

Abstract
β-Amyloid peptides that are cleaved from the amyloid precursor protein (APP) play a critical role in Alzheimer's disease (AD) pathophysiology. Here, we show that in Drosophila, the targeted expression of the key genes of AD, APP, theβ-site APP-cleaving enzyme BACE, and the presenilins led to the generation of β-amyloid plaques and age-dependent neurodegeneration as well as to semilethality, a shortened life span, and defects in wing vein development. Genetic manipulations or pharmacological treatments with secretase inhibitors influenced the activity of the APP-processing proteases and modulated the severity of the phenotypes. This invertebrate model of amyloid plaque pathology demonstrates Aβ-induced neurodegeneration as a basic biological principle and may allow additional genetic analyses of the underlying molecular pathways.
Keywords: Alzheimer, dementia, aging, neuropathology, degeneration, Drosophila
Introduction
Alzheimer's disease (AD) neuropathology is characterized by the accumulation and deposition of β-amyloid (Aβ) peptides in senile plaques, by the formation of neurofibrillary tangles (NFT) composed of abnormally phosphorylated forms of the microtubule binding protein tau, and by a progressive neuronal damage and cell loss (Gomez-Isla et al., 1996; Selkoe, 1999). The interrelation of Aβ and NFT in the development of AD neuropathologic lesions is a matter of debate. However, it has been shown that in AD-linked neurodegeneration, Aβ is an essential causative pathogenic factor. Tau-related neurotoxicity was recapitulated in transgenic Drosophila, demonstrating that important aspects of AD-related neuropathogenesis can be studied in the fruit fly (Wittmann et al., 2001; Jackson et al., 2002). Aβ-induced neurodegeneration and plaque formation in Drosophila would provide powerful additional evidence for a causative role of Aβ peptide toxicity in AD.
Aβ is generated by endoproteolysis of the amyloid precursor protein (APP) by β-secretase activity of the β-site APP-cleaving enzyme (BACE) and by subsequent intramembraneous γ-secretase cleavage that depends on a complex of presenilin 1 or 2 (PS1 or PS2), nicastrin, aph-1, and pen-2 (De Strooper and Annaert, 2000; Walter et al., 2001; Esler et al., 2002; Francis et al., 2002). Autosomal dominant mutations in APP, PS1, or PS2 accelerate the onset of neurodegeneration and the progression of the disease in familial AD (FAD). These FAD mutations are associated with altered APP processing and lead to an increased generation of amyloidogenic Aβ40 and Aβ42 peptides or to increased ratios of Aβ42/Aβtotal (Lemere et al., 1996; Scheuner et al., 1996; Tomita et al., 1997). Previous studies of Drosophila suggested pro-apoptotic functions of FAD-causing presenilin mutations and indicated that, although the genes required for a functional γ-secretase complex appeared to be highly conserved between Drosophila and vertebrates, the fly is lacking a homolog of human BACE (Fossgreen et al., 1998; Ye and Fortini, 1999; Adams et al., 2000).
To model β-amyloid plaque pathology and neurodegeneration in Drosophila, we expressed the genes involved in the pathophysiology of AD either separately or in combination in various neuronal and non-neuronal tissues by using the GAL4/UAS (transcriptional activator Gal4/Gal4 DNA binding motif, upstream activator sequence) system (Brand and Perrimon, 1993). Transgenic flies expressing the human APP695 gene (UAS-APP) and three fly lines expressing Drosophila presenilin (UAS-DPsn) point mutations that correspond to the FAD mutants N141I, L235P, and E280A were kindly provided by R. Paro (Center for Molecular Biology, Heidelberg, Germany) and E. Fortini (National Cancer Institute, Molecular Genetics Section, Frederick, MD), respectively (Fossgreen et al., 1998; Ye and Fortini, 1999). We generated UAS-BACE transgenic fly lines by P-element-mediated germline transformation and expressed APP, BACE, and DPsn in photoreceptor cells of the compound eye of Drosophila by using the eye-specific gmr-GAL4 driver line (Rubin and Spradling, 1982; Spradling and Rubin, 1982; Brand and Perrimon, 1993).
Materials and Methods
BACE transgenic flies. A 1.9 kb KpnI-XhoI restriction fragment of the human BACE (β-site APP-cleaving enzyme) cDNA containing the entire open reading frame was cloned into the plasmid pUAST (Brand and Perrimon, 1993). P-element-mediated germline transformation was performed as described (Rubin and Spradling, 1982; Spradling and Rubin, 1982). Three independent UAS-BACE insertion lines were used in our experiments (406, 437, 523).
APP and presenilin transgenic flies. The UAS-APP695II, UASAPP695III (Wt-34, Wt-35) and the UAS-DPsn+14 as well as UASDPsn-mutants (N141I, L235P, E280A) were kindly provided by R. Paro and E. Fortini (Fossgreen et al., 1998; Ye and Fortini, 1999). PsnC4, psnB3, and the psnK2 mutant alleles as well as the actin-GAL4 line were obtained from the Bloomington stock center. The gmr-GAL4 line from F. Pignoni was used to achieve the eye-specific expression of the transgenes.
Protein analyses. For Western blotting, fly heads and human normal brain tissue (Alzheimer Tissue Center, Northwestern University, Evanston, IL; A97-197 ITC; PMI 5 hr) were homogenized in 1× PBS, 5 mm EDTA, 0.5% Triton X-100, and a protease-inhibitor mix Complete (Roche Applied Science, Mannheim, Germany). Equal amounts of protein were separated by 10% SDS-PAGE, transferred to Immobilon membranes (Millipore, Bedford, MA), blocked in 5% low-fat milk for 2 hr at room temperature, and incubated with the monoclonal antibody (mAb) 22C11 (APP N terminal-specific; Chemicon, Temecula, CA) or polyclonal Ab (pAb) α-APP-C12 (APP C terminal-specific; kind gift from B. de Strooper, Center for Human Genetics, K.U. Leuven and Flanders Interuniversity Institute for Biotechnology, Leuven, Belgium). Bound antibodies were detected with goat anti-mouse peroxidase-conjugated (Dianova, Hamburg, Germany) or goat anti-rabbit peroxidase-conjugated (Vector Laboratories, Burlingame, CA) secondary antibodies. For immunoprecipitation, fly heads were homogenized as described above, and lysates were treated as described in the antibodies protocol guide from Clontech (Cambridge, UK). The following antibodies were used for immunoprecipitation: mAb 6E10 (α-Aβ5-10; Signet Pathology Systems, Dedham, MA), mAb 4G8 (α-Aβ17-24; Signet Pathology Systems), rabbit pAb α-APP-C12, and rabbit polyclonal antibodies Aβ1-42 and Aβ1-40 (QCB; Biosource International, Camarillo, CA). Samples were separated on 10-20% gradient Novex (Wadsworth, OH) Tris-Tricine gels (Invitrogen, San Diego, CA) and blotted onto Protran BA 79 Cellulosenitrate membranes (0.1 μm; Schleicher & Schuell, Dassel, Germany). Detection of β-amyloid or the APP C terminus was performed as described previously (Ida et al., 1996) using mAb 6E10 and goat anti-mouse peroxidase-conjugated secondary Ab (Dianova).
Immunohistochemistry and histology. For Immunostaining, adult flies were fixed in 4% paraformaldehyde for 3 hr, washed in 1× PBS, and transferred to 25% sucrose for an overnight incubation at 4°C. Flies were decapitated with a razor blade, and the heads were imbedded in Tissue Tek (Sakura, Tokyo, Japan) and snap frozen. Horizontal frozen sections (10 μm) were prepared on a cryostat. Immunostaining was done with the Vectastain Elite kit (Vector Laboratories) according to the instructions of the manufacturer. The following primary antibodies were used: 24B10 (α-chaoptin, 1:5), provided by the Developmental Studies Hybridoma Bank; mAb 4G8 (1:1000), and mAb 9G10 (αAβ42 C terminus that was generated by standard procedures; 1:500). The fly brain tissue sections were pretreated for 10 min in 70% formic acid to re-expose the epitope before 4G8 staining.
For thioflavin S staining, sections were counterstained for 5 min in Mayers Hemalum (Sigma, St. Louis, MO), rinsed for 10 min in tap water, and stained for 3 min in 1% thioflavin S (Sigma) watery solution. Slides were rinsed in several changes of distilled water, incubated for 15 min in 1% acetic acid, rinsed in tap water, and mounted in Vectashield mounting medium (Vector Laboratories). Slides were analyzed under a fluorescence microscopy (430 nm excitation, 550 nm emission).
For plastic sections, flies were fixed overnight in 5% glutaraldehyde at 4°C, decapitated with a razor blade, washed three times in PBS (5 min each), and treated with 2% osmium-tetraoxid on ice. Sections were again washed three times for 10 min each, sequentially dehydrated in 50, 70, 80, 95, and 3× 100% ethanol for 10 min each, incubated in 100% ethanol: epon 1:1 overnight, and embedded in epon. Plastic sections (1 μm) were cut on a microtome and stained with 1% toluidine blue, 1% borax. Ultra-thin sections (60 nm) were stained with aqueous uranyl acetate and lead citrate and examined using a CEM 902A (Zeiss, Thornwood, NY) electron microscope and analySIS software (SIS, Münster, Germany). Adult fly wings were removed and mounted in DPX (a mixture of distyrene, tricresyl phosphate, and xylene) mountant (Fluka, Buchs, Switzerland) for light microscopy.
Treatments with β- and γ-secretase inhibitors. The β-secretase inhibitor (Sinha et al., 1999) (Calbiochem, La Jolla, CA; 171601) or the γ-secretase inhibitor (Dovey et al., 2001) (Calbiochem; 565770) were used for treatment. The β-secretase inhibitor was added to the standard fly medium to a final concentration of 1, 5, 10, 20, and 50 nm in 0.001, 0.005, 0.01, 0.02, and 0.05% DMSO, respectively; for the γ-secretase inhibitor, a final concentration of 100 nm in 0.001% DMSO was used. Flies of the following genotypes were mated: w;actin-GAL4/Cyo;UASDPsn/Tm3 x w;UAS-APP/UAS-APP;UAS-BACE/UAS-BACE. The eggs were developed on fly medium supplemented with the inhibitors or with the vehicle (DMSO) through the larval stage until eclosion. The eclosion rate and extra vein phenotype of flies transgenic for w;actin-GAL4/UASAPP;UAS-BACE523/UAS-DPsn, w;actin-GAL4/UAS-APP;UAS-BACE406/UAS-DPsn, and w;actin-GAL4/+;UAS-DPsn/+ were scored. Adult transgenic flies were continuously treated at 29°C with or without 10 nm β-secretase inhibitor in 0.01% DMSO supplied with the fly food.
Results
Amyloidogenic processing of human APP in transgenic flies
In UAS-APP-expressing flies, antibodies against the N or C termini of human APP detected two differently glycosylated full-length forms of APP that comigrated with glycosylated APP695 from human brain tissue at ∼100 kDa (Fig. 1A, lanes 1, 3, 5). Transgenic coexpression of human BACE with APP resulted in the loss of higher glycosylated APP, together with the generation of a smaller 97 kDa N-terminal fragment that corresponded in molecular mass to the β-cleaved secreted ectodomain of APP (APPsβ) (Fig. 1A, lanes 2, 4). Thus, in Drosophila, human BACE appeared to cleave preferentially the higher glycosylated form of APP. In SDS protein extracts from double-transgenic flies, the monoclonal anti-Aβ Ab 6E10 (α-Aβ1-16) precipitated the β-cleaved C terminus of APP [β-C-terminal fragment (CTF)] (Fig. 1C) as well as SDS-stable Aβ dimers and oligomers (Fig. 1B, lane 3, rhomb). Flies transgenic for UAS-APP without BACE generated a slightly larger form of Aβ, δ-Aβ (δ-cleaved Aβ) (Fig. 1B, lane 4, asterisk). Like Aβ, δ-Aβ also formed SDS-stable dimers and oligomers. Identical bands were generated by immunoprecipitation with the mAb 4G8 (α-Aβ17-24) or with polyclonal Aβ40 or Aβ42 C terminal-specific antibodies that recognize only the free C terminus of Aβ40 or Aβ42, respectively (data not shown). Aβ and δ-Aβ were absent from nontransgenic w1118 flies and BACE transgenic flies (Fig. 1B, lanes 2, 9, 13) and were hardly detectable when only low levels of APP were expressed by the weak nervous system-specific elav-GAL4 line (Fig. 1B, lanes 5, 6). After extraction of fly proteins in the presence of 70% formic acid, Aβ immunoprecipitated from UAS-APP/UAS-BACE transgenic flies comigrated exactly with the size of the synthetic Aβ (4 kDa), whereas δ-Aβ from UAS-APP transgenic flies migrated with a slightly larger molecular mass (∼5 kDa) (Fig. 1B, right panel, rhombs and asterisk, respectively). Notably, in the presence of 70% formic acid, a β-CTF was immunoprecipitated from UAS-APP/UAS-BACE transgenic flies, whereas a δ-CTF could hardly be detected under these conditions in UAS-APP transgenic flies (Fig. 1B, lanes 10-12).
Figure 1.
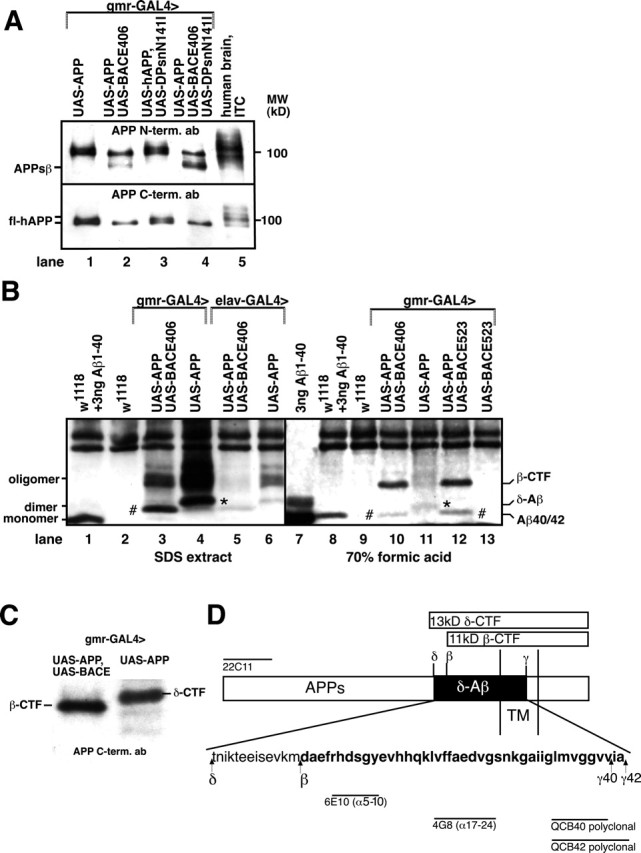
Amyloidogenic processing of APP in Drosophila. A, Equal amounts of protein extracts from transgenic flies at 2 d after eclosion were analyzed by Western blots, and full-length APP (fl-APP) was detected with APP-N-terminal (top panel) and APP-C-terminal (bottom panel) antibodies. Human brain extracts from the inferior temporal lobe (ITC) region were analyzed in parallel as control. B, Immunoprecipitation and immunoblotting of Aβ (rhomb) and the alternatively δ-cleaved δ-Aβ (asterisk) from extracts of transgenic and nontransgenicw1118 flies at 2 d after eclosion with and without the addition of synthetic Aβ1-40 using the Aβ-specific mAb 6E10. Fly proteins were extracted in SDS (left panel) or in 70% formic acid (right panel). C, Immunoprecipitation of the APP-C terminus by mAb 6E10 and detection by Western blotting with anti-APP-C12. D, Schematic representation of the amyloidogenic processing of APP at the secretase-cleavage sites. The amino acid sequence of the presumptive δ-Aβ peptide is indicated. The δ-secretase processing site was deduced from δ-secretase-processed APP695 found in rat primary neurons. Aβ sequence is indicated in bold letters. The epitopes of the antibodies that were used to characterize APP processing in transgenic flies are indicated by black bars.
To further characterize the δ-secretase processing of APP in Drosophila in the absence of BACE, we immunoprecipitated the C terminus of APP with mAb 6E10 and used a pAb against the last 12 C-terminal amino acids of full-length APP for detection on Western blots (Selkoe et al., 1988). In UAS-APP flies, 6E10 precipitated an APP C-terminal fragment with an apparent molecular mass of 13 kDa (δ-CTF), as compared with the expected 11 kDa β-CTF in UAS-APP/UASBACE-expressing flies (Fig. 1C).
Age-dependent neurodegeneration of photoreceptor cells in transgenic Drosophila
The neuropathology associated with amyloidogenic APP processing was characterized in the compound eye of Drosophila by staining cryostat sections with the photoreceptor cell-specific mAb 24B10. At day 2 after eclosion, no structural differences between UAS-BACE and UAS-APP single or UAS-APP/UAS-BACE double transgenic flies could be detected (Fig. 2A). At day 18, however, a severe degeneration of the retinal photoreceptors and their axonal projections to the first and second optic ganglion, in particular the lamina, was observed in UAS-APP/UAS-BACE transgenic flies (Fig. 2A). This degeneration of retinal photoreceptors was more pronounced in UASAPP transgenic flies, but completely absent in UAS-BACE-expressing control flies (Fig. 2A). At day 30, the neurodegeneration had progressed further as indicated by small degenerated retinas and a reduced volume of the underlying optic neuropil, the lamina. In addition, the photoreceptor cell projections to the medulla were heavily degenerated and stained only weakly.
Figure 2.
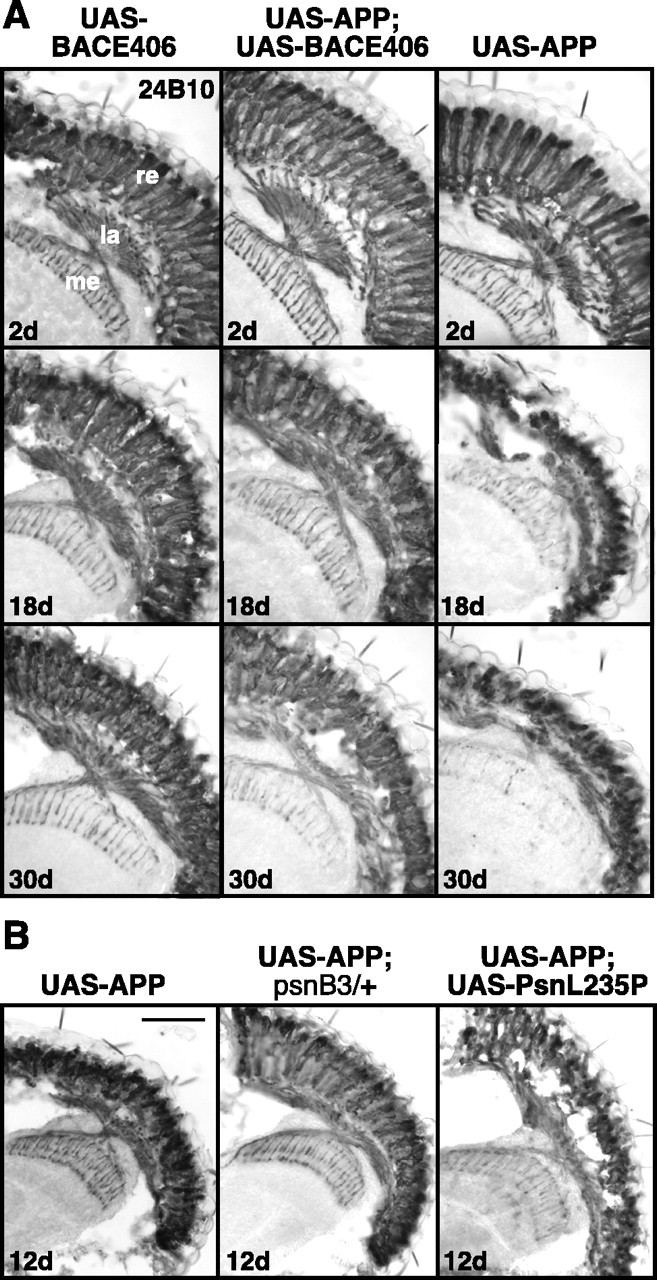
Age-dependent neurodegeneration in flies expressing the AD-associated transgenes under the control of the eye-specific gmr-GAL4 line. A, Staining of the photoreceptors in the retina (re) and their axonal projections to the lamina (la) and the medulla (me) on cryostat sections of different transgenic fly lines at days 2, 18, and 30. Photoreceptor degeneration in w; UAS-APP/+; gmr-GAL4/+ and w; UAS-APP/+; gmr-GAL4/UAS-BACE transgenic flies but not in w; gmr-GAL4/UAS-BACE transgenic control flies depends on age. The observed age-dependent degeneration of photoreceptor cells was not gender specific. B, Photoreceptor staining of transgenic flies at day 12. Loss of presenilin function in the w; UAS-APP/+; gmr-GAL4/psnB3 flies rescues the neurodegeneration; gain of presenilin function in w; UAS-APP/+; gmr-GAL4/UAS-DPsnL235P transgenic flies aggravates the phenotype. Scale bars: A, 45 μm; B, 50 μm.
Degeneration of photoreceptor cells is dependent on γ-secretase activity
The γ-secretase activity of the flies was modulated by coexpressing the Drosophila FAD-presenilin genes or by introducing loss of function alleles of the endogenous presenilin gene that reduces γ-secretase processing. Presenilin loss of function mutations psnC4, psnK2, psnB3, or one of three different human FAD mutants UAS-DPsnN141I, E280A, or L235P (Ye and Fortini, 1999; Ye et al., 1999) were expressed in w; UAS-APP/+; gmr-GAL4/+ transgenic background. At 12 d of age, the heterozygous presenilin loss of function mutants (w; UAS-APP/+; gmr-GAL4/psnB3) suppressed the degeneration of photoreceptor cells of UAS-APP-expressing flies, whereas coexpression of the presenilin FAD mutant L235P (w; UASAPP/+; gmr-GAL4/UAS-DPsnL235P) enhanced the degeneration (Fig. 2B). Similar results were obtained with two additional loss-of-function presenilin mutations psnK2 and psnC4 and with the FAD mutants UAS-DPsnN141I and UAS-DPsnE280A, and also when BACE was coexpressed (data not shown).
β-amyloid staining of plaques in the retina of transgenic Drosophila
Cryostat sections through eyes and optic lobes of adult double or single transgenic flies with accelerated degeneration of photoreceptor cells were stained with either thioflavin S or the β-amyloid-detecting mAb 4G8. Both thioflavin S and mAb 4G8 detected large globular deposits in the retinas of UAS-APP/UAS-DPsnL235P or UAS-APP/UAS-BACE/UAS-DPsnL235P flies (Fig. 3A-D). These large globular deposits also could be stained with the C-terminal end-specific Aβ-42 mAb 9G10 (data not shown). The deposits occurred age dependently with an onset at day 37 after eclosion in UAS-APP/UAS-DPsnL235P and UASAPP/UAS-BACE/UAS-DPsnL235P flies (Fig. 3J). In UAS-APP and in UAS-APP/UAS-BACE transgenic flies without additional DPsnL235P, the onset of plaque formation was strongly delayed and did not occur before day 68 after eclosion (Fig. 3J). In all of these fly lines, the neuronal degeneration preceded the occurrence of the amyloid plaques (Fig. 3J). Electron microscopy detected many “star-like” deposits of various sizes in the retina and neuropil regions of the first and second optic ganglion in UAS-APP/UAS-DPsnL235P and UAS-APP/UAS-BACE/UAS-DPsnL235P flies (Fig. 3E-H). These star-like deposits resembled the fibrillar EM ultrastructure of β-amyloid plaques in brains of human AD patients or APP-transgenic mice (Rockenstein et al., 2001) and were only found in EM sections of flies containing thioflavin S-positive plaques but not in flies without thioflavin S-positive plaques. The β-amyloid plaques that are formed by the deposition of either Aβ or δ-Aβ in UAS-APP/UAS-BACE/UAS-DPsnL235P or UAS-APP/UAS-DPsnL235P flies, respectively, shared a similar if not identical ultrastructure (Fig. 3G,H).
Figure 3.
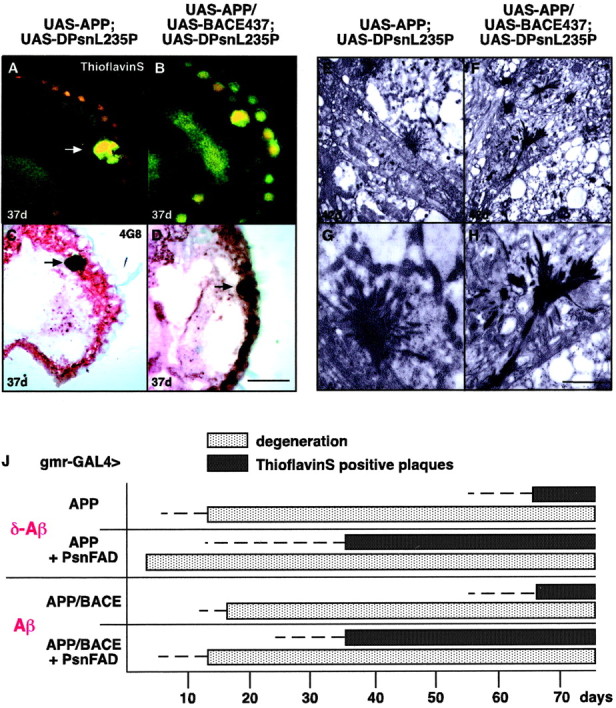
Amyloid-plaque deposition in the retina and neuropil region of transgenic flies expressing the indicated transgenes in the Drosophila compound eye. A-D, Thioflavin S (A, B) and 4G8 (C,D) staining of amyloid plaques deposited in the retina of w; UAS-APP/+; UAS-DPsnL235P/gmr-GAL4 transgenic flies (A, C) and in w; UAS-APP/UAS-BACE; UAS-DPsnL235P/gmr-GAL4 transgenic flies (B, D) from day 37 on. Large amyloid-plaque deposits were more evident in male flies, whereas female flies showed only small, more scattered, and diffuse thioflavin S-positive staining at 37 d. Inw; UAS-BACE/+; gmr-GAL4/+- and w; gmr-GAL4/UAS-DPsn-expressing control flies, no plaques could be detected by immunohistochemistry or electron microscopy at each time point investigated. Arrows indicate amyloid plaques. Scale bars: A, B, 45 μm; C, D, 50 μm.E-H, Ultrastructure of amyloid deposits in the retina of w; UAS-APP/+; UAS-DPsnL235P/gmr-GAL4 transgenic flies (E, G) and in neuropil regions of the optic lobes in w; UAS-APP/UAS-BACE; UAS-DPsnL235P/gmr-GAL4 transgenic flies (F, H). J, Progression of neurodegeneration and thioflavin S-positive plaque deposition. Dotted lines indicate the presence of thioflavin S-positive plaques and degenerating photoreceptor cells in <10% of the fly heads studied. Filled bars indicate detection of this phenotype in >80% of the fly heads studied. Scale bars: E, F, 15 μm; G, H, 9 μm.
Ubiquitous expression of APP, BACE, and Presenilin
Next, APP, BACE, and an additional copy of the DPsn gene were expressed ubiquitously in neuronal and non-neuronal cells of the fly by using actin-GAL4. Flies expressing APP alone or in combination with BACE and DPsn under the control of actin-GAL4 demonstrated semilethality and ectopic wing vein formation (Table 1, lines 3, 4; Fig. 4B-D, arrows). Only the UAS-DPsn and UAS-BACE/UAS-DPsn-expressing flies showed a normal survival rate and presented the correct wing vein pattern consisting of five longitudinal veins (L1-L5) and two cross veins [anterior and posterior cross veins (CVs)] (Fig. 4A; Table 1, lines 1, 2). Both the reduced viability and the wing vein phenotype were aggravated by the introduction of an additional presenilin copy, and they were rescued by presenilin loss of function mutations (psnC4) (Table 1, lines 4, 5; Fig. 4C,D). Replacing the human APP by its Drosophila homolog APPL, which does not contain an Aβ domain, rescued the lethality completely (Table 1, line 7), which implies an Aβ-related toxicity mechanism.
Table 1.
Ubiquitous expression of APP, BACE, and DPsn as a model system to identify genetic modifiers and validate pharmacologically active agents
|
Actin-GAL4> |
Ratio of flies expected |
Ratio of flies scored |
Wing phenotype |
|---|---|---|---|
| (1) UAS-DPsn/+ | 1:4 (0.25) | 1:4 (0.25) | Wild type |
| (2) UAS-BACE/UAS-DPsn | 1:4 (0.25) | 1:4 (0.25) | Wild type |
| (3) UAS-APP;UAS-BACE/+ | 1:4 (0.25) | 1:40 (0.025) | Ectopic veins (75%) |
| (4) UAS-APP;UAS-BACE/UAS-DPsn | 1:4 (0.25) | 1:97 (0.01) | Ectopic veins (100%) |
| (5) UAS-APP;UAS-BACE, psnC4/+ | 1:4 (0.25) | 1:13 (0.077) | Weak ectopic veins (70%) |
| (6) UAS-APP | 1:4 (0.25) | 1:127 (0.008) | Wild type |
| (7) UAS-APPL1;UAS-DPsn/+ |
1:4 (0.25) |
1:4 (0.25) |
Wild type |
|
|
Nonsupplemented |
Vehicle supplemented |
Inhibitor supplemented |
Wing phenotype |
Treatment |
|---|---|---|---|---|---|
| (8) UAS-APP;UAS-BACE523/UAS-DPsn | 1:17 (0.06) | 1:18 (0.06) | 1:6 (0.17) | Weak ectopic veins (100%) | 10 nM BACE inhibitor |
| (9) UAS-APP;UAS-BACE406/UAS-DPsn | 1:100 (0.01) | 1:97 (0.01) | 1:22 (0.045) | Ectopic veins (100%) | 100 nM γ-secretase inhibitor |
| (10) UAS-DPsn/+ |
1:2.6 (0.38) |
1:3 (0.33) |
1:2.7 (0.37) |
No ectopic veins |
10 nM BACE or 100 nM γ-secretase inhibitor |
Ratios of flies expressing the indicated transgenes under the control of the actin-GAL4 driver to nonexpressing flies. A comparison of expected and scored ratios indicates increased lethality during development of the fly. Lethality rate and wing phenotypes of transgenic flies (lines 1-7) and of transgenic larvae raised on nonsupplemented and vehicle supplemented versus BACE- or γ-secretase inhibitor supplemented medium (lines 8-10) are shown.
Figure 4.

Ectopic veins in wings of transgenic flies expressing APP, BACE, and DPsn ubiquitously under the control of the actin-GAL4 driver. A-C, w; actin-GAL4/+; UAS-BACE/UAS-DPsn transgenic flies presented the normal wing vein pattern (A), whereas w; actin-GAL4/UAS-APP; UAS-BACE/+ and w; actin-GAL4/UAS-APP; UAS-BACE/UAS-DPsn transgenic flies developed ectopic longitudinal and CVs (B, C, arrows). D, Introducing a loss-of-function presenilin allele (psnC4/+) rescued the wing vein phenotype.
β- and γ-secretase inhibitors to modulate Aβ formation
To determine whether these phenotypes are suitable for the validation of therapeutic drugs designed to reduce Aβ formation, the fly larvae were kept on a special fly medium supplemented either with BACE inhibitors or γ-secretase inhibitors (Sinha et al., 1999; Dovey et al., 2001). The eclosion rates of transgenic larvae that were raised on nonsupplemented medium and on medium supplemented with the respective inhibitors or the vehicle only were evaluated. Treatment of Act-GAL4;UAS-APP/+; UAS-BACE/UAS-DPsn larvae with the BACE inhibitor at concentrations ranging from 1 to 50 nm increased the survival rates in a dose-dependent manner with a maximum effectiveness at 10 nm. Although 1 nm BACE inhibitor had no effect on the survival rate of transgenic flies, 10 nm BACE inhibitor increased the survival rate by a factor of 3 (0.17 vs 0.06) (Table 1, line 8). In other words, the ratio of eclosed flies expressing the transgenes of interest to the total number of flies eclosed was 1:18 (0.06) in the group of larvae raised on the vehicle-supplemented food and 1:6 (0.17) in the 10 nm BACE inhibitor-treated group.
Prolonged treatment of adult flies with the BACE inhibitor markedly increased their life expectancy from 13 to 20 d in comparison with the vehicle-treated control flies (Fig. 5, gray and black triangles). Treatment of flies that do not express APP but only an additional copy of the Drosophila presenilin gene with either BACE inhibitor or vehicle did not alter their life span (Fig. 5, gray and black dots). Treatment of transgenic fly larvae with the γ-secretase inhibitor at a concentration of 100 nm increased their survival rates by a factor of 4.5 [ratio of inhibitor-treated flies, 1:22 (0.045), vs ratio of vehicle-treated flies, 1:97 (0.01)] (Table 1, line 9). The eclosion rates of Act-GAL4;UAS-DPsn/+ control flies were consistently at 0.33 and were not influenced by either the vehicle or inhibitor (Table 1, line 10). In addition, BACE inhibitor treatment reduced the ectopic wing vein phenotype in Act-GAL4;UAS-APP/+;UAS-BACE/UAS-DPsn transgenic flies, underscoring that it was related to aberrant APP processing during development.
Figure 5.

Increased median life span of BACE inhibitor-treated flies. Survival curves of w; act-GAL4/UAS-APP; UAS-BACE/UAS-DPsn and of w; act-GAL4/+; UAS-DPsn/+ transgenic adult flies after prolonged treatment with the BACE inhibitor (black triangles and circles, respectively) and of nontreated flies of the same genotypes (gray triangles and circles, respectively).
Discussion
Here, we describe the generation of a Drosophila model for AD-relatedβ-amyloidosis and neurodegeneration that is based on the accumulation and deposition of Aβ. In transgenic flies that express the AD-related genes APP, BACE, and presenilin, Aβ peptides processed from the human APP are detectable 2 d after eclosion. Most importantly, the generation of amyloidogenic Aβ peptides in the retina of transgenic flies is accompanied by an age-dependent neurodegeneration of the retinal photoreceptor cells and the deposition of β-amyloid-like plaque formations that also occur strictly dependent on aging. The degeneration of photoreceptor cells preceded the onset of β-amyloid plaque formation, indicating that β-amyloid precursors including Aβ oligomers or protofibrils mediated the toxic events. Coexpression of FAD presenilin mutants in UAS-APP and in UAS-APP/UAS-BACE transgenic flies accelerates Aβ-induced neurotoxicity in Drosophila. Age-dependent neurodegeneration and the onset of plaque deposition are prematurely induced in flies with the FAD presenilin mutants, thus resembling the effects of these mutations in human AD. This strongly suggests that the expression of human APP in flies induces analogous biochemical cascades that lead to cellular toxicity and degeneration in human AD.
The processing of human APP by BACE in transgenic flies is dependent on glycosylation. An interference of post-translational modifications with APP processing has been reported previously (Tomita et al., 1998; Galbete et al., 2000; Georgopoulou et al., 2001). The processing of human APP in transgenic flies lacking BACE leads to the generation of a slightly larger peptide, δ-Aβ, by cleavage aminoterminally to the usual β-cleavage site. A similar form of δ-cleavage of APP695 at Thr 584 was found previously in cultured rat neurons (Simons et al., 1996). However, the responsible secretase has not been characterized thus far (Simons et al., 1996).
Both δ-Aβ and Aβ generated through either δ- or β-cleavage aggregate to SDS-stable oligomers, suggesting that both cleavage events generate amyloidogenic products in Drosophila. Aβ oligomers are present in brains of AD patients, and there is growing evidence that Aβ oligomers are neurotoxic in vivo (Walsh et al., 2002; Wang et al., 2002; Gong et al., 2003). In human patients with AD, the density of β-amyloid plaques in the brain cortex is only weakly linked to the severity of dementia, whereas secreted monomeric and oligomeric forms of Aβ match the process of cell loss and impaired cognitive function more closely (Scheuner et al., 1996; Lue et al., 1999; McLean et al., 1999; Walsh et al., 2002). Therefore, the loss of photoreceptor cells in our Drosophila model could be attributable to toxic effects of the oligomeric forms of Aβ. Recent findings implicate an involvement of intracellular Aβ accumulation in the pathogenesis of AD disease (Oddo et al., 2003). Although we have no evidence for intracellular Aβ accumulation in our fly model of Alzheimer's disease, we cannot exclude the possibility that intracellular Aβ participates in the neurodegenerative process in our transgenic fly model. β-amyloid deposits localized outside the retina, despite expression of the transgenes in photoreceptor cells, suggest Aβ secretion by photoreceptor cells into their projection areas in addition to local secretion within the retina.
The accelerated degeneration of photoreceptor cells in flies generating the variant δ-Aβ peptide may indicate a more toxic effect of this peptide. Alternatively, it could be related to the absence of the N-terminal secreted part of APP (APPs) in these flies, because Western blots did not reveal a δ-cleaved APP-ectodomain analogous to APPsβ. APPs may act as a neuroprotective factor (Sisodia and Gallagher, 1998), and the absence of stable APPs in flies with δ-cleavage of APP may promote neurotoxic effects of δ-Aβ. In addition, a disruption of axonal transport mechanisms leading to axonal vesicle stalling may also contribute to the degenerative phenotypes in flies expressing full length APP695 (Gunawardena and Goldstein, 2001). However, the observation that coexpression of an additional copy of FAD presenilin mutants in either combination, UAS-APP or UAS-APP/UAS-BACE, caused more severe degenerative phenotypes along with earlier β-amyloid plaque deposition supports an Aβ-related toxicity mechanism.
The reason for the sexual dimorphism observed with respect to the occurrence of larger and more extensive β-amyloid plaques in male flies as compared with females is unknown but may be related to the smaller body weight and size of male flies.
Ubiquitous expression of APP, BACE, and DPsn resulted in semilethality and a visible wing phenotype. This phenotype could be used to screen for genes, drugs, or metabolites that modulate APP processing and have the potential to decrease Aβ-induced cellular degeneration. Both, γ- and β-secretase inhibitor treatment increased the survival rates of the UAS-APP/UAS-BACE/UAS-DPSn transgenic flies. γ-secretase cleavage is required not only for APP processing but also for cleavage of other proteins such as Notch in vertebrates and invertebrates (Fortini, 2002; De Strooper, 2003). The γ-secretase inhibitors available are not specific for the cleavage of APP. In Drosophila as well as in mice, loss-of-function mutants of the endogenous PS gene cause embryonic lethal Notch phenotypes (Shen et al., 1997; Ye et al., 1999). The absence of Notch phenotypes in our experiments indicates that the concentrations of the γ-secretase inhibitor used did not lead to a complete inhibition of γ-secretase cleavage and allowed sufficient endogenous DPsn activity required for normal development of the fly. However, this inhibition clearly prolonged the survival of the transgenic flies as compared with nontreated flies, suggesting that toxic β-amyloid accumulation could be prevented.
The targeted expression of human disease genes in Drosophila has been used previously as a model system to study the complex pathophysiology and potential treatment forms of human neurodegenerative diseases such as Parkinson's disease and amyotrophic lateral sclerosis as well as polyglutamine-repeat diseases (Feany, 2000; Fortini and Bonini, 2000). Our model closely mimics main characteristics of the human AD neuropathogenesis in the eye of the fly and demonstrates that Aβ-induced neurodegeneration is a conserved principle that acts even in flies. In particular, this system may be useful in the search of inhibitors of β- or γ-secretases that are currently regarded as a valid option for the treatment and prevention of AD.
Footnotes
This work was supported by a grant from the Bundesministerium für Bildung und Forschung (Leitprojekt), by National Centers of Competence in Research for Neural Plasticity and Repair, and by European Union Early Diagnosis of Alzheimer's Disease and Related Dementia to R.M.N., and by a grant from the Alzheimer Forschungs-Initiative e.V. to D.K. We thank R. Laas (Department of Neuropathology, University of Hamburg) for help in analyzing fly brain electron micrographs and S. Siegel for assistance in preparation of ultrathin sections for electron microscopy. We thank A. Kolot (Alzheimer Tissue Center, Northwestern University) for providing Alzheimer's disease and normal control brain tissue. We acknowledge J. Kumar, R. Paro, and M. E. Fortini for the gift of diverse fly stocks and B. de Strooper for providing the amyloid precursor protein-C-terminal antibody. The monoclonal antibody 24B10 developed by S. Benzer was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the National Institute of Child Health and Human Development, and maintained by the University of Iowa, Department of Biological Sciences.
Correspondence should be addressed to either Dr. Rita Reifegerste, Evotec Neurosciences GmbH, Schnackenburgallee 114, D-22525 Hamburg, Germany, E-mail: rita.reifegerste@evotecoai.com; or Dr. Roger M. Nitsch, University of Zurich, Division of Psychiatry Research, August Forel Strasse 1, CH-8008 Zurich, Switzerland, E-mail: nitsch@bli.unizh.ch.
Copyright © 2004 Society for Neuroscience 0270-6474/04/243899-08$15.00/0
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, et al. (2000) The genome sequence of Drosophila melanogaster. Science 287: 2185-2195. [DOI] [PubMed] [Google Scholar]
- Brand AH, Perrimon N (1993) Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118: 401-415. [DOI] [PubMed] [Google Scholar]
- De Strooper B (2003) Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron 38: 9-12. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W (2000) Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci 113: 1857-1870. [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, de Saint AP, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, Hu KL, Johnson-Wood KL, Kennedy SL, Kholodenko D, Knops JE, Latimer LH, Lee M, Liao Z, Lieberburg IM, Motter RN, et al. (2001) Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem 76: 173-181. [DOI] [PubMed] [Google Scholar]
- Esler WP, Kimberly WT, Ostaszewski BL, Ye W, Diehl TS, Selkoe DJ, Wolfe MS (2002) Activity-dependent isolation of the presenilin-gammasecretase complex reveals nicastrin and a gamma substrate. Proc Natl Acad Sci USA 99: 2720-2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany MB (2000) Studying human neurodegenerative diseases in flies and worms. J Neuropathol Exp Neurol 59: 847-856. [DOI] [PubMed] [Google Scholar]
- Fortini ME (2002) Gamma-secretase-mediated proteolysis in cell-surface-receptor signalling. Nat Rev Mol Cell Biol 3: 673-684. [DOI] [PubMed] [Google Scholar]
- Fortini ME, Bonini NM (2000) Modeling human neurodegenerative diseases in Drosophila: on a wing and a prayer. Trends Genet 16: 161-167. [DOI] [PubMed] [Google Scholar]
- Fossgreen A, Bruckner B, Czech C, Masters CL, Beyreuther K, Paro R (1998) Transgenic Drosophila expressing human amyloid precursor protein show gamma-secretase activity and a blistered-wing phenotype. Proc Natl Acad Sci USA 95: 13703-13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D (2002) aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 3: 85-97. [DOI] [PubMed] [Google Scholar]
- Galbete JL, Martin TR, Peressini E, Modena P, Bianchi R, Forloni G (2000) Cholesterol decreases secretion of the secreted form of amyloid precursor protein by interfering with glycosylation in the protein secretory pathway. Biochem J 348: 307-313. [PMC free article] [PubMed] [Google Scholar]
- Georgopoulou N, McLaughlin M, McFarlane I, Breen KC (2001) The role of post-translational modification in beta-amyloid precursor protein processing. Biochem Soc Symp 67: 23-36. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16: 4491-4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL (2003) Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA 100: 10417-10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunawardena S, Goldstein LS (2001) Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila Neuron 32: 389-401. [DOI] [PubMed] [Google Scholar]
- Ida N, Hartmann T, Pantel J, Schroder J, Zerfass R, Forstl H, Sandbrink R, Masters CL, Beyreuther K (1996) Analysis of heterogeneous A4 peptides in human cerebrospinal fluid and blood by a newly developed sensitive Western blot assay. J Biol Chem 271: 22908-22914. [DOI] [PubMed] [Google Scholar]
- Jackson GR, Wiedau-Pazos M, Sang T-K, Wagel N, Brown CA, Massachi S, Geschwind DH (2002) Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila Neuron 34: 509-519. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Lopera F, Kosik KS, Lendon CL, Ossa J, Saido TC, Yamaguchi H, Ruiz A, Martinez A, Madrigal L, Hincapie L, Arango JC, Anthony DC, Koo EH, Goate AM, Selkoe DJ (1996) The E280A presenilin 1 Alzheimer mutation produces increased A beta 42 deposition and severe cerebellar pathology. Nat Med 2: 1146-1150. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155: 853-862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46: 860-866. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM (2003) Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron 39: 409-421. [DOI] [PubMed] [Google Scholar]
- Rockenstein E, Mallory M, Mante M, Sisk A, Masliaha E (2001) Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42). J Neurosci Res 66: 573-582. [DOI] [PubMed] [Google Scholar]
- Rubin GM, Spradling AC (1982) Genetic transformation of Drosophila with transposable element vectors. Science 218: 348-353. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med 2: 864-870. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (1999) Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 399: 23-31. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T (1988) Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and non-neural tissues. Proc Natl Acad Sci USA 85: 7341-7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S (1997) Skeletal and CNS defects in Presenilin-1-deficient mice. Cell 89: 629-639. [DOI] [PubMed] [Google Scholar]
- Simons M, De Strooper B, Multhaup G, Tienari PJ, Dotti CG, Beyreuther K (1996) Amyloidogenic processing of the human amyloid precursor protein in primary cultures of rat hippocampal neurons. J Neurosci 16: 899-908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, John V, et al. (1999) Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402: 537-540. [DOI] [PubMed] [Google Scholar]
- Sisodia SS, Gallagher M (1998) A role for the beta-amyloid precursor protein in memory? Proc Natl Acad Sci USA 95: 12074-12076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM (1982) Transposition of cloned P elements into Drosophila germ line chromosomes. Science 218: 341-347. [DOI] [PubMed] [Google Scholar]
- Tomita S, Kirino Y, Suzuki T (1998) Cleavage of Alzheimer's amyloid precursor protein (APP) by secretases occurs after O-glycosylation of APP in the protein secretory pathway. Identification of intracellular compartments in which APP cleavage occurs without using toxic agents that interfere with protein metabolism. J Biol Chem 273: 6277-6284. [DOI] [PubMed] [Google Scholar]
- Tomita T, Maruyama K, Saido TC, Kume H, Shinozaki K, Tokuhiro S, Capell A, Walter J, Grunberg J, Haass C, Iwatsubo T, Obata K (1997) The presenilin 2 mutation (N141I) linked to familial Alzheimer disease (Volga German families) increases the secretion of amyloid beta protein ending at the 42nd (or 43rd) residue. Proc Natl Acad Sci USA 94: 2025-2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo Nature 416: 535-539. [DOI] [PubMed] [Google Scholar]
- Walter J, Kaether C, Steiner H, Haass C (2001) The cell biology of Alzheimer's disease: uncovering the secrets of secretases. Curr Opin Neurobiol 11: 585-590. [DOI] [PubMed] [Google Scholar]
- Wang HW, Pasternak JF, Kuo H, Ristic H, Lambert MP, Chromy B, Viola KL, Klein WL, Stine WB, Krafft GA, Trommer BL (2002) Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res 924: 133-140. [DOI] [PubMed] [Google Scholar]
- Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293: 711-714. [DOI] [PubMed] [Google Scholar]
- Ye Y, Fortini ME (1999) Apoptotic activities of wild-type and Alzheimer's disease-related mutant presenilins in Drosophila melanogaster. J Cell Biol 146: 1351-1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Lukinova N, Fortini ME (1999) Neurogenic phenotypes and altered Notch processing in Drosophila Presenilin mutants. Nature 398: 525-529. [DOI] [PubMed] [Google Scholar]