow). B, In hAPPhigh mice, GAP-43-IR in this layer further increased with age. C, Photomicrographs showing normal GAP-43-IR in the hippocampus of a 6-month-old NTG mouse and an hAPPlow TG littermate. D, Quantitation of GAP-43-IR in 6-month-old hAPPhigh mice revealed a 60% increase in the OML and a 13% increase in the MML (n = 10-13 mice/genotype). *p < 0.05; **p < 0.0001 versus NTG wild-type littermates. E, Quantitation of GAP-43-IR in the OML and MML revealed no aberrant sprouting in hAPPlow mice (n = 11-13 mice/genotype). mo, Months; IML, inner molecular layer.
Aberrant axonal sprouting in hAPPFAD mice is independent of Fyn kinase
Because genetic ablation of Fyn prevented additional SIPT reductions (Fig. 1C) and eliminated the inverse correlation between SIPT and Aβ levels in hAPPhigh mice (Fig. 1F), we examined whether ablation of Fyn also interfered with aberrant sprouting. Ablation of Fyn did not alter either the magnitude of sprouting (Fig. 3A) or the age at which sprouting became evident (data not shown). In 4- to 5-month-old mice, GAP-43 in the OML was unaffected by the fyn genotype but was increased by hAPPhigh on both fyn+/+ and fyn-/- backgrounds (p < 0.0001), and there was no GAP-43-related interaction between hAPPhigh and fyn. The magnitude of axonal sprouting in hAPPhigh mice did not correlate with hippocampal Aβ levels on fyn+/+ and fyn-/- backgrounds (Fig. 3B). Finally, overexpression of Fyn failed to elicit aberrant sprouting in hAPPlow/FYN doubly TG mice (Fig. 3C).
Figure 3.
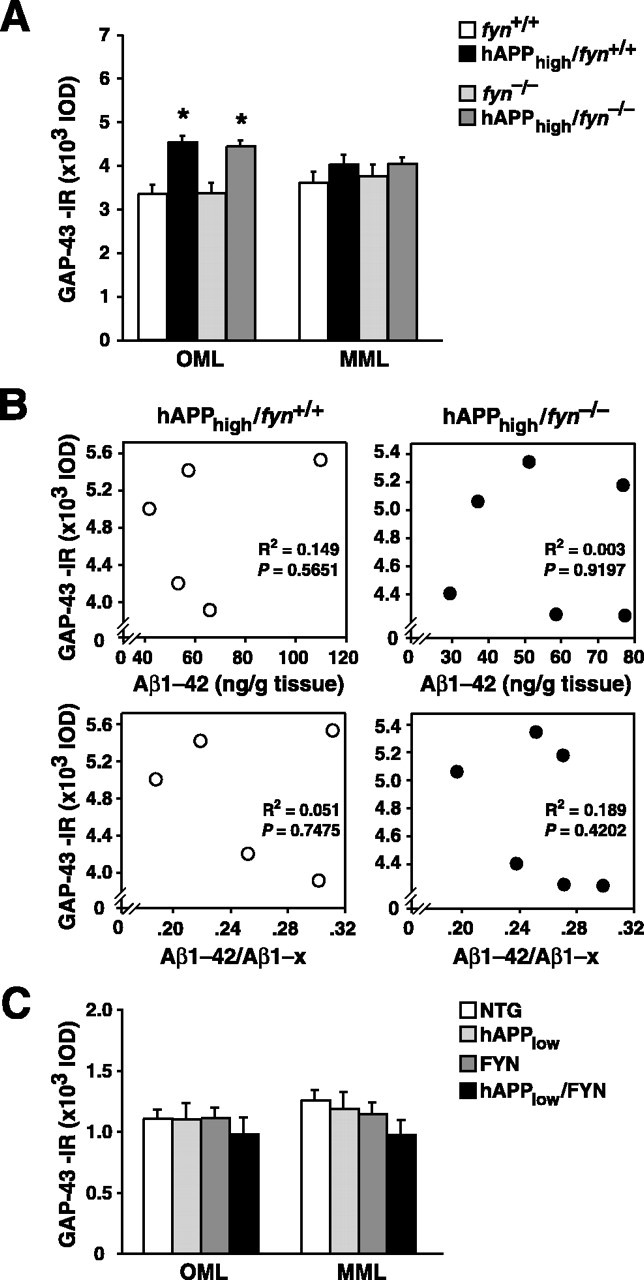
Genetic modulation of Fyn does not affect hAPP/Aβ-induced aberrant sprouting. A, Comparable increases in GAP-43-IR in the OML of hAPPhigh mice on the fyn+/+ and fyn-/- background (n = 5-8 mice/genotype; age, 4-5 months). *p < 0.005. B, GAP-43-IR did not correlate with hippocampal Aβ1-42 levels or Aβ1-42/Aβ1-x ratios in hAPPhigh mice on the fyn+/+ and fyn-/- backgrounds. C, Overexpressing Fyn did not increase GAP-43-IR levels in hAPPlow mice (n = 5-9 mice/genotype; age, 6-8 months).
Discussion
We have identified a mechanistic dichotomy in the cellular effects elicited by hAPPFAD/Aβ in vivo. hAPPFAD/Aβ induces both synaptotoxicity and aberrant growth processes. Our results suggest that only the former involves Fyn kinase-dependent signaling pathways.
A significant correlation between Aβ levels and SIPT levels was observed in hAPPhigh mice on the fyn+/+, but not fyn-/-, background, suggesting that Fyn is necessary for hAPPFAD/Aβ to affect SIPT levels. One caveat is that SIPT levels were lower in fyn-/- mice than in fyn+/+ mice even in the absence of hAPPFAD/Aβ expression, possibly relating to other hippocampal abnormalities (Grant et al., 1992; Kojima et al., 1997), which could create a “floor” effect in fyn-/- mice, preventing additional reductions by other insults. However, increased expression of Fyn exacerbated SIPT reductions in hAPPlow mice, providing additional direct evidence for a copathogenic role of Fyn in hAPPFAD/Aβ-induced synaptotoxicity.
Increased expression of Fyn by itself did not increase premature mortality in singly TG FYN mice, consistent with previous findings (Kojima et al., 1998). However, ablating Fyn decreased, whereas overexpressing Fyn increased premature mortality in hAPPFAD mice, suggesting that Fyn plays a key role in this disease manifestation. Because overexpression of Fyn was restricted to forebrain neurons (Kojima et al., 1998), our findings suggest a central mechanism. Although we did not witness seizures or the premature deaths during this study, we cannot exclude the involvement of epileptiform activities. hAPP mice on an FVB/N background were more prone to seizures and premature mortality than NTG littermates (Hsiao et al., 1995). Furthermore, Fyn phosphorylates the subunit 2B of the NMDA receptor, and FYN TG mice showed accelerated kindling, which was retarded by NMDA receptor antagonists (Kojima et al., 1998). Thus, increased expression of Fyn may lower the seizure threshold in hAPPFAD mice.
In contrast to synaptotoxicity and premature mortality, axonal sprouting was not altered by genetic modulation of Fyn in hAPPFAD mice. Aberrant sprouting is prominent in AD (Geddes et al., 1985; Masliah et al., 1991; Arendt, 2001) and has also been demonstrated in other lines of hAPPFAD mice (Phinney et al., 1999; Bronfman et al., 2000; Jaffar et al., 2001). hAPPhigh mice exhibited sprouting on both fyn+/+ and fyn-/- backgrounds, and the magnitude of sprouting increased with age. In contrast, no sprouting was observed in hAPPlow mice, even when Fyn was overexpressed. Thus, aberrant sprouting in hAPPFAD mice depends on hAPPFAD/Aβ levels and age but not on Fyn.
It is possible that Fyn-dependent synaptotoxicity is caused by Aβ, whereas Fyn-independent aberrant sprouting is caused by an alternative effect of the FAD mutation, another APP metabolite, or the precursor molecule itself. Consistent with this interpretation, SIPT reductions correlated with Aβ levels on the fyn+/+, but not fyn-/-, background, and SIPT reductions were worsened by overexpression of Fyn, whereas axonal sprouting did not correlate with Aβ levels on either background and was not worsened by overexpression of Fyn.
Alternatively, synaptotoxicity, premature death, and aberrant sprouting may all be caused by Aβ, but through mechanistically distinct pathways that do or do not involve Fyn. The lack of correlation between aberrant sprouting in the dentate gyrus and Aβ levels does not necessarily invalidate this interpretation because the sprouting stimulus could act on neurons in other regions that project to the molecular layer of the dentate gyrus. In that case, sprouting might correlate with Aβ levels in regions containing the cell bodies of the projections, a possibility that remains to be tested.
Although additional research is needed to define the precise interactions between Aβ and Fyn-related signaling pathways, there is evidence that integrins, cadherins, N-syndecan, FAK, paxillin, and tau-phosphorylating kinases might be involved (Shirazi and Wood, 1993; Kohmura et al., 1998; Lauri et al., 1999; Chavis and Westbrook, 2001; Williamson et al., 2002; Grace and Busciglio, 2003).
If Aβ affects Fyn itself, Fyn might be required to mediate some of the biological effects of Aβ. Notably, activation of Fyn can have different consequences in different brain regions. In the hippocampus, but not in the cerebral cortex, Fyn is targeted to the NR2B subunit of the NMDA receptor by the scaffolding protein RACK1 (Yaka et al., 2003). Peptides that disrupt interactions between RACK1, NR2B, and Fyn induce phosphorylation of NR2B and potentiate NMDA receptor-mediated currents (Yaka et al., 2002). Thus, increased Fyn activity might elicit excitotoxic neuronal injury in the hippocampus but not in other regions. If Aβ affects targets downstream of Fyn, Fyn might act primarily to prime or amplify Aβ-induced pathogenic cascades. These possibilities are not mutually exclusive and provide a framework of testable hypotheses.
In conclusion, our study demonstrates an important role of Fyn in hAPP/Aβ-dependent synaptic deficits and premature death. It implies that pharmacological modulation of Fyn or related signaling pathways might be of therapeutic benefit in AD.
Footnotes
This work was supported by National Institutes of Health Grants NS41787 and AG022074 (L.M.), the John Douglas French Alzheimer's Foundation (J.C.), C. Lester and Audrey Hogan (J.J.P.), and the Hillblom Center for the Biology of Aging (J.J.P). We thank Gary Howard and Stephen Ordway for editorial review of this manuscript and Denise McPherson for administrative assistance.
Correspondence should be addressed to Dr. Lennart Mucke, Gladstone Institute of Neurological Disease, P.O. Box 419100, San Francisco, CA 94141-9100. E-mail: lmucke@gladstone.ucsf.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244692-06$15.00/0
References
- Arendt T (2001) Disturbance of neuronal plasticity is a critical pathogenetic event in Alzheimer's disease. Int J Dev Neurosci 19: 231-245. [DOI] [PubMed] [Google Scholar]
- Benowitz LI, Perrone-Bizzozero NI (1991) The expression of GAP-43 in relation to neuronal growth, plasticity: when, where, how, and why? Prog Brain Res 89: 69-87. [DOI] [PubMed] [Google Scholar]
- Bi X, Gall CM, Zhou J, Lynch G (2002) Uptake and pathogenic effects of amyloid beta peptide 1-42 are enhanced by integrin antagonists and blocked by NMDA receptor antagonists. Neuroscience 112: 827-840. [DOI] [PubMed] [Google Scholar]
- Bronfman FC, Moechars D, Van Leuven F (2000) Acetylcholinesterase-positive fiber deafferentation and cell shrinkage in the septohippocampal pathway of aged amyloid precursor protein london mutant transgenic mice. Neurobiol Dis 7: 152-168. [DOI] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW (1999) Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/- mice: isoform-specific effects on neurodegeneration. J Neurosci 19: 4867-4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Yu G-Q, Shockley K, Huang Y, Jones B, Masliah E, Mallory M, Yeo T, Longo FM, Mucke L (2002) Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid β peptides but not on plaque formation. J Neurosci 22: 10539-10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson GA, Borchelt DR, Dake A, Turner S, Danielson V, Coffin JD, Eckman C, Meiners J, Nilsen SP, Younkin SG, Hsiao KK (1997) Genetic modification of the phenotypes produced by amyloid precursor protein overexpression in transgenic mice. Hum Mol Genet 6: 1951-1959. [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury Jr PT (2003) Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci 26: 267-298. [DOI] [PubMed] [Google Scholar]
- Chavis P, Westbrook G (2001) Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature 411: 317-321. [DOI] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol 27: 457-464. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Westerman M, Bui D, Bell K, Ashe KH, Sweatt JD (2001) β-Amyloid activates the mitogen-activated protein kinase cascade via hippocampal α7 nicotinic acetylcholine receptors: in vitro and in vivo mechanisms related to Alzheimer's disease. J Neurosci 21: 4125-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geddes JW, Monaghan DT, Cotman CW, Lott IT, Kim RC, Chui HC (1985) Plasticity of hippocampal circuitry in Alzheimer's disease. Science 230: 1179-1181. [DOI] [PubMed] [Google Scholar]
- Grace EA, Busciglio J (2003) Aberrant activation of focal adhesion proteins mediates fibrillar amyloid β-induced neuronal dystrophy. J Neurosci 23: 493-502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant SGN, O'Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER (1992) Impaired long-term potentiation, spatial learning, and hippocampal development in fyn mutant mice. Science 258: 1903-1910. [DOI] [PubMed] [Google Scholar]
- Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D (1999) Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet 29: 177-185. [DOI] [PubMed] [Google Scholar]
- Hsia A, Masliah E, McConlogue L, Yu G, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L (1999) Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci USA 96: 3228-3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao KK, Borchelt DR, Olson K, Johannsdottir R, Kitt C, Yunis W, Xu S, Eckman C, Younkin S, Price D, Iadecola C, Clark HB, Carlson G (1995) Age-related CNS disorder and early death in transgenic FVB/N mice over-expressing Alzheimer amyloid precursor proteins. Neuron 15: 1203-1218. [DOI] [PubMed] [Google Scholar]
- Jaffar S, Counts SC, Ma SY, Dadko E, Gordon MN, Morgan D, Mufson EJ (2001) Neuropathology of mice carrying mutant APPswe and/or PS1m146L transgenes: alterations in the p75NTR cholinergic basal forebrain septohippocampal pathway. Exp Neurol 170: 227-243. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L (1997) Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA 94: 1550-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37: 925-937. [DOI] [PubMed] [Google Scholar]
- Klein WL (2000) Aβ toxicity in Alzheimer's disease. In: Molecular mechanisms of neurodegenerative diseases (Chesselet M-F, ed), pp 1-49. Totowa, NJ: Humana.
- Kohmura N, Senzaki K, Hamada S, Kai N, Yasuda R, Watanabe M, Ishii H, Yasuda M, Mishina M, Yagi T (1998) Diversity revealed by a novel family of cadherins expressed in neurons at a synaptic complex. Neuron 20: 1137-1151. [DOI] [PubMed] [Google Scholar]
- Kojima N, Wang J, Mansuy IM, Grant SGN, Mayford M, Kandel ER (1997) Rescuing impairment of long-term potentiation in fyn-deficient mice by introducing Fyn transgene. Proc Natl Acad Sci USA 94: 4761-4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojima N, Ishibashi H, Obata K, Kandel ER (1998) Higher seizure susceptibility and enhanced tyrosine phosphorylation on N-methyl-d-aspartate receptor subunit 2B in fyn transgenic mice. Learn Mem 5: 429-445. [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL (1998) Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA 95: 6448-6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Kaukinen S, Kinnunen T, Ylinen A, Imai S, Kaila K, Taira T, Rauvala H (1999) Regulatory role and molecular interactions of a cell-surface heparan sulfate proteoglycan (N-syndecan) in hippocampal long-term potentiation. J Neurosci 19: 1226-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Hansen L, Alford M, Albright T, DeTeresa R, Terry R, Baudier J, Saitoh T (1991) Patterns of aberrant sprouting in Alzheimer's disease. Neuron 6: 729-739. [DOI] [PubMed] [Google Scholar]
- Masliah E, Ellisman M, Carragher B, Mallory M, Young S, Hansen L, De-Teresa R, Terry RD (1992) Three-dimensional analysis of the relationship between synaptic pathology and neuropil threads in Alzheimer disease. J Neuropathol Exp Neurol 51: 404-414. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR (1994) Synaptotrophic effects of human amyloid β protein precursor in the cortex of transgenic mice. Brain Res 666: 151-167. [DOI] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu G-Q, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L (2000) High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20: 4050-4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu G-Q, Raber J, Masliah E, Mucke L (2003) Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci USA 100: 9572-9577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phinney AL, Deller T, Stalder M, Calhoun ME, Frotscher M, Sommer B, Staufenbiel M, Jucker M (1999) Cerebral amyloid induces aberrant axonal sprouting and ectopic terminal formation in amyloid precursor protein transgenic mice. J Neurosci 19: 8552-8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, Wong D, Yu G-Q, Buttini M, Mahley RW, Pitas RE, Mucke L (2000) Alzheimer's disease: apolipoprotein E and cognitive performance. Nature 404: 352-354. [DOI] [PubMed] [Google Scholar]
- Rockenstein EM, McConlogue L, Tan H, Gordon M, Power M, Masliah E, Mucke L (1995) Levels and alternative splicing of amyloid β protein precursor (APP) transcripts in brains of transgenic mice and humans with Alzheimer's disease. J Biol Chem 270: 28257-28267. [DOI] [PubMed] [Google Scholar]
- Sabo S, Lambert MP, Kessey K, Wade W, Krafft G, Klein WL (1995) Interaction of β-amyloid peptides with integrins in a human nerve cell line. Neurosci Lett 184: 25-28. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D (2003) Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol 43: 545-584. [DOI] [PubMed] [Google Scholar]
- Shirazi SK, Wood JG (1993) The protein tyrosine kinase, fyn, in Alzheimer's disease pathology. NeuroReport 4: 435-437. [DOI] [PubMed] [Google Scholar]
- Small DH, Mok SS, Bornstein JC (2001) Alzheimer's disease and Aβ toxicity: from top to bottom. Nat Rev Neurosci 595: 595-598. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Hansen LA (1999) The neuropathology of Alzheimer disease and the structural basis of its cognitive alterations. In: Alzheimer disease, Ed 2 (Terry RD, Katzman R, Bick KL, Sisodia SS, eds), pp 187-206. Philadelphia: Lippincott Williams and Wilkins.
- Westerman MA, Cooper-Blacketer D, Mariash A, Kotilinek L, Kawarabayashi T, Younkin LH, Carlson GA, Younkin SG, Ashe KH (2002) The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 22: 1858-1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson R, Scales T, Clark BR, Gibb G, Reynolds CH, Kellie S, Bird IN, Varndell IM, Sheppard PW, Everall I, Anderton BH (2002) Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-β peptide exposure: involvement of Src family protein kinases. J Neurosci 22: 10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaar M, Zhai S, Pilch PF, Doyle SM, Eisenhauer PB, Fine RE, Gilchrest BA (1997) Binding of beta-amyloid to the p75 neurotrophin receptor induces apoptosis—a possible mechanism for Alzheimer's disease. J Clin Invest 100: 2333-2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Thornton C, Vagts AJ, Phamluong K, Bonci A, Ron D (2002) NMDA receptor function is regulated by the inhibitory scaffolding protein, RACK1. Proc Natl Acad Sci USA 99: 5710-5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaka R, Phamluong K, Ron D (2003) Scaffolding of Fyn kinase to the NMDA receptor determines brain region sensitivity to ethanol. J Neurosci 23: 3623-3632. [DOI] [PMC free article] [PubMed] [Google Scholar]