

Abstract
The accumulation of amyloid-β protein into plaques is a characteristic feature of Alzheimer's disease. However, the contribution of amyloid-β plaques to neuronal dysfunction is unknown. We compared intracellular recordings from neocortical pyramidal neurons in vivo in APP-Sw (Tg2576 transgenic mice overexpressing amyloid precursor protein with the Swedish mutation) transgenic mice to age-matched nontransgenic cohorts at ages either before or after deposition of cortical plaques. We show that the evoked synaptic response of neurons to transcallosal stimuli is severely impaired in cortex containing substantial plaque accumulation, with an average 2.5-fold greater rate of response failure and twofold reduction in response precision compared with age-matched nontransgenic controls. This effect correlated with the presence of amyloid-β plaques and alterations in neuronal process geometry. Responses of neurons in younger APP-Sw animals, before plaque accumulation, were similar to those in nontransgenic controls. In all cases, spontaneous membrane potential dynamics were similar, suggesting that overall levels of synaptic innervation were not affected by plaques. Our results show that plaques disrupt the synchrony of convergent inputs, reducing the ability of neurons to successfully integrate and propagate information.
Keywords: synaptic integration, subthreshold membrane potential, in vivo, Alzheimer's disease, amyloid-β plaques, neuronal geometry
Introduction
The amyloid hypothesis is central to the study of Alzheimer's disease (AD) (Hardy and Selkoe, 2002). The amyloid-β peptide accumulates in AD brains in a number of forms, ranging from soluble oligomeric entities to insoluble macroscopic senile plaques. Although the presence of amyloid-β plaques is a necessary criterion for the diagnosis of AD, the effect of amyloid-β remains uncertain. Several different mechanisms of action of amyloid-β have been proposed from in vitro and modeling studies, including alterations in ion channel properties (Hartley et al., 1999; Ye et al., 2003), increase in synaptic depression (Kamenetz et al., 2003), and decrease in long-term potentiation (LTP) (Chapman et al., 1999; Walsh et al., 2002; Kamenetz et al., 2003). These hypotheses predict changes in intrinsic membrane properties or synaptic properties of individual neurons exposed to amyloid-β. In contrast, morphological studies of postmortem human brain have emphasized the possibility that abnormal geometry of dendrites and axons could lead to a loss of synchronous activation of convergent afferents (Knowles et al., 1999; Buldyrev et al., 2000). Such an effect would be on the network as a whole rather than on individual cellular properties. Although a number of studies have measured effects of amyloid-β on individual neuronal function, the question of functional effects of amyloid-β on the cortical neuronal network has not been addressed at the cellular level. To test the hypothesis that amyloid-β affects synchronous activity of cortical neurons in vivo, we measured the functional properties of neurons in neocortex of Tg2576 transgenic mice [APP-Sw; Tg2576 (Hsiao et al., 1996; Hsiao, 1998)] overexpressing amyloid precursor protein with the Swedish mutation (APP-Sw). In the cortex of these mice, the amount of soluble amyloid-β, as well as insoluble, plaques increases in an age-dependent manner (Kawarabayashi et al., 2001). We compared measurements in neurons at two ages of APP+ mice and their transgene-negative littermates: 8-9 months, at which age soluble amyloid-β has accumulated but plaques have not yet formed, and at ≥14 months, at which age considerable plaque accumulation has occurred.
Materials and Methods
Surgery. Tg2576 (Hsiao et al., 1996) mice were used for this study. Mice were anesthetized with ketamine-xylazine (150 and 12 mg/kg, respectively) and placed in a custom-built stereotaxic device. Temperature was maintained at 37°C. The scalp was removed, and the skull was cleaned of tissue. Small holes were drilled for EEG electrodes and stimulating electrodes. A 3 × 3 mm craniotomy was opened, with the center directly contralateral to the position of the stimulating electrode. A bipolar stimulating electrode was inserted into the right cortical hemisphere 2-4 mm posterior to bregma and 2-4 mm lateral to midline. A cisternal drain was opened to reduce brain pulsations. Stimulating electrodes were bipolar Teflon-coated tungsten rods (0.008 inch uninsulated diameter) sharpened under microscopic control to a 20° angle and glued with dental acrylic. Uninsulated tips measured <0.5 mm, and distance between tips was 0.5 mm. Electrode impedance measured 0.5-0.8 MΩ in PBS.
Recording. Recording electrodes [1.5 mm outer diameter, glass (A-M Systems, Carlsborg, WA); resistance, 30-80 MΩ; filled with 4% biocytin (Sigma, St. Louis, MO) and, in some cases, 25 mm QX-314 dissolved in 1 m potassium acetate (Sigma)] were lowered into the cortex (as far as possible) directly contralateral to the stimulating electrode. When a neuron was impaled, stimuli were presented with increasing amplitude (n = 10; 0.2 Hz) until a consistent response was observed. Stimuli presented were 115% of this value. A high-impedance amplifier with active bridge circuitry (IR-283; Cygnus Technology, Delaware Water Gap, PA) was used to measure spontaneous and evoked membrane potentials. Data was directly acquired via an analog-to-digital board (National Instruments, Austin, TX) using custom software at 5 kHz. Spontaneous activity was measured for 5 min minimum per neuron. At least 30 stimuli per neuron were presented for response analysis.
Histology. After recording, the animals were given an overdose of ketamine-xyalzine or pentobarbital, a transcardial perfusion (4% paraformaldehyde in PBS) was performed, and the brains were postfixed for 24 hr. Sections containing recorded-stained neurons were reacted with avidin-biotin complex and diaminobenzidene (Horikawa and Armstrong, 1988) and thioflavine-S (Kelenyi, 1967) for plaque visualization. Adjacent sections were stained with SMI-32 (Campbell and Morrison, 1989) for measurement of neuritic curvature. Neuritic curvature ratios were calculated by dividing the path length of neuritic segments by the shortest end-to-end distance (Knowles et al., 1998).
Results
When recorded in anesthetized animals in vivo, the membrane potential of neocortical neurons spontaneously fluctuates between a relatively quiescent resting potential (“down state”) and a more depolarized state (“up state”), characterized by high-frequency activity centering around, or just negative to, action potential threshold (Steriade et al., 1993; Cowan and Wilson, 1994). The two states can be separated by as much as 20 mV (average, 11.33 ± 3.4 mV); from the down state, a considerable amount of depolarization (i.e., analog-to-digital large number of synaptic inputs) is required to reach threshold values. For an action potential to arise, the membrane potential must transition to the up state. The membrane potential fluctuations are caused by synchronized afferent activity. The afferents are primarily cortical in origin and can arise from neurons widely distributed over cortical areas (Timofeev et al., 2000).
Figure 1 shows an example of two pyramidal neurons recorded sequentially from the neocortex of a 14-month-old mouse stained by intracellular injection of biocytin. Dense-core plaques stained with thioflavine-S are clearly seen, and the high-magnification images show the distortion of the neuronal processes by the plaques.
Figure 1.

Amyloid-β plaques disrupt neuritic geometry without affecting overall levels of synaptic input. a, Two neocortical pyramidal neurons from a 15-month-old APP+ mouse injected with biocytin (Chapman et al., 1999). Thioflavine-S plaques are shown in yellow. Scale bar, 100 μm. b, Higher-magnification image of one of the plaques shown in a, with neurites deflected around the plaque. Scale bar, 20 μm. c, Another example of neuritic trajectory displacement by a thioflavine-S plaque. Scale bar, 20 μm. d, Spontaneous membrane potential fluctuations in 14-month-old nontransgenic (black trace; Vrest of -68 mV) and APP+ (red trace; Vrest of -71 mV) neocortical pyramidal neurons. Calibration: 20 mV, 500 msec.
All identified neurons had pyramidal morphology (Fig. 1a) and prominent dendritic spines. All recorded neurons had membrane potential dynamics and response to current pulses consistent with those of pyramidal neurons (McCormick et al., 1985; Nowak et al., 2003). Figure 1d shows spontaneous membrane potential recordings from a 14-month-old APP+ cortical neuron and an age-matched negative cohort. Although in vitro studies suggest that high levels of amyloid-β can impair NMDA-dependent synaptic transmission and increase synaptic depression (Kamenetz et al., 2003), no significant differences in membrane potential values or dynamics were observed between Tg+ and Tg- neurons in 8- to 10-month-old or ≥14-month-old mice (Table 1). These data indicate that the overall levels of synaptic input were not disrupted by amyloid-β accumulation.
Table 1.
Intrinsic and spontaneous membrane potential properties of mouse cortical neurons recorded in vivo
|
|
Tg− |
Tg+ |
||||
|---|---|---|---|---|---|---|
| Measurement |
8-10 months |
≥14 months |
8-10 months |
≥14 months |
||
| Down state | ||||||
| Membrane potential (mV) | −63.2 ± 4.6 (n = 8) | −61.8 ± 4.2 (n = 10) | −62.4 ± 3.8 (n = 8) | −63.5 ± 4.8 (n = 11) | ||
| Duration (msec) | 652.4 ± 237.6 (n = 8) | 769.0 ± 236.3 (n = 10) | 721.7 ± 311.4 (n = 8) | 684.8 ± 276.2 (n = 11) | ||
| Up state | ||||||
| Membrane potential (mV) | −52.8 ± 6.2 (n = 8) | −51.5 ± 4.9 (n = 8) | −53.1 ± 5.7 (n = 8) | −54.1 ± 5.2 (n = 8) | ||
| Duration (msec) | 424.0 ± 280.5 (n = 8) | 467.4 ± 322.1 (n = 10) | 390.8 ± 316.2 (n = 8) | 406.9 ± 388.5 (n = 11) | ||
| Spontaneous firing rate (Hz) |
14.0 ± 6.7 (n = 8) |
12.8 ± 6.0 (n = 10) |
11.6 ± 8.5 (n = 8) |
16 ± 9.3 (n = 11) |
||
To test the hypothesis that amyloid-β plaques physically disrupt the neuronal network, we recorded the responses of cortical neurons to contralateral cortical electrical stimulation. After determination of threshold for each neuron (see Materials and Methods), 25-35 superthreshold stimuli were given. Typical responses for ≥14-month-old nontransgenic neurons are shown in Figure 2a. Responses of the Tg- neurons were relatively stereotypical, depending on the membrane potential state of the postsynaptic neuron. When the stimuli arrived during the down state (black traces), the membrane potential typically transitioned to the up state, and one or more action potentials occurred, with a mean latency of the first action potential being 23.96 ± 6.06 msec (average ± SD within-neuron). When the stimuli arrived during the up state (gray traces), the average latency of the first spike was 22.59 ± 5.49 msec. Typically, the neuron fired a few times and the membrane potential then fell into the down state. Although not observed previously in mice, these responses are similar to those observed in the responses of cortical neurons in vivo to various electrical (Landry et al., 1984; Nunez et al., 1993; Cisse et al., 2003) and sensory (Moore and Nelson, 1998; Azouz and Gray, 1999; Zhu and Connors, 1999; Anderson et al., 2000; Sanchez-Vives et al., 2000; Stern et al., 2001) stimuli in rats and cats.
Figure 2.
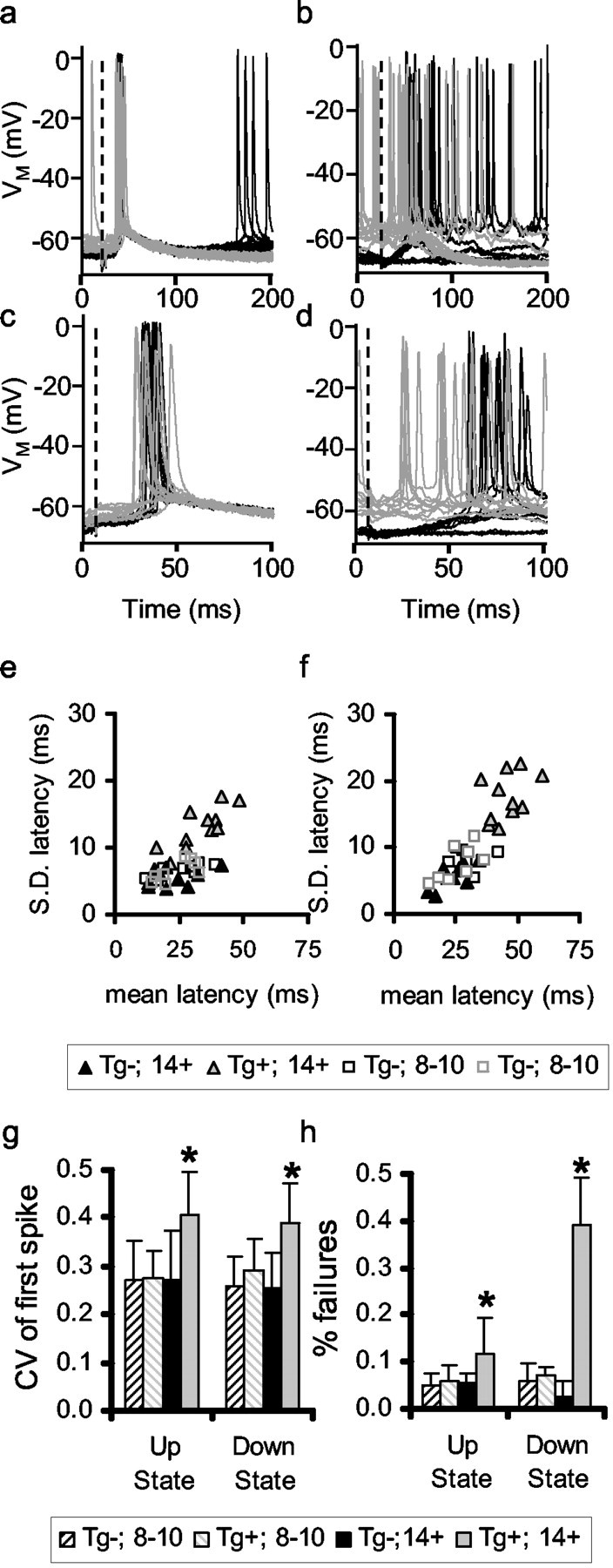
Evoked action potential jitter is greater in cortex with substantial amyloid-β plaque accumulation. a, b, Examples of responses to transcallosal stimuli in 14-month-old nontransgenic (a) and APP+ (b) mouse neocortical pyramidal neurons. Black traces are responses to stimuli occurring in the down state, and gray traces are responses to stimuli in the up state. c, d, Same traces as a and b at higher sweep speed. Dashed lines indicate stimulus. e, f, Average ± SD of first spike in response to transcallosal stimulus. Each point represents one neuron. e, Stimuli occurring in up state. f, Stimuli occurring in down state. g, h, Statistics of coefficient of variation (g) and response failures (h) for nontransgenic and transgenic 8- to 10-month-old and ≥14-month-old cortical neurons.
Transcallosal stimuli produced far less stereotypical responses in ≥14-month-old APP+ mice (Fig. 2b). The latency and pattern of the evoked action potentials in both states were significantly more variable in older APP+ cortical neurons than in APP- or younger APP+ neurons (two-way ANOVA; Tukey-Kramer post hoc comparisons; p ≤ 0.05). The latency of the first spike evoked from a stimulus in the up state was 33.00 ± 12.93 msec and from a stimulus evoked from the down state 45.64 ± 17.53 msec. The number of response failures in the transgenic neurons (no change observed within 100 msec) was on average approximately twofold greater when evoked from the up state and approximately fourfold greater in the down state. The greater failure rates for stimuli evoked from the down state presumably arise from the order-of-magnitude larger number of synchronized inputs needed to evoke a response in that state. Similar increase in response jitter and failure rate does not occur in 8- to 10-month-old APP+ cortical neurons, suggesting that the effect is not simply attributable to APP overexpression or formation of soluble oligomeric assemblies. The responses in 8- to 10-month-old APP+ cortical neurons did not differ from those in 8- to 10-month-old nontransgenic cohorts. Preliminary data from 4-month-old APP+ and nontransgenic animals were indistinguishable from the 8- to 10-month-old results. These data indicate that neurons in cortex containing substantial plaque accumulation do not respond to stimuli as precisely as age-matched nontransgenic or young transgenic animals.
We next examined the possibility that the reduced response reliability in older APP+ neurons is a result of the effects of amyloid-β on intrinsic, spike-generating mechanisms by measuring responses to stimuli in ≥14-month-old neurons injected with QX-314 (Connors and Prince, 1982). Examples of responses to contralateral stimuli are shown in Figure 3, a and b. Although individual responses (gray traces) of APP+ neurons can be approximately as large as those in nontransgenic neurons, the variation of timing, the large number of small responses, and failures substantially reduces the average response (heavy black trace). Separating the responses by membrane potential state (Fig. 3c-f) reveals the increase in variability in responses in both the up and down states of the membrane potentials of the APP+ neurons, as well as the increased number of failures. The results for the sample of APP+ and negative cohort neurons are summarized in the graphs in Figure 3, g and h. These results show that the source of the decreased response reliability of cortical neurons is not simply an effect of amyloid-β on the intrinsic electrical properties of cortical neurons but rather a result of reducing the temporal synchrony of afferent synapses. Because the afferent synapses must be relatively synchronized to raise the membrane potential from the down state to the up state and initiate action potentials from the up state (Stern et al., 1997), timing of the convergent inputs is critical for neuronal function. Because the synaptic response of a cortical neuron depends on the spatiotemporal convergence of several afferents, the distortion of the afferents in a heterogeneous manner may blur the temporal summation by the postsynaptic neuron.
Figure 3.

Increase in synaptic jitter is caused by reduction in input synchrony in ≥14-month-old APP+ cortical neurons. a, b, Individual traces (red) and averages (black) of responses to transcallosal stimuli in 14-month-old nontransgenic (a) and APP (b) neurons injected with QX-314. Dashed lines indicate stimulus. c-f, Responses to transcallosal stimuli in nontransgenic (c, e) and APP (d, f) neurons separated by state. Gray lines indicate individual responses, and heavy black line are averages. c, d, Responses to stimuli in up state; e, f, responses to stimuli in down state. g, h, Average subthreshold amplitudes (g) and probability of state transitions and failures (h) of responses of ≥14-month-old nontransgenic and APP cortical neurons. i, Synaptic jitter is not independent of neuritic curvature. Average curvature and synaptic jitter for 10 measured in 8- to 10-month-old (black; n = 4) and ≥14-month-old (white; n = 6) mice. Error bars denote SD.
We next asked whether morphological alterations could underlie this apparent failure of synaptic summation. One specific type of plaque, usually referred to as dense-core or thioflavine-S-positive plaques, comprises ∼10% of the total amyloid-β burden in neocortex (Hyman et al., 1993). Neuronal processes do not traverse dense-core plaques but are deflected around them, thus significantly distorting their morphology and presumably affecting their function (Masliah et al., 1993; Knowles et al., 1999; Le et al., 2001; D'Amore et al., 2003). The disruption of connectivity in the cortical network by distributed, insoluble protein aggregations has been suggested, on theoretical grounds, as an underlying mechanism of the cognitive dysfunctions associated with AD (Knowles et al., 1999). Although thioflavine-S-positive plaques fill only a small percentage of the cortical volume, they are sufficiently common that all of our identified neurons (n = 9) in ≥14-month-old APP+ mice had neuritic fields disrupted by these plaques (Fig. 1). These results complement previous observations using in vivo multiphoton microscopy that show that >50% of processes in the vicinity of plaques are morphologically altered (D'Amore et al., 2003). To test the hypothesis that plaque-associated changes in dendritic and axonal morphology could underlie the observed disruption of synaptic function, we measured average neuritic curvature ratios (see Materials and Methods) in SMI-32-stained sections from the younger and older APP+ mice used in this study to quantify the relationship between neuritic curvature and synaptic variability (Fig. 3i). We found a clear association between extent of average neuritic curvature and degree of response jitter (rS = -0.575; df = 10; p ≤ 0.05). These data indicate that the degree of synaptic dysfunction and spatial distortion of neurites are not independent.
Discussion
The synaptic dysfunction observed in this study is the first measure of an effect of amyloid-β plaques on neocortical synaptic transmission in single neurons in vivo. We propose that the increased synaptic variability seen here is not caused by molecular effects of amyloid-β on single synapses. Rather, we propose that the major effect of amyloid-β plaques is on the cortical network as a whole: the cause of the increased response variability is the reduced synchrony of converging synaptic inputs, which is at least partially caused by the physical distortion of the axonal and dendritic processes by insoluble protein aggregates. Spiking activity in the cortical network is to a large extent governed by coordinated synchronous presynaptic activity (Stern et al., 1997; Pare et al., 1998; Azouz and Gray, 1999; Destexhe and Pare, 1999). The overall level of spontaneous cortical activity in the transgenic mice is indistinguishable from that observed in nontransgenic mouse cortical neurons, which is consistent with the finding that no measurable synaptic loss is present in these models. Each pyramidal neuron in mouse neocortex receives ∼10,000-15,000 excitatory inputs (Braitenberg and Schüz, 1998). Apparently, in the presence of substantial plaque accumulation, enough of these inputs arrive synchronously to generate membrane potential dynamics. However, for a given signal to be reliably transmitted through a network of converging and diverging projections, a relatively large number of inputs must arrive at the neuron within a narrow time window. In a separate study, using simultaneous intracellular and multiple extracellular recordings, we estimated that ∼60 unit postsynaptic potentials (PSPs) arriving within 10 msec are required to elicit an action potential from the up state and ∼500 PSPs to cause a transition from the down to the up state (Leger et al., personal communication). Thus, ∼5% of the total excitatory synapses would need to be activated to cause a state transition, and an additional 0.6% of the total excitatory afferents would be needed to generate a spike from the up state. To maximize physiological relevance, in our experiments, we used the minimal levels of stimulation necessary to evoke responses in control neurons. The disruption of the evoked responses observed here suggests that the minimal levels of stimulation required for reliable information propagation through the network is significantly increased in plaque-containing cortex. In other words, it is not the number of synaptic afferents being disrupted by plaques but, rather, their synchrony. The desynchronization of the arriving PSPs would explain the increased numbers of transition failures from the down to up states, as well as the increased response jitter and failures from the up state in the aged transgenic animals. Because the transition from down to up state requires an order of magnitude more synchronous PSPs than does the generation of an action potential from the up state, we would expect that the responses from the down state would be more sensitive to disruption of synchrony by plaques (Fig. 3). The implications of this are that reliable cortical information processing is highly dependent on spatial structure, and, indeed, theoretical studies have shown that, on several levels, the connectivity of the brain is optimized for minimal neuritic length (Chklovskii, 2000; Chklovskii et al., 2002).
We do not discount the findings that soluble amyloid-β affects synaptic function. Although the synaptic dysfunction observed here occurs only in older mice in cortex with substantial numbers of plaques, the amount of soluble amyloid-β in cortex also increases as a function of age (Kawarabayashi et al., 2001). However, elevated soluble amyloid-β is also present at the younger age measured here. Increased oligomeric amyloid-β has been shown to alter hippocampal synaptic transmission (Chapman et al., 1999; Walsh et al., 2002; Kamenetz et al., 2003), especially reducing measures of plasticity, such as LTP. Although it is possible that soluble amyloid-β affects synaptic transmission only after reaching a certain threshold level, the presence of a correlation between the degrees of morphological alterations and electrophysiological impairments leads us to favor the hypothesis that the reduction in response precision observed in this study differs from those seen in previous in vitro electrophysiological studies and are at least partially attributable to the physical distortion of neuronal structure by insoluble amyloid-β.
Although the increased synaptic jitter is correlated with increases in neuritic curvature, a number of other factors may contribute to the alterations in synaptic physiology observed here. In particular, dystrophic axons and dendrites that envelop fibrillar amyloid have been observed in the cortical neuropil (Masliah et al., 1993, 1996; Holtzman et al., 2000; Dickson and Vickers, 2001; Brendza et al., 2003). These neurites are enlarged and may very well have altered physiological properties, such as axonal conduction velocities and synaptic release properties (Ferrer et al., 1998). The contribution of these dystrophic neurites to synaptic response properties has never been measured.
Synaptophysin immunoreactivity levels are normal at the ages of Tg2576 mice used in this study (Irizarry et al., 1997). However, the question of synaptic remodeling remains open: are neurites simply “stretched” when distorted by plaques and retain their specific connections, or do neurites reconnect to different afferents-efferents after distortion? Although the data in this study cannot directly address this question, clearly the physical disruption of neurites has a functional correlate. Indeed, it may be that the ability to remodel connections is essential for normal neuronal function and may underlie certain forms of synaptic plasticity (Stepanyants et al., 2002; Trachtenberg et al., 2002).
This study provides the first direct evidence that neocortical synaptic function in vivo is affected by amyloid-β plaques, suggesting a distinct role for insoluble plaques, which could act as a distributed lesion disrupting neural systems, contributing directly to AD dementia. This work also has important implications for possible therapeutic strategies for treatment of AD, in that clearing plaques may have different consequences than does reducing soluble amyloid-β on neuronal function (Lombardo et al., 2003).
Footnotes
This work was supported by a Pioneer award from the Alzheimer Association and National Institutes of Health Grant AG08487.
Correspondence should be addressed to Edward A. Stern, Massachusetts General Hospital, Department of Neurology, 16th Street, CNY Building 114, Room 2800, Charlestown, MA 02129. E-mail: estern@partners.org.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244535-06$15.00/0
References
- Anderson J, Lampl I, Reichova I, Carandini M, Ferster D (2000) Stimulus dependence of two-state fluctuations of membrane potential in cat visual cortex. Nat Neurosci 3: 617-621. [DOI] [PubMed] [Google Scholar]
- Azouz R, Gray CM (1999) Cellular mechanisms contributing to response variability of cortical neurons in vivo J Neurosci 19: 2209-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braitenburg V, Schüz A (1998) Cortex statistics and geometry of neuronal connectivity, Ed 2. Berlin: Springer.
- Brendza RP, O'Brien C, Simmons K, McKeel DW, Bales KR, Paul SM, Olney JW, Sanes JR, Holtzman DM (2003) PDAPP; YFP double transgenic mice: a tool to study amyloid-beta associated changes in axonal, dendritic, and synaptic structures. J Comp Neurol 456: 375-383. [DOI] [PubMed] [Google Scholar]
- Buldyrev SV, Cruz L, Gomez-Isla T, Gomez-Tortosa E, Havlin S, Le R, Stanley HE, Urbanc B, Hyman BT (2000) Description of microcolumnar ensembles in association cortex and their disruption in Alzheimer and Lewy body dementias. Proc Natl Acad Sci USA 97: 5039-5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, Morrison JH (1989) Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol 282: 191-205. [DOI] [PubMed] [Google Scholar]
- Chapman PF, White GL, Jones MW, Cooper-Blacketer D, Marshall VJ, Irizarry M, Younkin L, Good MA, Bliss TV, Hyman BT, Younkin SG, Hsiao KK (1999) Impaired synaptic plasticity and learning in aged amyloid precursor protein transgenic mice. Nat Neurosci 2: 271-276. [DOI] [PubMed] [Google Scholar]
- Chklovskii DB (2000) Optimal sizes of dendritic and axonal arbors in a topographic projection. J Neurophysiol 83: 2113-2119. [DOI] [PubMed] [Google Scholar]
- Chklovskii DB, Schikorski T, Stevens CF (2002) Wiring optimization in cortical circuits. Neuron 34: 341-347. [DOI] [PubMed] [Google Scholar]
- Cisse Y, Grenier F, Timofeev I, Steriade M (2003) Electrophysiological properties and input-output organization of callosal neurons in cat association cortex. J Neurophysiol 89: 1402-1413. [DOI] [PubMed] [Google Scholar]
- Connors BW, Prince DA (1982) Effects of local anesthetic QX-314 on the membrane properties of hippocampal pyramidal neurons. J Pharmacol Exp Ther 220: 476-481. [PubMed] [Google Scholar]
- Cowan RL, Wilson CJ (1994) Spontaneous firing patterns and axonal projections of single corticostriatal neurons in the rat medial agranular cortex. J Neurophysiol 71: 17-32. [DOI] [PubMed] [Google Scholar]
- D'Amore JD, Kajdasz ST, McLellan ME, Bacskai BJ, Stern EA, Hyman BT (2003) In vivo multiphoton imaging of a transgenic mouse model of Alzheimer disease reveals marked thioflavine-S-associated alterations in neurite trajectories. J Neuropathol Exp Neurol 62: 137-145. [DOI] [PubMed] [Google Scholar]
- Destexhe A, Pare D (1999) Impact of network activity on the integrative properties of neocortical pyramidal neurons in vivo. J Neurophysiol 81: 1531-1547. [DOI] [PubMed] [Google Scholar]
- Dickson TC, Vickers JC (2001) The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer's disease. Neuroscience 105: 99-107. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Marti E, Tortosa A, Blasi J (1998) Dystrophic neurites of senile plaques are defective in proteins involved in exocytosis and neurotransmission. J Neuropathol Exp Neurol 57: 218-225. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ (1999) Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 19: 8876-8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Ashe KH, Irizarry MC, Hyman BT (2000) Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Ann Neurol 47: 739-747. [PubMed] [Google Scholar]
- Horikawa K, Armstrong WE (1988) A versatile means of intracellular labeling: injection of biocytin and its detection with avidin conjugates. J Neurosci Methods 25: 1-11. [DOI] [PubMed] [Google Scholar]
- Hsiao K (1998) Strain dependent and invariant features of transgenic mice expressing Alzheimer amyloid precursor proteins. Prog Brain Res 117: 335-341. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Hyman BT, Marzloff K, Arriagada PV (1993) The lack of accumulation of senile plaques or amyloid burden in Alzheimer's disease suggests a dynamic balance between amyloid deposition and resolution. J Neuropathol Exp Neurol 52: 594-600. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT (1997) APPSw transgenic mice develop age-related A beta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965-973. [DOI] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R (2003) APP processing and synaptic function. Neuron 37: 925-937. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG (2001) Age-dependent changes in brain, CSF, and plasma amyloid (β) protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci 21: 372-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelenyi G (1967) Thioflavin S fluorescent and Congo red anisotropic stainings in the histologic demonstration of amyloid. Acta Neuropathol (Berl) 7: 336-348. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Gomez-Isla T, Hyman BT (1998) Abeta associated neuropil changes: correlation with neuronal loss and dementia. J Neuropathol Exp Neurol 57: 1122-1130. [DOI] [PubMed] [Google Scholar]
- Knowles RB, Wyart C, Buldyrev SV, Cruz L, Urbanc B, Hasselmo ME, Stanley HE, Hyman BT (1999) Plaque-induced neurite abnormalities: implications for disruption of neural networks in Alzheimer's disease. Proc Natl Acad Sci USA 96: 5274-5279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry P, Wilson CJ, Kitai ST (1984) Morphological and electrophysiological characteristics of pyramidal tract neurons in the rat. Exp Brain Res 57: 177-190. [DOI] [PubMed] [Google Scholar]
- Le R, Cruz L, Urbanc B, Knowles RB, Hsiao-Ashe K, Duff K, Irizarry MC, Stanley HE, Hyman BT (2001) Plaque-induced abnormalities in neurite geometry in transgenic models of Alzheimer disease: implications for neural system disruption. J Neuropathol Exp Neurol 60: 753-758. [DOI] [PubMed] [Google Scholar]
- Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT (2003) Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci 23: 10879-10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Mallory M, Deerinck T, DeTeresa R, Lamont S, Miller A, Terry RD, Carragher B, Ellisman M (1993) Re-evaluation of the structural organization of neuritic plaques in Alzheimer's disease. J Neuropathol Exp Neurol 52: 619-632. [DOI] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (1996) Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F beta-amyloid precursor protein and Alzheimer's disease. J Neurosci 16: 5795-5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA (1985) Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J Neurophysiol 54: 782-806. [DOI] [PubMed] [Google Scholar]
- Moore CI, Nelson SB (1998) Spatio-temporal subthreshold receptive fields in the vibrissa representation of rat primary somatosensory cortex. J Neurophysiol 80: 2882-2892. [DOI] [PubMed] [Google Scholar]
- Nowak LG, Azouz R, Sanchez-Vives MV, Gray CM, McCormick DA (2003) Electrophysiological classes of cat primary visual cortical neurons in vivo as revealed by quantitative analyses. J Neurophysiol 89: 1541-1566. [DOI] [PubMed] [Google Scholar]
- Nunez A, Amzica F, Steriade M (1993) Electrophysiology of cat association cortical cells in vivo: intrinsic properties and synaptic responses. J Neurophysiol 70: 418-430. [DOI] [PubMed] [Google Scholar]
- Pare D, Shink E, Gaudreau H, Destexhe A, Lang EJ (1998) Impact of spontaneous synaptic activity on the resting properties of cat neocortical pyramidal neurons in vivo. J Neurophysiol 79: 1450-1460. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, Nowak LG, McCormick DA (2000) Membrane mechanisms underlying contrast adaptation in cat area 17 in vivo. J Neurosci 20: 4267-4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepanyants A, Hof PR, Chklovskii DB (2002) Geometry and structural plasticity of synaptic connectivity. Neuron 34: 275-288. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F (1993) A novel slow (<1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci 13: 3252-3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern EA, Kincaid AE, Wilson CJ (1997) Spontaneous subthreshold membrane potential fluctuations and action potential variability of rat corticostriatal and striatal neurons in vivo. J Neurophysiol 77: 1697-1715. [DOI] [PubMed] [Google Scholar]
- Stern EA, Maravall M, Svoboda K (2001) Rapid development and plasticity of layer 2/3 maps in rat barrel cortex in vivo. Neuron 31: 305-315. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Grenier F, Bazhenov M, Sejnowski TJ, Steriade M (2000) Origin of slow cortical oscillations in deafferented cortical slabs. Cereb Cortex 10: 1185-1199. [DOI] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K (2002) Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature 420: 788-794. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416: 535-539. [DOI] [PubMed] [Google Scholar]
- Ye CP, Selkoe DJ, Hartley DM (2003) Protofibrils of amyloid beta-protein inhibit specific K+ currents in neocortical cultures. Neurobiol Dis 13: 177-190. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Connors BW (1999) Intrinsic firing patterns and whisker-evoked synaptic responses of neurons in the rat barrel cortex. J Neurophysiol 81: 1171-1183. [DOI] [PubMed] [Google Scholar]