

Abstract
Clearance of extracellular glutamate is essential for limiting the activity of metabotropic glutamate receptors (mGluRs) at excitatory synapses; however, the relative contribution of transporters found in neuronal and glial membranes to this uptake is poorly understood. Hippocampal interneurons located at the oriens-alveus border express mGluR1α, a metabotropic glutamate receptor that regulates excitability and synaptic plasticity. To determine which glutamate transporters are essential for removing glutamate at these excitatory synapses, we recorded mGluR1-mediated EPSCs from oriens-lacunosum moleculare (O-LM) interneurons in acute hippocampal slices. Stimulation in stratum oriens reliably elicited a slow mGluR1-mediated current in O-LM interneurons if they were briefly depolarized to allow Ca2+ entry before stimulation. Selective inhibition of GLT-1 [for glutamate transporter; EAAT2 (for excitatory amino acid transporter)] with dihydrokainate increased the amplitude of these responses approximately threefold, indicating that these transporters compete with mGluRs for synaptically released glutamate. However, inhibition of all glutamate transporters with TBOA (dl-threo-b-benzyloxyaspartic acid) increased mGluR1 EPSCs >15-fold, indicating that additional transporters also shape activation of these receptors. To identify these transporters, we examined mGluR1 EPSCs in mice lacking GLAST (for glutamate-aspartate transporter; EAAT1) or EAAC1 (for excitatory amino acid carrier; EAAT3). A comparison of responses recorded from wild-type and transporter knock-out mice revealed that the astroglial glutamate transporters GLT-1 and GLAST, but not the neuronal transporter EAAC1, restrict activation of mGluRs in O-LM interneurons. Transporter-dependent potentiation of mGluR1 EPSCs led to a dramatic increase in interneuron firing and enhanced inhibition of CA1 pyramidal neurons, suggesting that acute or prolonged disruption of transporter activity could lead to changes in network activity as a result of enhanced interneuron excitability.
Keywords: hippocampus, interneuron, glutamate transporter, GLT-1, GLAST, EAAC1, mGluR, EAAT3, EAAT2, EAAT1
Introduction
Glutamate transporters prevent the accumulation of extracellular glutamate that would otherwise lead to tonic activation of receptors, disrupted signaling, and excitotoxic damage. Because they are present at high densities near synapses (Lehre and Danbolt, 1998) and have a high affinity for glutamate, glutamate transporters also can influence the number of receptors that bind transmitter (Tong and Jahr, 1994; Diamond and Jahr, 1997) and the length of time that they are activated after release (Otis et al., 1996; Arnth-Jensen et al., 2002). However, the impact of transporter-mediated clearance varies dramatically among synapses; it depends on the synaptic structure, the amount of glutamate released, and the properties and distribution of glutamate receptors. Although the influence of transporters on the excitation of principal neurons has been examined in many brain regions (Danbolt, 2001), less is known about the contribution of transporters to clearance of glutamate at excitatory synapses on inhibitory interneurons (Carter and Regehr, 2000; Clark and Cull-Candy, 2002). In cerebellar Purkinje neurons, transporter inhibition profoundly enhances activation of perisynaptic metabotropic glutamate receptors (mGluRs) (Brasnjo and Otis, 2001), suggesting that these receptors must compete with transporters for glutamate as it diffuses out of the cleft. The slow turnover rate of transporters relative to rates of release may create a frequency filter, ensuring that mGluRs are activated only when glutamate is repeatedly released from the same site.
At least five different glutamate transporters are expressed in the CNS, which have been named GLAST [for glutamate-aspartate transporter; EAAT1 (for excitatory amino acid transporter)], GLT-1 (for glutamate transporter; EAAT2), EAAC1 (for excitatory amino acid carrier; EAAT3), EAAT4, and EAAT5 (Danbolt, 2001). GLAST and GLT-1 are expressed primarily by astroglial cells, EAAC1 and EAAT4 are expressed by neurons, and EAAT5 is found only in the retina (Danbolt, 2001). Although much has been learned about the distribution of these transporters, little is known about their relative contributions to the clearance of glutamate away from receptors at synapses. The occupancy of mGluRs in Purkinje neurons (Brasnjo and Otis, 2001) and NMDA receptors in hippocampal pyramidal neurons (Diamond, 2001) are increased when glutamate uptake into the these neurons is selectively disrupted, suggesting that postsynaptic transporters (e.g., EAAC1 and EAAT4) shield receptors. However, astrocytes are tightly associated with synapses along these perisynaptic regions (Spacek, 1985), and quantitative measurements of transporter densities (Lehre and Danbolt, 1998) and glutamate uptake (Rothstein et al., 1996) indicate that GLAST and GLT-1 are the most abundant glutamate transporters in brain tissue. Nevertheless, it is not known whether astroglial transporters also regulate the accessibility of mGluRs to synaptically released glutamate.
Here we show that mGluR1-mediated synaptic currents could be evoked in hippocampal oriens-lacunosum moleculare (OLM) interneurons if intracellular Ca2+ was elevated in these cells before stimulation. These synaptic mGluR1 currents were greatly potentiated by pharmacological inhibition of glutamate transporters, and recordings from mice deficient in glutamate transporters revealed that GLAST and GLT-1, but not EAAC1, were primarily responsible for shielding these perisynaptic receptors. Furthermore, inhibition of these transporters potentiated disynaptic inhibition of CA1 pyramidal cells, indicating that astroglial glutamate transporters may regulate hippocampal network excitability by regulating mGluR1 occupancy at interneuron synapses.
Materials and Methods
Glutamate transporter-deficient mice. The generation of EAAC1-/- mice and GLAST-/- mice have been described previously (Peghini et al., 1997; Watase et al., 1998). All comparisons between specific transporter knockout mice and wild-type mice were performed between littermates obtained from heterozygous matings. The genotype of all experimental animals was determined by PCR.
Slice preparation. Postnatal day 18 (P18) to P20 rats and P15-P20 mice were deeply anesthetized by halothane and killed by decapitation, in accordance with protocols approved by the Johns Hopkins University Animal Care and Use Committee. Hippocampi were removed and cut into 400 μm slices using a vibratome (VT1000S; Leica, Nussloch, Germany) in ice-cold saline solution containing the following (in mm): 110 choline chloride, 2.5 KCl, 7 MgSO4, 0.5 CaCl2, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 11.6 Na-ascorbate, and 3.1 Na-pyruvate (saturated with 95% O2-5% CO2). Slices were then incubated at 37°C for 30 min in artificial CSF (ACSF) composed of (in mm) 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose (saturated with 95% O2-5% CO2) and then allowed to recover for at least 30 min at room temperature (22-24°C) before experimentation.
Electrophysiological recordings. During experimentation, slices were superfused with ACSF that was heated to 33-35°C by passing the solution through a feedback controlled in-line heater (Warner Instruments, Hamden, CT) before entering the chamber. O-LM interneurons were visualized in stratum oriens using infrared differential interference contrast imaging with a 40× water immersion objective on an upright microscope (Axioskop 2FS; Zeiss, Thornwood, NY) equipped with a CCD camera (XC-73; Sony, Tokyo, Japan). O-LM interneuron whole-cell voltage-clamp recordings were made with an internal solution containing the following (in mm): 105 CsCH3O3S, 20 tetraethylammonium (TEA)-Cl, 20 HEPES, 10 EGTA (or 10 or 40 BAPTA, when noted), 1 MgCl2,2Na2-ATP, 0.2 Na-GTP, and 1 QX-314, pH 7.3. O-LM interneuron whole-cell current-clamp recordings were made with a solution containing the following (in mm): 120 KCH3O3S, 20 HEPES, 10 EGTA, 1 MgCl2, 2 Na2-ATP, and 0.2 Na-GTP, pH 7.3. Excitatory afferents were stimulated using a glass pipette filled with ACSF. Stimuli (20-35 μA) were generated using a constant-current isolated stimulator (DS3; Digitimer, Hertfordshire, UK), and a programmable pulse generator (Master-8; A.M.P.I., Jerusalem, Israel) was used to trigger a train of pulses (10 pulses at 100 Hz; 200 μsec duration for each pulse). Synaptic currents were recorded with a MultiClamp 700A amplifier (Axon Instruments, Foster City, CA), filtered at 3 kHz, amplified 5× or 10× (440; Brownlee, San Jose, CA), and then digitized at 10 kHz (Digidata 1322A; Axon Instruments). Data were analyzed off-line using pClamp 9 (Axon Instruments) and Origin (Microcal Software, Northampton, MA) software. The action potentials for waveform analysis were elicited by injecting just enough current to induce spontaneous firing. Action potential properties were determined as described previously (Zhang and McBain, 1995). IPSCs were recorded from CA1 pyramidal neurons using an internal solution containing the following (in mm): 100 CsCl, 20 TEA-Cl, 20 HEPES, 10 EGTA, 1 MgCl2, 2 Na2-ATP, and 0.2 Na-GTP, pH 7.3. The charge transfer associated with IPSCs was determined by integrating the charge over a window of 4 sec, starting 220 msec after the last stimulus artifact. Statistical significance was determined using the Student's t test (paired or unpaired, depending on the experiment), and mean values are expressed ± SEM.
Photolysis experiments were performed on an upright microscope (Axioskop FS2; Zeiss) to which the output of an argon ion laser (Stabilite 2017S; Spectra-Physics, Mountain View, CA) was coupled to the fluorescence light path using a multi-mode quartz fiber optic cable (Oz Optics, Carp, Ontario, Canada). The laser output was tuned to 333.6-363.8 nm and had an average output of ∼230 mW before entering the fiber. The UV beam was focused to a ∼50 μm spot on the preparation using a 40× water immersion objective (LUMPlanFl; Olympus Optical, Tokyo, Japan). This spot was centered on the soma of the recorded cell using a second targeting laser (632 nm), and photolysis was induced by opening a high-speed shutter (NM Laser Products, Sunnyvale, CA) for 1 msec. The caged compound, 4-methoxy-7-nitroindolinyl-L-glutamate (MNI l-glutamate) (125 μm; Tocris Cookson, Ellisville, MO) was dissolved in a HEPES-buffered solution composed of the following (in mm): 137 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, and 20 HEPES, pH 7.3. Any antagonists included in the bath solution were also included in solutions containing the caged compound. Solutions containing MNI-l-glutamate were superfused locally onto the slice through a wide-bore pipette placed near the recording area. Astrocyte recordings were performed with an internal solution containing (in mm): 130 CsCH3O3S, 20 HEPES, 10 EGTA, 1 MgCl2,2Na2-ATP, and 0.2 Na-GTP, pH 7.3. Whole-cell currents were recorded with an Axopatch 200B amplifier (Axon Instruments), filtered at 2 kHz, and digitized at 20 kHz.
Morphological reconstruction and immunocytochemistry. O-LM interneurons were filled with neurobiotin (0.2% w/v in intracellular solution; Vector Laboratories, Burlingame, CA) for 30 min during recordings and then incubated in oxygenated ACSF for 5-9 hr after removal of the recording electrode. Slices were then fixed overnight in 4% formaldehyde and 0.2% picric acid in PBS at 4°C and resectioned at 80 μm on a vibratome (VT1000S; Leica). These sections were processed using avidin-HRP reaction (Elite ABC; Vector Laboratories), developed using 3-3′-diaminobenzidine (DAB) (Peroxidase Substrate kit, Vector Laboratories), and intensified using DAB enhancing solution (Vector Laboratories). Morphological reconstruction was made with the aid of an upright Zeiss microscope and drawing tube.
Drugs. NBQX (2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f]quinoxaline), GYKI 52466 [1-(4-aminophenyl)-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine hydrochloride], SR-95531 [2-(3-carboxypropyl)-3-amino-6-(4-methoxyphenyl)pyridazinium bromide], bicuculline, RS-CPP [(RS)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid], MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], 7-chlorokynurenate, CGP 55845 [[1-(S)-(3,4-dichlorophenyl)ethyl]amino-2-(S)-hydroxypropyl-p-benzyl-phosphonic acid], LY 367385 [(S)-(+)-α-amino-4-carboxy-2-methylbenzene acetic acid], and SKF 96365 [1-2-(4-methoxyphenyl)-2-[3-(4-methoxyphenyl)propoxy]ethyl-1H-imidazole] were obtained from Tocris Cookson.
Results
Isolation of an mGluR1-mediated synaptic current in O-LM interneurons
To determine whether glutamate transporters are essential for clearing glutamate away from mGluRs at excitatory synapses on hippocampal interneurons, we recorded from O-LM interneurons (sometimes referred to as O-A interneurons) in the oriensalveus region of area CA1. mGluR1α is highly expressed by O-LM interneurons (Baude et al., 1993), and application of RS-1-aminocyclopentane-trans-1,3-dicarboxylic acid (trans-ACPD), a group I/II mGluR agonist, elicits an inward current in these cells (McBain et al., 1994).
The ability of mGluRs to induce opening of cation channels in these neurons provides a means to monitor mGluR activity using electrophysiological techniques. Putative O-LM interneurons had large, oval-shaped cell bodies with two prominent dendrites extending parallel to the pyramidal cell layer. Injection of depolarizing current induced a train of action potentials in these neurons that did not exhibit spike frequency adaptation, and each action potential during the train was followed by a large spike afterhyperpolarization (Fig. 1A, inset). Spontaneous action potentials recorded from these interneurons were 73.5 ± 3.2 mV in amplitude and 1.3 ± 0.1 msec in duration (n = 6; 33-35°C). These electrophysiological properties are in accordance with those reported for O-LM interneurons (Zhang and McBain, 1995). Reconstruction of neurobiotin-filled interneurons revealed that they had dendrites that extended parallel to the pyramidal cell layer and axons that projected through the pyramidal cell layer and ramified extensively in stratum-lacunosum moleculare (n = 9 of 9 cells), morphological features unique to O-LM interneurons (Freund and Buzsáki, 1996).
Figure 1.

Isolation of slow EPSCs mediated by mGluR1 in O-LM interneurons. A, Morphology of an O-LM interneuron that was filled with neurobiotin through the recording electrode. a, Alveus; so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; lm, lacunosum moleculare. Bottom inset shows a picture of this interneuron (scale bar, 200 μm), and the top inset shows the response of this neuron to injection of depolarizing current (300 pA). B, Left, Response of an O-LM interneuron to field stimulation in stratum oriens (black trace). The red trace shows the response of this interneuron to the same stimulus when the cell was depolarized (from -65 to 0 mV for 500 msec) 1 sec before stimulation. Right, Same traces displayed on a faster time scale. C, Inhibition of the slow EPSC recorded from an O-LM interneuron by the specific mGluR1 antagonist LY 367385 (50 μm). Responses in B and C were recorded in AMPA (15 μm NBQX and 25 μm GYKI 52466), NMDA (10 μm RS-CPP, 50 μm MK-801, and 20 μm 7-chlorokynurenate), and GABAA (5 μm SR 95531 and 20 μm bicuculline) receptor antagonists at a holding potential of -65 mV.
Although application of trans-ACPD elicits an inward current in O-LM interneurons (McBain et al., 1994; Gee et al., 2001), activation of an mGluR current in these cells after synaptic stimulation has not been reported previously. We found that brief, high-frequency stimulation in stratum oriens (100 Hz, 10 pulses, 200 μsec, 20-35 μA per pulse) elicited a slowly developing inward current in some O-LM interneurons (n = 7 of 20 cells) (Fig. 1B), which persisted when AMPA (15 μm NBQX and 25 μm GYKI 52466), NMDA (10 μm RS-CPP, 50 μm MK-801, and 20 μm 7-chlorokynurenate), and GABAA receptors (5 μm SR-95531 and 20 μm bicuculline) were blocked (n = 7 of 7 cells); evoked responses also were not affected by the GABAB antagonist CGP 55845 (n = 4 of 4 cells; data not shown). However, the low probability of observing this slow inward current limited our ability to examine transporter-receptor interactions. In cerebellar Purkinje neurons, mGluR1α-mediated currents are potentiated when the cytosolic concentration of Ca2+ is increased (Batchelor and Garthwaite, 1997), suggesting that this manipulation might similarly increase mGluR1-mediated currents in O-LM interneurons. As shown in Figure 1B, when O-LM interneurons were briefly depolarized to 0 mV (from -65 mV, 500 msec) just before stimulation to allow Ca2+ influx through voltage-gated Ca2+ channels, the amplitude of the evoked response was larger (without step, -29.8 ± 6.2 pA; with step, -112.2 ± 27.7 pA; n = 11; p < 0.01), and the rise time was faster (20-80% rise time: without step, 957 ± 282 msec; with step, 94 ± 9 msec; n = 3; p < 0.05), although it was only possible to measure the latter feature accurately when the stimulus artifact did not obscure the onset of the response (3 of 11 cells). Using this protocol, slow synaptic currents were observed in 27 of 28 O-LM interneurons in AMPA, NMDA, and GABAA receptor antagonists (see above). This synaptic current was inhibited 92.8 ± 1.1% (n = 9) by the selective mGluR1 antagonist LY 367385 (50 μm) (Fig. 1C), indicating that the response was dependent on activation of mGluR1. This response was also blocked by SKF 96365 (50 μm) (n = 4 of 4 cells), an antagonist of receptor-gated cation channels that also inhibits mGluR1α-mediated inward currents in Purkinje neurons (Kim et al., 2003), suggesting that the synaptic currents recorded from O-LM interneurons resulted from activation of nonselective cation channels.
To confirm that the step depolarization facilitated the mGluR1 response by increasing [Ca2+]i, we examined the size of these EPSCs when a high concentration of BAPTA (40 mm) was included in the pipette solution to rapidly chelate free Ca2+. As shown in Figure 2 A, responses recorded with this solution were significantly smaller than those recorded with the control solution (peak amplitude: 10 mm BAPTA, -121.2 ± 36.2 pA, n = 8; 40 mm BAPTA, -5.3 ± 4.5 pA, n = 5; p < 0.01), suggesting that elevation of [Ca2+]i was responsible for step-induced potentiation of the mGluR1 EPSC. To examine the dependence of this response on the step parameters and to maximize the amplitude of the mGluR1 EPSC, we varied both the step duration and the delay between the end of the step and the beginning of the stimulus (Fig. 2 B, C). The slow EPSC was close to its maximal amplitude when a 500 msec depolarization (from -65 to 0 mV) was applied 1 sec before stimulation; thus, this protocol was used for eliciting mGluR1 EPSCs in subsequent experiments.
Figure 2.

Ca2+ dependence of the mGluR synaptic current. A, mGluR1 EPSCs recorded from O-LM interneurons with an internal solution containing 10 or 40 mm BAPTA. The grouped data below shows the average size of the mGluR1 EPSC recorded under these two conditions. Numbers in parentheses indicate the number of experiments (**p < 0.01). B, mGluR1 EPSCs elicited after depolarizing steps (from -65 to 0 mV) of different durations, as indicated. C, mGluR1 EPSCs evoked with varying delay (as indicated) after the end of a 500 msec step depolarization to 0 mV. B and C were performed in the presence of 100 μm TBOA.
Inhibition of glutamate transporters enhances activation of mGluR1α
Ultrastructural studies indicate that mGluR1α receptors in O-LM interneurons are predominantly localized at the periphery of the postsynaptic density of type 1 excitatory synapses (Baude et al., 1993), in close proximity to ensheathing glial membranes (Spacek, 1985; Ventura and Harris, 1999). Despite the high affinity of mGluR1α (EC50 of ∼10 μm) (Conn and Pin, 1997) and the amplification of mGluR-mediated currents by second messengers, the amplitude of mGluR1 EPSCs were small compared with responses evoked by exogenous mGluR agonists (McBain et al., 1994), suggesting that these receptors may be exposed to only a fraction of the glutamate that is released. To determine whether glutamate transporters restrict activation of mGluR1 on O-LM interneurons, we examined whether glutamate transporter antagonists potentiated these synaptic currents. Application of 300 μm dihydrokainate (DHK), a concentration that results in selective antagonism of GLT-1 (Arriza et al., 1994), caused an approximately threefold increase in the amplitude of mGluR1 EPSCs (control, -97.5 ± 31.8 pA; with DHK, -365.0 ± 100.7 pA; n = 8; p < 0.01) (Fig. 3A,B); a similar effect was seen on the amount of charge transferred during mGluR1 EPSCs (control, 70.1 ± 40.0 pC; with DHK, 206.7 ± 64.7 pC; n = 8; p < 0.001) (Fig. 3A,B). These results indicate that GLT-1, a transporter that is present at a high density in astrocyte membranes (Lehre and Danbolt, 1998), restricts activation of these interneuron receptors. Subsequent application of a saturating dose of TBOA (dl-threo-b-benzyloxyaspartic acid) (100 μm), an antagonist that blocks all glutamate transporters, caused an additional, more dramatic enhancement of these slow EPSCs (Fig. 3A,B). The current enhanced by transporter antagonists was mGluR1 dependent, because the peak amplitudes of the synaptic responses in TBOA were inhibited 94.0 ± 2.2% (n = 7) by LY 367385 (50 μm) (charge transfer was inhibited 94.0 ± 2.1%). As shown in Figure 3C, TBOA dramatically increased the size of mGluR1-mediated EPSCs elicited in response to a given stimulation intensity. In the absence of transporter inhibition, mGluR1 EPSCs (elicited by high-frequency stimulation) were always smaller than AMPA EPSCs (elicited by single stimuli). Neither DHK nor TBOA induced an increase in holding current in the absence of stimulation (data not shown) (Arnth-Jensen et al., 2002), presumably because NMDA receptors were blocked in these experiments and the mGluR1-associated conductance depends on elevation of [Ca2+]i.
Figure 3.

Inhibition of glutamate transporters potentiates mGluR1 EPSCs.A, Evoked mGluR1 EPSCs recorded from O-LM interneurons with (traces labeled 2) or without (traces labeled 1) the step depolarization, under control conditions, in the presence of DHK (300 μm), TBOA (100 μm), or LY 367385 (50 μm) and TBOA. B, Grouped data showing the peak amplitude (open bars) and charge transfer (filleded bars) of the mGluR EPSC recorded under these different conditions. Numbers in parentheses indicate the number of experiments (**p < 0.01; ***p < 0.001). C, Plot of the peak amplitude of evoked AMPA EPSCs (single stimulus; ▪) (n = 5-12), mGluR1 EPSCs under control conditions (10 stimuli at 100 Hz; ○) (n = 5-13), and mGluR EPSCs in the presence of TBOA (10 stimuli at 100 Hz; •) (n = 8-13) in response to stimuli of different intensity. When absent, error bars were shorter than the points.
To determine the effect of transporter inhibition on interneuron excitation as a consequence of increased mGluR activation, we made current-clamp recordings from O-LM interneurons and measured their response to a similar stimulus. As shown in Figure 4, stimulation in the presence of TBOA led to a dramatic depolarization and sustained firing of action potentials that was not visible under control conditions (n = 5 of 5 cells). This effect was blocked by LY 367385 (50 μm) (n = 2 of 2 cells), indicating that this powerful excitatory response was dependent on mGluR1.
Figure 4.

Inhibition of glutamate transporters potentiates mGluR1-mediated depolarization of O-LM interneurons. Response of an O-LM interneuron to a train of stimuli (arrow) after a depolarizing current injection (200 pA) recorded under control conditions (left trace), in the presence of TBOA (100 μm; middle trace), and in the presence of TBOA and LY 367385 (50 μm; right trace). Transporter inhibition revealed an mGluR1-mediated response in this O-LM interneuron.
Astrocyte glutamate transporters shield interneuron mGluRs
mGluR1 EPSCs were potentiated more by TBOA than by DHK, suggesting that glutamate transporters other than GLT-1 also may be involved in clearing glutamate away from these receptors. Immunocytochemical localization studies indicate that three distinct transporters could participate in glutamate clearance at these synapses: GLT-1, GLAST, and EAAC1 (Rothstein et al., 1994; Lehre et al., 1995); a splice variant of GLT-1 (termed GLT-1b) also may be expressed (Reye et al., 2002) but has pharmacological properties similar to full-length GLT-1 (Utsunomiya-Tate et al., 1997). To determine the relative contribution of GLAST and EAAC1 to clearance at these synapses, we performed a similar set of experiments in mice deficient in EAAC1 or GLAST, because selective antagonists for these transporters have not yet been developed. If a particular transporter is involved in clearing glutamate away from mGluRs, then potentiation of mGluR1 EPSCs by DHK should be greater in mice lacking this transporter because, in the absence of this transporter, GLT-1 would be expected to contribute proportionally more to uptake. As shown in Figure 5, A and C, application of DHK (300 μm) caused an approximately onefold to twofold potentiation of mGluR1 EPSCs in EAAC1-/- mice, similar to that observed in wild-type littermates (fold increase, amplitude/charge transfer: EAAC1+/+, 1.34 ± 0.47/1.78 ± 0.68, n = 5; EAAC1-/-, 1.60 ± 0.21/0.96 ± 0.31, n = 6; p = 0.32/p = 0.16). In contrast, DHK (300 μm) produced a much greater enhancement of mGluR1 EPSCs in GLAST-/- mice than in wild-type littermates (Fig. 5B,C) (fold increase, amplitude/charge transfer: GLAST+/+, 1.57 ± 0.28/1.74 ± 0.58, n = 6; GLAST-/-, 7.25 ± 1.47/6.90 ± 1.07, n = 11; p < 0.01/p < 0.001). The size of mGluR1 EPSCs were not significantly different between transporter knock-out and wild-type littermates for EAAC1 (peak amplitude/charge transfer: EAAC1+/+, -113.9 ± 18.2 pA/84.8 ± 17.8 pC, n = 5; EAAC1-/-, -77.1 ± 25.9 pA/67.6 ± 15.2 pC, n = 6; p = 0.28/p = 0.48) and GLAST (peak amplitude/charge transfer: GLAST+/+, -149.7 ± 44.6 pA/109.8 ± 32.9 pC, n = 6; GLAST-/-, -111.9 ± 15.4 pA/64.0 ± 5.7 pC, n = 8; p = 0.45/p = 0.23). These results suggest that GLT-1 and GLAST, but not EAAC1, determine how much glutamate is available to activate mGluR1 receptors in O-LM interneurons.
Figure 5.

GLT-1 and GLAST, but not EAAC1, restrict the activation of mGluR1 receptors. A, Evoked mGluR1-mediated EPSCs recorded in O-LM interneurons from an EAAC1-/- mouse and a wild-type littermate under control conditions and in the presence of DHK (300 μm) or TBOA (100 μm). B, The same set of experiments as in A, performed on slices from a GLAST-/- mouse and a wild-type littermate. The arrow highlights the larger effect of DHK (300 μm) observed in the GLAST-/- mouse. C, Grouped data showing the effects of DHK and TBOA on the peak amplitude (left) and charge transfer (right) of mGluR1-mediated EPSCs in mice with different genotypes (as indicated). The effect of 1 mm DHK on mGluR1-mediated EPSCs was studied in GLAST-/- mice and wild-type littermates (**p < 0.01; ***p < 0.001). Numbers in parentheses indicate the number of experiments.
The effect of DHK was not equivalent to TBOA in GLAST-/- mice (fold increase over control in TBOA, peak amplitude/charge transfer: 11.12 ± 1.82/24.46 ± 2.84; n = 7; p = 0.06/p < 0.001), raising concerns that in the absence of GLAST another transporter (i.e., EAAC1 or another unidentified transporter) might also be involved in clearance. Alternatively, the discrepancy in potentiation by these two antagonists could be explained by differences in the ability of DHK and TBOA to inhibit GLT-1 (Bergles et al., 2002). To examine the efficacy of GLT-1 antagonism by DHK, we recorded glutamate transporter currents from stratum oriens astrocytes elicited through photolysis of caged l-glutamate (MNI-l-glutamate, 125 μm). As shown in Figure 6, a brief (1 msec) flash of UV light elicited an inward current in astrocytes that were exposed to MNI-l-glutamate. This current was not observed when MNI-l-glutamate was absent from the superfusing solution or when the UV beam was shuttered, and the response was blocked by TBOA (200 μm), indicating that this current was produced by the electrogenic cycling of glutamate transporters. In wild-type mice, DHK (300 μm) inhibited the peak amplitude of these responses by 54.3 ± 1.3% and slowed their decay (tau decay, single exponential fit: control, 14.7 ± 0.8 msec; DHK, 40.9 ± 3.0 msec; n = 10; p < 0.001), similar to effects seen on transporter currents evoked in astrocytes through synaptic release (Bergles and Jahr, 1997). The effects of 300 μm DHK in EAAC1-/- mice were similar to those seen in wild-type mice (percentage of inhibition by DHK, 56.8 ± 1.8%, n = 4, p = 0.295; tau decay in DHK, 53.8 ± 2.6 msec, n = 3, p < 0.05) (Fig. 6A,B). In contrast, DHK produced a greater inhibition of the peak amplitude (82.6 ± 0.7%; n = 8; p < 0.001) and a greater slowing of the decay of transporter currents in GLAST-/- mice (tau decay in DHK, 125.3 ± 8.9 msec; n = 8; p < 0.001) (Fig. 6A,B). However, this concentration of DHK (300 μm) did not completely block astrocyte transporter currents in GLAST-/- mice. Because only GLT-1 and GLAST are expressed by astrocytes, these results suggest that 300 μm DHK was not sufficient to block all GLT-1 activity, providing an explanation for the differential potentiation of mGluR1 EPSCs by TBOA and DHK in these mice. Increasing the concentration of DHK to 1 mm increased the inhibition of the transporter current to 91.6 ± 0.5% (n = 4; p < 0.001) (Fig. 6A,B) and increased the potentiation of mGluR1 EPSCs in GLAST-/- mice to the level of that observed with TBOA (Fig. 5C) (fold increase over control in 1 mm DHK vs 100 μm TBOA, peak amplitude/charge transfer: 10.94 ± 1.85/19.63 ± 3.66; n = 6; p = 0.47/p = 0.16), although 1 mm DHK had no greater effect than 300 μm DHK on the amplitude or charge transfer of mGluR1 EPSCs in wild-type mice (Fig. 5C) (fold increase, peak amplitude/charge transfer: 1 mm DHK, 2.03 ± 0.71/1.04 ± 0.60, n = 4; 100 μm TBOA, 11.57 ± 2.37/19.95 ± 5.23; p < 0.01/p < 0.01).
Figure 6.
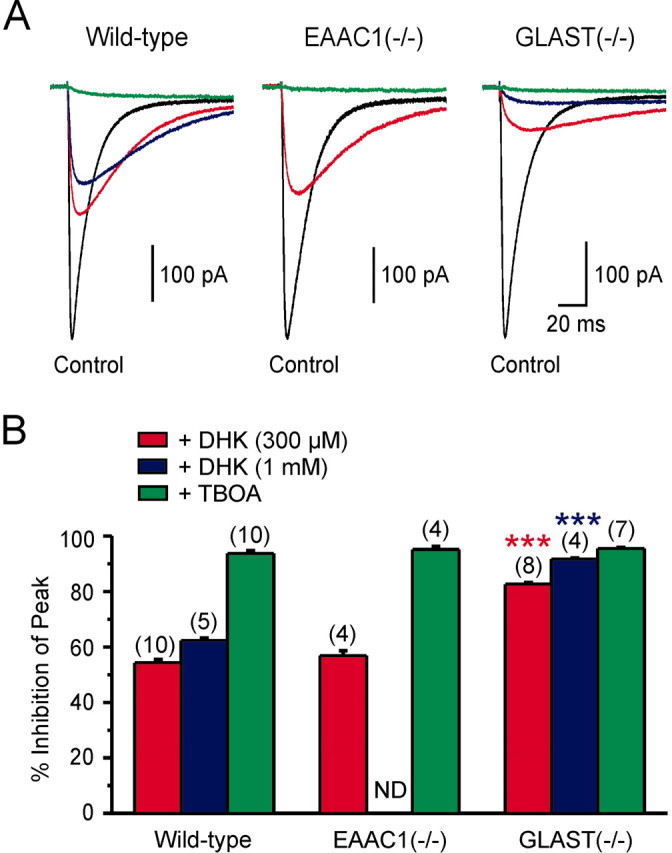
Differential effects of transporter antagonists on astrocyte transporter currents recorded from transporter deficient mice. A, Astrocyte transporter currents elicited by UV photolysis of MNI-l-glutamate (125 μm) recorded under control conditions (black traces), in the presence of DHK (300 μm, red traces; 1 mm, blue traces) or TBOA (200 μm; green traces) from mice with different genotypes (as indicated). B, Grouped data showing percentage of inhibition of the peak amplitude of these transporter currents by DHK and TBOA in wild-type, EAAC1-/-, and GLAST-/- mice (***p < 0.001 compared with wild-type). Numbers in parentheses indicate the number of experiments. ND, Not done.
Inhibition of glutamate transporters enhances inhibition of CA1 pyramidal neurons
The axons of O-LM interneurons ramify extensively in stratum-lacunosum moleculare where the distal dendrites of CA1 pyramidal cells are located, and results obtained from paired recordings indicate that GABA released from O-LM interneurons elicits GABAA receptor-mediated currents in pyramidal neurons (Maccaferri et al., 2000). The ability of astrocyte glutamate transporters to regulate mGluR1-dependent excitation of O-LM interneurons (Fig. 4) suggested that transporter inhibition might facilitate the release of GABA from these cells. To investigate this possibility, we recorded the response of CA1 pyramidal neurons to the same stratum oriens stimulation used to elicit mGluR1 responses in interneurons. These experiments were done in the presence of AMPA (15 μm NBQX and 25 μm GYKI 52466), NMDA (10 μm RS-CPP, 50 μm MK-801, and 20 μm 7-chlorokynurenate), and GABAB (1 μm CGP 55845) receptor antagonists to isolate GABAA receptor currents, and with internal solution containing a high [Cl-] to enlarge these responses. As shown in Figure 7A, stimulation in stratum oriens elicited rapid inward currents in pyramidal neurons, the onset of which was partially obscured by the train of stimuli. In the presence of TBOA (100 μm), a burst of IPSCs became visible after stimulation (Fig. 7B), which reached its peak amplitude several hundred milliseconds after the end of the stimulus, in accordance with the delayed onset and slow rise of mGluR1-mediated currents in O-LM interneurons (Fig. 1B). Note that a depolarizing step was not applied to O-LM interneurons during this experiment. This burst of IPSCs increased the charge transfer in response to stimulation 3.69 ± 0.84-fold (n = 12; p < 0.001) (Fig. 7E). This increase in IPSC frequency was blocked by LY 367385 (50-100 μm) (0.38 ± 0.24-fold increase; n = 8; p = 0.602), indicating that it required activation of mGluR1. In the presence of TBOA, all evoked responses were blocked by SR 95531 (5 μm) (n = 4 of 4 cells) (Fig. 7D,E), ruling out the possibility that the slow component was mediated by mGluRs on pyramidal cells themselves. These results indicate that inhibition of astrocyte glutamate transporters facilitates the inhibition of CA1 pyramidal neurons by potentiating mGluR1-mediated excitation of O-LM interneurons.
Figure 7.
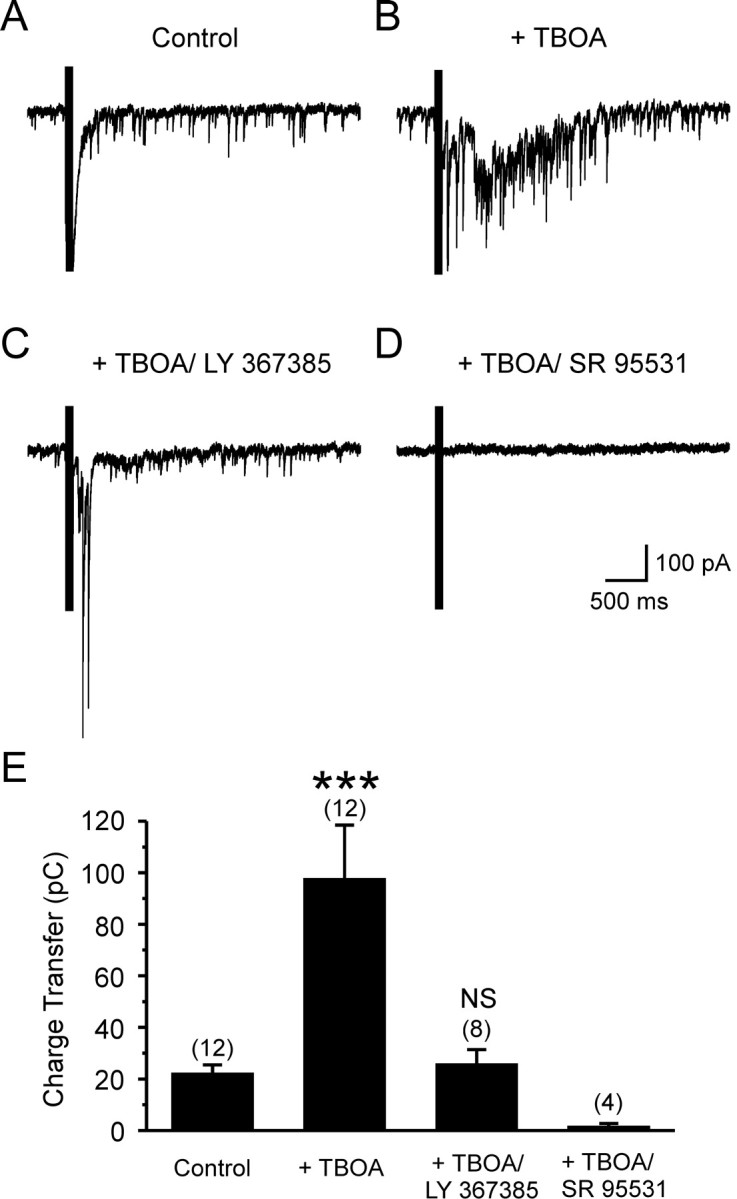
Inhibition of glutamate transporters induces an mGluR1-dependent delayed burst of IPSCs in CA1 pyramidal cells. A, Whole-cell recording from a CA1 pyramidal neuron showing the response to stimulation (10 stimuli at 100 Hz) in stratum oriens under control conditions. B, Response of this cell to the same stimulus in the presence of TBOA (100 μm). C, Response of this cell to the same stimulus in the presence of TBOA (100 μm) and the mGluR1 antagonist LY 367385 (100 μm). D, Response of this cell to the same stimulus in the presence of TBOA (100 μm) and the GABAA receptor antagonist SR 95531 (5 μm). E, Grouped data showing the charge transfer conveyed by IPSCs recorded from CA1 pyramidal neurons under different conditions, as indicated (LY 367385, 50-100 μm). The effect of SR 95531 on the burst of IPSCs was analyzed by performing a separate set of experiments (n = 4), in which SR 95531 was applied in the presence of TBOA but in the absence of LY 367385. (***p < 0.001). NS, Not significant. Numbers in parentheses indicate the number of experiments. Recordings were performed using a high Cl--based internal solution (Vm of -65 mV), and the ACSF contained the following (in μm): 15 NBQX, 25 GYKI 52466, 10RS-CPP, 50 MK-801, 207-chlorokynurenate, and 1 CGP 55845.
Discussion
In this study, we show that an mGluR1-dependent slow inward current can be elicited in hippocampal O-LM interneurons after stimulation of excitatory afferents in stratum oriens. This response was often revealed only when [Ca2+]i was elevated in interneurons at the time of stimulation, providing an explanation for why this current has not been observed previously (Gee et al., 2001). A similar Ca2+ dependence has been reported for postsynaptic mGluR1 activity at parallel fiber-Purkinje neuron synapses in the cerebellum (Batchelor and Garthwaite, 1997), suggesting that mGluR1 could serve as a coincidence detector to monitor presynaptic glutamate release and postsynaptic Ca2+ transients in both types of GABAergic neurons.
Regulation of mGluR1 activation by glial glutamate transporters
Inhibition of GLT-1 transporters caused an approximately twofold to threefold increase in the size of mGluR1 EPSCs in O-LM interneurons, whereas inhibition of all glutamate transporters caused a much larger potentiation, indicating that binding and/or uptake of glutamate by transporters restricts activation of mGluRs at these synapses. The relatively small enhancement of mGluR1 EPSCs by DHK suggests that GLT-1 is only a minor contributor to this regulation, particularly when considering that the potentiation by TBOA, and to a lesser extent by DHK, may have been underestimated, because transporter inhibition in TBOA may have decreased glutamate release by simultaneously enhancing activation of presynaptic mGluRs (Scanziani et al., 1998). These results seem at odds with the density of GLT-1 in the hippocampus, which is almost four times more abundant than GLAST (Lehre and Danbolt, 1998). This discrepancy could be explained if the relative densities of GLAST and GLT-1 are different at these particular synapses. It is also possible that the coupling between mGluR1 and cation channels is nonlinear, enabling the synaptic current to become disproportionately larger as more mGluRs are activated in TBOA. Nevertheless, the modest effect of DHK suggests that there are sufficient remaining GLAST transporters to compensate for the loss of GLT-1. This hypothesis is consistent with the findings that astrocyte transporter currents were inhibited only ∼50% by 300 μm DHK (Fig. 6) and that 1 mm DHK did not potentiate mGluR1 EPSCs more than 300 μm DHK in wild-type mice, although this higher concentration of DHK inhibited astrocyte transporter currents and potentiated mGluR1 EPSCs to a greater extent in GLAST-/- mice. Indeed, previous studies have shown that the pool of astrocyte transporters in stratum radiatum is not saturated after high-frequency stimulation, even when GLT-1 transporters are blocked with DHK (Diamond and Jahr, 2000). The decrease in expression of GLAST during development (Furuta et al., 1997) suggests that a proportionally larger role will be played by GLT-1 as the animal matures. Together, these results indicate that two astrocyte transporters, GLT-1 and GLAST, restrict activation of mGluR1 at excitatory synapses on O-LM interneurons.
Although immunolocalization studies indicate that GLT-1 is highly expressed by astrocytes (Rothstein et al., 1994; Lehre et al., 1995), in situ hybridization (Schmitt et al., 1996; Berger and Hediger, 1998) and immunolocalization studies (Chen et al., 2004) suggest that GLT-1 is also expressed by some CA3 pyramidal neurons. It is unlikely that the GLT-1 transporters contributing to uptake near these mGluRs reside in neuronal membranes, because O-LM interneurons in area CA1 receive primarily feedback excitation from CA1 pyramidal neurons rather than feedforward excitation from CA3 pyramidal neurons (Freund and Buzsáki, 1996), and glutamate transporter currents have not been observed in either CA1 or CA3 hippocampal pyramidal neurons (Bergles and Jahr, 1998).
The contribution of neuronal transporters to uptake at excitatory synapses
Recent studies suggest that neuronal glutamate transporters shield postsynaptic receptors at cerebellar (Brasnjo and Otis, 2001) and hippocampal (Diamond, 2001) synapses. EAAC1 is expressed by most neurons in the hippocampus (Rothstein et al., 1994), where it is found primarily in the somodendritic compartment (He et al., 2000). Although a detailed analysis of EAAC1 expression by interneurons has not been performed, EAAC1 mRNA is observed in stratum oriens interneurons (Berger and Hediger, 1998). Nevertheless, the similar potentiation of mGluR1 responses by DHK in wild-type and EAAC1-/- mice suggests that EAAC1 contributes little to clearance near these receptors. Although the amplitudes of mGluR1 EPSCs in GLAST-/- mice were increased to a similar extent by 300 μm DHK and 100 μm TBOA, these responses were more prolonged in TBOA, as shown by the enhanced charge transfer (Fig. 5). The Ki of TBOA is ∼10-fold lower than DHK at EAAT2 (Ki of TBOA, 5.7 μm; Ki of DHK, 79 μm) (Shimamoto et al., 1998) and unbinds from GLT-1 ∼10 times more slowly (Bergles et al., 2002). The persistent rise in extracellular glutamate resulting from high-frequency stimulation may force displacement of DHK, allowing GLT-1 to participate in clearance. Consistent with this possibility, 1 mm DHK increased the inhibition of astrocyte transporter currents and potentiated mGluR1 EPSCs to a similar extent as TBOA in GLAST-/- mice, suggesting that the discrepancy between the potentiation seen by DHK and TBOA in these mice is attributable, in part, to poorer antagonism of GLT-1 by DHK. However, it has been reported that DHK inhibits EAAC1 with a Ki of 1.12 mm (Dowd et al., 1996) (but see Arriza et al., 1994), raising the possibility that inhibition of EAAC1 also may have contributed to the increased potentiation seen in GLAST-/- mice in response to 1 mm DHK. Because 1 mm DHK did not produce greater potentiation than 300 μm DHK in wild-type animals, EAAC1, if involved, is likely to be redundant with GLAST. However, when analyzing complex phenomena in knock-out animals, there is the concern of compensation. Although a global upregulation of GLT-1 or GLAST is not observed in EAAC1-/- mice (Peghini et al., 1997), there could be local changes in the distribution or expression of these transporters at a subset of synapses. Notably, there was no significant change in inhibition of astrocyte transporter currents by DHK in EAAC1-/- mice (Fig. 6), suggesting that relative abundance of GLAST and GLT-1 is unaltered in astrocytes in these mice.
The prominent role of astrocyte transporters in lowering mGluR1 occupancy at these synapses contrasts with parallel fiber-Purkinje neuron synapses in the cerebellum, in which postsynaptic transporters appear to contribute significantly to clearance (Brasnjo and Otis, 2001). Purkinje neurons, unlike O-LM interneurons, express high levels of EAAT4, and recent studies indicate that EAAT4 is responsible for the postsynaptic transporter currents recorded from Purkinje neurons (Huang et al., 2004). These studies, and the present results, suggest that EAAC1 contributes little to perisynaptic clearance at these two excitatory synapses. Notably, EAAC1 is expressed by GABAergic interneurons, and glutamate accumulated by this transporter can be used for GABA synthesis (Rothstein et al., 1996; Sepkuty et al., 2002; Mathews and Diamond, 2003), suggesting that it may serve as a metabolic transporter rather than one specialized for removing synaptic glutamate. However, selective inhibition of postsynaptic transporters (presumably EAAC1) has been shown to slow the decay of evoked NMDA synaptic currents in CA1 pyramidal neurons (Diamond, 2001). Although the association of astroglia with synapses is essential for placing transporters near release sites (Iino et al., 2001; Oliet et al., 2001), the extent of this ensheathment varies dramatically among hippocampal synapses (Ventura and Harris, 1999), raising the possibility that EAAC1 may prevent spillover of transmitter at synapses that are devoid of astroglia.
Implications for neuronal excitability
Glutamate transporter inhibition potentiated mGluR1 activity in interneurons, increased their firing rate, and greatly enhanced the inhibition of CA1 pyramidal neurons. In cultured hippocampal slices, stimulation of mGluR1 and mGluR5 also have been shown to potentiate inhibition of CA3 pyramidal neurons by increasing interneuron excitability (Maccaferri and Dingledine, 2002; Mori and Gerber, 2002), and, in acute slices from guinea pig, tetanic stimulation in the presence of AMPA and NMDA receptor antagonists induces a transient increase in IPSPs in CA3 pyramidal neurons (Miles and Poncer, 1993), indicating a prominent role for mGluRs in regulating interneuron activity. Under physiological conditions, even small shifts in the membrane potential of interneurons caused by moderate mGluR1 activity could serve as a mechanism for short-term regulation of local circuit inhibition, because the timing of action potential firing in these cells is highly sensitive to the resting potential (Freund and Buzsáki, 1996). This modulation of O-LM interneuron mGluR1 activity by astrocyte glutamate transporters may fine tune the inhibition of perforant path inputs to CA1 pyramidal neurons (Kang et al., 1998), providing insight into the potential consequences of transporter dysregulation.
There is accumulating evidence that the activity of GLT-1 and GLAST are tightly regulated through changes in expression, trafficking, phosphorylation, and interactions with accessory proteins (Levy et al., 1995; Gegelashvili et al., 1997). Furthermore, their expression is closely correlated with neuronal activity (Rao et al., 1998) and is disrupted after trauma (Wong et al., 2003), in epilepsy (Danbolt, 2001), and in many neurological diseases. The results presented here suggest that alterations in transporter activity could profoundly impact the behavior of neuronal networks by regulating the excitability of interneurons, thereby contributing to the abnormal neuronal activity observed in disease states.
Footnotes
This work was supported by National Institutes of Health Grants NS041680 (J.D.R) and NS44261 (D.E.B.) and a grant from the Robert Packard Center for ALS Research at Johns Hopkins. D.E.B. is an Alfred P. Sloan Research Fellow. We thank Naoko Nishiyama for assistance with genotyping.
Correspondence should be addressed to Dwight Bergles, Department of Neuroscience, Johns Hopkins University, 725 North Wolfe Street, Wood Basic Science Building, Room 813, Baltimore, MD 21205. E-mail: dbergles@jhmi.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244551-09$15.00/0
References
- Arnth-Jensen N, Jabaudon D, Scanziani M (2002) Cooperation between independent hippocampal synapses is controlled by glutamate uptake. Nat Neurosci 5: 325-331. [DOI] [PubMed] [Google Scholar]
- Arriza JL, Fairman WA, Wadiche JI, Murdoch GH, Kavanaugh MP, Amara SG (1994) Functional comparisons of three glutamate transporter subtypes cloned from human motor cortex. J Neurosci 14: 5559-5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J (1997) Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature 385: 74-77. [DOI] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P (1993) The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron 11: 771-787. [DOI] [PubMed] [Google Scholar]
- Berger UV, Hediger MA (1998) Comparative analysis of glutamate transporter expression in rat brain using differential double in situ hybridization. Anat Embryol (Berl) 198: 13-30. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE (1997) Synaptic activation of glutamate transporters in hippocampal astrocytes. Neuron 19: 1297-1308. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Jahr CE (1998) Glial contribution to glutamate uptake at Schaffer collateral-commissural synapses in the hippocampus. J Neurosci 18: 7709-7716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergles DE, Tzingounis AV, Jahr CE (2002) Comparison of coupled and uncoupled currents during glutamate uptake by GLT-1 transporters. J Neurosci 22: 10153-10162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS (2001) Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron 31: 607-616. [DOI] [PubMed] [Google Scholar]
- Carter AG, Regehr WG (2000) Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci 20: 4423-4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C, Rosenberg PA (2004) The glutamate transporter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24: 1136-1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark BA, Cull-Candy SG (2002) Activity-dependent recruitment of extra-synaptic NMDA receptor activation at an AMPA receptor-only synapse. J Neurosci 22: 4428-4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Pin JP (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol 37: 205-237. [DOI] [PubMed] [Google Scholar]
- Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65: 1-105. [DOI] [PubMed] [Google Scholar]
- Diamond JS (2001) Neuronal glutamate transporters limit activation of NMDA receptors by neurotransmitter spillover on CA1 pyramidal cells. J Neurosci 21: 8328-8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (1997) Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17: 4672-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (2000) Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol 83: 2835-2843. [DOI] [PubMed] [Google Scholar]
- Dowd LA, Coyle AJ, Rothstein JD, Pritchett DB, Robinson MB (1996) Comparison of Na+-dependent glutamate transport activity in synaptosomes, C6 glioma, and Xenopus oocytes expressing excitatory amino acid carrier 1 (EAAC1). Mol Pharmacol 49: 465-473. [PubMed] [Google Scholar]
- Freund TF, Buzsáki G (1996) Interneurons of the hippocampus. Hippocampus 6: 347-470. [DOI] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ (1997) Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 17: 8363-8375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee CE, Woodhall G, Lacaille JC (2001) Synaptically activated calcium responses in dendrites of hippocampal oriens-alveus interneurons. J Neurophysiol 85: 1603-1613. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Danbolt NC, Schousboe A (1997) Neuronal soluble factors differentially regulate the expression of the GLT1 and GLAST glutamate transporters in cultured astroglia. J Neurochem 69: 2612-2615. [DOI] [PubMed] [Google Scholar]
- He Y, Janssen WG, Rothstein JD, Morrison JH (2000) Differential synaptic localization of the glutamate transporter EAAC1 and glutamate receptor subunit GluR2 in the rat hippocampus. J Comp Neurol 418: 255-269. [PubMed] [Google Scholar]
- Huang YH, Dykes-Hoberg M, Tanaka K, Rothstein JD, Bergles DE (2004) Climbing fiber activation of EAAT4 transporters and kainate receptors in cerebellar Purkinje cells. J Neurosci 24: 103-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M, Goto K, Kakegawa W, Okado H, Sudo M, Ishiuchi S, Miwa A, Takayasu Y, Saito I, Tsuzuki K, Ozawa S (2001) Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science 292: 926-929. [DOI] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M (1998) Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci 1: 683-692. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ (2003) Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature 426: 285-291. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18: 8751-8757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC (1995) Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci 15: 1835-1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy LM, Lehre KP, Walaas SI, Storm-Mathisen J, Danbolt NC (1995) Down-regulation of glial glutamate transporters after glutamatergic denervation in the rat brain. Eur J Neurosci 7: 2036-2041. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Dingledine R (2002) Control of feedforward dendritic inhibition by NMDA receptor-dependent spike timing in hippocampal interneurons. J Neurosci 22: 5462-5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Roberts JD, Szucs P, Cottingham CA, Somogyi P (2000) Cell surface domain specific postsynaptic currents evoked by identified GABAergic neurones in rat hippocampus in vitro. J Physiol (Lond) 524: 91-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews GC, Diamond JS (2003) Neuronal glutamate uptake contributes to GABA synthesis and inhibitory synaptic strength. J Neurosci 23: 2040-2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, DiChiara TJ, Kauer JA (1994) Activation of metabotropic glutamate receptors differentially affects two classes of hippocampal interneurons and potentiates excitatory synaptic transmission. J Neurosci 14: 4433-4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles R, Poncer JC (1993) Metabotropic glutamate receptors mediate a post-tetanic excitation of guinea-pig hippocampal inhibitory neurones. J Physiol (Lond) 463: 461-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Gerber U (2002) Slow feedback inhibition in the CA3 area of the rat hippocampus by synergistic synaptic activation of mGluR1 and mGluR5. J Physiol (Lond) 544: 793-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR, Piet R, Poulain DA (2001) Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292: 923-926. [DOI] [PubMed] [Google Scholar]
- Otis TS, Wu YC, Trussell LO (1996) Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci 16: 1634-1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peghini P, Janzen J, Stoffel W (1997) Glutamate transporter EAAC-1-deficient mice develop dicarboxylic aminoaciduria and behavioral abnormalities but no neurodegeneration. EMBO J 16: 3822-3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VL, Baskaya MK, Dogan A, Rothstein JD, Dempsey RJ (1998) Traumatic brain injury down-regulates glial glutamate transporter (GLT-1 and GLAST) proteins in rat brain. J Neurochem 70: 2020-2027. [DOI] [PubMed] [Google Scholar]
- Reye P, Sullivan R, Scott H, Pow DV (2002) Distribution of two splice variants of the glutamate transporter GLT-1 in rat brain and pituitary. Glia 38: 246-255. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13: 713-725. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF (1996) Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16: 675-686. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Gahwiler BH, Charpak S (1998) Target cell-specific modulation of transmitter release at terminals from a single axon. Proc Natl Acad Sci USA 95: 12004-12009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Asan E, Puschel B, Jons T, Kugler P (1996) Expression of the glutamate transporter GLT1 in neural cells of the rat central nervous system: non-radioactive in situ hybridization and comparative immunocytochemistry. Neuroscience 71: 989-1004. [DOI] [PubMed] [Google Scholar]
- Sepkuty JP, Cohen AS, Eccles C, Rafiq A, Behar K, Ganel R, Coulter DA, Rothstein JD (2002) A neuronal glutamate transporter contributes to neurotransmitter GABA synthesis and epilepsy. J Neurosci 22: 6372-6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T (1998) dl-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol 53: 195-201. [DOI] [PubMed] [Google Scholar]
- Spacek J (1985) Three-dimensional analysis of dendritic spines. III. Glial sheath. Anat Embryol (Berl) 171: 245-252. [DOI] [PubMed] [Google Scholar]
- Tong G, Jahr CE (1994) Block of glutamate transporters potentiates postsynaptic excitation. Neuron 13: 1195-1203. [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Endou H, Kanai Y (1997) Tissue specific variants of glutamate transporter GLT-1. FEBS Lett 416: 312-316. [DOI] [PubMed] [Google Scholar]
- Ventura R, Harris KM (1999) Three-dimensional relationships between hippocampal synapses and astrocytes. J Neurosci 19: 6897-6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K (1998) Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci 10: 976-988. [DOI] [PubMed] [Google Scholar]
- Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li W, Crino PB, Mennerick S, Yamada KA, Gutmann DH (2003) Impaired glial glutamate transport in a mouse tuberous sclerosis epilepsy model. Ann Neurol 54: 251-256. [DOI] [PubMed] [Google Scholar]
- Zhang L, McBain CJ (1995) Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol (Lond) 488: 661-672. [DOI] [PMC free article] [PubMed] [Google Scholar]