

Abstract
Ca2+-binding protein-1 (CaBP1) is a Ca2+-binding protein that is closely related to calmodulin (CaM) and localized in somatodendritic regions of principal neurons throughout the brain, but how CaBP1 participates in postsynaptic Ca2+ signaling is not known. Here, we describe a novel role for CaBP1 in the regulation of Ca2+ influx through Cav1.2 (L-type) Ca2+ channels. CaBP1 interacts directly with the α1 subunit of Cav1.2 at sites that also bind CaM. CaBP1 binding to one of these sites, the IQ domain, is Ca2+ dependent and competitive with CaM binding. The physiological significance of this interaction is supported by the association of Cav1.2 and CaBP1 in postsynaptic density fractions purified from rat brain. Moreover, in double-label immunofluorescence experiments, CaBP1 and Cav1.2 colocalize in numerous cell bodies and dendrites of neurons, particularly in pyramidal cells in the CA3 region of the hippocampus and in the dorsal cortex. In electrophysiological recordings of cells transfected with Cav1.2, CaBP1 greatly prolonged Ca2+ currents, prevented Ca2+-dependent inactivation, and caused Ca2+-dependent facilitation of currents evoked by step depolarizations and repetitive stimuli. These effects contrast with those of CaM, which promoted strong Ca2+-dependent inactivation of Cav1.2 with these same voltage protocols. Our findings reveal how Ca2+-binding proteins, such as CaM and CaBP1, differentially adjust Ca2+ influx through Cav1.2 channels, which may specify diverse modes of Ca2+ signaling in neurons.
Keywords: calcium, calmodulin, channel, postsynaptic, facilitation, inactivation
Introduction
Voltage-gated Ca2+ channels are major routes of Ca2+ entry in neurons and are crucial for many forms of synaptic plasticity because they couple membrane depolarization to Ca2+-dependent signal transduction. In the postsynaptic membrane, Cav1.2 (L-type) Ca2+ channels selectively trigger activity-dependent gene expression (Murphy et al., 1991; Bading et al., 1993; Ginty et al., 1993; Dolmetsch et al., 2001; Weick et al., 2003), mRNA targeting into dendrites (Tongiorgi et al., 1997), and depression of R-type Ca2+ channels in dendritic spines (Yasuda et al., 2003). The mechanism by which Ca2+ influx through Cav1.2 channels specifically activates these pathways remains unclear but may depend on the interaction of these channels with postsynaptic proteins that regulate their function and/or proximity to key effectors (Deisseroth et al., 1998; Davare et al., 2001).
Mounting experimental evidence supports a role for calmodulin (CaM) in regulating the signaling capability of postsynaptic Cav1.2 channels via two mechanisms. First, CaM limits excessive Ca2+ entry through Cav1.2 in a process known as Ca2+-dependent inactivation, which is mediated by CaM binding directly to the main α1 subunit of Cav1.2 (Peterson et al., 1999; Qin et al., 1999; Zühlke et al., 1999). Second, physical coupling between CaM and Cav1.2 is important for translocation of CaM to the nucleus and activation of transcription factors such as cAMP response element-binding protein and MEF-2 (myocyte enhancer factor 2) (Deisseroth et al., 1998; Dolmetsch et al., 2001). Such abilities of CaM to modulate and transduce Ca2+ signals have led to its proposed role as a multifunctional Ca2+ sensor (Chin and Means, 2000).
However, CaM is but one member of a family of Ca2+-binding proteins, some of which are expressed primarily in neurons (Burgoyne and Weiss, 2001; Haeseleer et al., 2002). Because of their structural similarity, CaM and these neuron-specific Ca2+-binding proteins (NCBPs) may interact with the same target molecules but mediate distinct forms of target regulation (Yang et al., 2002; Haynes et al., 2004). One such NCBP, Ca2+-binding protein-1 (CaBP1), interacts with Cav2.1 (P/Q-type) Ca2+ channels, which are also regulated directly by CaM. However, whereas CaM mediates Ca2+-dependent inactivation and facilitation of these channels (Lee et al., 1999, 2000; DeMaria et al., 2001), CaBP1 strongly enhances inactivation independent of Ca2+ (Lee et al., 2002).
Although CaBP1 colocalizes with Cav2.1 (P/Q-type) channels in a subset of presynaptic nerve terminals (Lee et al., 2002), antibodies recognizing CaBP1 and caldendrin, a splice variant of CaBP1, label primarily somatodendritic regions of neurons throughout the brain (Seidenbecher et al., 1998; Laube et al., 2002), a pattern that more closely parallels that of Cav1.2 (L-type) channels (Westenbroek et al., 1990; Hell et al., 1993). Therefore, we hypothesize that CaBP1 may play an important role in regulating postsynaptic Cav1.2 channels. We show that CaBP1 physically associates with Cav1.2 at the postsynaptic density (PSD) and competes with CaM for binding to the channel. However, compared with the inhibitory effects of CaM, CaBP1 greatly prolongs and facilitates Ca2+ currents conducted by Cav1.2. These distinct regulatory effects of CaBP1 may contribute to the diverse properties of Cav1.2 channels in neurons and determine how Ca2+ signals are encoded and decoded in the postsynaptic neuron.
Materials and Methods
Constructs and molecular biology. For electrophysiological and biochemical experiments, Cav1.2 subunits [α11.2 (rbcII), β2A, and α2δ (Ellis et al., 1988; Snutch et al., 1991; Perez-Reyes et al., 1992)] were expressed from the pcDNA3.1+ vector (Invitrogen, Carlsbad, CA). For the FLAG-α11.2 construct, a Kozak sequence and FLAG epitope (DYKDDDDK) were added to the N terminus of α11.2 by PCR of nucleotides 4-1272 of rbcII with a forward primer containing the additional sequences and the resulting PCR product cloned into HindIII sites in the parent plasmid (rbcII/pcDNA3.1+). FLAG-α11.2IQ-AA and FLAG-α11.2IQ-EE were generated by quick-change mutagenesis with primers incorporating nucleotide changes to substitute alanine or glutamate residues for I1624 and Q1625. The cDNA corresponding to the entire coding region of the short isoform of human CaBP1 (GenBank accession number AF169148) was subcloned into pcDNA3.1+ or pEGFP-N1 vector (BD Biosciences Clontech, Palo Alto, CA) (Haeseleer et al., 2000). For binding assays, constructs encoding fusion proteins of the C-terminal domain of α11.2 were generated by PCR and subcloned into pGEX 4T-1 (Amersham Biosciences, Piscataway, NJ) for glutathione S-transferase (GST)- or pTrcHisA (Invitrogen) for His-tagged proteins. The cDNA encoding neuronal Ca2+ sensor-1 (NCS-1) was generated by PCR with specific primers from rat brain cDNA and subcloned into pcDNA3.1+.
Cell culture and transfection. Human embryonic kidney HEK293T cells were maintained in DMEM with 10% fetal bovine serum at 37°C in a humidified atmosphere under 7% CO2. Cells were grown to ∼70-80% confluence and transfected with Gene Porter reagent (Gene Therapy Systems, San Diego, CA) according to protocols of the manufacturer.
Binding assays. CaBP1 used for binding assays was obtained by transfecting HEK293T cells plated on 150 mm dishes with a total of 5 μg of CaBP1 subcloned into pcDNA3.1+. Two days later, cells were homogenized in 1 ml of ice-cold lysis buffer (10 mm HEPES, 50 mm NaCl, 1 mm benzamidine, and 0.5% Triton X-100, pH 7.4), and membrane proteins were solubilized by rotating at 4°C for 30 min. Insoluble material was removed by ultracentrifugation at 100,000 × g for 30 min, and the supernatant was used immediately or aliquotted and stored at -80°C. This procedure separated CaBP1, which is primarily membrane associated in HEK293T cells (data not shown), and CaM, which is cytoplasmic and endogenously expressed by these cells. The absence of CaM, which would have complicated binding analyses with transfected CaBP1, was confirmed by Western blot of these solubilized membrane preparations with anti-CaM antibodies.
Fusion proteins containing fragments of the α11.2 C-terminal domain were expressed in BL21 Escherichia coli by isopropyl-β-d-thiogalactopyranoside induction and immobilized on glutathione- or Ni2+-agarose beads for GST or His fusion proteins, respectively. Purified CaM (5 μg; Sigma-Aldrich, St. Louis, MO) or lysates of cells transfected with CaBP1 were added to 50 μl of a 50% slurry of immobilized fusion protein and brought to a total volume of 1 ml with binding buffer ([Tris-buffered saline (TBS): 20 mm Tris, pH 7.3, and 150 mm NaCl], 0.1% Triton X-100, and protease inhibitors) containing either 2 mm CaCl2 or 10 mm EGTA. Binding reactions were incubated at 4°C, rotating, for 1 hr. The beads were washed three times with 1 ml of ice-cold binding buffer, and bound proteins were eluted, resolved by SDS-PAGE, and transferred to nitrocellulose. Bound CaBP1, CaM, or NCS-1 was detected by Western blot with rabbit polyclonal antibodies against CaBP1 [UW72, 1:2000 (Haeseleer et al., 2000)], monoclonal anti-CaM antibodies (1:1000; Up-state Biotechnologies, Waltham, MA), or rabbit polyclonal anti-NCS-1 antibodies (1:1000; Zymed, San Francisco, CA). Blots were processed with HRP-conjugated secondary antibodies (anti-rabbit IgG 1:4000, anti-mouse IgG 1:2000) and ECL reagents (Amersham Biosciences). For some experiments, His-tagged fusion proteins were detected by Western blotting with rabbit polyclonal anti-His antibodies (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA).
Coimmunoprecipitation assays. HEK293T cells were transfected with equimolar amounts of cDNAs encoding Cav1.2 subunits (FLAG-α11.2, β2A, and α2δ) with or without CaBP1. At least 48 hr later, cell lysates were prepared as described above and incubated with 40 μl of anti-FLAG M2 affinity gel (Sigma-Aldrich) for 1 hr, rotating at 4°C. After five washes with 1 ml of lysis buffer, proteins were eluted with sample buffer and subjected to SDS-PAGE, and coimmunoprecipitated proteins were detected by Western blotting with UW72 or monoclonal anti-FLAG antibodies (M2, 1:2000; Sigma-Aldrich), followed by secondary antibodies and ECL detection as described above.
For coimmunoprecipitation from rat brain, fractions containing the PSD were isolated by a modified method described previously (Carlin et al., 1980). These procedures, as well as those described for Immunohistochemistry, were done in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Freshly removed rat brains were homogenized in ice-cold buffer [homogenization buffer (HB): 4 mm HEPES, 1 mm EDTA, and 0.32 m sucrose, pH 7.4] with protease inhibitors (1 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin A, 17 μg/ml PMSF, 8 μg/ml calpain inhibitor I, 8 μg/ml calpain inhibitor II, and 1 mm benzamidine). After centrifugation at 1000 × g for 10 min, the membrane pellet was resuspended in HB and layered onto a sucrose step gradient (0.8 and 1.2 m), which was centrifuged at 150,000 × g for 1 hr. Material at the interface of the gradient was removed, centrifuged at 150,000 × g for 30 min, resuspended in HB containing 0.5% Triton X-100, incubated for 15 min, and centrifuged at 35,000 × g for 20 min. The final pellet containing the PSD fraction was resuspended in HB and stored at -80°C. Before an experiment, PSD fractions were pelleted and solubilized in HB containing 1% Triton X-100 for 20 min at 4°C with continuous stirring, and insoluble material was removed by centrifugation at 150,000 × g for 30 min. The presence of PSD-95 in the resulting supernatant, which was detected by Western blotting with anti-PSD-95 antibodies (1:2000; Affinity Bioreagents, Golden, CO) was used to confirm that postsynaptic density proteins were solubilized by this procedure. For coimmunoprecipitations, the supernatant (1 ml) was incubated for 1.5 hr, rotating at 4°C with 10 μg of rabbit polyclonal antibodies against α1C (Alomone Labs, Jerusalem, Israel) or purified rabbit IgG (Jackson ImmunoResearch, West Grove, PA) as a control. Immune complexes were isolated by incubating with 3-5 mg of preswollen Protein A-Sepharose for 2 hr at 4°C. The beads were washed three times with 0.1% Triton X-100 in TBS and subjected to SDS-PAGE, and immuno-precipitated proteins were detected by Western blotting with α1C (1:200) or UW72 antibodies as described above.
Double-label immunohistochemistry. Adult male Sprague Dawley rats were anesthetized deeply (4% chloral hydrate, i.p.) and intracardially perfused with 3% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.3. The brains were removed and cryoprotected overnight at 4°C in 30% sucrose, 0.1 m PB. Tissue sections (30-40 μm) were cut on a freezing sliding microtome, rinsed in TBS, and blocked in TBS containing 5% normal goat serum (NGS) and 0.025% Triton X-100 [blocking buffer (BB)]. All antibodies were diluted in TBS containing 2.5% NGS and 0.025% Triton X-100, and sections were rinsed three times for 20 min each after each step. Blocked brain sections were incubated with anti-α1C antibodies (1:100; generously provided by Dr. Johannes Hell, University of Iowa, Iowa City, IA) overnight at 4°C and, after rinsing in TBS, with 1:400 rhodamine-conjugated donkey anti-rabbit Fab fragments (Jackson ImmunoResearch) for 60 min. Before double labeling, sections were again blocked at 37°C for 60 min with BB, 120 min with goat anti-rabbit Fab fragments (1:50; Jackson ImmunoResearch), 30 min in 2% avidin-TBS, and 30 min in 2% biotin-TBS. Sections were then incubated overnight at 4°C with UW72 (1:1000), the next day with biotinylated donkey anti-rabbit IgG (1:1000; Jackson ImmunoResearch), and subsequently with FITC-avidin D in TBS (1:1600; Vector Laboratories, Burlingame, CA). The double-labeled sections were mounted and coverslipped with Vectashield (Vector Laboratories) for viewing under a Zeiss (Oberkochen, Germany) LSM510 Meta confocal microscope.
Electrophysiological recordings. HEK293T cells plated on 35 mm dishes were transfected with a total of 5 μg of DNA including 0.3 μg of a pEGFP-N1 or CaBP1/pEGFPN1 for fluorescent detection of transfected cells. At least 48 hr after transfection, whole-cell patch-clamp recordings of transfected cells were acquired with a HEKA Elektronik (Lambrecht/Pfalz, Germany) EPC-9 patch-clamp amplifier. Data acquisition and leak subtraction using a P/-4 protocol were done with Pulse software (HEKA Electronik). Extracellular recording solutions contained the following (in mm): 150 Tris, 2 MgCl2, and 10 CaCl2 or BaCl2. Intracellular solutions consisted of the following (in mm): 140 N-methyl-d-glucamine, 10 HEPES, 2 MgCl2, 2 Mg-ATP, and 5 EGTA. The pH of intracellular and extracellular recording solutions was adjusted to 7.3 with methanesulfonic acid. Electrode resistances were typically 1-2MΩ in the bath solution, and series resistance was ∼2-4 MΩ, compensated up to 70%. In recordings of Ba2+ currents, voltage protocols were adjusted by -10 mV to compensate for the corresponding shift in voltage dependence of activation with substitution of extracellular Ba2+ for Ca2+. The time course of ICa decay was fit by Aslow[exp(-t/τslow)] + Afast[exp(-t/τfast)], where t is time, Aslow and Afast are the amplitudes of the slow and fast exponentials, respectively, at t = 0, and τslow and τfast are the time constants of the decay of the two processes. Data were analyzed using Igor software (WaveMetrics, Lake Oswego, OR), and graphs and statistical analysis were done with Sigma Plot (SPSS, Chicago, IL).
Results
CaBP1 binds to the C-terminal domain of α11.2
Ca2+-dependent inactivation of Cav1.2 depends on CaM binding to multiple sequence elements in the C-terminal domain of α11.2 (Soldatov et al., 1998; Zühlke and Reuter, 1998; Peterson et al., 1999; Qin et al., 1999; Zühlke et al., 1999, 2000). These sites (A, C, and IQ) (Fig. 1A) mediate binding to either Ca2+-free (apo)CaM or Ca2+-bound CaM and are in the proximal portion of the cytoplasmic C-terminal domain just downstream of an EF-hand motif, also implicated in Ca2+-dependent inactivation (Zühlke and Reuter, 1998; Pate et al., 2000; Peterson et al., 2000; Romanin et al., 2000; Pitt et al., 2001; Erickson et al., 2003; Tang et al., 2003). On the basis of its similarity with CaM, we hypothesized that CaBP1 might also interact with these C-terminal CaM-binding sites. To test this, we generated a GST fusion protein including these regions (CT1) (Fig. 1A) and analyzed binding to CaBP1 in pull-down assays. CaBP1 was brought down with CT1 but not a more distal region of the C-terminal domain used as a negative control (CT2) (Fig. 1A,B). CaBP1 binding to CT1 was stronger in the presence of Ca2+ but was not abolished when EGTA was included in the assay, indicating that CaBP1 binding to α11.2 is partially Ca2+ independent. This result was confirmed in reverse experiments in which GST-tagged CaBP1, but not GST, pulled down His-tagged CT1, but not CT2, both with and without Ca2+ (Fig. 1C). This interaction with CaBP1 was specific in that a related NCBP, NCS-1, did not bind CT1 under the same conditions (Fig. 1D).
Figure 1.
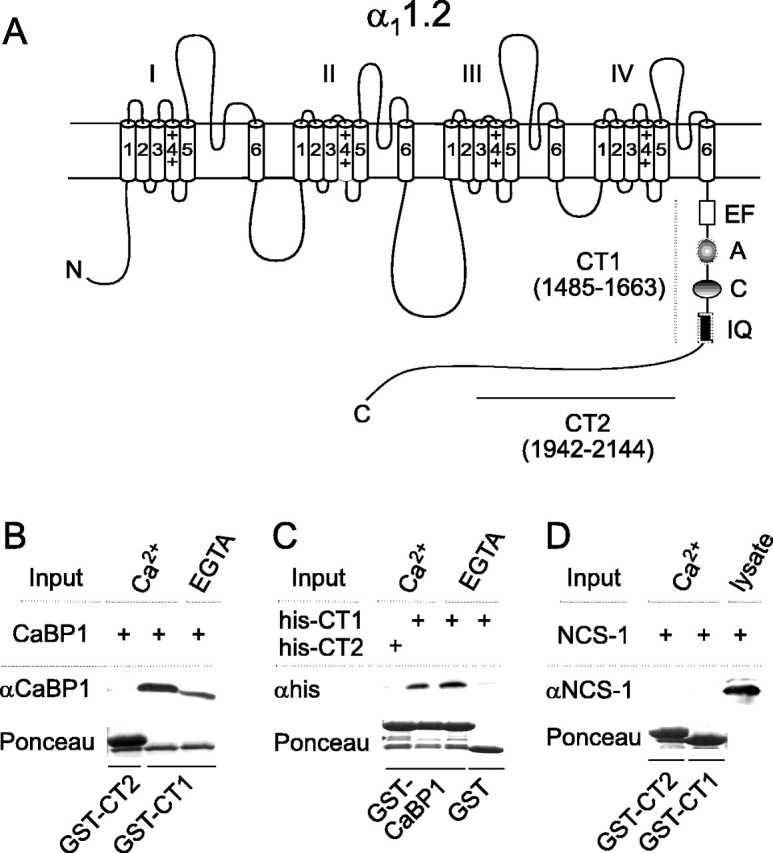
CaBP1 interacts with the C-terminal domain of the α1 subunit of Cav1.2. A, Schematic of α11.2 with C-terminal domain showing CT1 and CT2 regions used for GST fusion constructs in binding experiments. Amino acid boundaries are indicated in parentheses. CT1 included sequences involved in Ca2+-dependent inactivation of Cav1.2: the EF-hand (EF) and CaM-interacting sites A, C, and the IQ domain (IQ). N, N terminus of α11.2. B, Binding of CaBP1 to CT1 but not CT2. GST-CT1 or CT2 fusion proteins were immobilized on glutathione-agarose beads and incubated with lysates from cells transfected with CaBP1 in the presence of 2 mm Ca2+ or 10 mm EGTA. Bound CaBP1 was detected by Western blot. Ponceau staining of the blot indicated levels of GST fusion protein used in each binding assay. C, Binding of His-tagged CT1 to GST-tagged CaBP1. GST or GST-CaBP1 was immobilized on glutathione-agarose beads and in cubated with purified His-CT1 or His-CT2. Bound fusion proteins were detected by Western blot with anti-His antibodies. D, NCS-1 does not bind to CT1. Binding assay was performed as in B, except that lysates from cells transfected with NCS-1 were used and immunoblotting was done with rabbit polyclonal antibodies against NCS-1. NCS-1 was detected in transfected cell lysates (last lane) but not in samples pulled down by GST-CT1 or CT2.
To further refine the binding determinants for CaBP1, we compared CaBP1 interactions with GST fusion proteins containing various subsets of the A, C, and IQ sites (Fig. 2A). As shown in Figure 2B, CaBP1 was pulled down by all fusion proteins that contained the C and/or IQ sites (CT1, CT5, CT6, and CT7) but not CT4, which had only the A site. An identical pattern of binding was observed for CaM (Fig. 2B). However, previous studies show binding of CaM to peptides but not fusion proteins containing the A site, perhaps because of the lower affinity of CaM binding to this site, which may not be detectable in our pull-down assay (Pate et al., 2000; Romanin et al., 2000; Pitt et al., 2001; Tang et al., 2003). Therefore, although we cannot rule out the possibility that CaBP1 also interacts with low affinity at the A site, our results confirm that both C and IQ sites contain sequences important for binding CaBP1 and CaM.
Figure 2.
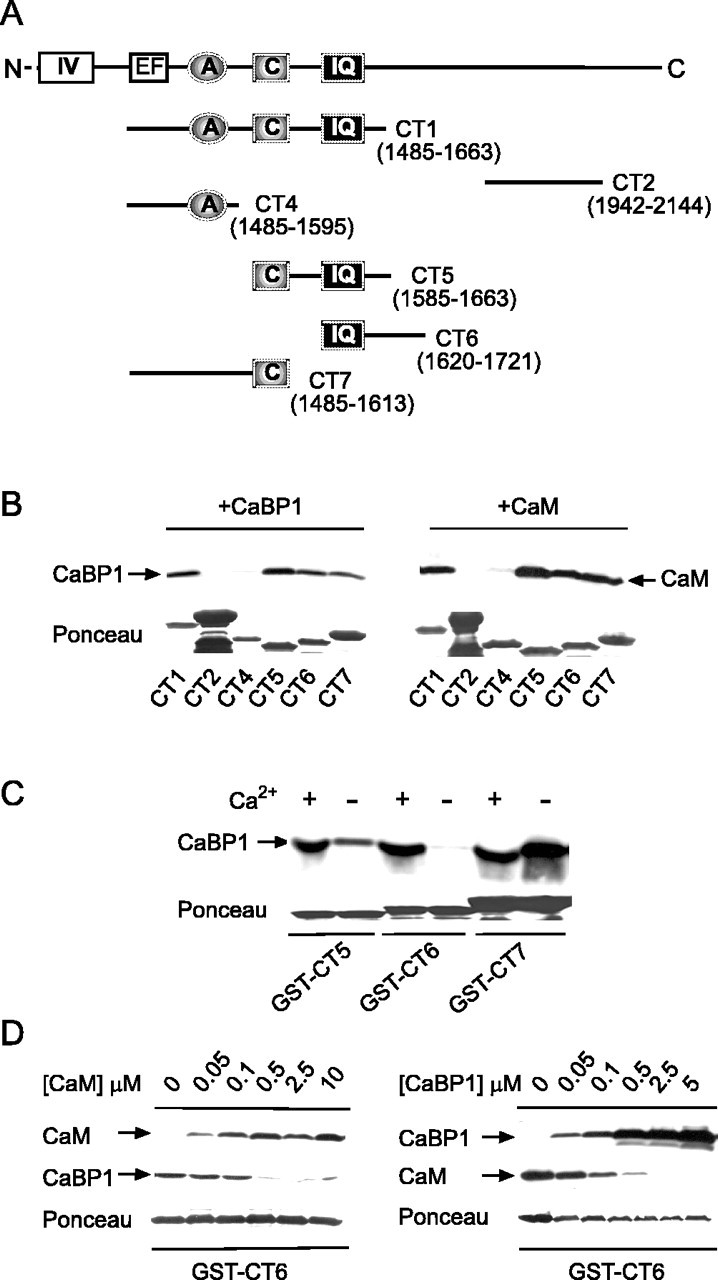
CaBP1 binds to putative CaM-binding sites in the C-terminal domain of α11.2. A, Schematic of C-terminal domain of α11.2 showing regions tested for binding CaM or CaBP1 in binding assays. B, Binding of CaM and CaBP1 to multiple sites in α11.2. GST-tagged α11.2 fragments shown in A were incubated with CaBP1 from transfected cells (left) or purified CaM (right) in the presence of 2 mm Ca2+. Bound proteins were detected by Western blotting with specific antibodies. Integrity and levels of GST-tagged proteins were confirmed by Ponceau staining. C, Ca2+-dependent and -independent binding of CaBP1. Binding of CaBP1 to GST-CT5, GST-CT6, or GST-CT7 was performed as in B and compared when assay was done with 2 mm Ca2+ (+) or 10 mm EGTA (-). D, Competitive binding of CaM and CaBP1 to CT6. Binding of CaBP1 or CaM to GST-CT6 was performed as in B with 2 mm Ca2+ and compared in the presence of increasing concentrations of purified CaM (left) or CaBP1 (right).
Current models of CaM interactions with Cav1.2 involve constitutive association of CaM with the A and C sites with Ca2+ binding to CaM promoting interactions with the IQ, which then transduces Ca2+-dependent inactivation (Pate et al., 2000; Pitt et al., 2001) (but see Romanin et al., 2000; Erickson et al., 2003; Tang et al., 2003). To determine whether the same was true for CaBP1, Ca2+-dependent binding of CaBP1 to these sites was analyzed with Ca2+ (2 mM) or EGTA (10 mM) in the assay. In these experiments, CaBP1 binding to the C site was Ca2+ independent, whereas binding to CT6, which contained the IQ, occurred only in the presence of Ca2+(Fig. 2C). Moreover, binding of CaM and CaBP1 to the IQ was mutually exclusive in that CaM effectively displaced CaBP1 binding to CT6 at a concentration between 100 and 500 nM, and CaBP1 competed with CaM for binding to CT6 at a similar range of concentrations (Fig. 2D). These results confirm that similar molecular determinants mediate binding of CaBP1 and CaM. As has been reported for CaM, CaBP1 may be tethered to the C site at resting Ca2+ concentrations, with elevations in Ca2+ promoting conformational changes in CaBP1 that permit binding to IQ.
Although CaBP1 effectively competed with CaM for interaction with the GST fragment containing the IQ (Fig. 2D), it was important to determine whether CaBP1 could associate with the intact channel in cells in which CaM is endogenously expressed at high concentrations. Therefore, we tested whether CaBP1 could coimmunoprecipitate with FLAG-tagged α11.2 (FLAG-α11.2) in transfected HEK293T cells. As shown in Figure 3A, FLAG antibodies coimmunoprecipitated CaBP1 from cells cotransfected with FLAG-α11.2 but not from cells transfected with CaBP1 or FLAG-α11.2 alone. Moreover, the association of CaBP1 and FLAG-α11.2 did not require Ca2+, which is consistent with the Ca2+-independent binding of CaBP1 to the C site (Fig. 2B). These results confirm that CaBP1 is constitutively associated with Cav1.2 channels in cells, which may involve displacing CaM from binding sites in the cytoplasmic C-terminal domain of α11.2.
Figure 3.

Coimmunoprecipitation of CaBP1 and α11.2 from transfected cells and rat brain. A, Cells transfected with Cav1.2 (α11.2-FLAG, β2A, and α2δ), Cav1.2 plus CaBP1, or CaBP1 alone were subject to lysis and immunoprecipitation using anti-FLAG antibodies. Experiments were done in the presence of 2 mm Ca2+ or 10 mm EGTA. Immunoprecipitated proteins were detected by Western blotting with antibodies recognizing the FLAG epitope (top) or CaBP1 (bottom). B, Immunopurification of a complex containing both α11.2 and CaBP1 from PSD fractions of rat brain. PSD proteins were isolated on sucrose gradients, solubilized, and incubated with α11.2 antibodies. Immunoprecipitated proteins were detected by Western blotting with antibodies against α11.2 (top) or CaBP1 (bottom). Immunoblotting of the PSD fraction was also performed with antibodies against PSD-95 (right).
CaBP1 forms a postsynaptic complex with Cav1.2
Previous immunohistochemical studies of CaBP1 localization in the brain indicated a cellular and subcellular distribution (Seidenbecher et al., 1998; Laube et al., 2002) that overlapped considerably with that for Cav1.2 (Westenbroek et al., 1990; Hell et al., 1993). However, initial attempts to coimmunoprecipitate Cav1.2 and CaBP1 from whole brain homogenates were unsuccessful, perhaps because of a preferential interaction of both CaBP1 and Cav1.2 at the PSD, in which both proteins are enriched (Hell et al., 1996; Seidenbecher et al., 1998). Because many protein complexes in the PSD are difficult to extract with standard detergent concentrations (Lau et al., 1996), it is possible that interactions between Cav1.2 and CaBP1 at the PSD may have been undetectable in whole brain lysates attributable to their insolubility. Therefore, we focused on whether CaBP1 and Cav1.2 channels associated in PSD fractions that were solubilized and checked for expression of postsynaptic markers such as PSD-95 by immunoblotting (Fig. 3B). From these fractions, anti-α11.2 antibodies, but not rabbit IgG used as a control, consistently coimmunoprecipitated the short isoform of CaBP1, which was the isoform used in our biochemical studies (Fig. 3B). The isolation of a postsynaptic complex containing CaBP1 and Cav1.2 indicates that the molecular interactions between these proteins we characterized in vitro may be physiologically relevant in vivo.
To determine the extent to which CaBP1/Cav1.2 interactions occur in the brain, we used double-label immunofluorescence for colocalizing both proteins in sections of rat brain. In general, CaBP1 and Cav1.2 were strongly colocalized in numerous cell groups throughout the brain. In the hippocampus, labeling for CaBP1 was prominent in the soma and dendrites of CA3 pyramidal cells and almost completely overlapped with that for Cav1.2 (Fig. 4A-C). In the dorsal cortex, CaBP1 and Cav1.2 were colocalized in pyramidal cells, particularly in layer V. However in these neurons, Cav1.2 labeling was restricted to the soma and proximal dendrites, whereas CaBP1 labeling extended far into the distal dendrites (Fig. 4D-F). Furthermore, whereas Cav1.2 immunoreactivity was excluded from the nucleus, all neurons that were labeled with the CaBP1 antibody showed strong immunoreactivity in the nuclear as well as cytoplasmic compartments, which may signify a role for CaBP1 also in regulating gene expression. In the cerebellum, weak-to-moderate labeling for both proteins was detected in Purkinje cell bodies and proximal dendrites (Fig. 4G-I). In addition, intense labeling for CaBP1 was found in interneurons resembling basket and stellate cells in the molecular layer and in the axon terminals and large Pinceaux synapses formed from basket cells onto Purkinje cell bodies (Fig. 4G,I) (Sotelo and Llinas, 1972). In contrast, no Cav1.2 labeling was detected in these interneurons or presynaptic terminals (Fig. 4H,I). These cellular and subcellular distinctions between CaBP1 and Cav1.2 immunostaining exclude the possibility that the extensive colocalization in the hippocampus and the cortex was attributable to nonspecific reactivity of the secondary antibodies used in the double-labeling procedure. Although CaBP1 may have activities independent of interactions with Cav1.2, the coimmunoprecipitation of CaBP1 with Cav2.1, and their colocalization in many of the same cells, suggest that the two may interact in a subset of principal neurons in the brain.
Figure 4.
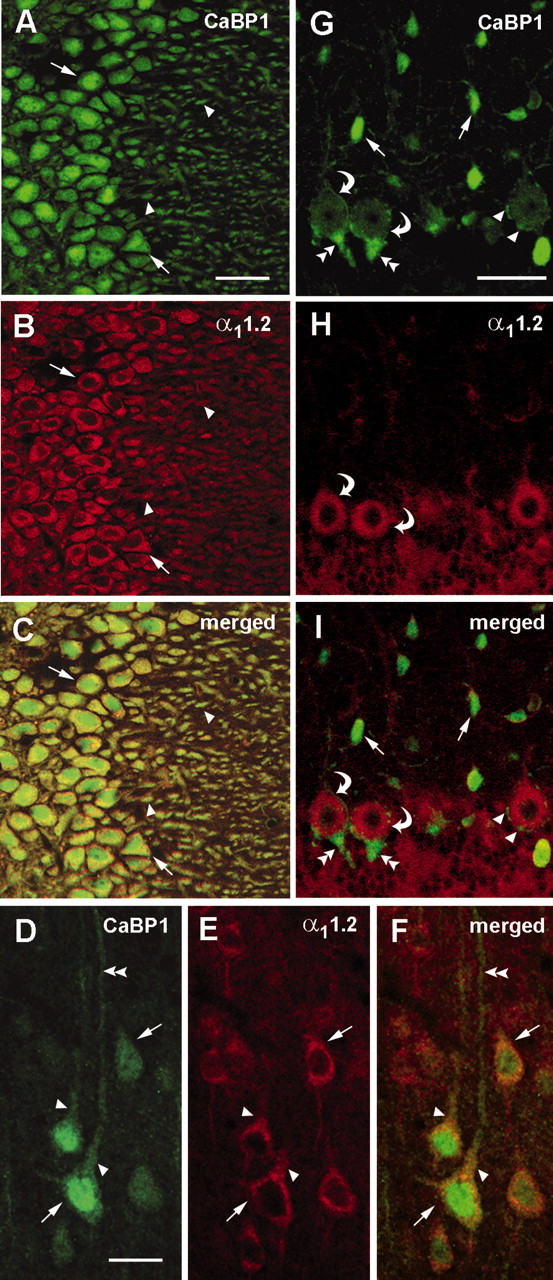
Colocalization of CaBP1 and α11.2 in rat brain. A-I, Confocal images of rat brain sections sequentially double labeled with antibodies against CaBP1 and α11.2. Immunofluorescence was viewed under optics for fluorescein(for CaBP1; A,D,G) or rhodamine (for α11.2;B, E, H). Regions of colocalization appear yellow in the merged images (C, F, I). Extensive colocalization of CaBP1 and α11.2 was detected in cell bodies (arrows) and dendrites (arrowheads) of neurons in the CA3 region of the hippocampus (A-C). In layer V of the cerebral cortex (D-F), CaBP1 and α11.2 colocalized in pyramidal cell soma (arrows) and proximal dendrites (arrowheads) but not more distal dendrites (double arrowheads). In the cerebellum (G-I), CaBP1 and α11.2 were colocalized in Purkinje cell bodies (curved arrows), but only CaBP1 was found in interneurons in the molecular layer (arrows) and in small perisomatic synapses (arrowheads) and large Pinceaux synapses (double arrowheads) onto Purkinje cell bodies. Scale bars: A-C, G-I, 100 μm; D-F, 50 μm.
CaBP1 prolongs Cav1.2 Ca2+ currents and does not support Ca2+-dependent inactivation
Because CaBP1 and CaM share similar binding determinants in α11.2, CaBP1 could simply replicate the modulatory effects of CaM. Alternatively, CaBP1 may differentially regulate the channel as we showed for Cav2.1 (P/Q-type) channels (Lee et al., 2002). To distinguish between these possibilities, we analyzed inactivation of Ca2+ currents through Cav1.2 channels transfected in HEK293T cells in whole-cell patch-clamp recordings. Inactivation was quantitatively compared as the residual current amplitude at the end of the test pulse normalized to the peak current (Ires/Ipk). In these experiments, Cav1.2 Ca2+ currents (ICa) inactivated rapidly and with a biphasic time course during a 1 sec step depolarization, as a result of endogenous Ca2+/CaM interacting with the channel (Peterson et al., 1999; Qin et al., 1999; Zühlke et al., 1999) (Fig. 5A). In contrast, cotransfection with CaBP1 caused ICa to inactivate more slowly, resulting in a significant increase in Ires/Ipk compared with in cells transfected with Cav1.2 alone [0.63 ± 0.05 (n = 11) for Cav1.2 plus CaBP1 vs 0.32 ± 0.02 (n = 17) for Cav1.2 alone; p < 0.001] (Fig. 5A). Biexponential fits of ICa inactivation revealed a significant effect of CaBP1 in slowing the fast time constant (τfast = 2.9 ± 1.5 sec for Cav1.2 plus CaBP1 vs 0.4 ± 0.03 sec for Cav1.2 alone; p < 0.05) and decreasing the fraction of channels inactivating with τfast (Ffast = 0.27 ± 0.04 for Cav1.2 plus CaBP1 vs 0.60 ± 0.03 for Cav1.2 alone; p < 0.001) (Fig. 5B). In addition, CaBP1 nearly doubled the slow time constant (τslow = 9.5 ± 1.6 sec for Cav1.2 plus CaBP1 vs 4.6 ± 0.6 sec for Cav1.2 alone; p < 0.001) but did not affect the fraction of channels inactivating with τslow (Fslow = 0.22 ± 0.04 for Cav1.2 plus CaBP1 vs 0.19 ± 0.01 for Cav1.2 alone; p = 0.4) (Fig. 5B).
Figure 5.

CaBP1 prolongs Ca2+ currents through Cav1.2 channels and does not support Ca2+-dependent inactivation. A, Effect of CaBP1 on Cav1.2 Ca2+ currents evoked by step depolarizations. Traces represent Ca2+ currents evoked by a 1 sec step from -80 to +10 mV in whole-cell patch-clamp recordings of HEK293T cells transfected with Cav1.2 subunits with or without CaBP1. Ires /Ipk was determined by dividing the residual current amplitude at the end of the pulse by the peak current amplitude. Extracellular solution contained 10 mm Ca2+, and intracellular solution contained 5 mm EGTA. Data represent mean ± SEM for Cav1.2 (n = 17) and Cav1.2 plus CaBP1 (n = 11) (*p < 0.001). B, Impact of CaBP1 on fast and slow inactivation. Ca2+ currents obtained in A were fit with a double-exponential function. The fast and slow time constants (τfast,τslow) and fraction of channels showing fast and slow components of inactivation (Ffast, Fslow) were averaged and shown for cells transfected with Cav1.2 alone or Cav1.2 plus CaBP1 (*p < 0.001). C, Comparison of Ca2+-dependent inactivation in cells transfected with Cav1.2 alone or cotransfected with CaBP1. Traces show currents evoked by 1 sec depolarizing step from -80 to +10 mV for ICa (black trace) or 0 mV for IBa (gray trace) to compensate for -10 mV voltage shift for IBa. Extracellular solution contained either 10 mm Ca2+ or Ba2+.[ICa - IBa] represents the difference between Ires /Ipk for ICa and the mean Ires /Ipk for IBa for Cav1.2 channels alone (n = 17; open bar) or Cav1.2 plus CaBP1 (n = 11; hatched bar) (*p < 0.001). D, U-Shaped voltage dependence of Ires /Ipk for ICa in cells transfected with Cav1.2 alone but not in cells cotransfected with CaBP1. Ires /Ipk was determined for ICa evoked by 1 sec test pulses from -80 mV to various voltages, averaged, and plotted against test voltage for Cav1.2 alone (open circles; n = 9) or Cav1.2 plus CaBP1 (filled circles; n = 8).
These effects of CaBP1 on slowing inactivation of ICa are similar to results obtained with CaM mutants unable to bind Ca2+. These CaM mutants interact with the tethering site on the channel but are unable to support Ca2+-dependent inactivation (Peterson et al., 1999; Zühlke et al., 1999). To determine whether a similar mechanism could account for the effects of CaBP1, we compared inactivation with Ca2+ and Ba2+ as the charge carriers, because Ba2+ does not substitute favorably for Ca2+ in binding to EF-hand motifs and therefore blocks Ca2+-dependent effects of CaM and other Ca2+ sensors (Wang, 1985). In cells transfected with Cav1.2 alone, Ca2+/CaM-dependent inactivation was evident as a sizeable difference in Ires/Ipk for ICa compared with IBa [(ICa - IBa) =-0.46 ± 0.02; n = 17] (Fig. 5C). In comparison, in cells cotransfected with CaBP1, this difference in ICa and IBa inactivation was nearly abolished [(ICa - IBa) = -0.06 ± 0.05; n = 11; p < 0.01] (Fig. 5C). In addition, for Cav1.2 alone, a plot of Ires/Ipk versus test voltage was U-shaped, with maximal inactivation at +5 mV, which was near the peak of the I-V curve, as would be expected of a process depending on incoming Ca2+ (Brehm et al., 1980) (Fig. 5D). In contrast, the extent of ICa inactivation in cells cotransfected with CaBP1 did not change appreciably with different test voltages (Fig. 5D). These results are similar to those obtained previously with the dominant-negative CaM mutants (Peterson et al., 1999; Zühlke et al., 1999) and demonstrate a novel effect of CaBP1 on prolonging Ca2+ currents and opposing Ca2+-dependent inactivation of Cav1.2.
CaBP1 causes Ca2+-dependent facilitation and prevents inactivation during repetitive stimuli
To characterize the significance of CaBP1 regulation of Cav1.2 channels during more physiological stimuli, we evoked Cav1.2 currents with trains of short (5 msec) depolarizations using both Ca2+ and Ba2+ as permeant ions. With this voltage protocol, inactivation of ICa proceeds rapidly and causes an ∼50% reduction in current amplitude by the end of the train (Fig. 6A). This strong inactivation is Ca2+ dependent because the amplitude of IBa remains relatively constant throughout the protocol. Consistent with the prolonged currents during the 1 sec step depolarizations (Fig. 5A,C), neither ICa nor IBa inactivated appreciably during the train in cells cotransfected with CaBP1 (Fig. 6B). Moreover, CaBP1 caused an initial facilitation in ICa current amplitude, which was Ca2+ dependent in that no similar change in IBa was observed (Fig. 6B,C). The facilitated ICa was maximal by the third pulse in the train and declined rapidly to initial levels by the sixth pulse. Ca2+-dependent regulation of Cav1.2 with and without CaBP1 was quantitatively expressed as the difference in ICa and IBa [(ICa - IBa)] during the first (0-0.1 sec) and last (0.9-1 sec) periods of the stimulus train. CaBP1 caused a significant increase (∼6%) in ICa compared with IBa in the first 100 msec, in contrast to ∼25% decrease in ICa relative to IBa over this same time period with Cav1.2 alone (Fig. 6D). The effect of CaBP1 was especially dramatic in the last 100 msec, in which Ca2+-dependent inactivation of Cav1.2 caused ICa to decay to nearly 50% of the amplitude of IBa, whereas cells cotransfected with CaBP1 showed virtually no difference in ICa and IBa amplitudes (Fig. 6D).
Figure 6.
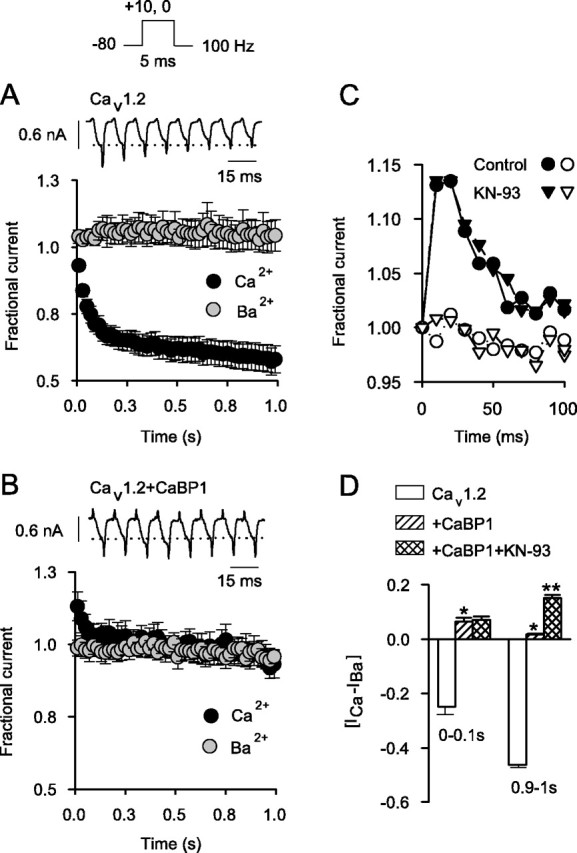
CaBP1 blocks Ca2+-dependent inactivation and causes facilitation during repetitive stimuli. A, Ca2+-dependent inactivation of Cav1.2 during trains of depolarizations. ICa and IBa were evoked by 5 msec test pulses from -80 to +10 mV for ICa or 0 mV for IBa as shown in voltage protocol above. Shown are representative traces of ICa during the first eight pulses. Dotted line indicates initial current amplitude. Fractional current, plotted below, represents test current amplitude normalized to that for the first pulse in the train and plotted against time for ICa(black circles; n = 11) and IBa (gray circles; n=5). Points represent the mean ± SEM, and every other point is plotted. B, Loss of Ca2+-dependent inactivation but gain of ICa facilitation with CaBP1. Current traces (top) and fractional current (bottom) were obtained as in A, except that recordings were from cells cotransfected with CaBP1 (n = 5 for ICa; n = 4 for IBa). C, Ca2+-dependent facilitation in cells cotransfected with CaBP1 is not prevented by KN-93. Facilitation of ICa (filled symbols) but not IBa (open symbols) is shown for cells cotransfected with Cav1.2 plus CaBP1 recorded with control intracellular solution (circles) or that containing the CaM kinase II inhibitor KN-93 (2 μm; triangles). Results were obtained as in A; and data from single representative cells were plotted for the first 100 msec of the train. D, Effects of CaBP1 on Ca2+-dependent modulation of Cav1.2. [ICa - IBa] represents the difference between the average fractional current for ICa and IBa for the first 10 (0-0.1 sec) or last 10 (0.9-1 sec) pulses. Shown are results obtained from cells transfected with Cav1.2 alone (n = 11), Cav1.2 plus CaBP1 (n = 5), and cells cotransfected with Cav1.2 plus CaBP1 that were treated with KN-93 (n = 5) (*p < 0.05 compared with Cav1.2 alone; **p < 0.01 compared with Cav1.2 plus CaBP1).
Ca2+-dependent facilitation caused by CaBP1 could result from a direct interaction with Cav1.2 or through an indirect pathway, such as through the activation of CaM kinase II, which causes Ca2+-dependent facilitation of cardiac Cav1.2 channels (Xiao et al., 1994; Yuan and Bers, 1994; Dzhura et al., 2000). To rule out the latter mechanism, we tested the effect of the CaM kinase II inhibitor KN-93 on ICa and IBa in cells cotransfected with Cav1.2 and CaBP1. KN-93 did not significantly reduce the initial facilitation of ICa [(ICa - IBa) = 0.06 ± 0.01 (n = 5) for control vs 0.07 ± 0.01 (n = 5) for KN-93; p = 0.58] and actually augmented the difference in ICa and IBa in the later part of the train in cells cotransfected with CaBP1 [ICa - IBa) = 0.02 ± 0.002 for control vs 0.15 ± 0.01 for KN-93; p < 0.01] (Fig. 6C,D). This later effect of KN-93 suggests that CaM kinase II may oppose the effects of CaBP1 on ICa facilitation. Alternatively, enhanced Ca2+-dependent facilitation with KN-93 may be related to nonspecific actions independent of CaM kinase II, which have been reported previously (Smyth et al., 2002). Regardless of a potential modulatory role for CaM kinase II, these results clearly show that CaM kinase II activation is not required for CaBP1-dependent facilitation of Cav1.2, which may rely instead on direct Ca2+-dependent interactions of CaBP1 with Cav1.2.
The IQ mediates facilitation of Cav1.2 Ca2+ currents by CaBP1
CaM binding to the IQ is critical for Ca2+-dependent inactivation of Cav1.2 (Qin et al., 1999; Romanin et al., 2000; Zühlke et al., 2000; Erickson et al., 2003). To determine whether facilitation of ICa in cells cotransfected with CaBP1 similarly depended on the IQ, we analyzed channels in which the first two residues of this site (I1624 and Q1625) were replaced with either alanine (Cav1.2IQ-AA) or glutamate (Cav1.2IQ-EE). Previous studies showed that the IQ-AA mutation significantly impairs Ca2+-dependent inactivation and, interestingly, unmasks a second effect of Ca2+/CaM in causing facilitation in trains of depolarizations given at significantly lower frequencies (3.3 Hz) than that used in Figure 6 (100 Hz) (Zühlke et al., 2000). This Ca2+/CaM-dependent facilitation would complicate analyses of the effect of the IQ-AA mutation on CaBP1-dependent facilitation but was absent in recordings with the 100 Hz stimulus protocol, which favored inactivation of ICa rather than facilitation (Fig. 7A). Unlike its impact on Ca2+/CaM-dependent modulation of Cav1.2, the IQ-AA mutation was only partially effective in preventing the functional effects of CaBP1. During the first 100 msec of the train, overt facilitation of ICa was not observed in cells cotransfected with Cav1.2IQ-AA and CaBP1, in that the amplitude of ICa did not increase above initial levels (compare Figs. 6B, 7A, first five pulses). However, CaBP1 still caused a significant increase in ICa during both the first 10 pulses [0.99 ± 0.01 (n = 6) for Cav1.2IQ-AA plus CaBP1 vs 0.90 ± 0.01 (n = 5) for Cav1.2IQ-AA alone; p < 0.05] and last 10 pulses of the stimulus train (1.05 ± 0.004 for Cav1.2IQ-AA plus CaBP1 vs 0.68 ± 0.003 for Cav1.2IQ-AA alone; p < 0.001) (Fig. 7A,B). Binding analyses showed that the IQ-AA mutation weakened, but did not abolish, CaBP1 binding to the IQ (Fig. 7A), which may explain the residual modulation of Cav1.2IQ-AA by CaBP1.
Figure 7.

Essential role for the IQ in CaBP1 binding and modulation of Cav1.2 during repetitive stimuli. A, Effect of alanine substitutions in the IQ on binding of CaBP1 and modulation of Cav1.2. Top panels show CaBP1 binding to GST-tagged fragments containing the IQ with or without IQ-AA substitutions (CT6, CT6IQ-AA). Binding was done with 2 mm Ca2+ as described in Figure 2B. Bottom panel shows pattern of ICa evoked by repetitive stimuli in cells transfected with Cav1.2IQ-AA alone (open circles) or cotransfected with CaBP1 (filled circles). Data were obtained with voltage protocols and plotted as in Figure 6. Points represent mean ± SEM (n = 4-6), with every second point plotted. B, Fractional current obtained in A was averaged for the first 10 (0-0.1 sec) and last 10 (0.9-1 sec) pulses in the train and shown for Cav1.2IQ-AA transfected alone (open bars) or cotransfected with CaBP1 (filled bars) (*p < 0.005 compared with Cav1.2IQ-AA). C, IQ to EE substitutions prevent binding and inhibit functional effects of CaBP1. Top panel shows loss of CaBP1 binding to GST-tagged CT6 with IQ-EE substitutions. Bottom panel shows ICa, obtained as in A, in cells transfected with Cav1.2 channels containing the IQ-EE mutation alone (n = 5; open circles) or cotransfected with CaBP1 (n = 6; filled circles). D, Fractional current for data plotted in C was averaged as in B for Cav 1.2IQ-EE alone (open bars) or Cav1.2IQ-EE plus CaBP1 (filled bars) (* p < 0.05 compared with Cav1.2IQ-EE).
In contrast to these results, the IQ-EE mutation completely prevented binding of CaBP1 to the IQ (Fig. 7C). Moreover, this mutation significantly suppressed the facilitatory actions of CaBP1 on ICa evoked in both the initial and later periods of the 100 Hz stimulus train (Fig. 7C,D). During the first 100 msec, there was no significant difference in the fractional amplitude of ICa in cells transfected with Cav1.2IQ-EE alone (0.93 ± 0.01; n = 5) and in cells cotransfected with CaBP1 (0.95 ± 0.01; n = 6; p = 0.22). The IQ-EE mutation nearly abolished the effect of CaBP1 on prolonging ICa during the train, although there was still a small but statistically significant difference between the average fractional current during the last 100 msec in cells transfected with Cav1.2IQ-EE alone (0.77 ± 0.01) and in cells cotransfected with CaBP1 (0.85 ± 0.002; p < 0.01). These findings are consistent with a model in which low-affinity interactions of CaBP1 with the IQ, which are lost in Cav1.2IQ-AA (Fig. 7A), trigger the initial phase of facilitation, whereas high-affinity binding of CaBP1, which is spared by the IQ-AA mutation (Fig. 7A) but eliminated by IQ-EE (Fig. 7C), could mediate the effects of CaBP1 on prolonging Cav1.2 Ca2+ currents during a train of depolarizations. Thus, a complex mechanism accounts for how CaBP1 stabilizes the open state of Cav1.2 channels, involving dynamic interactions of CaBP1 with multiple determinants in the C-terminal domain of α11.2.
Discussion
Our results provide new insights into the role of CaBP1 in the regulation of neuronal Ca2+ signaling. CaBP1 binds to the same sites as CaM in the C-terminal domain of the α1 subunit of Cav1.2 but has opposite effects on channel function. Whereas CaM enhances inactivation of Cav1.2 in a Ca2+-dependent manner, CaBP1 causes overt Ca2+-dependent facilitation and prolongs Ca2+ currents by inhibiting inactivation. The association and colocalization of CaBP1 and Cav1.2 postsynaptically in neurons throughout the brain implicate Ca2+ channel regulation by CaBP1 as an important determinant of voltage-gated Ca2+ influx in neurons.
Molecular basis for opposing regulation of Cav1.2 by CaBP1 and CaM
Several lines of evidence imply that CaBP1 is constitutively associated with α11.2 via the C site at resting Ca2+ levels, with Ca2+ influx through the channel promoting a Ca2+-dependent shift of CaBP1 to the IQ and subsequent facilitation of Cav1.2. First, CaBP1 coimmunoprecipitates with the intact channel under Ca2+-free conditions, and CaBP1 binding only to the C site is Ca2+ independent (Figs. 2, 3). Second, Ca2+-dependent facilitation caused by CaBP1 occurs within the first 10-100 msec in repetitive pulse protocols and is prevented by mutations in the IQ that also prevent binding of CaBP1 (Figs. 6B, 7C,D). These results implicate the IQ as an important effector site mediating CaBP1-dependent facilitation, the rapid onset of which is consistent with a secondary interaction of preassociated CaBP1 with the IQ. It is important to note that CaM is endogenously expressed in our transfected cells, which raises the additional possibility that CaM and CaBP1 binding to separate sites simultaneously in α11.2 could jointly modulate Cav1.2. This seems unlikely given that CaBP1 displaces CaM binding to the IQ (Fig. 2D), and preassociated CaBP1 rather than free CaM would presumably have the advantage in modulating the channel via interactions with the IQ. Although additional experiments will be required to fully exclude the involvement of CaM, our current findings support a model in which CaBP1 enhances Cav1.2 currents through sequential interactions at the tethering region (C site) and IQ.
Because CaBP1 does bind to the same sites in α11.2 as CaM, a major question is how CaBP1 causes such distinct regulation of Cav1.2. One possibility is that CaBP1 simply acts as a dominant negative in blocking the effects of endogenous CaM. Although dominant-negative CaM mutants prevent Ca2+-dependent inactivation of Cav1.2 (Peterson et al., 1999; Zühlke et al., 1999), they do not cause overt facilitation of Cav1.2 Ca2+ currents like that in cells transfected with CaBP1 (Fig. 6B,C). Therefore, we propose that CaBP1 acts not simply by displacing endogenous CaM but rather induces different conformational changes during binding α11.2 compared with CaM. Although CaBP1 and CaM are ∼50% similar at the amino acid level, CaBP1 differs in that it is N-terminally myristoylated, a modification that mediates its association with the plasma membrane (Haynes et al., 2004). It is possible, then, that myristoylated CaBP1 may facilitate interactions of the α11.2 C-terminal domain with the plasma membrane that suppress inactivation. CaBP1 also has mutations in EF-hand 2 that prevent Ca2+ binding and an extra α-helical turn in the linker between N-and C-terminal lobes (Haeseleer et al., 2000). These differences collectively could permit interactions of CaBP1 with molecular contacts within the C-terminal domain of α11.2 that directly stabilize channel opening rather than inactivation.
Neuronal Ca2+ binding proteins as functional subunits of voltage-gated Ca2+ channels
Increasing evidence supports a role for NCBPs, such as CaBP1, in diversifying the properties of voltage-gated Ca2+ channels in neurons (Wang et al., 2001; Weiss and Burgoyne, 2001; Lee et al., 2002; Tsujimoto et al., 2002; Weiss and Burgoyne, 2002). Given that CaM binding and regulation appears to be a common mechanism among different Ca2+ channel classes (Liang et al., 2003), NCBPs, like CaBP1, may contribute to the heterogenous properties of these channels in neurons through direct interactions with the α1 subunit. In some cases, the modulatory effect of an NCBP may be similar to that of CaM (Tsujimoto et al., 2002). In others, differences in how the NCBP interacts with the CaM-binding site may lead to alternate forms of channel regulation. For example, we showed previously that CaBP1 also interacts with the CaM-binding domain of Cav2.1 (P/Q-type) channels, but unlike CaM, has strong inhibitory effects on Cav2.1 currents (Lee et al., 2002). This provides an interesting contrast to our present results showing a facilitatory effect of CaBP1 on Cav1.2 channels, suggesting fundamental differences in the structure and function of CaM/CaBP1-binding sites in Cav1.2 compared with Cav2.1 channels.
Physiological significance of CaBP1 in regulating the L-type conductance in neurons
Cav1.2 channels in many neurons show little inactivation during sustained depolarizations, leading to their original classification as L-type for their “long-lasting” conductance (Nowycky et al., 1985). Because most characterizations of these channels used Ba2+ as a charge carrier, it is not clear to what extent they undergo rapid Ca2+-dependent inactivation similar to Cav1.2 channels in the heart (Yue et al., 1990; Neely et al., 1994). Although prominent Ca2+-dependent inactivation typifies L-type channels in thalamocortical neurons (Meuth et al., 2002), Cav1.2 currents carried by Ca2+ ions in a variety of neuronal cell types are prolonged and slowly inactivating (Fisher and Bourque, 1995; Avery and Johnston, 1996; von Gersdorff and Matthews, 1996; Beck et al., 1997). Furthermore, L-type channels with properties similar to those we observed in cells cotransfected with Cav1.2 and CaBP1 have been characterized in neurons in which we found CaBP1 and Cav1.2 to be strongly colocalized. Slowly inactivating L-type Ca2+ currents have been described in neocortical pyramidal neurons (Brown et al., 1993; Sayer et al., 1993), and L-type channels in some hippocampal neurons exhibit sustained activation after tetanic stimulation (Schjött and Plummer, 2000). In contrast, pyramidal cells specifically from the CA1 region of the hippocampus, in which CaBP1 is not expressed (Laube et al., 2002), possess L-type Ca2+ channels with strong Ca2+-dependent inactivation (Kay, 1991). Although facilitation of neuronal L-type channels by cAMP- and voltage-dependent mechanisms is well documented (Parri and Lansman, 1996; Song and Surmeier, 1996; Davare et al., 2001; Meuth et al., 2002), our findings are the first to show that Cav1.2 channels undergo Ca2+-dependent facilitation mediated by CaBP1, which may contribute to the observed phenotypic diversity of L-type channels in neurons (Forti and Pietrobon, 1993; Kavalali and Plummer, 1994).
L-Type Ca2+ channels have long been implicated in mechanisms of activity-dependent synaptic plasticity. Ca2+ influx through postsynaptic L-type channels can promote long-term potentiation (LTP) or long-term depression (LTD) at various synapses (Grover and Teyler, 1990; Huang and Malenka, 1993; Christie et al., 1996). How Ca2+ influx through L-type channels specifically activates pathways leading to LTP or LTD is not clear but may depend on the nature of the Ca2+ signal itself. For example, LTD at mossy fiber-CA3 pyramidal cell synapses requires significant increases in postsynaptic Ca2+ mediated by L-type channels localized to the proximal dendrites of CA3 neurons (Lei et al., 2003), in which we found Cav1.2 channels to strongly colocalize with CaBP1 (Fig. 4A-C). At these and other synapses, CaBP1 interactions with Cav1.2 may play an important role in amplifying postsynaptic Ca2+ signals, thus regulating the strength and direction of activity-dependent synaptic change.
Footnotes
This work was supported by National Institutes of Health Grants NS044922 and AG021723 (A.L.) and the Whitehall Foundation (A.L.). We are grateful to Drs. Howard Rees and Laura Volpicelli-Daley for assistance with immunohistochemistry and confocal microscopy, Dr. Johannes Hell for α11.2 antibodies, Dr. Greg Hockerman for advice and sharing of cDNAs, and Dr. Criss Hartzell for comments on this manuscript.
Correspondence should be addressed to Amy Lee, Department of Pharmacology, Emory University School of Medicine, 5123 Rollins Research Building, 1510 Clifton Road, Atlanta, GA 30322. E-mail: alee@pharm.emory.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244698-11$15.00/0
H.Z. and S.-A.K. contributed equally to this work.
References
- Avery RB, Johnston D (1996) Multiple channel types contribute to the low-voltage-activated calcium current in hippocampal CA3 pyramidal neurons. J Neurosci 16: 5567-5582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME (1993) Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science 260: 181-186. [DOI] [PubMed] [Google Scholar]
- Beck H, Steffens R, Heinemann U, Elger CE (1997) Properties of voltage-activated Ca2+ currents in acutely isolated human hippocampal granule cells. J Neurophysiol 77: 1526-1537. [DOI] [PubMed] [Google Scholar]
- Brehm P, Eckert R, Tillotson D (1980) Calcium-mediated inactivation of calcium current in Paramecium. J Physiol (Lond) 306: 193-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown AM, Schwindt PC, Crill WE (1993) Voltage dependence and activation kinetics of pharmacologically defined components of the high-threshold calcium current in rat neocortical neurons. J Neurophysiol 70: 1530-1543. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Weiss JL (2001) The neuronal calcium sensor family of Ca2+-binding proteins. Biochem J 353: 1-12. [PMC free article] [PubMed] [Google Scholar]
- Carlin RK, Grab DJ, Cohen RS, Siekevitz P (1980) Isolation and characterization of postsynaptic densities from various brain regions: enrichment of different types of postsynaptic densities. J Cell Biol 86: 831-845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin D, Means AR (2000) Calmodulin: a prototypical calcium sensor. Trends Cell Biol 10: 322-328. [DOI] [PubMed] [Google Scholar]
- Christie BR, Magee JC, Johnston D (1996) Dendritic calcium channels and hippocampal long-term depression. Hippocampus 6: 17-23. [DOI] [PubMed] [Google Scholar]
- Davare MA, Avdonin V, Hall DD, Peden EM, Burette A, Weinberg RJ, Horne MC, Hoshi T, Hell JW (2001) A β2 adrenergic receptor signaling complex assembled with the Ca2+ channel Cav1.2. Science 293: 98-101. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW (1998) Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature 392: 198-202. [DOI] [PubMed] [Google Scholar]
- DeMaria CD, Soong T, Alseikhan BA, Alvania RS, Yue DT (2001) Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411: 484-489. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294: 333-339. [DOI] [PubMed] [Google Scholar]
- Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME (2000) Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol 2: 173-177. [DOI] [PubMed] [Google Scholar]
- Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A, Schwartz A, Harpold MM (1988) Sequence and expression of mRNAs encoding the α1 and α2 subunits of a DHP-sensitive calcium channel. Science 241: 1661-1664. [DOI] [PubMed] [Google Scholar]
- Erickson MG, Liang H, Mori MX, Yue DT (2003) FRET two-hybrid mapping reveals function and location of L-type Ca2+ channel CaM preassociation. Neuron 39: 97-107. [DOI] [PubMed] [Google Scholar]
- Fisher TE, Bourque CW (1995) Voltage-gated calcium currents in the magnocellular neurosecretory cells of the rat supraoptic nucleus. J Physiol (Lond) 486: 571-580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti L, Pietrobon D (1993) Functional diversity of L-type calcium channels in rat cerebellar neurons. Neuron 10: 437-450. [DOI] [PubMed] [Google Scholar]
- Ginty DD, Kornhauser JM, Thompson MA, Bading H, Mayo KE, Takahashi JS, Greenberg ME (1993) Regulation of CREB phosphorylation in the suprachiasmatic nucleus by light and a circadian clock. Science 260: 238-241. [DOI] [PubMed] [Google Scholar]
- Grover LM, Teyler TJ (1990) Two components of long-term potentiation induced by different patterns of afferent activation. Nature 347: 477-479. [DOI] [PubMed] [Google Scholar]
- Haeseleer F, Sokal I, Verlinde CLMJ, Erdjument-Bromage H, Tempst P, Pronin AN, Benovic JL, Fariss RN, Palczewski K (2000) Five members of a novel Ca2+-binding protein (CaBP) subfamily with similarity to calmodulin. J Biol Chem 275: 1247-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeseleer F, Imanishi Y, Sokal I, Filipek S, Palczewski K (2002) Calcium-binding proteins: intracellular sensors from the calmodulin superfamily. Biochem Biophys Res Commun 290: 615-623. [DOI] [PubMed] [Google Scholar]
- Haynes LP, Tepikin AV, Burgoyne RD (2004) Calcium-binding protein 1 is an inhibitor of agonist-evoked inositol 1,4,5-trisphosphate-mediated calcium signaling. J Biol Chem 279: 547-555. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA (1993) Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel α1 subunits. J Cell Biol 123: 949-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Breeze LJ, Wang KKW, Chavkin C, Catterall WA (1996) N-methyl-d-aspartate receptor-induced proteolytic conversion of postsynaptic class C L-type calcium channels in hippocampal neurons. Proc Natl Acad Sci USA 93: 3362-3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Malenka RC (1993) Examination of TEA-induced synaptic enhancement in area CA1 of the hippocampus: the role of voltage-dependent Ca2+ channels in the induction of LTP. J Neurosci 13: 568-576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR (1994) Selective potentiation of a novel calcium channel in rat hippocampal neurones. J Physiol (Lond) 480: 475-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR (1991) Inactivation kinetics of calcium current of acutely dissociated CA1 pyramidal cells of the mature guinea-pig hippocampus. J Physiol (Lond) 437: 27-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LF, Mammen A, Ehlers MD, Kindler S, Chung WJ, Garner CC, Huganir RL (1996) Interaction of the N-methyl-d-aspartate receptor complex with a novel synapse-associated protein, SAP102. J Biol Chem 271: 21622-21628. [DOI] [PubMed] [Google Scholar]
- Laube G, Seidenbecher CI, Richter K, Dieterich DC, Hoffmann B, Landwehr M, Smalla KH, Winter C, Bockers TM, Wolf G, Gundelfinger ED, Kreutz MR (2002) The neuron-specific Ca2+-binding protein caldendrin: gene structure, splice isoforms, and expression in the rat central nervous system. Mol Cell Neurosci 19: 459-475. [DOI] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, Catterall WA (1999) Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 339: 155-159. [DOI] [PubMed] [Google Scholar]
- Lee A, Scheuer T, Catterall WA (2000) Ca2+/calmodulin-dependent facilitation and inactivation of P/Q-type Ca2+ channels. J Neurosci 20: 6830-6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA (2002) Differential modulation of Cav2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci 5: 210-217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S, Pelkey KA, Topolnik L, Congar P, Lacaille JC, McBain CJ (2003) Depolarization-induced long-term depression at hippocampal mossy fiber-CA3 pyramidal neuron synapses. J Neurosci 23: 9786-9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan B, Yue DT (2003) Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 39: 951-960. [DOI] [PubMed] [Google Scholar]
- Meuth S, Pape HC, Budde T (2002) Modulation of Ca2+ currents in rat thalamocortical relay neurons by activity and phosphorylation. Eur J Neurosci 15: 1603-1614. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM (1991) L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron 7: 625-635. [DOI] [PubMed] [Google Scholar]
- Neely A, Olcese R, Wei X, Birnbaumer L, Stefani E (1994) Ca2+-dependent inactivation of a cloned cardiac Ca2+ channel α1 subunit (α1C) expressed in Xenopus oocytes. Biophys J 66: 1895-1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowycky MC, Fox AP, Tsien RW (1985) Three types of neuronal calcium channel with different calcium agonist sensitivity. Nature 316: 440-443. [DOI] [PubMed] [Google Scholar]
- Parri HR, Lansman JB (1996) Multiple components of Ca2+ channel facilitation in cerebellar granule cells: expression of facilitation during development in culture. J Neurosci 16: 4890-4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate P, Mochca-Morales J, Wu Y, Zhang JZ, Rodney GG, Serysheva II, Williams BY, Anderson ME, Hamilton SL (2000) Determinants for calmodulin binding on voltage-dependent Ca2+ channels. J Biol Chem 275: 39786-39792. [DOI] [PubMed] [Google Scholar]
- Perez-Reyes E, Castellano A, Kim HSBP, Baggstrom E, Lacerda AE, Wei X, Birnbaumer L (1992) Cloning and expression of a cardiac/brain β subunit of the L-type calcium channel. J Biol Chem 267: 1792-1797. [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Yue DT (1999) Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron 22: 549-558. [DOI] [PubMed] [Google Scholar]
- Peterson BZ, Lee JS, Mulle JS, Wang Y, DeLeon M, Yue D (2000) Critical determinants of Ca2+-dependent inactivation within a EF-hand motif of L-type Ca2+ channels. Biophys J 78: 1906-1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt GS, Zühlke RD, Hudmon A, Schulman H, Reuter H, Tsien RW (2001) Molecular basis of calmodulin tethering and Ca2+-dependent inactivation of L-type Ca2+ channels. J Biol Chem 276: 30794-30802. [DOI] [PubMed] [Google Scholar]
- Qin N, Olcese R, Bransby M, Lin T, Birnbaumer L (1999) Ca2+-induced inhibition of the cardiac Ca2+ channel depends on calmodulin. Proc Natl Acad Sci USA 96: 2435-2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, Abernethy DR, Soldatov NM (2000) Ca2+ sensors of L-type Ca2+ channel. FEBS Lett 487: 301-306. [DOI] [PubMed] [Google Scholar]
- Sayer RJ, Brown AM, Schwindt PC, Crill WE (1993) Calcium currents in acutely isolated human neocortical neurons. J Neurophysiol 69: 1596-1606. [DOI] [PubMed] [Google Scholar]
- Schjött JM, Plummer MR (2000) Sustained activation of hippocampal Lp-type voltage-gated calcium channels by tetanic stimulation. J Neurosci 20: 4786-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidenbecher CI, Langnaese K, Sanmarti-Vila L, Boeckers TM, Smalla KH, Sabel BA, Garner CC, Gundelfinger ED, Kreutz MR (1998) Caldendrin, a novel neuronal calcium-binding protein confined to the somatodendritic compartment. J Biol Chem 273: 21324-21331. [DOI] [PubMed] [Google Scholar]
- Smyth JT, Abbott AL, Lee B, Sienaert I, Kasri NN, De Smedt H, Ducibella T, Missiaen L, Parys JB, Fissore RA (2002) Inhibition of the inositol trisphosphate receptor of mouse eggs and A7r5 cells by KN-93 via a mechanism unrelated to Ca2+/calmodulin-dependent protein kinase II antagonism. J Biol Chem 277: 35061-35070. [DOI] [PubMed] [Google Scholar]
- Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM (1991) Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron 7: 45-47. [DOI] [PubMed] [Google Scholar]
- Soldatov NM, Oz M, O'Brien KA, Abernethy DR, Morad M (1998) Molecular determinants of L-type Ca2+ channel inactivation—segment exchange analysis of the carboxyl-terminal cytoplasmic motif encoded by exons 40-42 of the human α1C subunit gene. J Biol Chem 273: 957-963. [DOI] [PubMed] [Google Scholar]
- Song WJ, Surmeier DJ (1996) Voltage-dependent facilitation of calcium channels in rat neostriatal neurons. J Neurophysiol 76: 2290-2306. [DOI] [PubMed] [Google Scholar]
- Sotelo C, Llinas R (1972) Specialized membrane junctions between neurons in the vertebrate cerebellar cortex. J Cell Biol 53: 271-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Halling DB, Black DJ, Pate P, Zhang JZ, Pedersen S, Altschuld RA, Hamilton SL (2003) Apocalmodulin and Ca2+/calmodulin-binding sites on the Cav1.2 channel. Biophys J 85: 1538-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tongiorgi E, Righi M, Cattaneo A (1997) Activity-dependent dendritic targeting of BDNF and TrkB mRNAs in hippocampal neurons. J Neurosci 17: 9492-9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto T, Jeromin A, Saitoh N, Roder JC, Takahashi T (2002) Neuronal calcium sensor 1 and activity-dependent facilitation of P/Q-type calcium currents at presynaptic nerve terminals. Science 295: 2276-2279. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G (1996) Calcium-dependent inactivation of calcium current in synaptic terminals of retinal bipolar neurons. J Neurosci 16: 115-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CL (1985) A note on Ca2+ binding to calmodulin. Biochem Biophys Res Commun 130: 426-430. [DOI] [PubMed] [Google Scholar]
- Wang C-Y, Yang F, He X, Chow A, Du J, Russell JT, Lu B (2001) Ca2+-binding protein frequenin mediates GDNF-induced potentiation of Ca2+ channels and transmitter release. Neuron 32: 99-112. [DOI] [PubMed] [Google Scholar]
- Weick JP, Groth RD, Isaksen AL, Mermelstein PG (2003) Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. J Neurosci 23: 3446-3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JL, Burgoyne RD (2001) Voltage-independent inhibition of P/Q-type Ca2+ channels in adrenal chromaffin cells via a neuronal Ca2+ sensor-1-dependent pathway involves Src family tyrosine kinase. J Biol Chem 276: 44804-44811. [DOI] [PubMed] [Google Scholar]
- Weiss JL, Burgoyne RD (2002) Sense and sensibility in the regulation of voltage-gated Ca2+ channels. Trends Neurosci 25: 489-491. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Ahlijanian MK, Catterall WA (1990) Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature 347: 281-284. [DOI] [PubMed] [Google Scholar]
- Xiao R-P, Cheng H, Lederer WJ, Suzuki T, Lakatta EG (1994) Dual regulation of Ca2+/calmodulin-dependent kinase II activity by membrane voltage and by calcium influx. Proc Natl Acad Sci USA 91: 9659-9663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, McBride S, Mak DO, Vardi N, Palczewski K, Haeseleer F, Foskett JK (2002) Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca2+ release channels. Proc Natl Acad Sci USA 99: 7711-7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda R, Sabatini BL, Svoboda K (2003) Plasticity of calcium channels in dendritic spines. Nat Neurosci 6: 948-955. [DOI] [PubMed] [Google Scholar]
- Yuan W, Bers DM (1994) Ca-dependent facilitation of cardiac Ca2+ current is due to Ca2+-calmodulin-dependent protein kinase. Am J Physiol 267: H982-H993. [DOI] [PubMed] [Google Scholar]
- Yue DT, Backx PH, Imredy JP (1990) Calcium-sensitive inactivation in the gating of single calcium channels. Science 250: 1735-1738. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Reuter H (1998) Ca2+-sensitive inactivation of L-type Ca2+ channels depends on multiple cytoplasmic amino acid sequences of the α1C subunit. Proc Natl Acad Sci USA 95: 3287-3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zühlke RG, Pitt GS, Deisseroth K, Tsien RW, Reuter H (1999) Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399: 159-161. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Tsien RW, Reuter H (2000) Ca2+-sensitive inactivation and facilitation of L-type Ca2+ channels both depend on specific amino acid residues in a consensus calmodulin-binding motif in the α1C subunit. J Biol Chem 275: 21121-21129. [DOI] [PubMed] [Google Scholar]