

Abstract
Whole-cell patch-clamp recordings were performed on acute brain slices of male rats to investigate the ability of the neurons of the median preoptic nucleus (MnPO) to detect fluctuation in extracellular osmolarity and sodium concentration ([Na+]out). Local application of hypotonic and hypertonic artificial CSF hyperpolarized and depolarized the neurons, respectively. Similar responses obtained under synaptic isolation (0.5 μM TTX) highlighted the intrinsic ability of the MnPO neurons to detect changes in extracellular osmolarity and [Na+]out. Manipulating extracellular osmolarity, [Na+]out, and [Cl-]out showed in an independent manner that the MnPO neurons responded to a change in [Na+]out exclusively. The specific Na+ response was voltage insensitive and depended on the driving force for Na+ ions, indicating that a sustained background Na+ permeability controlled the membrane potential of the MnPO neurons. This specific response was not reduced by Gd3+, amiloride, or benzamil, ruling out the participation of mechanosensitive cationic channels, specific epithelial Na+ channels, and Phe-Met-Arg-Phe-gated Na+ channels, respectively. Combination of in situ hybridization, using a riboprobe directed against the atypical Na+ channel (NaX), and immunohistochemistry, using an antibody against neuron-specific nuclei protein, revealed that a substantial population of MnPO neurons expressed the NaX channel, which was characterized recently as a concentration-sensitive Na+ channel. This study shows that a neuronal population of the MnPO acts as functional Na+ sensors and that the NaX channel might represent the molecular basis for the extracellular sodium level sensing in these neurons.
Keywords: lamina terminalis, sodium homeostasis, salt appetite, sodium sensors, hydromineral homeostasis, NaX, sodium channel
Introduction
Constant physiological osmolarity of plasma and CSF requires detectors that continuously monitor sodium ion concentration ([Na+]) and hydration within these extracellular fluids. One major challenge in neurobiology of the hydromineral homeostasis is to reveal the existence, at the level of the brain, of specific sensors involved in the control of the water and mineral balance. Fluctuations in plasma and CSF [Na+] not only trigger short-term adjustments, such as the release of antidiuretic and natriuretic hormones from the neurohypophysis (Bourque et al., 1994; Hussy et al., 2000; Voisin and Bourque, 2002), but also long-term regulation that controls thirst and specific appetite for salt (Weisinger et al., 1979, 1982; Denton et al., 1996). It has been hypothesized that specific brain Na+ sensors initiate sodium intake (Weisinger et al., 1979; Denton et al., 1996) as well as natriuresis (Cox et al., 1987; Denton et al., 1996), and the recent discovery of coincident detectors of extracellular fluid osmolarity and [Na+] in the supraoptic nucleus (SON) established the cellular basis for Na+ detection in this nucleus (Voisin et al., 1999; Voisin and Bourque, 2002). However, physiological experiments indicated the presence of Na+ sensors in the periventricular region of the brain (Cox et al., 1987; Park et al., 1989; Denton et al., 1996), and recent molecular biology data combined with behavioral studies revealed that Na+ sensors located in the preoptic region were associated with salt intake (Watanabe et al., 2000; Hiyama et al., 2002). The location of the median preoptic nucleus (MnPO) along the third ventricle, as well as the absence of tight junctions between ciliated cells that form the ventricular ependyma separating the CSF from the MnPO, make this nucleus a strategic locus for detection of CSF [Na+] (Landas and Philipps, 1987; McKinley et al., 1987; Oldfield and McKinley, 1995). Functionally, neuronal subpopulations of the MnPO have been shown to display an osmosensitive profile (Honda et al., 1990; Travis and Johnson, 1993; Aradachi et al., 1996). However, the influence of osmotic challenges has been controversial (Honda et al., 1990; Travis and Johnson, 1993), and the intrinsic ability of these neurons to detect changes in environmental osmolarity and [Na+] has yet to be determined. Moreover, the adjustment of the extra-cellular osmolarity with NaCl did not distinguish between the presence of osmoreceptors or Na+ sensors in this nucleus.
Using a hypothalamic slice preparation and manipulating the extracellular osmolarity and [Na+] separately, we investigated the possibility that neurons of the MnPO might act as intrinsic osmoreceptors, Na+ sensors, or Na+-dependent osmoreceptors. We also investigated the cellular mechanism underlying the ability of the MnPO neurons to detect a change in the extracellular osmolarity and [Na+].
Materials and Methods
All experiments were performed in accordance with the guidelines established by the Canadian Council on Animal Care and were approved by the Animal Care Committee of Centre Hospitalier de l'Université Laval.
Brain slice preparation. Male Wistar rats (100-150 gm) were deeply anesthetized with a ketamine-xylasine solution (43.7 and 1.25 mg/kg, respectively) injected intraperitoneally. After decapitation, the brain was quickly removed and immersed in ice-cold (2°C) artificial CSF (aCSF) continuously bubbled with a gas mixture (95% O2-5% CO2) and containing the following (in mm): 123 NaCl, 3.1 KCl, 10 d-glucose, 2.4 CaCl2, 1.3 MgCl2, 26 NaHCO3, and 1.2 NaHPO4, pH 7.4 (298-300 mOsm/l). A block of tissue containing the forebrain region was mounted on a metallic plate, immersed in the ice-cold oxygenated aCSF, and placed on a vibratome stage (VT 1000S; Leica, Nussloch, Germany). One to two slices (350-μm-thick) containing the MnPO were generated in the sagittal plane, an anatomic orientation that allows optimum access to the MnPO. The slice was placed into an oxygenated holding chamber for 30 min at room temperature (20-24°C). After this recovery period, the slice was transferred to a submersion-type recording chamber (Warner Instruments, Hamden, CT) and continuously perfused at 2 ml/min with oxygenated isotonic aCSF containing the following (in mm): 145 NaCl, 3.1 KCl, 2.4 CaCl2, 1.3 MgCl2, and 10 HEPES; pH 7.4 with 10 NaOH. Osmolarity was verified with a vapor pressure osmometer (Wescor, Logan, UT) and then adjusted to 300 mOsm/l by adding mannitol.
Data acquisition and analysis. Whole-cell patch-clamp recordings were mainly performed in neurons of the ventral part of the MnPO (vMnPO), which extends from the anterior commissure down to the vascular organ of the lamina terminalis. The precise boundary of the vMnPO was, however, hard to define all along the structure in the sagittal brain slice; thus, electrophysiological recordings obtained from neurons located in the area immediately adjacent to the anterior commissure were reported. Additional recordings were also performed in the dorsal part of the MnPO, in a region adjacent to the anterior commissure.
A tight gigaohm seal on neurons of the MnPO was obtained under visual control using the near infrared differential interference contrast principle. Microelectrodes were pulled from thin-walled (outer diameter, 1.5 mm; inner diameter, 1.1 mm) borosilicate glass (G75150T-4; Warner Instruments) on a vertical puller (model PIP5; Heka Elektronik, Mahone Bay, Nova Scotia, Canada). They had a final tip resistance of 4-5.5 MΩ and were filled with an intracellular solution containing the following (in mm): 130 K-gluconate, 6 NaCl, 10 HEPES, 0.1 Na+-GTP, and 2 Na+-ATP, pH 7.2 with 1 KOH. Osmolarity was measured with a vapor pressure osmometer and adjusted to 300 mOsm/l by adding sorbitol.
Whole-cell recordings were performed in either current-clamp or voltage-clamp mode, using an EPC 9 amplifier (Heka Elektronik). The fast capacitance electrode was first compensated, and appropriate whole-cell and series-resistance compensation was applied after rupture of the cell membrane by applying negative pressure within the patch pipette. Liquid junction potential was evaluated (14 mV), and membrane potential was corrected accordingly.
Electrophysiological signals (voltage and current) were digitalized (sampling rate, 24 kHz) and stored on tape using a digital audiotape recorder (model DTR-200; Bio-Logic, Grenoble, France). Current and voltage traces were also continuously displayed on-line, using a chart recorder (model TA240; Gould Instrument Systems, Valley View, OH). Intensity-to-voltage relationships were generated and analyzed off-line using Pulse and Pulsefit software (Heka Elektronik). All electrophysiological experiments were performed at room temperature (20-24°C).
Drugs and solutions. To study the effect of osmotic and natriuric challenges on the excitability of MnPO neurons, modified aCSF containing different concentrations of Na+ and Cl- ions, as well as different osmolarity, was delivered in the vicinity of the MnPO by using a rapid solution changer and manifold (model RSC-160; Bio-Logic). aCSF osmolarity was assessed by using a vapor pressure osmometer (Wescor), and adjustment to the appropriate osmolarity values was performed by adding mannitol to aCSF after pH adjustment, when necessary. Composition and osmolarity of the various aCSF used for that study are detailed in Table 1.
Table 1.
Composition of the different aCSFs used in the study
|
|
Hypotonic aCSF |
Hypertonic aCSF |
Hyperosmotic isonatriuric aCSF |
Iso-osmotic hyponatriuric aCSF |
Hypotonic isochloride aCSF |
Hypertonic isochloride aCSF |
Isonatriuric hyperchloride aCSF |
|---|---|---|---|---|---|---|---|
|
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
| NaCl (mm) | 100 | 170 | 150 | 100 | 100 | 150 | 150 |
| Choline chloride (mm) | 50 | 20 | |||||
| Na-gluconate (mm) | 20 | ||||||
| KCl (mm) | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 | 3.1 |
| CaCl2 (mm) | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 | 2.4 |
| MgCl2 (mm) | 1.3 | 1.3 | 1.3 | 1.3 | 1.3 | 1.3 | 1.3 |
| HEPES (mm) | 10 | 10 | 10 | 10 | 10 | 10 | 10 |
| Mannitol | + | − | +++ | ++ | − | − | − |
| Osmolarity (mOsm/l) |
270 |
330 |
330 |
300 |
280 |
330 |
330 |
aCSF of different composition and osmolarity were transiently and locally applied over the region of the ventral MnPO to investigate how extracellular osmolarity and extracellular concentration of sodium and chloride ions might alter the excitability of the ventral MnPO neurons. After pH adjustment with NaOH (10 mm), aCSF osmolarity was measured with a vapor pressure osmometer, and mannitol was added to aCSF, when necessary. Note that final aCSF [Na+] took into account Na+ ions added for pH adjustment (~5 mm). The constancy of aCSF osmolarity during the experiment was verified, and no time-dependent change in osmolarity was observed. Plus signs indicate the amount of mannitol that has been added to the solution to reach the osmolarity indicated in the last row of the table. A minus sign indicates that no mannitol has been added to reach the indicated osmolarity value.
Drugs expected to affect neurons of the vMnPO in a steady-state manner were directly added to the extracellular solution and bath applied.
Gadolinium and TTX, as well as amiloride and benzamil, were purchased from Sigma (St. Louis, MO).
Statistical analysis. All results are expressed as means ± SEM. Statistical comparisons were performed using paired Student's t test for absolute values (cell input resistance); p < 0.05 was considered significant. Results expressed as percentages (pharmacological data) were considered to be nonparametric data. They were analyzed using the Mann-Whitney U test. Statistical significance was determined at p < 0.05.
Immunohistochemistry and in situ hybridization protocols. A combination of immunohistochemistry (IHC) for neuron-specific nuclei protein (NeuN) and in situ hybridization (ISHH) immunohistochemistry for NaX mRNA was performed to allow simultaneous visualization of NeuN immunoreactivity and NaX mRNA in the same tissue sections that contained the MnPO. To match the anatomical observations with the electrophysiological recordings, IHC and ISHH experiments were performed on rats of the same age as those used for the electrophysiological study (100-150 gm). Brain sections (30 μm) were processed using the avidin-biotin bridge method with hydrogen peroxide as a substrate. The IHC reaction was performed in RNase-free condition using a mouse monoclonal antibody (MAB377; Chemicon, Temecula, CA) that recognized NeuN at a concentration of 1:5000. After the IHC procedure, the brain sections were processed for ISHH using a 35S-labeled atypical Na+ channel NaX cRNA probe for mRNA. The NaX cRNA probe was generated from a rat cDNA contained in a pBS(SK-) plasmid from Stratagene (La Jolla, CA) (a kind gift from Dr. Waxman, Yale University, West Haven, CT) (Black et al., 1994). The insert corresponds to the cDNA sequence encoding a portion of domain 4: nucleotides 880-1308 of the sequence M96578 (Gautron et al., 1992) or nucleotides 4671-5119 of the sequence Y09164 (Akopian et al., 1997). To produce the antisense probe, DNA was digested with EcoRI and T7 polymerase. Radioisotope-labeled sense cRNA copies (control) were also prepared to verify the specificity of NaX probe. To produce the sense probe, DNA was digested with BglII and T3 polymerase. Hybridization with the sense probe did not reveal any positive signal in rat brain (see Fig. 7).
Figure 7.

NaX channel is expressed in the median preoptic nucleus and subfornical organ. The line drawings, adapted from the brain atlas of Swanson (1992), represent coronal sections of rat brain through the subfornical organ (A, left) and the median preoptic nucleus (B, left). The autoradiograms illustrate the distribution of NaX mRNA in the subfornical organ (A, right) and in the ventral portion of median preoptic nucleus (B, right). Note that in situ hybridization performed with the sense probe did not reveal any signal in both the SFO and MnPO (A, B, middle, white arrows). These pictures were taken from autoradiogram of 30 μm coronal sections hybridized with 35S-labeled NaX riboprobe through corresponding sections. 3rd V, Third ventricle; LV, lateral ventricle.
The experimental procedure for IHC and ISHH is briefly described, as follows. Brain sections were washed in sterile DEPC-treated 0.05 mm potassium PBS (KPBS) and incubated at 4°C with NeuN antibody mixed in sterile KPBS, 0.4% Triton X-100, 1% heparin sodium salt United States Pharmacopeia, and 1% bovine serum albumin. Approximately 18 hr after incubation with NeuN antibody, brain sections were rinsed in sterile KPBS and incubated with a mixture of KPBS-heparin and biotinylated secondary antibody for 120 min. The sections were then rinsed with KPBS and incubated at room temperature for 60 min with an avidin-biotin-peroxidase complex (Vectastain Elite; Vector Laboratories, Burlingame, CA). After several rinses in sterile KPBS, the brain sections were immersed in a mixture containing sterile KPBS, the chromagen 3,3′-diaminobenzidine tetrahydrochloride (0.5 mg/ml), and hydrogen peroxide (0.003%). Thereafter, hybridization histochemical localization of NaX mRNA was performed using a 35S-labeled NaX cRNA probe. Protocols for riboprobe synthesis, hybridization, and autoradiographic localization of NaX mRNA signal were adapted from Simmons et al. (1989) and Dumont et al. (1999). Immunoreactive sections for NeuN were mounted onto poly-l-lysine-coated slides and were dessicated under vacuum overnight, fixed in 4% paraformaldehyde for 30 min, and digested by proteinase K (10 μg/ml in 100 mm Tris HCl, pH 8.0) and 50 mm EDTA, at 37°C for 25 min. The brain sections were then rinsed in sterile DEPC water, followed by a solution of 100 mm triethanolamine (TEA), pH 8.0, acetylated in 0.25% acetic anhydride in 100 mm TEA, and dehydrated through graded concentrations of alcohol (50, 70, 95, and 100%). After vacuum drying for a minimum of 2 hr, 90 μl of hybridization mixture (107 cpm/ml) was spotted on each slide, sealed under a coverslip, and incubated at 55°C overnight (∼15-20 hr) in a slide warmer. Coverslips were then removed, and the slides were rinsed in 4× SSC at room temperature. Sections were digested by RNase A (20 μg/ml, 37°C, 30 min), rinsed in descending concentrations of SSC (2×,1×, and 0.5×), washed in 0.1× SSC for 30 min at 60°C (1× SSC: 150 mm NaCl and 15 mm trisodium citrate buffer, pH 7.0), and dehydrated through graded concentrations of alcohol. After being dried for 2 hr under vacuum, the sections were defatted in xylene and dipped in NTB2 nuclear emulsion (diluted 1:1 with distilled water; Eastman Kodak, Rochester, NY). Slides were exposed for 8 weeks, developed in D19 developer (Eastman Kodak) for 3.5 min at 14-15°C, and fixed in rapid fixer (Eastman Kodak) for 5 min. Thereafter, brain tissue was rinsed in running distilled water for 1-2 hr, dehydrated through graded concentrations of alcohol, cleared in xylene, and coverslipped with a mixture of distyrene, tricresyl phosphate, and xylene (DPX).
Results
In the first set of experiments, vMnPO neurons were maintained around their resting potential (range, -63 to -60 mV) or were slightly depolarized with current injection to generate a regular spiking activity (range, -55 to -45 mV). Baseline for stable membrane potential was established for 2 to 4 min under isotonic aCSF. Hypotonic aCSF (Table 1, column 1) was then locally applied for 1 min over the region of the vMnPO. Transient application of the hypotonic solution triggered membrane hyperpolarization, with a concomitant abolition of the firing activity in cells that were sufficiently depolarized to fire spikes during the control period. Membrane hyperpolarization amounted to 6.0 ± 0.4 mV (n = 20) and was fully reversed after 4-5 min. Local application of hypertonic aCSF (Table 1, column 2) produced a transient depolarization of the cell (3.9 ± 0.2 mV; n = 22) that was often accompanied by a sustained discharge of action potentials. Neurons returned to their initial membrane potential after reexposure to isotonic condition, and the action potentials stopped. As illustrated in Figure 1A, individual neurons of the vMnPO were able to respond to opposite changes in extracellular tonicity. In 6 of the 20 neurons that displayed a hypotonicity-induced hyper-polarization (top trace), a subsequent application of a hypertonic stimulus was tested after the recovery period. In all of the neurons tested, the hypertonic aCSF induced a transient depolarization (bottom trace). The ability of vMnPO neurons to respond to consecutive hypotonic stimuli was then tested. In 11 neurons of the vMnPO held at -60 mV, two consecutive local applications of hypotonic aCSF hyperpolarized the neurons by 7 ± 0.9 and 7 ± 0.6 mV, respectively. Furthermore, in four other neurons, three consecutive hypotonic challenges induced membrane hyperpolarization amounting to 6.9 ± 1.1, 6.9 ± 1.1, and 7.8 ± 0.9 mV, respectively.
Figure 1.

Intrinsic osmosensitivity is a unique property of the MnPO neurons. A, Depolarization of a ventral MnPO neuron with current injection triggered a regular spiking activity. Local and transient (1min) application of hypotonic aCSF (270 mOsm/l; 100 mm NaCl) over the vMnPO region hyperpolarized the neuron with a concomitant abolition of the spike discharges (top trace). The same neuron was held below the spike threshold, and local application of hypertonic aCSF (330 mOsm/l; 170 mm NaCl) depolarized the neuron that discharged a prolonged burst of spikes (bottom trace). Details of the spiking activity was illustrated before (*) and after (**) application of the modified aCSF. B, TTX (0.5 μm) was bath applied to block synaptic inputs onto the recorded vMnPO neuron. Under this steady-state synaptic blockage, transient and local application of a hypotonic aCSF hyperpolarized the neuron (top trace). In contrast, local application of a hypertonic aCSF induced a depolarization (bottom trace). C, Local application of hypertonic aCSF did not alter the membrane potential of this vMnPO neuron, which was considered as a non-osmoresponsive neuron. D, Local application of hypotonic aCSF did not change the membrane potential of the neurons located in a region adjacent to the MnPO, the median septum. Note that spike amplitude has been truncated in the traces presented here. These results indicate that detection of the extracellular tonicity involved intrinsic properties of a specific neuronal population of the vMnPO. HP, Holding potential.
The application of hypotonic or hypertonic aCSF was done locally, i.e., over the MnPO area. However, spillover of the modified aCSF over the adjacent region, which could modify the excitability of upstream neuronal cells projecting to the vMnPO neurons, could not be excluded. Therefore, the intrinsic ability of the MnPO neurons to detect changes in aCSF tonicity was verified in a series of experiments in which synaptic transmission was fully abolished by perfusing a potent blocker of the voltage-gated Na+ channel within the recording chamber (0.5 μM TTX). Under this steady-state condition, local application of hypotonic and hypertonic stimuli hyperpolarized and depolarized the MnPO neurons by 5.5 ± 0.5 mV (n = 7) and 3.9 ± 0.4 mV (n = 5), respectively (Fig. 1B). Consecutive applications of hypotonic and hypertonic aCSF were tested in three neurons, and all of them displayed hyperpolarization and depolarization in response to the appropriate stimulus.
Of a total of 75 neurons tested under standard condition and in the presence of TTX, 72% of the neurons (n = 54) showed a significant change in membrane potential in response to the local change in extracellular tonicity; these were considered to be osmoresponsive. The remaining neurons did not respond to osmotic stimuli, as illustrated in Figure 1C, for a hypertonic challenge; these were considered to be non-osmoresponsive. The specificity of the MnPO to contain osmoresponsive neurons was then verified. Ten neurons located in the median septum, a region rostral to the MnPO in the sagittal brain slice, were tested for similar changes in extracellular tonicity. These neurons were either maintained around their resting membrane potential or slightly depolarized with current injection. None of these neurons showed a membrane hyperpolarization or depolarization in response to local application of a hypotonic or a hypertonic stimulus, respectively. A typical example of a non-osmoresponsive septal neuron is illustrated in Figure 1D.
vMnPO neurons detect [Na+]out rather than extracellular osmolarity
Changing the extracellular Na+ salt concentration ([NaCl]out)to alter the osmotic pressure suggests that the osmoresponsive vMnPO neurons were able to detect either variation of the extra-cellular osmolarity or the extracellular concentration of Na+ and/or Cl- ions ([Na+]out and [Cl-]out, respectively). In the next series of experiments, we investigated the possibility that the vMnPO neurons acted as osmoreceptors by detecting alteration of the extracellular osmolarity without changing the [NaCl]out. To test this hypothesis, we applied a hyperosmotic stimulus while maintaining [Na+]out at 150 mM, the standard concentration found in the extracellular environment of central neurons. Local application of hyperosmotic-isonatriuric solution (Table 1, column 3) on vMnPO neurons maintained at their resting potential did not change the membrane potential of the neuron (n = 5) (Fig. 2A, top trace). It should be noted that the effect of a hypo-osmotic-isonatriuric aCSF was not tested because aCSF containing 150 mM NaCl could not be hypo-osmotic. In the next series of experiments, we varied [Na+]out while keeping constant the osmolarity of the aCSF that was directly applied over the vMnPO region. Neurons of the vMnPO were maintained at their resting potential or slightly depolarized to generate action potential discharges. Decreasing [Na+]out from 150 to 100 mM while maintaining osmolarity at 300 mOsm/l (by adding D-mannitol) (Table 1, column 4) induced a reversible membrane hyperpolarization of 6.2 ± 0.3 mV (n = 36) (Fig. 2A, middle trace). The amplitude of the hyperpolarization was similar to that which was obtained with a hypo-osmotic-hyponatriuric (hypotonic) aCSF (6.0 ± 0.4 mV; n = 20) (Fig. 2A, bottom trace). This observation suggests that osmolarity had no additional effect on the neuronal response induced by hyponatremia. It should be noted that the effect of increased [Na+]out in iso-osmotic solution was not investigated, because [Na+]out >150 mM was always associated with hyperosmolarity.
Figure 2.
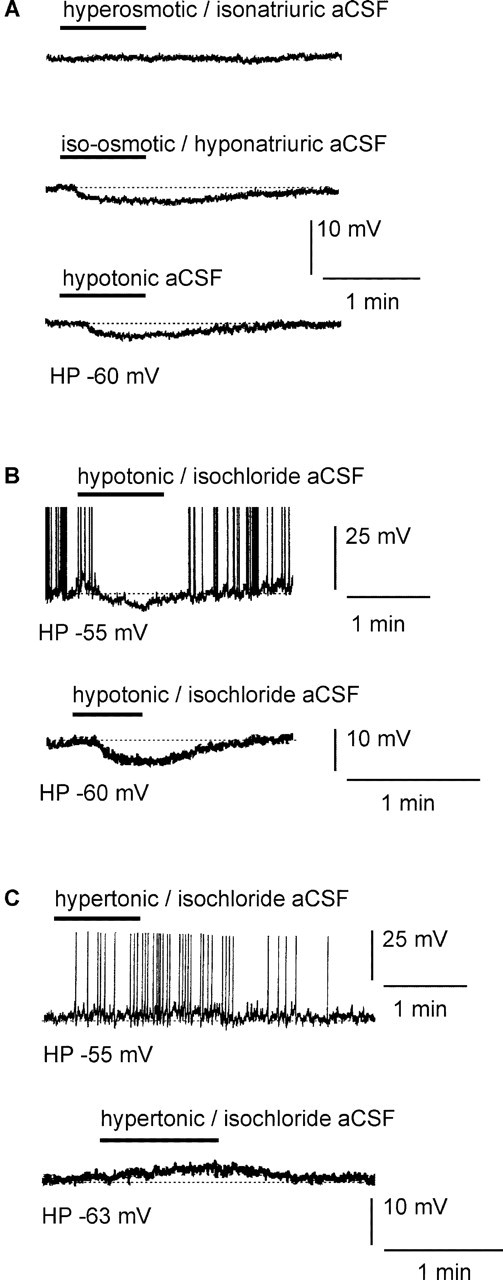
Ventral MnPO neurons detect changes in extracellular Na+ level but not osmolarity. A, Local application of a hyperosmotic-isonatriuric stimulus (330 mOsm/l; 150 mm NaCl) had no apparent effect on the excitability of the vMnPO neuron (top trace). In contrast, local application of an iso-osmotic-hyponatriuric aCSF (300 mOsm/l; 100 mm NaCl) hyperpolarized the vMnPO neuron (middle trace). The amplitude of the hyperpolarization was similar to the one obtained with a hypotonic aCSF (270 mOsm/l; 100 mm NaCl; bottom trace). B, Traces obtained from two different vMnPO neurons. The top trace was obtained with slight injection of depolarizing current to make the neuron fire (holding potential, -55 mV). The bottom trace was obtained from a neuron maintained around its resting potential (holding potential, -60 mV). In these neurons, transient and local application of hypotonic-isochloride aCSF (270 mOsm/l; 100 mm NaCl and 50mm choline chloride) drove the membrane potential to hyperpolarization. C, Two neurons maintained at different membrane potential[holding potential, -55 mV (top trace); holding potential, -63 mV (bottom trace)]. Local application of hypertonic-isochloride aCSF (330 mOsm/l; 150mm NaCl and 20 mm Na+-gluconate) onto these neurons triggered depolarization of the cells. Spike amplitude has been truncated in all of the panels presented. These results show that neurons of the vMnPO did respond to a change in extracellular [Na+] and not to an alteration of the extracellular osmolarity or [Cl-]. HP, Holding potential.
This series of results obtained by varying osmolarity and [NaCl]out independently indicated that vMnPO neurons did detect a change in [NaCl]out rather than a change in extracellular osmolarity. In our experimental protocol, varying [Na+]out induced a concomitant change in [Cl-]out. We tested the possibility that the vMnPO neurons responded to a change in [Cl-]out rather than to a change in [Na+]out. First, hypotonic-isochloride aCSF (Table 1, column 5) was obtained by adding 50 mM choline chloride to the hypotonic aCSF. As illustrated in Figure 2B (top trace), local application of the hypotonic-isochloride aCSF induced a hyperpolarization that amounted to 6.6 ± 0.4 mV (n = 4), a response similar to the one obtained previously with a low Na+ and low Cl- hypotonic aCSF (6.3 ± 0.3 mV). Second, local application of a hypertonic-isochloride aCSF (Table 1, column 6) depolarized the vMnPO neurons by 3.7 ± 0.7 mV (n = 5) (Fig. 2B, bottom trace). In contrast, raising [Cl-]out without changing [Na+]out (Table 1, column 7) had no effect on the membrane potential of the vMnPO neurons (n = 5). These results show that alteration of the [Na+]out did influence the membrane potential of the vMnPO neurons, indicating that these neurons were able to detect a change in [Na+]out.
Cellular mechanisms underlying detection of CSF [Na+]
If Na+ ions are the major current carrier associated with the observed changes in membrane potential of vMnPO neurons, iso-osmotic variation in [Na+]out might change the membrane potential with graded magnitude, revealing the Na+ specificity of the response. As illustrated in Figure 3A, local application of different iso-osmotic-hyponatriuric stimuli (300 mOsm/l; 142, 130, and 100 mM NaCl) hyperpolarized the vMnPO neurons by 3.2 ± 0.3 mV (n = 5), 4.1 ± 0.3 mV (n = 4), and 6 ± 0.4 mV (n = 20), respectively. In addition, the hyperpolarization induced by local application of iso-osmotic-hyponatriuric aCSF was not accompanied with a change in input resistance (1.35 ± 0.15 GΩ under control and 1.37 ± 0.15 GΩ under the iso-osmotic-hyponatriuric aCSF; n = 12; p = 0.18) (Fig. 3B). Similarly, no change in input resistance was observed during the response to hypernatremia (1.33 ± 0.17 GΩ in control vs 1.33 ± 0.18 GΩ under hypernatriuric aCSF; n = 8; p = 0.83) (Fig. 3C).
Figure 3.

The specific response to a natriuric challenge depended on the driving force for Na+ ions and was not associated with a change in input resistance. A, In the same vMnPO neuron, local application of iso-osmotic-hyponatriuric aCSF with a reduction of 8, 20, and 50 mm in the [Na+]out compared with regular aCSF (150 mm) induced a cellular response of graded amplitude. B, Local application of iso-osmotic-hyponatriuric aCSF (300 mOsm/l; 100 mm NaCl) induced membrane hyperpolarization that was not accompanied with a change in input resistance. Input resistance was tested with 800 msec hyperpolarizing current steps (-8 pA) tested before and during the cellular response. Note that positive current was transiently injected to compensate for the hypotremia-induced hyperpolarization. Typical representation of the passive membrane response to the current step is an average of five consecutive traces. C, Same neuron as in B. Local application of hypernatriuric aCSF (330 mOsm/l; 170 mm NaCl) induced a depolarization that was not accompanied by a change in input resistance. Input resistance was assessed with identical current steps as those depicted in B. Negative current was transiently injected to the cell to compensate for the hypernatremia-induced depolarization during evaluation of the input resistance. HP, Holding potential.
In the next series of experiments, we used the voltage-clamp mode to further characterize the cellular mechanism(s) underlying detection of CSF [Na+]. vMnPO neurons were clamped at their resting potential (-60 mV). In the presence of TTX (0.5 μM) and TEA-Cl (10 mM), local application of iso-osmotic-hyponatriuric aCSF triggered a reversible outward current that amounted to +5.9 ± 0.4 pA (n = 26) (Fig. 4A). Intensity-to-voltage relationships obtained with a ramp protocol ranging from -100 to -10 mV were tested before, during, and after the application of the iso-osmotic-hyponatriuric aCSF (rate of change fixed at 16 mV/sec; n = 13) (Fig. 4B). To test the action of an iso-osmotic-hypernatriuric aCSF, vMnPO neurons were bathed with the iso-osmotic-hyponatriuric aCSF (300 mOsm/l; 100 mM NaCl) for 5-10 min (control). Local application of iso-osmotic-hypernatriuric aCSF (300 mOsm/l; 150 mM NaCl) compared with control induced a reversible inward current of -5.7 ± 0.4 pA (n = 9) (Fig. 5A). Similar intensity-to-voltage relationships were triggered before, during, and after the application of the iso-osmotic-hypernatriuric aCSF (Fig. 5B). These two experimental protocols, performed on the same vMnPO neurons when possible (n = 6), allowed the isolation of the Na+-evoked current, which showed no voltage dependency between -100 and -10 mV (Figs. 4C, 5C). The inward direction of the current observed during application of iso-osmotic-hypernatriuric aCSF materialized influx of Na+ ions within the neuron through a background permeability, whereas the outward current observed during application of iso-osmotic-hyponatriuric aCSF reflected a reduction of an Na+ influx.
Figure 4.

Transient application of iso-osmotic-hyponatriuric aCSF on vMnPO neurons triggered a voltage-independent outward current. A, A vMnPO neuron was recorded under the voltage-clamp mode and maintained at a potential of -60 mV. Local application of iso-osmotic-hyponatriuric aCSF (300 mOsm/l; 100 mm NaCl) triggered an outward current. B, Three I-V relationships from -100 to -10 mV (16 mV/sec) were elicited under control (labelA), iso-osmotic-hyponatriuric aCSF (labelB), and back to iso-osmotic-isonatriuric aCSF (label C). The specific current (Δ current) evoked by hyponatremia was isolated by digital subtraction of the I-V relationships recorded in A and B. Note the absence of voltage dependency over the range of potentials tested. TTX (0.5 μm) and TEA-Cl (20 mm) were present in isotonic aCSF, as well as in iso-osmotic-hyponatriuric aCSF, to block voltage-gated Na+ and K+ currents. For these experiments, extracellular osmolarity (300 mOsm/l) was achieved with or without adding mannitol to aCSF (iso-osmotic-hyponatriuric aCSF and control, respectively). C, Mean ramp current obtained from the protocol described in B (n = 6 MnPO neurons), showing a parallel shift of the current in the outward direction during transient application of iso-osmotic-hyponatriuric aCSF (top). Mean control current is represented by open circles, and mean current recorded during application of modified aCSF is represented by filled circles. Digital subtraction of the traces shown in the top panel resolved the I-V relationship of the mean current evoked by application of iso-osmotic-hyponatriuric aCSF (bottom). HP, Holding potential.
Figure 5.
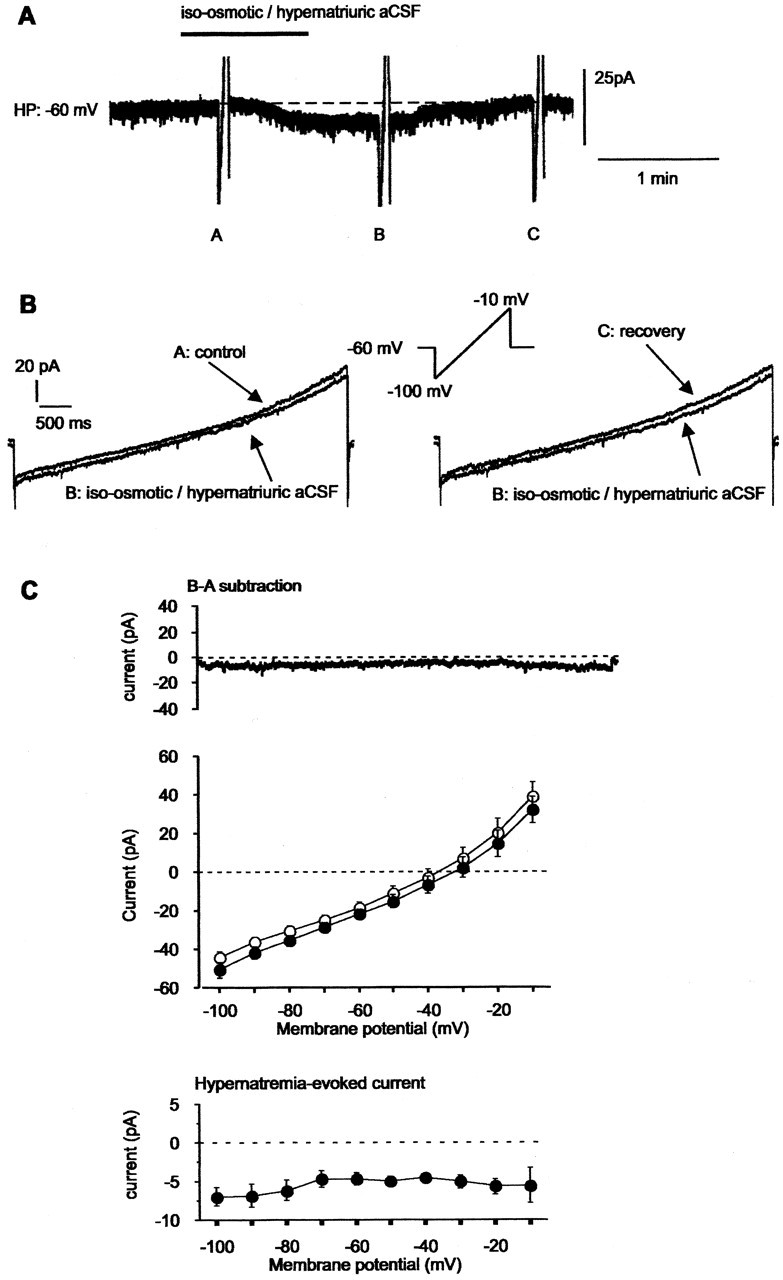
Transient application of iso-osmotic-hypernatriuric aCSF on vMnPO neurons triggered a voltage-independent inward current. A, A vMnPO neuron was maintained at a membrane potential of -60 mV. Local application of iso-osmotic-hypernatriuric aCSF (300 mOsm/l; 150 mm NaCl) compared with control solution (300 mOsm/l; 100 mm NaCl) triggered an inward current. B, I-V relationships from -100 to -10 mV (16 mV/sec) were elicited before (label A), during (label B), and after (label C) the application of iso-osmotic-hypernatriuric aCSF. The specific current (Δ current) evoked by hypernatremia was isolated by digital subtraction of the I-V relationships recorded in A and B. TTX (0.5 μm) and TEA-Cl (20 mm) were present in control aCSF, as well as in iso-osmotic-hypernatriuric aCSF to block voltage-gated Na+ and K+ currents. For these experiments, extracellular osmolarity (300 mOsm/l) was achieved by adding mannitol to the control aCSF and by removing glucose from the iso-osmotic-hypernatriuric aCSF. C, Mean ramp current obtained from the protocol described in B and obtained from the six MnPO neurons illustrated in Figure 4C. Transient application of iso-osmotic-hypernatriuric aCSF evoked an inward shift of the current (top). Mean current recorded under control is represented by open circles, and mean current recorded during application of modified aCSF is represented by filled circles. Digital subtraction of the traces shown in the top panel resolved the I-V relationship of the mean current evoked by application of iso-osmotic-hypernatriuric aCSF (bottom). HP, Holding potential.
The presence of such a constitutive Na+ permeability was investigated in the dorsal part of the MnPO. Ten neurons located above the anterior commissure were bathed in an iso-osmotic aCSF, and transient application of iso-osmotic-hyponatriuric aCSF triggered an outward current in 7 of the 10 cells tested (70%), the amplitude of which amounted to +4.7 ± 1.2 pA (data not shown).
Finally, we investigated the pharmacology of the ionic channels underlying the intrinsic background Na+ permeability of MnPO neurons. Na+ ion flow through stretch-inactivated channels (SICs) has been shown to constitute the cellular basis for Na+ sensing in the supraoptic neuroendocrine cells (Voisin et al., 1999). Because the open probability of these channels was greatly reduced by the lanthanide trivalent gadolinium (Gd3+), we investigated the involvement of mechanosensitive channels in the response induced by hypotonic (Table 1, column 1) and hypertonic (Table 1, column 2) stimuli. In the presence of TTX (0.5 μM), local application of hypotonic and hypertonic aCSF hyper-polarized and depolarized the vMnPO neurons, respectively, as demonstrated previously. After recovery, Gd3+ (100 μM) was bath applied for 5-6 min before a second local application of hypotonic and hypertonic aCSF that also contained 100 μM Gd3+. The amplitude of both the hyperpolarization and depolarization was not reduced in the presence of Gd3+ (Fig. 6A), and the percentage of the responses induced by hypotonic and hypertonic aCSF remained unchanged (99.9 ± 6.7% in control vs 109.1 ± 7.1% in the presence of Gd3+; n = 12; p > 0.99). Then, we tried to antagonize the responses by using blockers for specific Na+ channels that are constitutively active. Bath application of amiloride (3 μM) remained without effect on the hyperpolarization induced by hypotonic aCSF (Fig. 6B), thus unaffecting the percentage of the response (99.9 ± 9.6% in control vs 104 ± 8.7% in the presence of amiloride; n = 8; p > 0.99) (Fig. 6C). In addition, bath application of benzamil (30 μM), an analog of amiloride, did not reduce the amplitude of the hypotonicity-induced response, for which the percentage remained constant (99.9 ± 10.9% in control vs 99.9 ± 9.1% in the presence of benzamil; n = 3; p > 0.99) (Fig. 6C). Similar inefficacy of these molecules was also observed for the depolarization induced by hypertonic aCSF.
Figure 6.
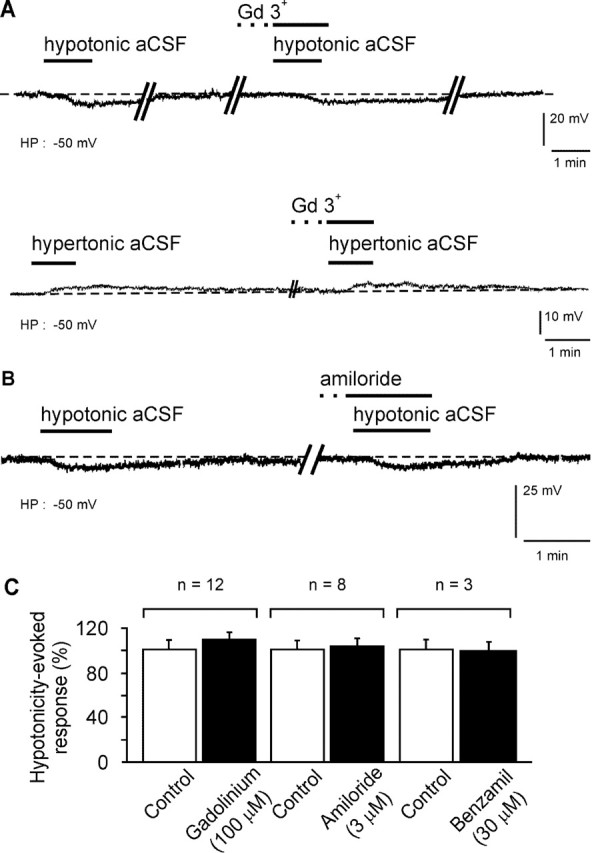
The pharmacology of the specific Na+ response ruled out the involvement of mechanosensitive cationic channels, as well as amiloride-sensitive Na+ channels. A, Typical response of a neuron of the vMnPO to local application of hypotonic (270 mOsm/l; 100 mm NaCl) and hypertonic (330 mOsm/l; 170 mm NaCl) aCSF, recorded in the presence of TTX in the bath (0.5 μm). Note that bath application of Gd3+ before the hypotonic and hypertonic stimuli did not reduce the amplitude of the hyperpolarization and depolarization of the vMnPO neurons, respectively. B, In a second set of vMnPO neurons, steady-state application of amiloride, a blocker of specific brain Na+ channels, remained ineffective for blocking the hyperpolarization induced by local application of a hypotonic aCSF. C, Histograms summarizing the pharmacology of the Na+ response mediated by local application of a hypotonic stimulus. The action of gadolinium, amiloride, and benzamil was tested on 12, 8, and 3 neurons, respectively. HP, Holding potential.
Together, our results indicate that neurons of the MnPO display a sustained permeability through which Na+ flux depends on the electrochemical gradient of Na+ ions. The pharmacological profile of the sustained Na+ permeability remains, however, to be determined.
Neurons of the MnPO express the atypical Na+ channel NaX
Recently, the atypical Na+ channel NaX has been demonstrated to act as a concentration-sensitive Na+ channel in dissociated subfornical neurons (Hiyama et al., 2002). Despite that the pharmacology of the NaX current has not been determined yet, this current shares numerous properties with the sustained Na+ current that we report here. We, therefore, performed in situ hybridization experiments using a riboprobe directed against the NaX mRNA to possibly identify the Na+ background permeability to the NaX channel. Results obtained with in situ hybridization indicated that NaX mRNA are expressed in discrete regions of the hypothalamus. As illustrated in Figure 7A, NaX mRNA are strongly expressed in the subfornical organ (SFO), the neurons of which having been demonstrated to express NaX channels (Watanabe et al., 2000; Hiyama et al., 2002). Note that in situ hybridization performed with the sense riboprobe did not reveal any staining in the SFO region. These results, therefore, did validate the specificity of the riboprobe used in the present study. More importantly, our data revealed that NaX mRNA are constitutively expressed in the MnPO (0.18 ± 0.03 μCi/gm; n = 7) (Fig. 7B), in which we identified neuronal populations that act as genuine Na+ sensors. Interestingly, the SON that contains coincident detectors of CSF [Na+] and osmotic pressure (Voisin et al., 1999) did not show any staining that is indicative of NaX mRNA-containing cells (data not shown). This latter result indicates that neuroendocrine cells of the SON did not express detectable levels of the atypical Na+ channel mRNA.
The atypical Na+ channel NaX has been first cloned from rat cortical astrocytes and then in neurons (Gautron et al., 1992). To determine whether the NaX channel gene is encoded by neuronal populations in the MnPO, we performed immunohistochemistry for NeuN combined with in situ hybridization immunohistochemistry for NaX mRNA to allow simultaneous visualization of NeuN immunoreactivity and NaX mRNA in the same tissue sections that contained the MnPO (Fig. 8). We found prominent double-labeled cells localized in both the dorsal and ventral portions of the MnPO, clearly indicating that neurons of the MnPO express the NaX channel (Fig. 8A,D,E). However, adjacent brain regions, such as the septum (Fig. 8C) or the bed nucleus of the stria terminalis (Fig. 8G,H), did not show any apparent Nax mRNA-labeled neurons.
Figure 8.

Neuronal populations of the median preoptic nucleus express NaX. A-H, Bright-field photomicrographs obtained from two different frontal sections of the brain that include the MnPO. B, F, A general view of the selected area (MnPO), together with adjacent control regions [lateral septum and bed nucleus of the stria terminalis (BnST)]. A,D,E, Photomicrographs at different magnifications showing the distribution of both the immunoreactivity for neuron-specific nuclei protein and NaX mRNA at the level of the MnPO. The arrows in A and E illustrate double-labeled cells that are in the same focal plan for the Nax channel mRNA (black silver grains) and neuron-specific nuclei protein (immunoreactivity in brown). C, G, H, Photomicrographs of control regions taken at different magnifications. Note the absence of double-labeled neurons in surrounding nuclei, as shown for the septum (C) and the bed nucleus of the stria terminalis (H). Scale bars: A, C, E, H, 20 μm; B, F, 400 μm; D, G, 50 μm. 3V, Third ventricle; LV, lateral ventricle; ac, anterior commissure.
Discussion
The present study indicates that fluctuations in extracellular [Na+] directly affect the excitability of a neuronal population of the MnPO by changing its membrane potential. The cellular mechanism involved in Na+ detection was based on a background Na+ permeability active at the resting membrane potential. This permeability is likely identified to the NaX channel, as strongly suggested by the expression of this atypical Na+ channel by MnPO neuronal population.
Osmoreceptors or specific Na+ sensors in the MnPO?
In vivo, extracellular electrophysiological recordings showed that a subset of MnPO neurons projecting to the supraoptic neuroendocrine cells increased their firing rate during systemic injection of hypertonic saline (McKinley et al., 1992; Aradachi et al., 1996) and during application of hypertonic saline directly into the MnPO (Honda et al., 1990). These studies certainly demonstrated the osmoresponsiveness of a population of MnPO neurons projecting to the SON and suggested the osmosensitivity of these neurons. Here we showed that, under complete synaptic isolation of the ventral region of the MnPO, extracellular hypertonicity and hypotonicity depolarized and hyperpolarized a majority of neurons, highlighting the innate ability of a single vMnPO neuron to respond to opposite changes in the extracellular tonicity (osmosensitivity). Although our results were in agreement with previous in vivo studies (Honda et al., 1990; McKinley et al., 1992; Aradachi et al., 1996), they were in contradiction, at least partially, with in vitro results reporting that hyperosmolality was associated with a decreased spike activity in few vMnPO neurons (Travis and Johnson, 1993). However, these authors also reported that hypo-osmolality decreased, or increased the electrical activity, depending on the vMnPO neuron tested. These contrasting data might arise from the presence of two separate neuronal populations in the vMnPO that respond to a similar osmotic challenge with an opposite change in their membrane potential. The effects of consecutive applications of hyponatriuric and hypernatriuric aCSF in a significant number of neurons have only revealed a stereotyped response: hyponatriuric aCSF always triggered membrane hyperpolarization, or outward current, whereas hypernatriuric aCSF triggered depolarization, or inward current. These results almost rule out the presence of two different populations with opposite capabilities. Alternatively, different vMnPO neurons might receive opposite synaptic inputs from upstream osmosensitive neurons. Application of hypertonic or hypotonic aCSF was done locally, i.e., over the vMnPO region, thus limiting the stimulation of upstream osmosensitive neurons. Moreover, neuronal responses were reproduced under synaptic isolation, excluding opposite synaptic inputs to the vMnPO.
Determining whether vMnPO neurons, as well as neurons located in the dorsal part of the nucleus, were osmoreceptors or Na+ sensors was of primary interest in the characterization of the role of the MnPO in hydromineral homeostasis. In the previous studies, the osmotic challenge was always achieved by manipulating [Na+]out, in either the extracellular solution (Travis and Johnson, 1993) or the extracellular fluid compartment (Honda et al., 1990). Here, we independently manipulated [Na+]out and extracellular osmolarity to evaluate the ability of the vMnPO neurons to discriminate between aCSF osmolarity and aCSF [Na+]. Our results showed that local application of hypertonic aCSF (330 mOsm/l; 170 mM NaCl) depolarized vMnPO neurons. Surprisingly, vMnPO cells did not respond to extracellular hyperosmolarity (330 mOsm/l) when [Na+]out was kept constant (150 mM NaCl). Because it is generally accepted that deviation of 20 mOsm/l from the set point corresponds to an approximate change of [Na+]out by 10 mM, we did expect that setting the extracellular osmolarity to 330 mOsm/l with mannitol would mimic a response obtained with a [Na+]out amounting to 165-170 mM. This was not the case. Furthermore, we showed that vMnPO neurons did respond to hyponatremia without a change in extracellular osmolarity.
Our results clearly indicate that a subpopulation of MnPO neurons located in both parts of the nucleus are specifically endowed with the detection of [Na+]out and, thus, favor the role of MnPO neurons as cerebral Na+ sensors, whose existence have long been hypothesized in periventricular tissue (Cox et al., 1987; Park et al., 1989; Denton et al., 1996). We now provide evidence for functional Na+ sensors in the MnPO that likely belong to a complex neuronal network of interconnected structures within the lamina terminalis (Honda et al., 1990; Oldfield et al., 1991, 1992; McKinley et al., 1999; McKinley et al., 2003). This network might function as a central osmoregulatory unit that might control vasopressin and may be oxytocin secretion, together with the innate Na+ and osmolarity sensitivity of magnocellular neurons (MCNs) (Voisin et al., 1999; Voisin and Bourque, 2002).
The cellular mechanism underlying Na+ detection in the MnPO neurons
The association of the background Na+ permeability with the high input resistance of the MnPO neurons (input resistance, >1 GΩ) might represent the cellular mechanism of sodium sensing in this region of the lamina terminalis. Indeed, a small increase in the leak Na+ current ([Na+]out above 150 mM) appears sufficient to depolarize the MnPO neurons, leading to spiking activity. Conversely, a slight reduction of the leak Na+ current ([Na+]out below 150 mM) hyperpolarizes the neurons, thus reducing their excitability.
One possibility was that the sustained Na+ permeability, and thus the Na+ sensing ability, might be attributable to the expression of SICs, highly permeable to Na+, as shown in the hypothalamic MCNs (Voisin et al., 1999). However, the involvement of the SICs in the vMnPO neurons was unlikely, as demonstrated by the inability of Gd3+ to abolish the responses to hypotonic and hypertonic aCSF. Although sensitivity to Gd3+ was often considered to be an intrinsic property of mechanoreceptors, a different class of mechanosensitive channels expressed in corneal epithelial cells, or in colon DRG cells, were insensitive to Gd3+ (Watanabe et al., 1997; Su et al., 2000). In the study by Su et al. (2000), the open probability of the mechanosensitive channels was reduced by amiloride, a pharmacological agent that was also shown to block constitutively active specific Na+ channels (Horisberger, 1998), such as the epithelial Na+ channels involved in Na+ homeostasis (Garty and Palmer, 1997). However, hyperpolarization induced by the hypotonic stimulus was not antagonized by either amiloride or benzamil, a potent amiloride analog. This ruled out the involvement of such amiloride-sensitive Na+ channels in the cellular mechanism of Na+ detection used by the MnPO neurons.
The unsuccessful pharmacological characterization of the natriuric responses suggests that detection of CSF [Na+] in MnPO neurons was mediated by specific channels highly permeable to Na+ but with unknown pharmacology. An attractive possibility might be the presence of the NaX channel on the MnPO neurons, a specific Na+ channel that has been shown to mediate sensing of Na+ levels in the CNS (Hiyama et al., 2002). Several electrophysiological properties, such as TTX resistance, voltage independence, and insensitivity to [Cl-]out and to a change in extracellular osmolarity, are shared with the Na+ current that we report here. These similarities might favor the existence of such a channel in the MnPO neurons.
MnPO neurons encode the atypical sodium channel NaX gene
To verify the latter possibility, we used a riboprobe directed against the NaX mRNA in combination with the immunohistochemical detection of NeuN. This anatomical study revealed that a substantial neuronal population of the MnPO expressed NaX mRNA, supporting the possibility that the background Na+ current flows through the atypical Na+ channel NaX. In addition, the anatomical data shed new light on the cellular mechanisms that are likely involved in Na+ detection by specialized brain areas. Previous studies have shown that the Nav2/NaG gene, recently renamed NaX gene (Goldin et al., 2000), was highly expressed in the circumventricular organs (CVOs) that lack a regular blood-brain barrier (Watanabe et al., 2000; Hiyama et al., 2002). The use of high cellular resolution techniques such as in situ hybridization and immunohistochemistry showed that additional structures located along the third ventricle, such as the MnPO and the periventricular nucleus (data not shown), also express NaX channel. These results emphasize the role of these structures in the detection of Na+ concentration fluctuation within the CSF. In addition, the presence of such channels in the CVOs and periventricular regions, but not in the SON, indicate the diversity of molecular mechanisms dedicated to the detection of osmotic and mineral changes in the structures that constitute the interface between the brain and the fluid compartments (CSF and blood).
Physiological significance
A majority of results reported in our study were obtained with a drastic change in Na+ concentration, and one might reasonably question the role of such Na+ sensors in the context of a highly regulated body Na+ concentration in vivo. However, we showed that a physiological reduction of ∼5% in aCSF [Na+] (150-142 mM NaCl) was able to initiate a neuronal response, i.e., a membrane hyperpolarization. This important result is in the range of CSF [Na+] that is known to induce salt appetite. Indeed, in vivo manipulation of CSF osmotic pressure that reduced CSF by 10-15 mM NaCl triggered a robust sodium intake (Weisinger et al., 1979). Under normal conditions, the human body can maintain [Na+]out between 145 and 135 mM as a physiological range. More drastic alteration of Na+ levels as those used in this study have been, however, reported (Moritz and Ayus, 2002). Hyponatremia is defined as a serum Na+ level inferior to 135 mM, mainly attributable to electrolyte disorders, and might lead to encephalopathy. Severe hyponatremia (107 mM NaCl) has been reported after pituitary tumor (Lin et al., 2002), and cerebral salt wasting syndrome induces acute hyponatremia (120 mM NaCl), usually consecutive to viral infection (Ti et al., 1998; Singh et al., 2002).
Footnotes
This study was supported by the Canadian Institute for Health Research and the Heart and Stroke Foundation of Quebec. D.M. received an award from the Fond de la Recherche en Santé du Québec in partnership with the Quebec Hypertension Society. We thank S. Laforest for excellent technical assistance with IHC and ISHH techniques.
Correspondence should be addressed to Dr. Didier Mouginot, Centre de Recherche en Neurosciences, Centre de Recherche du Centre Hospitalier de l'Université Laval, RC9800, 2705, Boulevard Laurier, Sainte-Foy, Quebec G1V 4G2, Canada. E-mail: dmougino@crchul.ulaval.ca.
Copyright © 2004 Society for Neuroscience 0270-6474/04/243974-11$15.00/0
References
- Akopian AN, Souslova V, Sivilotti L, Wood JN (1997) Structure and distribution of a broadly expressed atypical sodium channel. FEBS Lett 400: 183-187. [DOI] [PubMed] [Google Scholar]
- Aradachi H, Honda K, Negoro H, Kubota T (1996) Median preoptic neurones projecting to the supraoptic nucleus are sensitive to haemodynamic changes as well as to rise in plasma osmolality in rats. J Neuroendocrinol 8: 35-43. [DOI] [PubMed] [Google Scholar]
- Black JA, Yokoyama S, Waxman SG, Oh Y, Zur KB, Sontheimer H, Higashida H, Ransom BR (1994) Sodium channel mRNAs in cultured spinal cord astrocytes: in situ hybridization in identified cell types. Brain Res Mol Brain Res 23: 235-245. [DOI] [PubMed] [Google Scholar]
- Bourque CW, Oliet SHR, Richard D (1994) Osmoreceptors, osmoreception and osmoregulation. Front Neuroendocrinol 15: 231-274. [DOI] [PubMed] [Google Scholar]
- Cox PS, Denton DA, Mouw DR, Tarjan E (1987) Natriuresis induced by localized perfusion within the third cerebral ventricle of sheep. Am J Physiol 252: R1-R6. [DOI] [PubMed] [Google Scholar]
- Denton DA, McKinley M, Weisinger RS (1996) Hypothalamic integration of body fluid regulation. Proc Natl Acad Sci USA 93: 7397-7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont EC, Rafrafi S, Laforest S, Drolet G (1999) Involvement of central angiotensin receptors in stress adaptation. Neuroscience 93: 877-884. [DOI] [PubMed] [Google Scholar]
- Garty H, Palmer LG (1997) Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359-396. [DOI] [PubMed] [Google Scholar]
- Gautron S, Dos Santos G, Pinto-Henrique D, Koulakoff A, Gros F, Bernwald-Netter Y (1992) The glial voltage-gated sodium channel: cell- and tissue-specific mRNA expression. Proc Natl Acad Sci USA 89: 7272-7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldin AL, Barchi RL, Caldwell JH, Hofmann F, Howe JR, Hunter JC, Kallen RG, Mandel G, Meisler MH, Netter YB, Noda M, Tamkun MM, Waxman SG, Wood JN, Catterall WA (2000) Nomenclature of voltage-gated sodium channels. Neuron 28: 365-368. [DOI] [PubMed] [Google Scholar]
- Hiyama TY, Watanabe E, Ono K, Inenaga K, Tamkun MM, Yoshida S, Noda M (2002) Na(x) channel involved in CNS sodium-level sensing. Nat Neurosci 5: 511-512. [DOI] [PubMed] [Google Scholar]
- Honda K, Negoro H, Dyball RE, Higuchi T, Takano S (1990) The osmoreceptor complex in the rat: evidence for interactions between the supraoptic and other diencephalic nuclei. J Physiol (Lond) 431: 225-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horisberger JD (1998) Amiloride-sensitive Na channels. Curr Opin Cell Biol 10: 443-449. [DOI] [PubMed] [Google Scholar]
- Hussy N, Deleuze C, Desarmenien M, Moos F (2000) Osmotic regulation of neuronal activity: a new role for taurine and glial cells in a hypothalamic neuroendocrine structure. Prog Neurobiol 62: 113-134. [DOI] [PubMed] [Google Scholar]
- Landas S, Philipps MI (1987) Comparative anatomy of the organum vasculosum of the lamina terminalis. In: Circumventricular organs and body fluids (Gross P, ed), pp 131-156. Boca Raton, FL: CRC.
- Lin SH, Hung YH, Lin YF (2002) Severe hyponatremia as the presenting geature of clinically non-functional pituitary adenoma with hypopituitarism. Clin Nephrol 57: 85-88. [DOI] [PubMed] [Google Scholar]
- McKinley M, Gerstberger R, Mathai M, Oldfield B, Schmid H (1999) The lamina terminalis and its role in fluid and electrolyte homeostasis. J Clin Neurosci 6: 289-301. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, Clevers J, Denton DA, Oldfield BJ, Penschow J, Rundgren M (1987) Fine structure of the organum vasculosum of the lamina terminalis. In: Circumventricular organs and body fluids (Gross P, ed), pp 111-130. Boca Raton, FL: CRC.
- McKinley MJ, Bicknell RJ, Hards D, McAllen RM, Vivas L, Weisinger RS, Oldfield BJ (1992) Efferent neural pathways of the lamina terminalis subserving osmoregulation. Prog Brain Res 91: 395-402. [DOI] [PubMed] [Google Scholar]
- McKinley MJ, McAllen RM, Davern P, Giles ME, Penschow J, Sunn N, Uschakov A, Oldfield BJ (2003) The sensory circumventricular organs of the mammalian brain. Adv Anat Embryol Cell Biol 172: 1-127. [DOI] [PubMed] [Google Scholar]
- Moritz ML, Ayus JC (2002) Disorders of water metabolism in children: hyponatremia and hypernatremia. Pediatr Rev 23: 371-380. [PubMed] [Google Scholar]
- Oldfield BJ, McKinley MJ (1995) Circumventricular organs. In: The rat nervous system, Ed 2 (Paxinos G, ed), pp 391-405. Sydney: Academic.
- Oldfield BJ, Miselis RR, McKinley MJ (1991) Median preoptic nucleus projections to vasopressin-containing neurones of the supraoptic nucleus in sheep. A light and electron microscopic study. Brain Res 542: 193-200. [DOI] [PubMed] [Google Scholar]
- Oldfield BJ, Hards DK, McKinley MJ (1992) Neurons in the median pre-optic nucleus of the rat with collateral branches to the subfornical organ and supraoptic nucleus. Brain Res 586: 86-90. [DOI] [PubMed] [Google Scholar]
- Park R, Denton DA, McKinley MJ, Pennington G, Weisinger RS (1989) Intracerebroventricular saccharide infusions inhibit thirst induced by systemic hypertonicity. Brain Res 493: 123-128. [DOI] [PubMed] [Google Scholar]
- Simmons DM, Arriza JL, Swanson LW (1989) A complete protocol for in situ hybridization. J Histotechnol 12: 169-181. [Google Scholar]
- Singh S, Bohn D, Carlotti AP, Cusimano M, Rutka JT, Halperin ML (2002) Cerebral salt wasting: truth, fallacies, theories and challenges. Crit Care Med 30: 2575-2579. [DOI] [PubMed] [Google Scholar]
- Su X, Wachtel RE, Gebhart GF (2000) Mechanosensitive potassium channels in rat colon sensory neurons. J Neurophysiol 84: 836-843. [DOI] [PubMed] [Google Scholar]
- Swanson LW (1992) Brain maps: structure of the rat brain. Amsterdam: Elsevier.
- Ti LK, Kang SC, Cheong KF (1998) Acute hyponatremia secondary to cerebral salt wasting syndrome in a pationet with tuberculous meningitis. Anaesth Intensive Care 26: 420-423. [DOI] [PubMed] [Google Scholar]
- Travis KA, Johnson AK (1993) In vitro sensitivity of median preoptic neurons to angiotensin II, osmotic pressure and temperature. Am J Physiol 264: R1200-R1205. [DOI] [PubMed] [Google Scholar]
- Voisin DL, Bourque CW (2002) Integration of sodium and osmosensory signals in vasopressin neurons. Trends Neurosci 25: 199-205. [DOI] [PubMed] [Google Scholar]
- Voisin DL, Chakfe Y, Bourque CW (1999) Coincident detection of CSF Na+ and osmotic pressure in osmoregulatory neurons of the supraoptic nucleus. Neuron 24: 453-460. [DOI] [PubMed] [Google Scholar]
- Watanabe SI, Tanizaki M, Kaneko A (1997) Two types of stretch-activated channels coexist in the rabbit corneal epithelial cell. Exp Eye Res 64: 1027-1035. [DOI] [PubMed] [Google Scholar]
- Watanabe E, Fujikawa A, Matsunaga H, Yasoshima Y, Sako N, Yamamoto T, Saegusa C, Noda M (2000) Nav2/NaG channel is involved in control of salt-intake behavior in the CNS. J Neurosci 20: 7743-7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisinger RS, Considine P, Denton DA, McKinley MJ (1979) Rapid effect of change in cerebrospinal fluid sodium concentration on salt appetite. Nature 280: 490-491. [DOI] [PubMed] [Google Scholar]
- Weisinger RS, Considine P, Denton DA, Leksell L, McKinley MJ, Mouw DR, Muller AF, Tarjan E (1982) Role of sodium concentration of the cerebrospinal fluid in the salt appetite of sheep. Am J Physiol 242: R51-R63. [DOI] [PubMed] [Google Scholar]