

Abstract
GABA-mediated fast-hyperpolarizing inhibition depends on extrusion of chloride by the neuron-specific K-Cl cotransporter, KCC2. Here we show that sustained interictal-like activity in hippocampal slices downregulates KCC2 mRNA and protein expression in CA1 pyramidal neurons, which leads to a reduced capacity for neuronal Cl- extrusion. This effect is mediated by endogenous BDNF acting on tyrosine receptor kinase B (TrkB), with down-stream cascades involving both Shc/FRS-2 (src homology 2 domain containing transforming protein/FGF receptor substrate 2) and PLCγ (phospholipase Cγ)-cAMP response element-binding protein signaling. The plasmalemmal KCC2 has a very high rate of turnover, with a time frame that suggests a novel role for changes in KCC2 expression in diverse manifestations of neuronal plasticity. A downregulation of KCC2 may be a general early response involved in various kinds of neuronal trauma.
Keywords: BDNF, neurotrophic factors, intracellular chloride, activity-dependent gene expression, GABAergic transmission, epilepsy
Introduction
There is ample evidence that K-Cl cotransport is responsible to a large extent for neuronal Cl- extrusion and hence for the generation of Cl--dependent hyperpolarizing postsynaptic currents mediated by ionotropic GABAA and glycine receptors (Thompson and Gähwiler, 1989; Kaila, 1994; Payne et al., 2003). Expression of the neuron-specific K-Cl cotransporter KCC2 is required for the generation of the inwardly directed Cl- electrochemical gradient in CA1 pyramidal neurons of the rat hippocampus (Rivera et al., 1999) and several other types of neurons (Lu et al., 1999; Williams et al., 1999; DeFazio et al., 2000; Kakazu et al., 2000; Ganguly et al., 2001; Hubner et al., 2001).
Interestingly, short- and long-term shifts from hyperpolarizing to depolarizing (sometimes even excitatory) GABAA-mediated responses have been observed under various experimental conditions, including tetanic stimulation, neuronal trauma, GABA uptake block, and axotomy, as well as pharmacologically induced epileptiform activity (Kapur and Coulter, 1995; Avoli, 1996; van den Pol et al., 1996; Kaila et al., 1997; Jackson et al., 1999; Voipio and Kaila, 2000; Nabekura et al., 2002). In addition, spontaneous network activity associated with depolarizing postsynaptic GABAA responses has been seen in human neocortical tissue resected for treatment of temporal lobe epilepsy (Avoli et al., 1995; Köhling et al., 1998; Cohen et al., 2002).
Activation of tyrosine receptor kinase B (TrkB) receptors by BDNF leads to autophosphorylation of tyrosine residues within the intracellular domain of the receptor, creating docking sites for proteins such as src homology 2 domain containing transforming protein (Shc) and FGF receptor substrate 2 (FRS-2) and phospholipase Cγ (PLCγ) that activate second messengers and other downstream effectors. These include the cAMP response element-binding protein (CREB), which couples to the transcriptional machinery in the nucleus and thereby controls the expression of a wide variety of genes linked to long-term changes in neuronal plasticity (Finkbeiner et al., 1997; Bibel and Barde, 2000; Minichiello et al., 2002).
We have shown recently that KCC2 in the hippocampus is downregulated after kindling-induced seizures in vivo and also by applying exogenous BDNF or neurotrophin 4 in vitro (Rivera et al., 2002). These data, although suggestive, did not demonstrate a causal link between enhanced neuronal activity and TrkB-mediated downregulation of KCC2. In the present work, we provide direct evidence to show that in hippocampal CA1 pyramidal neurons, KCC2 is downregulated by continuous, synchronous neuronal activity via endogenous BDNF-TrkB signaling, which recruits both Shc and PLCγ-coupled intracellular pathways. The plasmalemmal KCC2 has a very high rate of turnover, which provides a time frame that is compatible with a role for changes in KCC2 expression in various manifestations of neuronal plasticity.
Materials and Methods
Hippocampal slices. Hippocampal slices from Wistar rats (Rivera et al., 2002) and the mutant trkBPLC/PLC, trkBSHC/SHC, and trkBW/W mice (Minichiello et al., 2002) were prepared using a Vibratome (Technical Products International). The standard physiological solution contained (in mm): 124 NaCl, 3.0 KCl, 2.0 CaCl2, 25 NaHCO3, 1.1 NaH2PO4, 2.0 MgSO4, and 10 d-glucose, and was equilibrated with 95% O2 and 5% CO2 to yield a pH of 7.4 at the experimental temperature, +32°C. Slices were left to recover at room temperature for ∼1 hr and thereafter transferred into a submerged-type (double-sided perfusion) chamber. MgSO4 was omitted in the 0-Mg2+ solution. 4-Aminopyridine (AP) (applied at 100 μm), 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline (NBQX) (10 μm), and dl-AP-5 (40 μm) were obtained from Tocris Cookson.
Interictal-like activity is generated in the absence of Mg2+ in slices kept under both interface and submerged conditions (Anderson et al., 1986; Mody et al., 1987; Beau and Alger, 1998). Because it turned out to be much easier to obtain quantitatively reproducible data under submerged conditions for the large number of slices required in the RT-PCR measurements and Western blots, as well as immunohistochemical and biotinylation experiments (see below), all experiments were done on submerged slices.
Electrophysiology. Field potentials were recorded using glass microelectrodes filled with 150 mm NaCl. Intracellular recordings were made from rat CA1 pyramidal cells using microelectrodes filled with 0.5 m K-acetate plus 0.5 m KCl, pH 6.6-6.8 (resistance 135-220 MΩ). Recordings were discarded if electrodes showed rectification or time-dependent changing of electrode resistance during current pulses. In a few experiments, 50 mm N-ethyl bromide quaternary salt (QX-314) was added to the microelectrode filling solution. An Axoclamp 2B amplifier (Axon Instruments, Foster City, CA) was used in either bridge or discontinuous current passing mode. Cells with a stable resting membrane potential of at least -55 mV (see Results) and input resistance of 38-126 MΩ were used in the measurements. Inhibitory postsynaptic responses were evoked in the presence of the ionotropic glutamate antagonists by stimuli (16-28 V, 60-100 μsec, frequency 1/10 or 1/15 Hz) delivered via a bipolar tungsten electrode positioned close (≤ 500 μm) to the recording electrode (Davies et al., 1990). Responses with spikes interfering with the IPSPA amplitude measurement were discarded from EIPSP-A analysis.
RT-PCR. RT-PCR analysis was performed as described previously (Rivera et al., 1999). Briefly, total RNA from the slices was extracted with Trizol (Sigma, St. Louis, MO) using a modified version of the protocol by Chomczynski and Sacchi (1987). Total RNA was reverse transcribed using random hexamer primers and SuperscriptII reverse transcriptase (Invitrogen, Gaithersburg, MD). Oligodeoxynucleotide primers were synthesized over regions specific for light neurofilament (NF-L), neuron-specific enolase (NSE), or KCC2 cDNA. For NF-L, forward (5′-GCACATCTCCAGCGTGCGCAG-3′) and reverse (5′-GGATCTGAGCCTGCAGCTCGG-3′) primers were used to amplify a 120 bp fragment. For KCC2, forward (5′-CTCAACAACCTGACGGACTG-3′) and reverse (5′-GCAGAAGGACTCCATGATGCCTGCG-3′) primers were used to amplify a 399 bp fragment (bp 4-402 in KCC2). For NSE, forward (5′-CTCTACCAGGACTTTGTCCGG-3′) and reverse (5′-GGGACAGCAAGAAAGAGGCT-3′) primers were used to amplify a 600 bp fragment.
Immunoblotting. Crude membrane fractions were prepared from slices in homogenization buffer (250 mm sucrose, 10 mm Tris, 10 mm HEPES, 1 mm EDTA; pH adjusted to 7.2 at +24°C, containing protease inhibitors by differential centrifugation). Membrane proteins were separated by SDS-PAGE, electrophoretically transferred to polyvinylidene difluoride or nitrocellulose membranes, and immunoblotted as described previously (Payne et al., 1996) using affinity-purified rabbit anti-KCC2 polyclonal antibodies (B2C; 1:1000), rabbit anti-phospho-CREB (pCREB) (1:1000; Upstate Biotechnology, Lake Placid, NY), and rabbit anti-α-tubulin (1: 10000; Sigma). Bands were developed using ECL-plus (Amersham, Arlington Heights, IL) on x-ray films as well as on a phosphoimager BAS-1500 (Fujifilm) and analyzed with TINA software (Tamro).
Immunohistochemistry. Slices were washed three times in PBS (0.1 m phosphate buffer, pH 7.4, 0.9% NaCl) and immersion-fixed in 4% PFA overnight. They were then processed into paraffin and sectioned (10-14 μm). The sections were dewaxed in xylene and rehydrated through a graded series of aqueous ethanol. The slices were washed three times in PBS and then placed in BSA/PBS (PBS containing 0.2% Triton X-100, 2% BSA) for 2 hr at room temperature. Sections were incubated at +4°C for 48 hr with primary antibody (Ab) diluted in BSA/PBS. Rabbit anti-KCC2 (B2C) Ab was diluted 1:200 (Payne et al., 1996), and rabbit anti-pCREB was diluted 1:200 (Upstate Biotechnology). After three PBS washes they were incubated for 2 hr with FITC-conjugated or tetramethylrhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit IgG (1:200; Jackson ImmunoResearch, West Grove, PA). Sections were then washed three times in PBS and once in distilled water and mounted on Superfrost Plus (Menzel-Glaser) slides with 4,6-diamidino-2-phenylindol (DAPI) (Sigma) containing gelvatol (Biomedia). For assessment of acute cell death (Poirier et al., 2000), DAPI-positive nuclei were counted in five frames of 100 × 200 μm randomly placed on the CA1 stratum pyramidale.
Regional and experimentally induced differences in immunofluorescence intensity were quantified with TINA software. The intensity was integrated over a given area in a slice (see Fig. 2 A), and when the experimental data (see Fig. 2 B) were pooled, each data point was based on at least three sections from a given slice. Confocal immunofluorescence microscopy was performed with a Bio-Rad MRC-1024 confocal microscope system (Bio-Rad, Hercules, CA) equipped with an argon-krypton ion laser (American Laser Corporation) and an Axiovert 135M using a 63× oil numerical aperture 1.4 objective (Carl Zeiss, Oberkochen, Germany). TRITC-stained samples were imaged by excitation at 568 nm and with a 598-621 nm bandpass emission filter. Optical sections were taken at every 0.6 μm.
Figure 2.
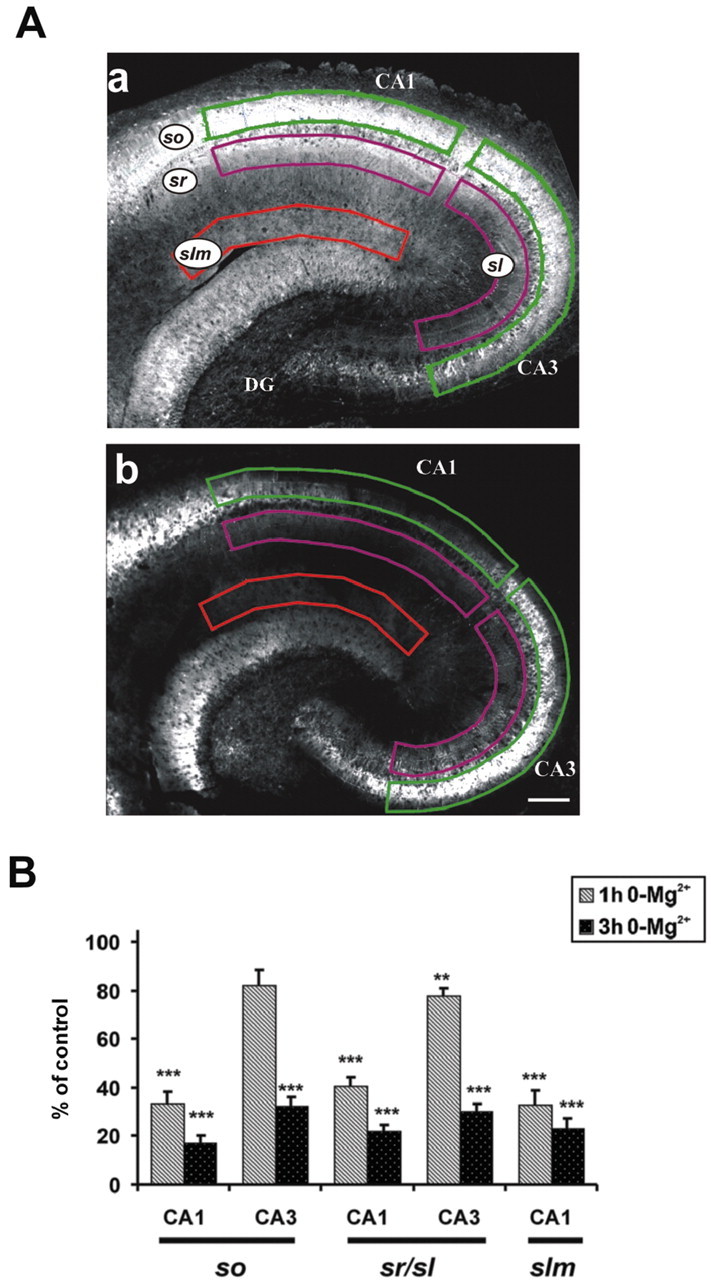
Area-specific decrease of KCC2 immunofluorescent staining in slices exposed to the 0-Mg2+ solution. A, Slices superfused for 1 hr in control solution (Aa) and in the Mg2+-free solution (Ab). Note the pronounced reduction in staining intensity in CA1. so, Stratum oriens; sr, stratum radiatum; sl, stratum lucidum; slm, stratum lacunosum-moleculare; DG, dentate gyrus). B, Plot of optical intensities from the distinct areas marked in Aa and Ab after 1 and 3 hr of 0-Mg2+ superfusion. (t test; ***p < 0.001; n = 10.) Scale bar, 400 μm.
In situ hybridization. A 1039 bp mouse KCC2 expressed sequence tag clone (AA982489), corresponding to nucleotides 4605-5566 of the full-length rat KCC2 cDNA (Payne et al., 1996), was used for the synthesis of labeled cRNA KCC2 probe (Hiltunen et al., 1996). Free-floating in situ hybridization was performed on 100 μm sections obtained from cutting thick acute hippocampal slices (350 μm) and using digoxigenin-labeled riboprobes as described (Nieto et al., 1996).
Biotinylation assay of membrane surface KCC2. To examine the rate of decay of plasmalemmal KCC2, we used a recently developed method for membrane surface biotinylation in acute slice preparations (Thomas-Crusells et al., 2003). Slices were placed in an experimental chamber and incubated on ice in the standard physiological solution (95% O2/5% CO2) containing biotin-sulfo-N-hydroxysuccinimide (100 μm; Sigma). After 1 hr, slices were rinsed with cold physiological solution containing 1 μm lysine (Sigma) to stop the reaction. Thereafter, the temperature was raised to +32°C, and the slices were exposed to the various experimental conditions (see Fig. 4), collected, and lysed at fixed time points in a solution containing 1% Triton X-100, 0.1% SDS, 1 mm EDTA, 50 mm NaCl, 20 mm Tris-HCl, pH 7.5, and protease inhibitors (Roche, Hertfordshire, UK). The lysates were precipitated with streptavidin-Sepharose beads (Roche) by overnight incubation at +4°C followed by a brief centrifugation. The beads were washed with PBS and stored at -70°C until use. The samples were run through a 10% ready-gel SDS-PAGE (Bio-Rad), transferred to nitrocellulose membrane (Hybond ECL; Amersham Biosciences), and blotted at +4°C overnight with rabbit anti-rat KCC2 (B2C) primary Ab (1:2000) and mouse anti-human transferrin receptor primary Ab (1:2000; Zymed Laboratories, San Francisco, CA). Incubation with the secondary Ab (goat anti-rabbit IgG-peroxidase, 1:4000, and goat anti-mouse IgG-peroxidase, 1:4000; Sigma) was for 1 hr at room temperature. For calculation of the rate of KCC2 decay, the density of the bands was analyzed with the TINA software, and the data were fitted by regression analysis to the equation f(t) = y0 + ae-t/τ, where a is the amplitude of the decline of biotinylated KCC2 protein, y0 is the apparent baseline level in the present experiments, t is the time from the onset of the experiment, and τ is the time constant of the exponential decay.
Figure 4.

Decay of cell surface biotinylated KCC2. Aa, Western blot of streptavidin-precipitated B-KCC2 showing a very fast rate of fall of B-KCC2 protein under control conditions that was enhanced further by omission of extracellular Mg2+. The level of plasmalemmal B-TfR does not show any significant change within 90 min. Ab, The application of NBQX (10 μm) and dl-AP-5 (40 μm) blocked the enhancing effect of 0-Mg2+ on the rate of decline of B-KCC2. B, Plot of normalized B-KCC2 and B-TfR optical band intensities from experiments such as shown in A.
Statistics. The data are presented as mean ± SEM. Statistical analysis was performed using nonpaired Student's t test. Statistical significance was defined as p < 0.01 (**) and p < 0.001 (***).
Results
Effect of interictal-like activity on KCC2 mRNA and protein levels
Exposure of hippocampal slices to a solution devoid of Mg2+ (0-Mg2+) resulted in the generation of continuous interictal-like activity (Anderson et al., 1986; Mody et al., 1987), which commenced at ∼10-15 min and showed a progressive increase in frequency during the 1-3 hr recording sessions (Fig. 1A). As shown by RT-PCR, there was a progressive fall in KCC2 mRNA after 1-3 hr in the Mg2+-free solution when compared with control slices or slices exposed to the 0-Mg2+ solution for 30 min only (Fig. 1B) (n = 4).
Figure 1.

Interictal-like activity leads to a downregulation of KCC2 protein and mRNA levels in acute hippocampal slices. A, Extracellular field potentials in CA1 stratum pyramidale at 0.5, 1, and 3 hr after withdrawal of Mg2+. B, RT-PCR shows that KCC2 mRNA is downregulated, whereas NF-LmRNA remains constant. C, Western blots show activity-dependent downregulation of the KCC2 protein (∼150 kDa band) after 1-3 hr in 0-Mg2+. KCC2 mRNA and protein are not downregulated in slices in which 0-Mg2+-induced activity was blocked with 10 μm NBQX and 40 μm dl-AP-5 (B, C), showing an activity-specific effect.α-Tubulin was used to monitor the amount of total protein. D, Statistical representation of optical densities from five experiments such as those in C, expressed as percentage of control (means ± SEM; t test; ***p < 0.001; **p < 0.01).
In Western blot analyses, the ∼150 kDa band that corresponds to the glycosylated form of KCC2 protein (Rivera et al., 1999; Williams et al., 1999) showed a pronounced fall during 0-Mg2+ exposure for 1-3 hr (Fig. 1C). A slight decline with a rate of ∼0.2%/min was seen under control conditions. To address whether the prompt downregulation of KCC2 was caused by the withdrawal of Mg2+ per se or by the consequent discharge activity, we blocked the activity by using NBQX plus AP-5 (Anderson et al., 1986; Mody et al., 1987). Under these conditions, KCC2 mRNA as well as protein levels were retained at a level that was indistinguishable from that obtained from parallel controls (Fig. 1B,C).
Figure 1D shows a compilation of quantitative data obtained in Western blots from experiments terminated at 1-3 hr. The relative fall of KCC2 protein in slices with interictal-like activity versus time-matched controls was 22 ± 7% (p < 0.01) at 1 hr, 36 ± 5% (p < 0.001) at 1.5 hr, and 61 ± 5% at 3 hr (p < 0.001; n = 5).
Effect of interictal-like activity on area-specific KCC2 expression patterns
Figure 2A shows the distribution of KCC2 immunofluorescent staining in slices superfused with standard (n = 10) or 0-Mg2+ solution (n = 10) for 1 hr. After 1 hr under control conditions (Fig. 2Aa), the KCC2 staining pattern is similar to the one observed in perfused brain sections (Gulyas et al., 2001). The immunoreactivity of the plasmalemmal KCC2 in the dendrites of principal cells is mainly responsible for the diffuse staining found in the neuropil (Gulyas et al., 2001). We found that, also in acute slices, the intensity of the diffuse neuropil staining in CA1 is strongest in stratum oriens (Fig. 2Aa) close to stratum pyramidale and decreases along the stratum radiatum. The staining intensity increases again close to the stratum lacunosummoleculare border. In CA3, KCC2 staining was high in stratum oriens, but in stratum lucidum, the overall staining was less intense than in the apical dendritic regions in CA1.
In agreement with the data from Western blots, we observed a clear fall in the KCC2 staining intensity in slices exposed to the 0-Mg2+ solution (Fig. 2Ab). This decrease was most conspicuous in the CA1 region, where the pyramidal neurons are well known for their high vulnerability to a wide variety of pathological insults (Schmidt-Kastner and Freund, 1991).
To compare in more quantitative terms the effects of 0-Mg2+ exposure on the expression patterns of KCC2, we measured the area-specific intensity profiles of the immunofluorescence signal (Fig. 2A). The intensity of each region in 0-Mg2+ slices was compared with the corresponding region in parallel control slices. A statistical quantification is provided in Figure 2B, which clearly indicates that after 1 hr in 0-Mg2+ (n = 10), the expression of KCC2 in CA1 was downregulated on average by >60% relative to controls (n = 10) in all layers examined. The corresponding changes in CA3 were much smaller: ∼20%. Superfusion of the slices (control, n = 9; 0-Mg2+, n = 9) for 3 hr in the 0-Mg2+ solution led to a further decrease in KCC2 protein levels in all regions examined (Fig. 2B).
Because no significant difference in the number of DAPI-positive nuclei (see Materials and Methods) was found in the CA1 region between control slices (n = 9) and those exposed for 3 hr to 0-Mg2+ (16 ± 2 vs 14 ± 3 nuclei per 100 × 200 μm frame; n = 9), the reduced intensity in KCC2-positive staining observed in the CA1 region cannot be explained on the basis of an acute 0-Mg2+-induced cell death.
Interictal-like activity induced by the convulsant drug 4-AP (100 μm) for 1 hr in acute hippocampal slices (data not shown) (Avoli, 1996; Lamsa and Kaila, 1997) also induced downregulation of KCC2 (Fig. 3). Free-floating nonradioactive in situ hybridization showed a general downregulation of KCC2 mRNA as compared with control slices (n = 4). A more pronounced effect was observed in the CA3 and dentate gyrus (Fig. 3A). Parallel Western blot analyses showed a clear downregulation of KCC2 protein levels (35 ± 9%; p < 0.001) in response to 4-AP exposure (Fig. 3B) (n = 4). These data provide further evidence for the view that the activity-dependent effects observed in 0-Mg2+ are not attributable to the withdrawal of magnesium per se.
Figure 3.

Exposure to the convulsant drug, 4-AP, leads to downregulation of KCC2. A, Nonradioactive free-floating in situ hybridization showing the distribution and expression intensity of KCC2 mRNA in acute hippocampal slices superfused in the absence (left) and presence (right) of 100 μm 4-AP for 1 hr. B, Western blot analysis from slices treated as in A show strong 4-AP-induced downregulation of KCC2 protein levels (two parallel experiments). α-Tubulin was used to monitor the amount of total protein. Scale bar, 900 μm.
Rapid degradation of cell-surface biotinylated KCC2 protein
Under steady state, the rate of incorporation of KCC2 molecules into the cell surface membrane must equal the rate of their removal and subsequent degradation. To gain information on the turnover of plasmalemmal KCC2, we used Western blot analyses of the surface-biotinylated KCC2 protein (B-KCC2) (Thomas-Crusells et al., 2003). Biotinylated transferrin receptors (B-TfRs) were used as controls because they show a very low turnover rate (Mellman, 1996).
A quantification of the average density of the individual bands obtained in the Western blots (Fig. 4A) indicated a very fast decay of B-KCC2 protein under control conditions, with a time constant (τ) of 19 ± 1 min (n = 7) (Fig. 4B). In the above experiments, a baseline B-KCC2 level of ∼20% was seen (Fig. 4B). The slow kinetics of this protein fraction was not studied further.
An additional interesting observation here was that, in 0-Mg2+, the rate of the decay of B-KCC2 was increased further, to yield a τ of 10 ± 3 min (p < 0.01; n = 5). This effect was fully attributable to the interictal-like activity and not to the omission of Mg2+, because in experiments with ionotropic glutamate antagonists the rate of B-KCC2 decay in 0-Mg2+ was identical if not slightly lower (τ = 25 ± 3 min; n = 5) than that prevailing under control conditions (Fig. 4Ab,B). Notably, the B-TfR levels remained virtually constant.
These experiments show that KCC2 molecules, once incorporated into the plasma membrane, display a very short lifetime. This fast turnover is further accelerated in an activity-dependent manner, which will make the plasmalemmal pool of KCC2 molecules even more susceptible to concomitant downregulation at the transcriptional level (Fig. 1B).
Interictal-like activity leads to a fall in neuronal Cl- extrusion capacity
The activity-induced fall in KCC2 expression should be paralleled by a decrease in the neuronal Cl- extrusion capacity (Rivera et al., 1999). This prediction was studied in experiments on pharmacologically isolated GABAA receptor-mediated IPSPs (IPSPAs). IPSPAs were evoked in the presence of NBQX plus AP-5, which also terminated the 0-Mg2+-induced activity immediately before the intracellular measurements. We used microelectrodes containing 0.5 m Cl- because it is inappropriate to examine changes in the efficacy of ion extrusion in the absence of an ionic load (Bevensee and Boron, 1998; Jarolimek et al., 1999; Payne et al., 2003).
The IPSPA had a fast initial component, with a delay of 3-7 msec and peak value at 8-13 msec from the stimulus, followed by a more negative phase lasting tens of milliseconds (Fig. 5A-C) (Davies et al., 1990). These features of the “compound” IPSPA evoked by simultaneous mass stimulation of somatic and dendritic inhibitory afferents (see Materials and Methods) are readily understood because (1) the experimentally induced Cl- load used was somatic, whereas extrusion of Cl- is mainly dendritic (Gulyas et al., 2001), and (2) the fastest inhibitory inputs activated in the present experiments are likely to be basket cells that target the somata of pyramidal neurons (Buhl et al., 1995). We therefore used two time windows when determining the reversal potential of IPSPA (EIPSP-A) from responses recorded under steady-state conditions (Fig. 5C, insets). The mean driving force of IPSPAs (defined here as EIPSP-A - Vm) at 7-9 and 20-25 msec after the stimulus pulse was -4.4 ± 1.5 and -7.9 ± 1.9 mV in control slices (n = 8) and 9.0 ± 1.3 and 6.1 ± 1.4 mV after 0-Mg2+-induced activity (n = 11) (Fig. 5D). The differences are statistically significant: p = 0.000004 for control versus 0-Mg2+ at 7-9 msec, and p = 0.000015 for control versus 0-Mg2+ at 20-25 msec. The rather depolarizing reversal potentials were not affected by activation of voltage-gated Na+ channels because adding QX-314 (50 mm) in the microelectrode filling solution caused a block of action potentials in the target cell, with no distinguishable effect on EIPSP-A (n = 3); therefore, these data were pooled. Finally, the dependence of the depolarizing shift of EIPSP-A on the interictal-like activity was verified in experiments in which EIPSP-A values similar to those obtained under control conditions were seen after a 3-4 hr exposure to the Mg2+-free solution containing the glutamate antagonists (n = 3) (Fig. 5D).
Figure 5.

0-Mg2+-induced activity leads to a decrease in the efficacy of Cl- extrusion in CA1 pyramidal neurons. The IPSPA reversal potential (EIPSP-A) was measured in the presence of NBQX plus AP-5 using the 0.5 m Cl--containing microelectrodes from control slices (A) and after a 1-3 hr period of 0-Mg2+-induced activity (B). Pharmacologically isolated IPSPAs were evoked by electrical stimulation (asterisk) at various levels of membrane potential (Vm; superimposed top traces) set by constant current pulses (bottom traces). The amplitudes of the IPSPAs measured within two time windows (the time of stimulus taken as 0) from the recordings in A and B have been plotted against membrane potential in C for detection of EIPSP-A. The insets in C show individual IPSPAs evoked from the resting membrane potential (r.p.) level, and the arrowheads indicate the two successive time windows used for EIPSP-A analysis (only the beginning of stimulus artifact is shown in the insets to indicate the timing of stimulation). D shows a summary of the results under steady-state conditions in the presence of the electrode-induced Cl- load. Data are means ± SEM of driving forces of IPSPs (EIPSP-A - Vm) obtained from control cells (n = 8), from cells exposed to 0-Mg2+ in the presence of NBQX plus AP-5 (to block interictal-like activity; n = 3), and from cells (n = 11) of slices that had undergone a 1-3 hr period of 0-Mg2+-induced activity before the intracellular recordings.
TrkB activation by endogenous BDNF mediates activity induced downregulation of KCC2
We have shown recently that KCC2 expression and, consequently, neuronal Cl- extrusion are downregulated after exposure to exogenous BDNF (Rivera et al., 2002). BDNF may induce hyperexcitability and has been postulated to be involved in epileptogenesis (Binder et al., 2001). With this in mind we hypothesized that activity-dependent downregulation of KCC2 as demonstrated in the present experiments would be mediated by endogenously released BDNF acting on TrkB receptors. To test this assumption, we superfused acute hippocampal slices with 0-Mg2+ solution for 3 hr in the presence or absence of the tyrosine kinase inhibitor K252a, which prevents TrkB activation by inhibiting autophosphorylation of the receptor. As shown in Figure 6, Aa and Ab, KCC2 mRNA (n = 8) as well as protein levels (110 ± 5%) (n = 8) remained at control levels in the presence of K252a. Control experiments showed that K252a had no detectable effect on the 0-Mg2+-induced interictal-like activity (n = 3). We then performed similar experiments in which we scavenged endogenously released BDNF with TrkB receptor bodies (TrkB-Fc). We also found here (Fig. 6B) that the downregulation of KCC2 was attenuated, although the effect of TrkB-Fc was smaller (82 ± 7%; p < 0.01; n = 6). These results clearly show that TrkB activation by endogenous BDNF is needed for the activity-induced downregulation of KCC2.
Figure 6.

Endogenous BDNF mediates the activity-dependent downregulation of KCC2 in hippocampal slices. Aa, RT-PCR analysis showing block of 0-Mg2+-induced KCC2 downregulation by the tyrosine kinase inhibitor K252a. NSE was used to confirm equal loading of total RNA. Ab, K252a was also able to block downregulation of KCC2 protein levels as evidenced by Western blot analysis. α-Tubulin was used to monitor an equal amount of total protein per lane. B, Scavenging endogenous BDNF with TrkB-Fc also inhibited the 0-Mg2+-induced downregulation of KCC2 mRNA (Ba) and protein (Bb) levels.
0-Mg2+-induced activity leads to CREB phosphorylation
A known consequence of TrkB signaling is activation of the transcription factor CREB (Finkbeiner et al., 1997; Bibel and Barde, 2000; Minichiello et al., 2002). We examined regional CREB activation in slices that had been superfused with 0-Mg2+ solution using an Ab that specifically recognizes CREB phosphorylated at Ser-133 (pCREB). We observed a distinct increase in CREB activation in the CA1 region and dentate gyrus in slices superfused for 1 hr with 0-Mg2+ solution (Fig. 7Aa,Ab) (n = 4). After 3 hr in 0-Mg2+ solution, an increase in Ser-133 phosphorylation levels was also found in CA3 (Fig. 7Ac,Ad) (n = 4). Interestingly, parallel Western blot analyses (Fig 7B) showed that 0-Mg2+-induced CREB activation (176 ± 15%; p < 0.001; n = 4) was fully blocked (91 ± 8%; n = 4) if not reversed by scavenging endogenous BDNF with TrkB-Fc, indicating that CREB activation in this paradigm is mediated at least partly by TrkB activation and may have a role in the downregulation of KCC2 expression in the hippocampus.
Figure 7.

TrkB is a major mediator of 0-Mg2+-induced CREB phosphorylation in hippocampal slices. A, Immunofluorescent staining showing 0-Mg2+-induced increase in CREB phosphorylation. After 1 hr in 0-Mg2+ solution, slices showed robust increase in anti-pCREB nuclear immunoreactivity in the CA1 region and dentate gyrus as compared with parallel control slices. After 3 hr in 0-Mg2+ solution, the level of activated CREB increased further and reached the CA3 region. Scale bar, 900 μm. B, Comparison of KCC2 and pCREB expression by Western blot analysis of 0-Mg2+-superfused (3 hr) hippocampal slices in the presence or absence of TrkB-Fc showing that scavenging of endogenous BDNF blocks CREB phosphorylation.α-Tubulin was used to monitor the amount of total protein.
Activation of TrkB PLCγ and Shc docking sites are required for downregulation of KCC2
BDNF-TrkB signaling is mediated by well known intracellular signaling cascades, including the PLCγ-CaM kinase pathway as well as the phosphoinositide 3 kinase and Ras/MAPK (mitogen-activated protein kinase) pathways. These cascades are selectively activated by the recruitment of adaptor proteins such as PLCγ that bind to TrkB phosphotyrosine 816, and by Shc/FRS-2, which binds to TrkB phosphotyrosine 515 (Finkbeiner et al., 1997; Bibel and Barde, 2000; Minichiello et al., 2002). To investigate the signaling cascades triggered by BDNF via TrkB involved in the mechanism for downregulation of KCC2, we made use of a recently developed transgenic mice targeted mutation in the PLCγ docking site of TrkB (trkBPLC/PLC mice), where Tyr816 was replaced by Phe and a strain with a similar point mutation (Tyr515Phe) in the Shc binding site (trkBSHC/SHC mice) (Minichiello et al., 1998, 2002). Acute hippocampal slices were treated with 100 ng/ml BDNF for 3-4 hr. In control trkBw/w slices (5 mice; 10 slices per mouse and treatment) (Minichiello et al., 2002), BDNF application induced, as could be expected, a downregulation of KCC2 protein levels (Fig. 8B, left). In the trkBSHC/SHC mutant slices (5 mice; 10 slices per mouse and treatment), KCC2 protein levels were not significantly affected by BDNF, indicating that activation of the Shc-site cascade is necessary for KCC2 downregulation (Fig. 8B, middle). A striking observation was that in the trkBPLC/PLC mutant slices (5 mice; 10 slices per mouse and treatment), KCC2 protein levels were elevated by the exposure to BDNF, indicating that when acting in the absence of the PLCγ cascade, the Shc pathway upregulates KCC2 (Fig. 8B, right).
Figure 8.
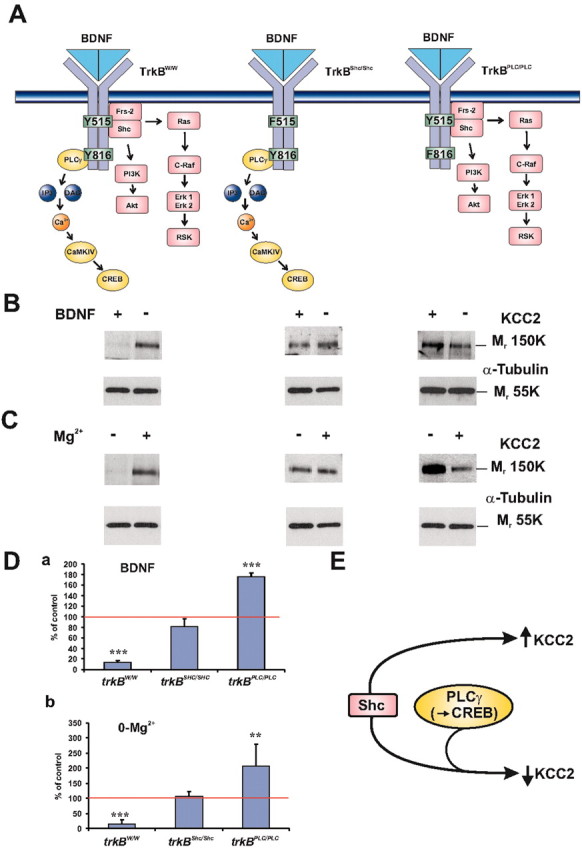
Both PLCγ and Shc docking sites of TrkB are required for BDNF-TrkB-mediated KCC2 downregulation, whereas the Shc site acting in isolation mediates an upregulation of KCC2. A, Schematic representation of the mutant TrkB receptors with a point mutation at either the tyrosine 515 or Shc site in the juxtamembrane region (Minichiello et al., 1998) or at the tyrosine 816 or PLCγ site in the C-terminal region. The trkBW/W is a control-strategy receptor for the trkBPLC/PLC mutant receptor (Minichiello et al., 2002). In the trkBPLC/PLC and trkBSHC/SHC mutant receptors, tyrosine (Y816) and tyrosine (Y515) were replaced by phenylalanine (F816) and phenylalanine (F515), respectively. B, Western blot analysis of BDNF-exposed (3-4 hr) acute slices from the point-mutant mice. In the control trkBW/W mice, BDNF induced downregulation of KCC2 as expected. On the contrary, KCC2 was not downregulated in the trkBSHC/SHC mutants, indicating that the Shc/FRS-2-coupled pathway is important for KCC2 downregulation. Strikingly, KCC2 was upregulated by BDNF in the trkBPLC/PLC mutant mice. C, Western blot analysis of slices from the knock-in mice exposed for 3 hr to the 0-Mg2+ solution shows effects similar to those mediated by BDNF: the 0-Mg2+-induced KCC2 downregulation was blocked in the trkBSHC/SHC mutant slices, and a clear upregulation was seen in trkBPLC/PLC. α-Tubulin was used to monitor the amount of total protein. D, Plot of band intensities from experiments in B and C (means ± SEM; t test; ***p < 0.001; **p < 0.01). E, Scheme interpreting the data obtained with the trkB mutant mice. Activation of both Shc and PLCγ cascades is required for trkB-mediated downregulation of KCC2, whereas activation of the Shc pathway in the absence of PLCγ activation leads to an upregulation of KCC2.
Additional evidence for the conclusion (see above) that endogenous BDNF mediates the activity-dependent downregulation of KCC2 was obtained from experiments in which slices from TrkB mutant mice were exposed to the 0-Mg2+ solution for 3 hr (4 mice; 10 slices per mouse and treatment) (Fig. 8C). A block of this downregulation was evident in Western blots from slices from the trkBSHC/SHC mice, whereas an increase in KCC2 protein took place in the trkBPLC/PLC tissue. The quantitative data from the above BDNF and 0-Mg2+ experiments are shown in Figure 8D.
Discussion
The main novel findings of the present work include, first, a dramatic downregulation of KCC2 that is strictly dependent on neuronal hyperactivity. At the functional level, this is shown to compromise neuronal Cl- extrusion required for GABAergic postsynaptic hyperpolarization. Second, the surprisingly high turnover rate of membrane-bound KCC2 points to changes in KCC2 expression as a novel mechanism in the control of neuronal plasticity via changes in the efficacy of inhibition. Interestingly, a recent report has shown a Ca2+-dependent decrease in K-Cl cotransport activity associated with coincident presynaptic and postsynaptic spiking (Woodin et al., 2003). Whether this effect shares mechanisms of the kind studied here is an interesting question to be examined in future studies. In addition, our results provide an explanation for the fast qualitative change in GABAergic transmission from hyperpolarizing to depolarizing that has been reported to take place during neuronal trauma (Kapur et al., 1995; van den Pol et al., 1996; Kaila et al., 1997; Nabekura et al., 2002; Toyoda et al., 2001; Payne et al., 2003). Third, the near-complete block by TrkB-Fc of activity-induced KCC2 downregulation in a widely used in vitro preparation implies an important role for endogenous BDNF in the postsynaptic modulation of the ionic mechanisms underlying GABAergic events. Finally, the analysis of transgenic mice (trkBSHC/SHC and trkBPLC/PLC) carrying point mutations that selectively disable intracellular signaling cascades triggered by PLCγ or Shc/FRS-2 docking to TrkB indicated the presence of distinct TrkB-mediated cascades resulting in KCC2 downregulation and upregulation (PLCγ plus Shc; Shc in isolation). The requirement of the joint operation of these two pathways for KCC2 downregulation is in itself intriguing. On the other hand, the upregulation mediated by the Shc pathway in isolation serves as an obvious starting point for future work on the molecular mechanisms underlying the ontogenetic upregulation of KCC2 and the associated “developmental switch” in GABAergic transmission (Rivera et al., 1999; Aguado et al., 2003; Ludwig et al., 2003).
Activity-dependent downregulation of KCC2 expression
In a recent study we reported that KCC2 mRNA and protein levels were significantly lower in the hippocampus in vivo after kindling-induced seizures (Rivera et al., 2002), suggesting that the expression of KCC2 would be downregulated by pathophysiological activity. Direct evidence supporting this hypothesis was obtained in experiments on hippocampal slices exposed to 0-Mg2+, in which the interictal-like spontaneous activity (Anderson et al., 1986; Mody et al., 1987) led to downregulation of KCC2 that was blocked by pharmacological inhibition of neuronal activity.
Immunofluorescent KCC2 staining in slices exposed to the 0-Mg2+ solution showed that the downregulation of KCC2 displayed an intriguing spatiotemporal profile. A salient feature here was the marked fall in KCC2 levels in the CA1 dendritic region that was much faster than in CA3. This is an interesting finding also in the light of recent evidence that suppression of dendritic inhibition is strongly proconvulsant (Cossart et al., 2001).
The experiments on cell-surface biotinylated KCC2 showed that the plasmalemmal pool of KCC2 has a strikingly fast turnover rate, which provides a time window for modulatory actions at the level of synaptic transmission similar to that of glutamate receptor subunits (Ehlers, 2000; Lin et al., 2000; Thomas-Crusells et al., 2003). The rate of decay of the surface-biotinylated KCC2 protein was increased further in an activity-dependent manner. Obviously, this activity-dependent increase in the turnover of the plasmalemmal KCC2 transporters enhances their susceptibility to downregulation that takes place at the transcriptional level.
Interictal-like activity leads to a fall in the efficacy of neuronal Cl- extrusion
The prediction that the fall in plasmalemmal KCC2 is paralleled by an activity-dependent decrease in the efficacy of Cl- extrusion by pyramidal neurons was verified in experiments in which EIPSP-A was measured in neurons subjected to a tonic chloride load with a 0.5 m Cl--containing sharp microelectrode. Control cells turned out to be able to maintain a mean level of EIPSP-A more negative than resting Vm, despite the load, whereas neurons that had undergone 0-Mg2+-induced hypersynchronous activity had a depolarizing reversal potential, showing that the capacity of the afflicted cells to deal with Cl- loads had been strongly compromised. Notably, a similar impairment of neuronal Cl- extrusion was achieved with application of exogenous BDNF on acute hippocampal slices (Rivera et al., 2002). These results also shed light on previous findings from in vivo and in vitro models of epilepsy as well as from human epileptic tissue that have demonstrated a positive shift in the driving force of GABAA-mediated inhibition, which is sometimes large enough to change the postsynaptic response from hyperpolarizing to depolarizing (Tasker et al., 1992; Avoli et al., 1995; Kapur and Coulter, 1995; Kohling et al., 2000; Cohen et al., 2002).
Molecular mechanism of TrkB-mediated KCC2 downregulation
BDNF-TrkB-mediated signaling appears to be a major link between neuronal activity and changes in protein expression required for long-term plasticity such as long-term potentiation (LTP) and epileptogenesis (Kokaia et al., 1995; Thoenen, 1995; Binder et al., 1999; Croll et al., 1999; Lu and Chow, 1999; Lu and Gottschalk, 2000).
The results presented here show clearly that 0-Mg2+-induced spontaneous activity is able to induce BDNF release and activation of TrkB receptors, which subsequently leads to downregulation of KCC2. This was evident from the experiments in which 0-Mg2+-induced KCC2 downregulation was inhibited by blocking TrkB with K252a and by scavenging endogenous BDNF with TrkB-Fc. The fact that the downregulation was inhibited slightly more efficiently by K252a may suggest that other endogenous trophic factors in addition to BDNF make a small contribution to this process.
Scavenging endogenous BDNF also suppressed the increase in CREB activation that was observed in slices exposed to the 0-Mg2+ solution. The temporal and spatial distribution observed by immunofluorescent staining for pCREB (Fig. 7) is consistent with the idea that CREB activation is required for downregulation of KCC2 as seen in KCC2 immunostainings (Fig. 2). The robust increase in CREB phosphorylation observed in the CA1 region after 1 hr showed a good match not only with the downregulation of KCC2 in the same region but also with the pattern of previously reported BDNF-induced CREB activation in the hippocampus (Finkbeiner et al., 1997).
Two major intracellular cascades have been shown to be involved in plasticity-related effects of BDNF in the hippocampus. Using two strains of mice with a single amino acid-targeted mutation of the TrkB gene that abolish binding of either PLCγ or Shc/FRS-2 to the TrkB receptor, respectively, Minichiello et al. (2002) showed that PLCγ-CREB signaling is necessary for LTP, whereas TrkB-coupled activation of the Shc pathway is not required (Ernfors and Bramham, 2003). We used the same transgenic mice (trkBPLC/PLC and trkBSHC/SHC) to identify the mechanisms required for BDNF-TrkB-mediated downregulation of KCC2. Perhaps somewhat unexpectedly, we found that both the PLCγ and Shc/FRS-2-activated signaling cascades are required for KCC2 downregulation caused by the 0-Mg2+ solution and by exogenous BDNF (n = 5). Furthermore, with deletion of the PLCγ-coupled pathway, both 0-Mg2+ and BDNF triggered an intriguing upregulation of KCC2 (n = 5).
Taken together, the data obtained using slices from the trk-BPLC/PLC and trkBSHC/SHC mice suggest that the Shc pathway is crucial for both the downregulation and upregulation of KCC2. Downregulation of KCC2 appears to take place if the Shc pathway is activated in conjunction with the PLCγ cascade, whereas an upregulation is triggered by the Shc pathway acting in the absence of the PLCγ cascades. A particularly interesting question for future work is whether such data will shed light on the mechanisms that control the developmental upregulation of KCC2 (Rivera et al., 1999), which is known to be crucial for the maturation of functional postsynaptic inhibition in the CNS.
A recent report by Aguado et al. (2003) showed that in the late prenatal hippocampus, BDNF may have an upregulatory effect on KCC2 mRNA expression. The apparently opposite actions of BDNF on KCC2 expression in immature versus mature neurons is likely to reflect the activation of the distinct TrkB-linked signaling pathways (Fig. 8). It is interesting to note that a similar development-related qualitative difference in BDNF action has been reported for GABAA receptor trafficking (Mizoguchi et al., 2003). Finally, the present results suggest that a downregulation of KCC2 may be a general early response involved in various kinds of neuronal trauma (Kapur and Coulter, 1995; van den Pol et al., 1996; Kaila et al., 1997; Toyoda et al., 2001; Nabekura et al., 2002; Payne et al., 2003).
Footnotes
This work was supported by the Academy of Finland and by the Sigrid Jusélius Foundation. M. S., K. K., and C. R. are Fellows of the Biocentrum Helsinki. We thank Marjo Heikura and Miika Palviainen for excellent technical assistance.
Correspondence should be addressed to Dr. Claudio Rivera, Institute of Biotechnology, P.O. Box 56, University of Helsinki, FIN-00014 Finland. E-mail: Claudio.Rivera@helsinki.fi.
Z. Emri's present address: Chemical Research Center, H-1025 Budapest, Hungary.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244683-09$15.00/0
References
- Aguado F, Carmona MA, Pozas E, Aguilo A, Martinez-Guijarro FJ, Alcantara S, Borrell V, Yuste R, Ibanez CF, Soriano E (2003) BDNF regulates spontaneous correlated activity at early developmental stages by increasing synaptogenesis and expression of the K+/Cl- co-transporter KCC2. Development 130: 1267-1280. [DOI] [PubMed] [Google Scholar]
- Anderson WW, Lewis DV, Swartzwelder HS, Wilson WA (1986) Magnesium-free medium activates seizure-like events in the rat hippocampal slice. Brain Res 398: 215-219. [DOI] [PubMed] [Google Scholar]
- Avoli M (1996) GABA-mediated synchronous potentials and seizure generation. Epilepsia 37: 1035-1042. [DOI] [PubMed] [Google Scholar]
- Avoli M, Louvel J, Drapeau C, Pumain R, Kurcewicz I (1995) GABAA-mediated inhibition and in vitro epileptogenesis in the human neocortex. J Neurophysiol 73: 468-484. [DOI] [PubMed] [Google Scholar]
- Beau FE, Alger BE (1998) Transient suppression of GABAA-receptor-mediated IPSPs after epileptiform burst discharges in CA1 pyramidal cells. J Neurophysiol 79: 659-669. [DOI] [PubMed] [Google Scholar]
- Bevensee MO, Boron WF (1998) Thermodynamics and physiology of cellular pH regulation. In: pH and brain function (Kaila K, Ransom BR, eds), pp 173-194. New York: Wiley.
- Bibel M, Barde YA (2000) Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev 14: 2919-2937. [DOI] [PubMed] [Google Scholar]
- Binder DK, Routbort MJ, Ryan TE, Yancopoulos GD, McNamara JO (1999) Selective inhibition of kindling development by intraventricular administration of TrkB receptor body. J Neurosci 19: 1424-1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE (2001) BDNF and epilepsy: too much of a good thing? Trends Neurosci 24: 47-53. [DOI] [PubMed] [Google Scholar]
- Buhl EH, Cobb SR, Halasy K, Somogyi P (1995) Properties of unitary IPSPs evoked by anatomically identified basket cells in the rat hippocampus. Eur J Neurosci 7: 1989-2004. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N (1987) Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem 162: 156-159. [DOI] [PubMed] [Google Scholar]
- Cohen I, Navarro V, Clemenceau S, Baulac M, Miles R (2002) On the origin of interictal activity in human temporal lobe epilepsy in vitro. Science 298: 1418-1421. [DOI] [PubMed] [Google Scholar]
- Cossart R, Dinocourt C, Hirsch JC, Merchan-Perez A, De Felipe J, Ben Ari Y, Esclapez M, Bernard C (2001) Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4: 52-62. [DOI] [PubMed] [Google Scholar]
- Croll SD, Suri C, Compton DL, Simmons MV, Yancopoulos GD, Lindsay RM, Wiegand SJ, Rudge JS, Scharfman HE (1999) Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience 93: 1491-1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Davies SN, Collingridge GL (1990) Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 424: 513-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ (2000) Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J Neurosci 20: 8069-8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28: 511-525. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Bramham CR (2003) The coupling of a trkB tyrosine residue to LTP. Trends Neurosci 26: 171-173. [DOI] [PubMed] [Google Scholar]
- Finkbeiner S, Tavazoie SF, Maloratsky A, Jacobs KM, Harris KM, Greenberg ME (1997) CREB: a major mediator of neuronal neurotrophin responses. Neuron 19: 1031-1047. [DOI] [PubMed] [Google Scholar]
- Ganguly K, Schinder AF, Wong ST, Poo M (2001) GABA itself promotes the developmental switch of neuronal GABAergic responses from excitation to inhibition. Cell 105: 521-532. [DOI] [PubMed] [Google Scholar]
- Gulyas AI, Sik A, Payne JA, Kaila K, Freund TF (2001) The KCl cotransporter, KCC2, is highly expressed in the vicinity of excitatory synapses in the rat hippocampus. Eur J Neurosci 13: 2205-2217. [DOI] [PubMed] [Google Scholar]
- Hiltunen JO, Arumae U, Moshnyakov M, Saarma M (1996) Expression of mRNAs for neurotrophins and their receptors in developing rat heart. Circ Res 79: 930-939. [DOI] [PubMed] [Google Scholar]
- Hubner CA, Stein V, Hermans-Borgmeyer I, Meyer T, Ballanyi K, Jentsch TJ (2001) Disruption of KCC2 reveals an essential role of K-Cl cotransport already in early synaptic inhibition. Neuron 30: 515-524. [DOI] [PubMed] [Google Scholar]
- Jackson MF, Esplin B, Capek R (1999) Inhibitory nature of tiagabine-augmented GABAA receptor-mediated depolarizing responses in hippocampal pyramidal cells. J Neurophysiol 81: 1192-1198. [DOI] [PubMed] [Google Scholar]
- Jarolimek W, Lewen A, Misgeld U (1999) A furosemide-sensitive K+-Cl- cotransporter counteracts intracellular Cl- accumulation and depletion in cultured rat midbrain neurons. J Neurosci 19: 4695-4704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K (1994) Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol 42: 489-537. [DOI] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J (1997) Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci 17: 7662-7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakazu Y, Uchida S, Nakagawa T, Akaike N, Nabekura J (2000) Reversibility and cation selectivity of the K(+)-Cl(-) cotransport in rat central neurons. J Neurophysiol 84: 281-288. [DOI] [PubMed] [Google Scholar]
- Kapur J, Coulter DA (1995) Experimental status epilepticus alters gamma-aminobutyric acid type A receptor function in CA1 pyramidal neurons. Ann Neurol 38: 893-900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohling R, Vreugdenhil M, Bracci E, Jefferys JG (2000) Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J Neurosci 20: 6820-6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokaia M, Ernfors P, Kokaia Z, Elmer E, Jaenisch R, Lindvall O (1995) Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol 133: 215-224. [DOI] [PubMed] [Google Scholar]
- Köhling R, Lucke A, Straub H, Speckmann EJ, Tuxhorn I, Wolf P, Pannek H, Oppel F (1998) Spontaneous sharp waves in human neocortical slices excised from epileptic patients. Brain 121: 1073-1087. [DOI] [PubMed] [Google Scholar]
- Lamsa K, Kaila K (1997) Ionic mechanisms of spontaneous GABAergic events in rat hippocampal slices exposed to 4-aminopyridine. J Neurophysiol 78: 2582-2591. [DOI] [PubMed] [Google Scholar]
- Lin JW, Ju W, Foster K, Lee SH, Ahmadian G, Wyszynski M, Wang YT, Sheng M (2000) Distinct molecular mechanisms and divergent endocytotic pathways of AMPA receptor internalization. Nat Neurosci 3: 1282-1290. [DOI] [PubMed] [Google Scholar]
- Lu B, Chow A (1999) Neurotrophins and hippocampal synaptic transmission and plasticity. J Neurosci Res 58: 76-87. [PubMed] [Google Scholar]
- Lu B, Gottschalk W (2000) Modulation of hippocampal synaptic transmission and plasticity by neurotrophins. Prog Brain Res 128: 231-241. [DOI] [PubMed] [Google Scholar]
- Lu J, Karadsheh M, Delpire E (1999) Developmental regulation of the neuronal-specific isoform of K-Cl cotransporter KCC2 in postnatal rat brains. J Neurobiol 39: 558-568. [PubMed] [Google Scholar]
- Ludwig A, Li H, Saarma M, Kaila K, Rivera C (2003) Developmental upregulation of KCC2 in the absence of GABAergic and glutamatergic transmission. Eur J Neurosci 18: 3199-3206. [DOI] [PubMed] [Google Scholar]
- Mellman I (1996) Endocytosis and molecular sorting. Annu Rev Cell Dev Biol 12: 575-625. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R (1998) Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron 21: 335-345. [DOI] [PubMed] [Google Scholar]
- Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M (2002) Mechanism of TrkB-mediated hippocampal long-term potentiation. Neuron 36: 121-137. [DOI] [PubMed] [Google Scholar]
- Mizoguchi Y, Ishibashi H, Nabekura J (2003) The action of BDNF on GABAA currents changes from potentiating to suppressing during maturation of rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 548: 703-709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody I, Lambert JD, Heinemann U (1987) Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol 57: 869-888. [DOI] [PubMed] [Google Scholar]
- Nabekura J, Ueno T, Okabe A, Furuta A, Iwaki T, Shimizu-Okabe C, Fukuda A, Akaike N (2002) Reduction of KCC2 expression and GABAA receptor-mediated excitation after in vivo axonal injury. J Neurosci 22: 4412-4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Patel K, Wilkinson DG (1996) In situ hybridization analysis of chick embryos in whole mount and tissue sections. Methods Cell Biol 51: 219-235. [DOI] [PubMed] [Google Scholar]
- Payne JA, Stevenson TJ, Donaldson LF (1996) Molecular characterization of a putative K-Cl cotransporter in rat brain. A neuronal-specific isoform. J Biol Chem 271: 16245-16252. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K (2003) Cation-chloride co-transporters in neuronal communication, development and trauma. Trends Neurosci 26: 199-206. [DOI] [PubMed] [Google Scholar]
- Poirier JL, Capek R, De Koninck Y (2000) Differential progression of dark neuron and fluoro-jade labeling in the rat hippocampus following pilocarpine-induced status epilepticus. Neuroscience 97: 59-68. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999) The K+/Cl- co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251-255. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia K, Airaksinen MS, Voipio J, Kaila K, Saarma M (2002) BDNF-induced TrkB activation downregulates the K+-Cl- cotransporter KCC2 and impairs neuronal Cl- extrusion. J Cell Biol 159: 747-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF (1991) Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 40: 599-636. [DOI] [PubMed] [Google Scholar]
- Tasker JG, Peacock WJ, Dudek FE (1992) Local synaptic circuits and epileptiform activity in slices of neocortex from children with intractable epilepsy. J Neurophysiol 67: 496-507. [DOI] [PubMed] [Google Scholar]
- Thoenen H (1995) Neurotrophins and neuronal plasticity. Science 270: 593-598. [DOI] [PubMed] [Google Scholar]
- Thomas-Crusells J, Vieira A, Saarma M, Rivera C (2003) A novel method for monitoring surface membrane trafficking on hippocampal acute slice preparation. J Neurosci Methods 125: 159-166. [DOI] [PubMed] [Google Scholar]
- Thompson SM, Gähwiler BH (1989) Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl- in hippocampal CA3 neurons. J Neurophysiol 61: 512-523. [DOI] [PubMed] [Google Scholar]
- Toyoda H, Ohno K, Yamada J, Ikeda M, Okabe A, Sato K, Hashimoto K, Fukuda A (2001) Induction of NMDA and GABAA receptor-mediated Ca2+ oscillations with KCC2 mRNA downregulation in injured facial motoneurons. J Neurophysiol 89: 1353-1362. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Obrietan K, Chen G (1996) Excitatory actions of GABA after neuronal trauma. J Neurosci 16: 4283-4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voipio J, Kaila K (2000) GABAergic excitation and K+-mediated volume transmission in the hippocampus. Prog Brain Res 125: 329-338. [DOI] [PubMed] [Google Scholar]
- Williams JR, Sharp JW, Kumari VG, Wilson M, Payne JA (1999) The neuron-specific K-Cl cotransporter, KCC2. Antibody development and initial characterization of the protein. J Biol Chem 274: 12656-12664. [DOI] [PubMed] [Google Scholar]
- Woodin MA, Ganguly K, Poo MM (2003) Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl- transport activity. Neuron 39: 807-820. [DOI] [PubMed] [Google Scholar]