

Abstract
The N-ethylmaleimide-sensitive factor (NSF) and soluble NSF attachment protein (SNAP) are cytosolic factors that promote vesicle fusion with a target membrane in both the constitutive and regulated secretory pathways. NSF and SNAP are thought to function by catalyzing the disassembly of a SNAP receptor (SNARE) complex consisting of membrane proteins of the secretory vesicle and target membrane. Although studies of NSF function have provided strong support for this model, the precise biochemical role of SNAP remains controversial. To further explore the function of SNAP, we have used mutational and transgenic approaches in Drosophila to investigate the effect of altered SNAP dosage on neurotransmitter release and SNARE complex metabolism. Our results indicate that reduced SNAP activity results in diminished neurotransmitter release and accumulation of a neural SNARE complex. Increased SNAP dosage results in defective synapse formation and a variety of tissue morphological defects without detectably altering the abundance of neural SNARE complexes. The SNAP overexpression phenotypes are enhanced by mutations in other secretory components and are at least partially overcome by co-overexpression of NSF, suggesting that these phenotypes derive from a specific perturbation of the secretory pathway. Our results indicate that SNAP promotes neurotransmitter release and SNARE complex disassembly but inhibits secretion when present at high abundance relative to NSF.
Keywords: Drosophila, mutant, secretion, synapse, SNAP, NSF, SNARE
Introduction
Vesicle fusion with a target membrane in the constitutive and regulated secretory pathways requires a core set of proteins conserved from yeast to mammals (Jahn and Sudhof, 1999; Klenchin and Martin, 2000; Lin and Scheller, 2000; Wickner and Haas, 2000; Sollner, 2003). Among these proteins are the cytosolic N-ethylmaleimide-sensitive factor (NSF) and the soluble NSF attachment protein (SNAP) and membrane proteins of the target and vesicle membranes known as SNAP receptors (SNAREs). SNARE proteins assemble into a stable complex after vesicle docking, and the formation of this complex is thought to either activate docked vesicles for fusion or catalyze the vesicle fusion reaction. Biochemical studies have shown that SNAP binds to the SNARE complex and facilitates the recruitment of NSF to this complex (Clary and Rothman, 1990). NSF is a hexameric ATPase that catalyzes disassembly of the SNARE complex on hydrolysis of ATP in either a prefusion priming step or after vesicle fusion to regenerate a pool of uncomplexed SNAREs (Jahn and Sudhof, 1999; Klenchin and Martin, 2000; Lin and Scheller, 2000; Wickner and Haas, 2000; Sollner, 2003).
Although in vitro studies indicate that SNAP and NSF act in concert to catalyze disassembly of a SNARE complex, in vivo studies of NSF and SNAP have produced conflicting results regarding the functions of these components. For example, studies in Drosophila and yeast have shown that SNARE complex abundance in these organisms is inversely correlated with NSF activity (Sogaard et al., 1994; Littleton et al., 1998; Tolar and Pallanck, 1998; Mohtashami et al., 2001). These observations are consistent with expectations from in vitro studies of NSF function and strongly support the involvement of NSF in SNARE complex disassembly. In contrast, SNARE complex abundance in yeast is correlated with SNAP activity, thus challenging the proposed role of this factor in SNARE complex disassembly (Sogaard et al., 1994; Wang et al., 2000). Although increased NSF activity does not detectably affect secretion (Golby et al., 2001), studies in yeast have shown that increased SNAP activity inhibits membrane fusion, and this inhibition can be overcome by a coordinate increase in NSF activity (Wang et al., 2000). These findings have led to the suggestion that SNAP may promote the formation or stability of SNARE complexes residing in the same membrane, thereby inhibiting fusion at a high concentration by precluding the formation of trans-SNARE complexes between membranes. However, this model appears to conflict with findings in the squid and crayfish systems, in which it has been shown that neurotransmitter release is enhanced by increased SNAP activity (He et al., 1999; Wang et al., 2000).
To further explore the effects of altered SNAP dosage on neurotransmitter release and SNARE complex metabolism, we have initiated a genetic and molecular analysis of the Drosophila SNAP gene. Our studies indicate that reduced SNAP activity results in attenuation of neurotransmitter release and increased SNARE complex abundance. Although moderate increases in SNAP expression do not detectably affect neurotransmitter release or the abundance of neural SNARE complexes, high levels of SNAP expression result in reduced viability, tissue morphological defects, and severely attenuated neurotransmission owing to defective synapse formation. The SNAP overexpression phenotypes are enhanced by loss-of-function mutations in other secretory components and suppressed by co-overexpression of NSF. Our results indicate that SNAP plays a positive role in vesicle fusion by disassembling SNARE complexes but also possesses inhibitory properties that are dependent on the relative abundance of NSF.
Materials and Methods
Fly strains and maintenance. Drosophila stocks were maintained on standard cornmeal agar and raised at 25°C, unless otherwise indicated. The Df(3L)rdgC-co2, Df(3L)ri-79C, Tm6B-green fluorescent protein (GFP), syx1AΔ229, w-; P{w+mC = GAL4-ninaE.GMR}, w-;P{[w+] elavC155-GAL4}, and heat shock protein (HSP)-GAL4 stocks were obtained from the Bloomington Stock Center. The Ok6-GAL4 stock was provided by Dr. Edward Giniger (Fred Hutchinson Cancer Research Center, Seattle, WA).
Generation and characterization of SNAP mutants. To identify deletions that remove the SNAP gene, a collection of stocks bearing deletions mapping to polytene region 77-78 were obtained. Polytene chromosomes prepared from these stocks were subjected to in situ hybridization with a probe corresponding to the SNAP gene (Pardue, 2000). This analysis led to the identification of the Df(3L)rdgC-co2 deletion, which removes the SNAP gene, and the overlapping Df(3L)ri-79C deletion, which fails to remove SNAP. To generate SNAP alleles, a chromosome bearing the Df(3L)rdgC-co2 deletion was used in an F2 screen for recessive lethal mutations mapping to the deletion interval. This screen was performed by feeding 25 mm ethylmethanesulfonate (EMS; Grigliatti, 1998) to males bearing an isogenic chromosome 3 marked with the recessive mutation scarlet [designated iso(3)st]. Mutagenized males were then mated to TM3/TM6B females, and balanced offspring from this cross were individually mated to Df(3L)rdgC-co2 st/TM6C females. Stocks bearing recessive lethal mutations mapping to the Df(3L)rdgC-co2 deletion interval were established by recovering TM6C offspring. A single SNAP allele was recovered from this screen and designated SNAPl65 (see Results).
Representative mutations corresponding to a collection of recessive lethal complementation groups obtained in a screen using a chromosome bearing the Df(3L)rdgC-co2 deletion were kindly provided by Dr. Mark Fortini's (University of Pennsylvania, Philadelphia, PA) laboratory (Lukinova et al., 1999). These mutations were individually crossed to a stock bearing the SNAPl65 mutation. Two mutations, designated M4 and P2, representing complementation groups l(3)77ABa and l(3)77ABc, respectively, failed to complement the SNAPl65 mutation. Additional complementation testing revealed that the M4 mutation represents an allele of the l(3)77ABc complementation group.
Lethal phase analysis of the SNAP alleles was performed by placing the SNAP mutations in trans to a GFP-labeled TM6B balancer chromosome and crossing to a Df(3L)rdgC-co2/TM6B-GFP stock. Non-GFP embryos from this cross were monitored until lethality occurred. DNA extracted from these non-GFP progeny was subjected to PCR amplification and sequencing using primers specific to the SNAP gene to identify the SNAP mutations. Sequencing of the SNAP gene recovered from the l(3)77ABcM3 and l(3)77ABcO1 mutants revealed that these mutants bear identical SNAP mutations. Given that these mutations likely represent clonal derivatives of one another, only the l(3)77ABcM3 allele was used in this study.
Transgenic lines. To generate a SNAP genomic DNA transgenic construct, genomic clones encoding SNAP were identified using a SNAP cDNA to screen a Drosophila genomic DNA library according to established procedures (Maniatis et al., 1989). One of the clones recovered from this screen, s14, was subcloned into pBluescript in two steps. First, a 2.6 kb SacI-HindIII fragment of s14 bearing the C-terminal SNAP coding sequences and 3′ untranslated region was ligated to the SacI-HindIII sites of pBluescript (see Fig. 1 A). This construct was then digested with SacI, and a 6.3 kb SacI subclone of s14 bearing the N-terminal coding sequences of SNAP and ∼5 kb of upstream sequence was ligated into this SacI site. Restriction mapping was used to confirm the proper orientation of the 6.3 kb SacI fragment with respect to the 2.6 kb SacI-HindIII fragment. The resulting 8.9 kb genomic DNA fragment, bearing the entire SNAP coding sequence and ∼5 kb of upstream sequence, was excised from pBluescript using the BssHII and KpnI sites flanking this DNA fragment and inserted into the AscI and KpnI sites of pCasper. This transgene was introduced into the Drosophila germ line using established procedures (Spradling and Rubin, 1982). A single transgenic line was generated and designated P[SNAP+].
Figure 1.

The Drosophila SNAP mutations. A, The SNAP transcript is shown above a genomic DNA fragment that was used to rescue the SNAP mutant phenotypes. The locations of mutations likely to affect transcript splicing are shown above the SNAP transcript (capitalized sequences represent exons), and those affecting the SNAP coding sequence are shown below the transcript. B, Western blot analysis of head protein extracts from Canton-S (wild-type) flies using the anti-SNAP antiserum revealed a band of ∼35 kDa that is not detected by the preimmune serum. The size of this band is in good agreement with the expected size of SNAP protein (33 kDa). Furthermore, the intensity of this band is decreased by approximately half in lanes corresponding to a protein extract from a strain that is heterozygous for the Df(3L)rdgC-co2 deletion (def/+), which removes the SNAP gene, and significantly increased in lanes from transgenic flies overexpressing SNAP protein (HSP-GAL4/UAS-SNAP-1, designated HS-UAS-SNAP-1). C, Levels of SNAP protein present in head extracts from heterozygous SNAP mutants, SNAPM4 homozygotes, and SNAPM4 hemizygotes relative to a control [iso(3)st]. Genotype abbreviations: iso(3)st, isogenic parental chromosome; def/+, Df(3L)rdgC-co2/+; l65/+, SNAPl65/+; I1/+, SNAPI1/+; M3/+, SNAPM3/+; G8/+, SNAPG8/+; P2/+, SNAPP2/+; M4/+, SNAPM4/+; M4/M4, SNAPM4/SNAPM4; M4/def, SNAPM4/Df(3L)rdgC-co2. Error bars indicate SEM.
To generate a UAS-responsive SNAP transgene, the SNAP cDNA clone dS2 was excised from pBluescript using the XhoI and SmaI restriction sites flanking this cDNA (Ordway et al., 1994). This cDNA was then ligated to an XhoI- and XbaI-digested pUAST vector after converting the XbaI restriction site in pUAST to a blunt end using DNA polymerase. This construct was used to generate germ line transformant flies using established procedures (Spradling and Rubin, 1982). A single transgenic line was generated and designated UAS-SNAP-1. The UAS-SNAP-5 line was established from the UAS-SNAP-1 line by introducing an exogenous source of transposase to induce transposition of the SNAP transgene and selecting for flies with eye color intensity differing from that of the original UAS-SNAP-1 strain. The UAS-dNSF1 (73C) and UAS-dNSF2 (23A) transgenic lines used in this work were described previously (Golby et al., 2001).
Preparation of SNAP antiserum.A KpnI restriction site was engineered into the N-terminal SNAP coding sequence of the SNAP cDNA clone dS2 by altering the sequence of SNAP codon three from GAC to ACC. The SNAP coding sequence was excised from this modified dS2 cDNA as a KpnI-PstI fragment and inserted into the pQE30 vector (Qiagen, Valencia, CA) for use in producing a His6-tagged SNAP fusion protein. Recombinant SNAP protein was expressed from this vector and purified according to established procedures (Qiagen). Purified SNAP protein was subjected to preparative SDS gel electrophoresis, and a polyacrylamide gel fragment containing the recombinant protein was used to immunize rabbits through the assistance of the animal facility at the University of Wisconsin (Madison, WI).
Western blot analyses. Analysis of SNARE complex levels was performed using a monoclonal antibody (mAb) to syntaxin1A (Fujita et al., 1982) as previously described (Tolar and Pallanck, 1998). To minimize experimental variability arising from Western blot analysis, all experimental samples analyzed were loaded adjacent to samples prepared from Canton-S flies (wild-type). SNARE complex and syntaxin monomer abundance in each sample was quantified using an Eastman Kodak Co. (Rochester, NY) Image Station 440. The fold change in the ratio of SNARE complex abundance to syntaxin monomer abundance was determined by comparing this ratio in experimental and Canton-S samples loaded adjacent to one another. The mean fold change and SEM were determined from eight independent loadings per sample.
Protein samples for Western blot analysis using the anti-SNAP antiserum were prepared in a manner similar to those used in SNARE complex analysis but were boiled for 5 min before gel electrophoresis. Samples were subjected to electrophoresis and Western blotting as described (Tolar and Pallanck, 1998) using the anti-SNAP antiserum at a 1:5,000 dilution. A mouse anti-actin antiserum (Sigma, St. Louis, MO) was used to normalize protein loading. The ratio of SNAP abundance to actin abundance was determined as described above for SNARE complex quantitation.
Confocal microscopy. Third instar larvae were prepared and stained as described (Bellen and Budnik, 2000). Samples were incubated with SNAP antiserum (1:2,000) and imaged using a Bio-Rad (Hercules, CA) MRC600 confocal microscope. These samples were co-stained with FITC-conjugated anti-HRP (Jackson ImmunoResearch, West grove, PA) or anti-endophilin1A (Verstreken et al., 2002) and anti-VAP-33 antisera [both kindly provided by Hugo Bellen (Baylor College of Medicine, Houston, TX); Pennetta et al., 2002]. Preimmune serum was used at the same concentration as the SNAP immune serum to control for antibody specificity. To examine neuromuscular junction (NMJ) structure in the SNAPM4/Df(3L)rdgC-co2 and Ok6-gal4 UAS-SNAP-5 larvae, samples were incubated with the 22C10 mAb (Hummel et al., 2000) and antisyt [kindly provided by Troy Littleton (Massachusetts Institute of Technology Department of Biology, Cambridge, MA); Littleton et al., 1993].
Electrophysiology. All larval electrophysiological recordings were made in hemolymph-like solution (HL3) (Stewart et al., 1994) containing 1 mm CaCl2. The motor neurons innervating abdominal segment 3 (Budnik et al., 1990) were stimulated with 0.3 msec pulses using a voltage sufficient to reliably elicit a response from both axons 1 and 2 (Lnenicka and Keshishian, 2000). All intracellular recordings were made from body wall muscle 6 of segment 3, and only recordings from cells with a resting membrane potential (RMP) exceeding -60 mV were analyzed. The amplitude of an excitatory junctional potential (EJP) subsequent to the first pulse in a 10 Hz train (EJPs summate at 10 Hz in HL3 containing 1 mm CaCl2) was defined as the baseline before the first pulse (RMP) subtracted from the RMP at the peak of the EJP. All measurements were made from at least six cells and four different animals in each genotype. Average EJP amplitude was calculated from a minimum of 10 EJPs evoked at 0.1 Hz from each cell. EJPs were corrected for nonlinear summation using the procedure of McLachlan and Martin (1981), as adapted by Feeney et al. (1998). Average miniature EJP (mEJP amplitude) was calculated from a minimum of 40 mEJPs from each cell. Quantal content is calculated by dividing the corrected EJP amplitude by the mEJP amplitude.
Electroretinogram (ERG) recordings were performed as described Pak et al. (1969). The absolute amplitude (millivolts) of the ON and OFF transients were measured from a series of light and dark pulses of 1 sec duration each. The amplitude of at least five ON and OFF transients was averaged for each fly tested, and at least eight animals were used for each genotype analyzed. The mean and SE were calculated from the average ON and OFF transient amplitude.
Scanning electron microscopy. Adult flies were dehydrated by 15 min incubations in a graded ethanol series and then hexamethyldisilazane-treated, mounted on stubs, gold-palladium sputter-coated, and examined with scanning electron microscopy.
Transmission electron microscopy. Third instar larvae were prepared as described (Bellen and Budnik, 2000) and fixed in Karnovsky's fixative containing 1 mm MgCl. Nerve terminals from muscle 6 and 7 segments 2 and 3 were identified and imaged. Only vesicles lying within a box extending 100 nm to either side of the t-bar structure and extending 200 nm into the nerve terminal from the plasma membrane were analyzed in this study. The average vesicle size per 200 × 200 nm box was calculated per release site for each genotype analyzed. Synaptic vesicle distribution was measured by counting the number of synaptic vesicles at 20 nm intervals extending inward from the plasma membrane and then calculating the percentage of total vesicles per interval. At least 18 release sites were analyzed per genotype, and the mean and SEM are plotted for both vesicle size and distribution.
Results
Generation of recessive lethal SNAP mutations
To generate SNAP mutations, we conducted an F2 screen for recessive lethal mutations mapping to the Df(3L)rdgC-co2 deletion, which removes the SNAP gene. From a screen of ∼7000 EMS-mutagenized third chromosomes, 43 mutations were recovered that were lethal in trans to Df(3L)rdgC-co2. Mating these mutants to a stock bearing the Df(3L)ri-79C deficiency chromosome, which partially overlaps with Df(3L)rdgC-co2 but does not remove the SNAP gene, eliminated 28 mutants as SNAP candidates. To test whether any of the remaining 15 mutations represent SNAP alleles, all of the candidate SNAP mutants were crossed to stocks bearing the Df(3L)rdgC-co2 deletion and an 8.9 kb SNAP genomic rescue construct designated P[SNAP+] (Fig. 1A). This transgenic construct was able to rescue the recessive lethal phenotype of only a single mutation, designated SNAPl65. The SNAPl65 mutation complemented all other candidate SNAP mutations, indicating that it is the sole SNAP allele derived from this screen.
To identify a larger set of SNAP alleles, the SNAPl65 mutant was crossed to a collection of mutants obtained from an identical screen for recessive lethal mutations in the Df(3R)rdgC-co2 interval that was performed independently by another laboratory (Lukinova et al., 1999). This complementation analysis led to the identification of five mutations that failed to complement one another and the SNAPl65 allele. The recessive lethal phenotypes of all of these mutants were fully rescued by the P[SNAP+] transgene, confirming that these mutations are alleles of SNAP. These five mutations, previously designated l(3)77AbcI1, l(3)77AbcM3, l(3)77AbcG8, l(3)77AbcP2, and l(3)77AbaM4 (Lukinova et al., 1999), will hereafter be referred to as SNAPI1, SNAPM3, SNAPG8, SNAPP2, and SNAPM4, respectively, to designate their identities as SNAP alleles.
To further characterize these six SNAP mutants, the SNAP gene was recovered from these mutants and the SNAP coding regions and intron-exon boundaries were sequenced. Additionally, Western blot analysis using an antiserum that specifically recognizes Drosophila SNAP protein was used to investigate the effects of these mutations on protein abundance (Fig. 1B). Finally, lethal phase analysis was performed to investigate the severity of the SNAP mutations and to identify the developmental stages that require SNAP function.
Four of the SNAP alleles identified in this analysis, SNAPM3, SNAPI1, SNAPP2, and SNAPM4, were found to have mutations affecting the SNAP coding sequence (Fig. 1A). The SNAPM3 mutation alters the presumptive SNAP start codon from ATG to ATA and severely reduces SNAP protein abundance when heterozygous with a wild-type SNAP allele, indicating that this mutation abrogates translational initiation from the normal start site. Embryos bearing the SNAPM3 mutation in trans to the Df(3L)rdgC-co2 display mild cuticle secretion defects, fail to undergo tracheal inflation, and die late in embryogenesis. The severe phenotype of SNAPM3 mutants and substantial reduction of SNAP protein conferred by the SNAPM3 mutation in trans to a wild-type SNAP allele suggest that SNAPM3 represents a null or dominant negative allele of the SNAP gene.
The SNAPI1 and SNAPP2 mutations result in premature stop codons in the SNAP coding sequence. The SNAPI1 mutation resides approximately two-thirds of the way into the SNAP coding sequence, whereas the SNAPP2 mutation resides in the penultimate codon. The embryonic lethality and apparent absence of a truncated SNAP derivative produced by the SNAPI1 allele suggest that this mutation represents a null allele of SNAP. However, SNAPP2 heterozygotes appear to produce nearly normal amounts of SNAP protein, indicating that truncation of the SNAP C-terminal amino acid affects SNAP function without affecting protein stability (Fig. 1C).
The SNAPM4 mutation alters a highly conserved alanine residue located at position 59 to a threonine residue. Western blot analysis of SNAPM4 heterozygotes, homozygotes, and hemizygotes [SNAPM4/Df(3L)rdgC-co2] indicates that this mutation results in a 30-40% reduction in SNAP protein levels. The SNAPM4 mutation resides in a region of SNAP that is implicated in SNARE protein interactions and thus likely also exerts its effect through altered SNARE complex binding. The SNAPM4 mutation is semilethal, producing a low percentage of adult survivors in trans to the Df(3L)rdgC-co2 deletion. Hemizygous SNAPM4 adult escapers are uncoordinated, exhibit increasingly frequent seizures, and typically die within 3 d of eclosion. Homozygous SNAPM4 mutants produce viable adults that lack the severe behavioral phenotypes observed in the SNAPM4 hemizygotes. The milder phenotype displayed by the SNAPM4 homozygotes relative to the SNAPM4 hemizygotes indicates that this mutation is a hypomorphic allele of SNAP.
The remaining SNAP mutations, SNAPl65 and SNAPG8, reside in functionally important SNAP intron sequences. The SNAPl65 allele alters the first nucleotide in the third intron of the SNAP gene from guanine to adenine. This residue is invariably a guanine in all eukaryotic introns, and the SNAPl65 mutation likely abolishes splicing of this intron (Snustad and Simmons, 2000). The early lethal phase of SNAPl65 mutants suggests that this mutation results in complete loss of SNAP activity. The SNAPG8 allele affects a moderately conserved site in the first intron of SNAP. Although Western blot analysis indicates that this mutation severely affects SNAP protein production, lethal phase analysis suggests that this mutation is not a null allele of SNAP (Fig. 1).
Altering SNAP dosage attenuates synaptic transmission
Previous work has shown that Drosophila SNAP transcripts are present throughout development and are enriched in the embryonic nervous system (Ordway et al., 1994); however, the spatial distribution of SNAP protein in Drosophila has not previously been addressed. As a prelude to electrophysiological analysis of SNAP mutants, immunocytochemical analysis of the larval NMJ was conducted using an anti-SNAP antiserum. Results of this analysis revealed strong SNAP immunoreactivity co-localizing with a neuronal marker at the larval NMJ (Fig. 2A-C). The staining pattern produced by the anti-SNAP antiserum appears to be specific because preimmune serum from the same rabbit fails to stain the NMJ (Fig. 2D,E).
Figure 2.

SNAP protein localizes to the presynaptic nerve terminal. Double labeling of the NMJ of larval muscles 6 and 7 with an anti-SNAP antiserum (A) and the neuronal marker anti-HRP (B) shows extensive overlap of immunoreactivity (C). Preimmune serum from rabbits immunized with SNAP protein fails to label these structures (D, E). Double-labeling experiments with anti-SNAP antiserum (F) and an antiserum to the presynaptic protein endophilin (G) reveals coincidental co-immunoreactivity (H). A single confocal section through synaptic boutons labeled with anti-SNAP antiserum (I) and antiserum to the presynaptic and postsynaptic membrane-associated protein VAP-33 (J) reveals a lack of co-localization between these markers (K). Scale bars, 5 μm.
To investigate whether the SNAP staining pattern reflects presynaptic or postsynaptic localization, the pattern of SNAP immunoreactivity was compared with those of endophilin, which localizes to intracellular regions of the presynaptic terminal (Verstreken et al., 2002), and VAP-33, which localizes to both the presynaptic and postsynaptic membranes (Pennetta et al., 2002). Confocal microscopy revealed strong colocalization of SNAP and endophilin (Fig. 2F-H) but little with VAP-33 (Fig. 2I-K), indicating that SNAP protein localizes predominantly or exclusively to the intracellular presynaptic nerve terminal.
To explore a possible role of SNAP in presynaptic neurotransmitter release, electrophysiological studies were performed on SNAP mutant embryos. Because of a defect in the structural integrity of motor neurons detected upon dissection of the putative null alleles of SNAP, only the severe hypomorphic SNAPG8 and SNAPP2 alleles were used in this analysis. Results of these studies failed to detect differences in the amplitude of EJP responses between SNAP mutants and control animals, possibly reflecting the sizeable maternal contribution of SNAP protein masking a loss of SNAP function (data not shown). In an effort to circumvent this potential complication, additional electrophysiological recordings were performed in third instar larvae and adults using the hypomorphic SNAPM4 allele (Fig. 3). Electrophysiological studies of SNAPM4 hemizygous larvae (M4/def) showed that the EJP amplitude was not significantly different from that measured in wild-type control larvae (elavC155-w1118; Fig. 3A). However, the average amplitude of mEJPs was significantly increased in SNAPM4 hemizygotes (Fig. 3C), which masked a reduced quantal content in these mutant larvae (Fig. 3D). The P[SNAP+] transgene completely rescued the reduced quantal content phenotype of SNAPM4 hemizygotes (Fig. 3D), demonstrating that this defect results from a loss of SNAP activity. Repetitive stimulation at 10 Hz revealed no difference in facilitation or synaptic fatigue in SNAPM4 mutants and controls (Fig. 3F). No difference was detected in the mEJP frequency in SNAPM4 hemizygotes relative to controls (data not shown).
Figure 3.
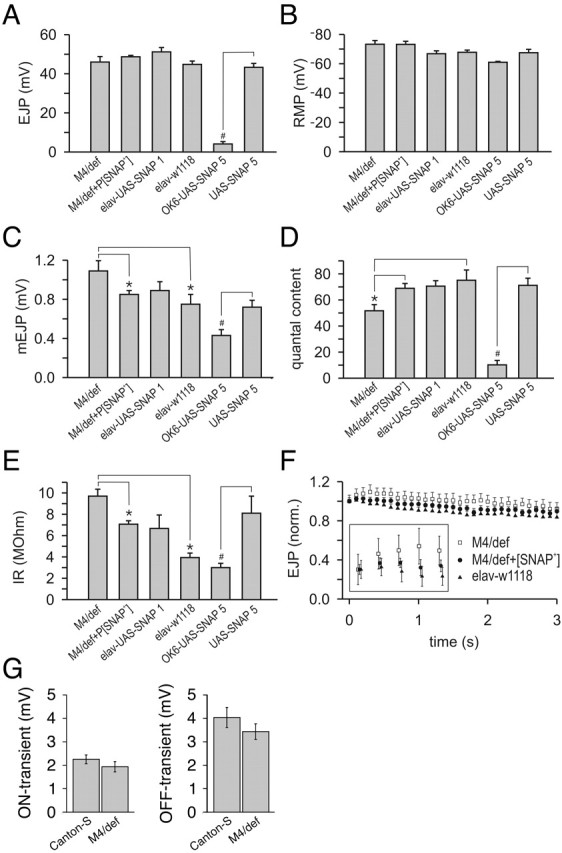
Altering SNAP dosage results in attenuation of synaptic transmission. A, Average amplitude of EJPs plotted for each of six larval genotypes: SNAPM4/Df(3L)rdgC-co2 (M4/def), SNAPM4/Df(3L)rdgC-co2 P[SNAP+] (M4/def+P[SNAP+]), elavC155-GAL4;UAS-SNAP-1/+ (elav-UAS-SNAP1), elavC155-GAL4 w1118 (elav-w1118), OK6-GAL4/UAS-SNAP-5 (OK6-UAS-SNAP5), and UAS-SNAP-5/UAS-SNAP-5 (UAS-SNAP5). B, Average RMP for each genotype. C, Average amplitude of mEJPs for each genotype. D, Average quantal content for each genotype. E, Input resistance of each genotype. F, Average EJP amplitude during repetitive stimulation at 10 Hz. F, inset, Detail of the first five EJP amplitudes of the 10 Hz train. G, Average absolute amplitudes of the ON and OFF transients plotted for SNAPM4/Df(3L)rdgC-co2 (M4/def) and wild-type controls (Canton-S). Error bars indicate SEM. *,#Significant differences at p < 0.05; *one-way ANOVA followed by a Student-Newman-Keuls pair wise multiple-comparison test; #unpaired (2-tailed) t test.
To investigate the effect of reduced SNAP function on neurotransmitter release during the adult stage of development, ERG recordings were performed on SNAPM4 hemizygotes. The ON and OFF transients of the ERG reflect synaptic transmission between photoreceptor terminals and second-order neurons in the lamina and medulla (Burg et al., 1993), and the amplitude of these events represents a crude measure of synaptic transmission. Although not statistically significant from controls, the ON and OFF transient amplitudes of the ERG were found to be mildly reduced in SNAPM4 hemizygotes relative to controls (Fig. 3G). Use of the eyeless-GAL4 UAS-FLP system (Stowers and Schwarz, 1999) to generate mosaic animals bearing eyes that are completely homozygous for a null allele of SNAP resulted in the ablation of eye tissue, indicating that SNAP function is required for general cell viability (data not shown).
Because previous work has produced conflicting results on the effect of increased SNAP activity on exocytosis, we sought to explore the role of SNAP overexpression on neurotransmitter release. To perform this experiment, a full-length SNAP cDNA was subcloned into the GAL4-responsive PUAST plasmid, and transgenic lines were generated using this construct. Two independent transgenic lines, designated UAS-SNAP-1 and UAS-SNAP-5, were used in this experiment. Third instar larvae bearing the UAS-SNAP-1 transgene and the elavC155-GAL4 driver, which expresses in all neurons (Robinow and White, 1988), were subjected to electrophysiological analysis at the NMJ. Overexpression of SNAP from the UAS-SNAP-1 line failed to significantly affect any of the parameters of transmitter release measured (Fig. 3A-D). Use of elavC155-GAL4 to express SNAP protein from the UAS-SNAP-5 transgene resulted in embryonic lethality precluding electrophysiological studies at the NMJ of third instar larvae. However, use of the Ok6-GAL4 driver, which expresses GAL4 only in motor neurons (Aberle et al., 2002), in conjunction with the UAS-SNAP-5 transgene, allowed survival until the third instar larval stage of development. Although third instar larvae bearing the Ok6-GAL4 and the UAS-SNAP-5 transgene are viable, they are nearly paralyzed and display severe electrophysiological defects (Fig. 3A,C,D,E), indicating that high levels of SNAP expression inhibit synaptic transmission.
Altered SNAP dosage affects SNARE complex metabolism and synapse morphology
To investigate the mechanisms by which altered SNAP dosage affects neurotransmitter release, we investigated SNARE complex abundance, NMJ morphology, and synaptic ultrastructure in SNAP mutants and SNAP-overexpressing lines. SNARE complex abundance was analyzed by subjecting adult head extracts to Western blot analysis using an antiserum to the neuronal SNARE protein syntaxin (Fig. 4A). The amount of syntaxin incorporated into 73 kDa SNARE complexes relative to the syntaxin monomer was increased in flies heterozygous for a SNAP deficiency or heterozygous for any of the SNAP mutations identified in this work, demonstrating that SNAP dosage is limiting for optimal SNARE complex disassembly (Fig. 4B). The SNAPP2 allele confers the greatest increase in SNARE complex/SNARE monomer ratio among the SNAP heterozygotes analyzed in this work. The finding that the SNARE complex/SNARE monomer ratio of SNAPP2 heterozygotes exceeds that of SNAP deficiency heterozygotes suggests that the SNAPP2 mutation represents a dominant negative allele. Previous analysis has shown that a mutationally altered version of mammalian SNAP that bears an amino acid substitution at the site immediately adjacent to the codon affected by the SNAPP2 mutation dominantly inhibits exocytosis in adrenal chromaffin cells (Barnard et al., 1997; Graham and Burgoyne, 2000). This derivative is thought to act by inhibiting the activation of NSF, and the dominant negative behavior of the SNAPP2 allele may result from a similar inhibitory effect on NSF. The finding that SNAPG8 heterozygotes also manifest a larger than expected SNARE complex/SNARE monomer ratio, raises the possibility that this mutation is also a dominant negative allele of SNAP. However, the mechanism by which the SNAPG8 mutation would result in a dominant negative allele is unclear.
Figure 4.

SNARE complex abundance is increased in SNAP mutants. Protein extracts from adult heads were subjected to Western blot analysis with an mAb that recognizes the SNARE protein syntaxin. A, Representative Western blot depicting the SNARE complex and syntaxin monomer in a wild-type control sample (Canton-S) and animals bearing the SNAPM4 mutation in trans to the Df(3L)rdgC-co2 deletion (M4/def). Note the increase in SNARE complex abundance and decrease in syntaxin monomer abundance in the hemizygous SNAPM4 mutant relative to Canton-S. B, The ratio of SNARE complex abundance to syntaxin monomer abundance is plotted relative to iso(3)st (for details, see Materials and Methods). Genotype abbreviations are as defined in Figures 1 and 3 but also include the following: M4/def + elav-UAS-SNAP1, elavC155;UAS-SNAP-1/+;SNAPM4/Df(3L)rdgC-co2; elav-UAS-SNAP1, elavC155;UAS-SNAP-1/+. All of the genotypes shown manifest significant differences from the iso(3)st control with the exception of I1/+ and elav-UAS-SNAP-1 by unpaired (2-tailed) t tests (p < 0.05). Additionally, M4/def + P[SNAP+] and M4/def + elav-UAS-SNAP1 manifest significant differences from the M4/def alone using the above test. Error bars indicate SEM.
Although the ratios of SNARE complex level to syntaxin monomer level were increased in all of the mutants, the highest ratios were observed in SNAPM4 homozygotes and hemizygotes (Fig. 4A,B). SNAPM4 homozygotes and hemizygotes display fourfold and sixfold increased ratios of SNARE complex to monomer, respectively. The increase in SNARE complex seen in SNAPM4 hemizygotes is substantially rescued by introduction of the P[SNAP+] transgene or by using the elavC155-GAL4 driver in conjunction with the UAS-SNAP-1 transgene to ectopically express SNAP in a SNAPM4 mutant background (Fig. 4). These results demonstrate that SNAP functions to disassemble SNARE complexes in vivo and suggest that the electrophysiological phenotypes seen in SNAPM4 mutants derive directly from reduced SNARE complex disassembly.
Previous studies have produced conflicting results concerning the effects of NSF and SNAP overexpression on SNARE complex abundance. Elevated NSF expression in the Drosophila nervous system has been reported to greatly reduce the amount of SNARE complex (Golby et al., 2001), whereas elevated SNAP levels in yeast increase the abundance of SNARE complexes (Wang et al., 2000). To examine the effects of SNAP overproduction on neuronal SNARE complexes, the UAS-SNAP-1 transgene was expressed using the pan-neuronal elavC155-GAL4 driver, and SNARE complex abundance in an adult head extract was quantified (Fig. 4B). In this configuration, SNAP overexpression failed to detectably affect the ratio of SNARE complex to monomer relative to controls, indicating that, unlike NSF, endogenous SNAP protein levels are not limiting for optimal SNARE complex disassembly.
To determine whether defects in NMJ formation or maintenance underlie the electrophysiological phenotypes observed on alteration of SNAP dosage, confocal microscopy was used to examine larval NMJ morphology in SNAP mutants and SNAP-overexpressing lines. This analysis was conducted with antibodies specific to the axonal filament-associated protein futsch (Hummel et al., 2000) and the synaptic vesicle protein synaptotagmin (Littleton et al., 1993). Although NMJ morphology in SNAPM4 hemizygotes was indistinguishable from that of wild-type controls (Fig. 5B), animals overexpressing SNAP from the UAS-SNAP-5 transgene using the Ok6-GAL4 driver displayed dramatic reductions in axonal branching and had few or no synaptic boutons (Fig. 5D). The synaptic structural alterations resulting from SNAP overexpression appeared more severe in the posterior hemisegments, with most of muscles 6 and 7 in the third hemisegment being completely devoid of synapses. Although nerve terminal morphology is greatly perturbed by SNAP overexpression, axonal regions that are more proximal to the cell body appear unaltered, suggesting that SNAP overexpression preferentially affects synapse formation or maintenance. These results indicate that the physiological phenotypes resulting from SNAP overexpression derive primarily, if not exclusively, from defects in synapse formation, maintenance, or both. Synaptic structural alterations were not observed on overexpression of SNAP protein from the UAS-SNAP-1 transgene, consistent with the lack of electrophysiological phenotypes associated with over-expression of SNAP protein from this transgene (data not shown).
Figure 5.

SNAP overexpression disrupts neuromuscular junction morphology. The larval NMJs of muscles 6 and 7 were subject to immunohistochemistry using antisera to the axonal filament protein futch (green) and the synaptic vesicle protein synaptotagmin (red). NMJ morphology in SNAPM4 hemizygotes (B) is similar to that in control larvae (A). Use of the Ok6-GAL4 line to drive overexpression of SNAP protein from the UAS-SNAP-5 transgene results in a dramatically reduced synaptic bouton number (D) relative to a control (C). Scale bars: A, B,10 μm; C, D, 50 μm.
Given the findings that reduced SNAP activity results in reduced quantal content and increased mEJP amplitude, we investigated whether these phenotypes derive from altered synaptic vesicle diameter, distribution, or abundance. Results of ultrastructural analysis indicate that the diameter of synaptic vesicles, the distribution of synaptic vesicles, and the abundance of synaptic vesicles in the nerve terminal in SNAPM4 hemizygotes are not significantly different from those of wild-type controls (Fig. 6A-F). Although we do not detect a statistically significant difference in the diameter of synaptic vesicles in SNAPM4 mutants and controls, given the mild increase in mEJP amplitude in SNAPM4 mutants, we cannot definitively exclude the possibility that this increase in quantal size derives from a correspondingly small increase in synaptic vesicle size.
Figure 6.
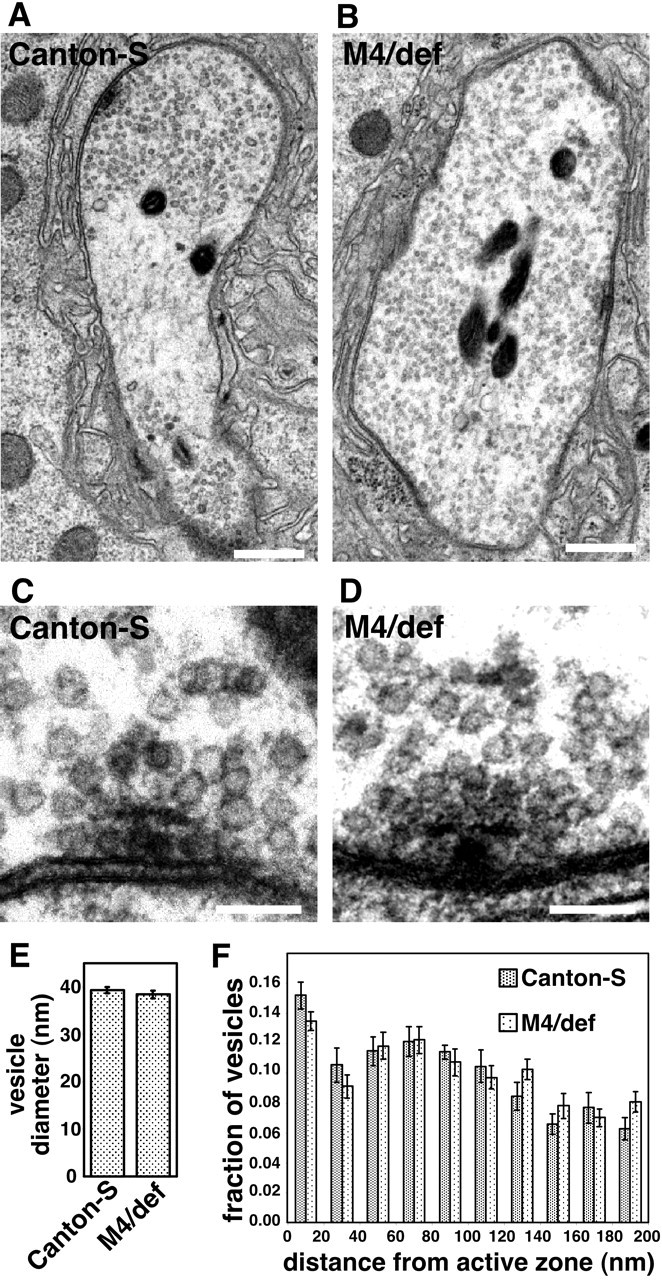
Synaptic vesicle size, distribution, and number are unaltered in SNAPM4 mutants. A, B, Representative electron micrographs of the larval NMJ at muscle 6 and 7 segment 3 from Canton-S control larvae (A) and SNAPM4/Df(3L)rdgC-co2 hemizygotes (M4/def; B). C, D, Higher-magnification images of synaptic vesicles surrounding the t-bar structures located at sites of neurotransmitter release in control larvae (C) and larvae hemizygous for the SNAPM4 mutation (D). The average diameter of synaptic vesicles in SNAPM4 hemizygotes is not significantly different from that of Canton-S using unpaired (2-tailed) t tests (E). The distribution of synaptic vesicles within 200 nm of release sites (F) is grossly similar in SNAPM4 hemizygotes and Canton-S. The total numbers of synaptic vesicles within 200 nm of release sites in Canton-S (33.0 ± 1.8) and SNAPM4 hemizygotes (35.4 ± 1.8) are also within experimental error of one another. Scale bars: A, B, 100 nm; C, D, 10 nm.
SNAP overexpression inhibits secretion
Although ectopic expression of SNAP using the elavC155-GAL4 driver in conjunction with the UAS-SNAP-1 transgene failed to reveal a phenotype in larvae, adults of this genotype manifest wing inflation defects (Fig. 7A). This phenotype is indicative of reduced neurohormonal secretion, and similar phenotypes have been observed after expression of neurotoxic proteins using the elavC155-GAL4 driver (Nie et al., 1999). Overexpression of SNAP in the compound eye using the GMR-GAL4 driver in conjunction with the UAS-SNAP-1 transgene results in mildly rough eyes containing patches of necrotic tissue (Fig. 7D). Higher magnification of these eyes using scanning electron microscopy revealed slight furrowing along the perimeter of the ommatidia (Fig. 7D, bottom panel). Use of the GMR-GAL4 driver to express SNAP protein from the stronger UAS-SNAP-5 insertion resulted in almost complete ablation of the eye, indicating that these phenotypes are correlated with levels of SNAP expression (Fig. 7E).
Figure 7.
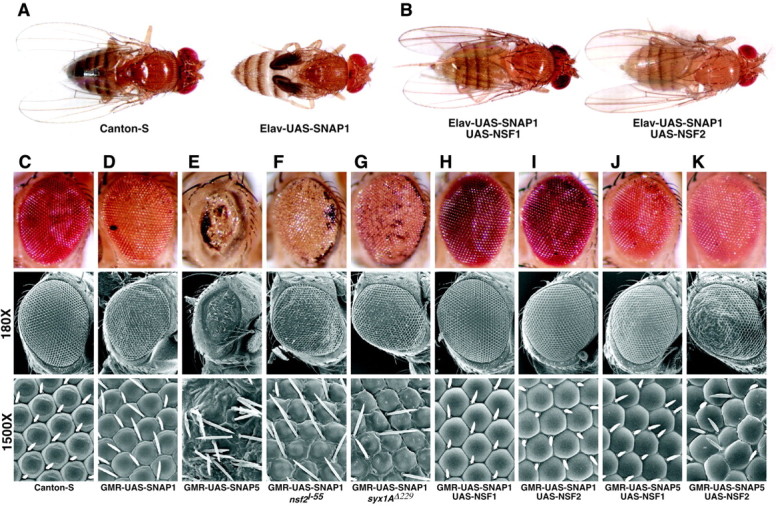
SNAP overexpression phenotypes are modified by altered dosage of other secretory components. Pan-neuronal expression of the UAS-SNAP-1 transgene using the elavC155-GAL4 driver results in a wing inflation defect (A), and co-expression of either dNSF1 or dNSF2 completely suppresses this phenotype (B). GMR-GAL4-driven expression of UAS-SNAP-1 produces a mildly rough eye showing patches of necrosis, mild furrowing at ommatidial borders, and progressive degeneration (D) with respect to wild type (C). GMR-GAL4-driven expression of the stronger UAS-SNAP-5 results in near total ablation of the eye (E). The presence of a single copy of the dNSF2l55 mutation (F) or the syxΔ229 mutation (G) in the GMR-GAL4 UAS-SNAP-1 background resulted in enhancement of the phenotypes resulting from ectopic expression of SNAP in the eye, including increased necrosis around the perimeter of the compound eye and more dramatic furrowing at ommatidial junctions (bottom panels). Phenotypes associated with GMR-GAL4-driven expression of UAS-SNAP-1 and UAS-SNAP-5 are suppressed by co-expression of either dNSF1 or dNSF2 (H-K). Genotype abbreviations include the following: Elav-UAS-SNAP1, elavC155-GAL4/+;UAS-SNAP-1/+; Elav-UAS-SNAP1 UAS-NSF1, elavC155-GAL4/+;UAS-SNAP-1/UAS-NSF1(73C); Elav-UAS-SNAP1 UAS-NSF2, elavC155-GAL4/+;UAS-SNAP-1/UAS-NSF2(23A); GMR-UAS-SNAP1, GMR-GAL4/UAS-SNAP-1; GMR-UAS-SNAP5, GMR-GAL4/UAS-SNAP-5; GMR-UAS-SNAP1 nsf2l-55, GMR-GAL4 UAS-SNAP-1/nsf2l-55; GMR-UAS-SNAP1 syx1AΔ229, GMR-GAL4 UAS-SNAP-1/syx1AΔ229; GMR-UAS-SNAP1 UAS-NSF1, GMR-GAL4 UAS-SNAP-1/UAS-dNSF1(73C); GMR-UAS-SNAP1 UAS-NSF2, GMR-GAL4 UAS-SNAP-1/UAS-dNSF2(23A); GMR-UAS-SNAP5 UAS-NSF1, GMR-GAL4/UAS-dNSF1(73C) UAS-SNAP-5; GMR-UAS-SNAP5 UAS-NSF2, GMR-GAL4 UAS-dNSF2(23A)/UAS-SNAP-5.
The phenotypes resulting from SNAP overexpression may result from a specific perturbation of the secretory pathway or, alternatively, from the interference of excess SNAP protein with an unrelated biological process. To distinguish between these possibilities, we altered the gene dosage of secretory components that are known to physically interact with SNAP in a background of SNAP overexpression and investigated whether these perturbations modified the SNAP overexpression phenotypes. Introduction of heterozygous loss-of-function alleles of dNSF2 (Golby et al., 2001) or syntaxin1A (Littleton et al., 1998) into a SNAP overexpression background resulted in enhancement of the UAS-SNAP-1 eye overexpression phenotype (Fig. 7F,G). This enhancement is manifested as increased necrotic tissue around the perimeter of the compound eye and greater furrowing and overlapping of ommatidia (compare Fig. 7D).
Experiments in yeast have shown that the inhibition of vesicle fusion resulting from increased SNAP activity is overcome by increasing NSF levels. To test whether increased NSF activity can also overcome the phenotypes resulting from increased Drosophila SNAP expression, SNAP protein was co-expressed with either of the two Drosophila NSF proteins, dNSF1 and dNSF2. Co-expression of either dNSF1 or dNSF2 completely suppressed the phenotypes imparted by the UAS-SNAP-1 transgene and partially suppressed the phenotypes conferred by the stronger UAS-SNAP-5 transgene (Fig. 7B,H-K). Control experiments involving co-overexpression of the SNAP transgene and a GFP or β-gal UAS-responsive transgene failed to modify the SNAP overexpression phenotypes, indicating that suppression is specific to NSF and is not merely the result of titrating GAL4 protein (data not shown). The sensitivity of the SNAP overexpression phenotypes to perturbation of other secretory components strongly suggests that these phenotypes derive from excess SNAP specifically interfering with the secretory pathway.
Discussion
In an effort to clarify the role of SNAP in membrane trafficking, we explored the effects of altered SNAP dosage on neurotransmitter release and SNARE complex metabolism in Drosophila. In the course of our studies, we generated a collection of SNAP mutants that display lethality at a broad range of developmental stages, indicating that SNAP function is essential for viability and is required throughout development. Detailed analysis of a hypomorphic SNAP allele revealed that SNAP plays a positive role in the regulated fusion of synaptic vesicles at the synapse, presumably by promoting SNARE complex disassembly. SNAP overexpression results in severe tissue morphological defects, loss of viability, and aberrant synapse formation, maintenance, or both. Although SNAP overexpression does not detectably affect SNARE complex metabolism, the finding that the SNAP overexpression phenotypes can be modified by altering the dosage of other secretory components indicates that these phenotypes derive directly from secretory defects.
Many of the results obtained in this study support current models of SNAP function. In particular, our findings that reduced SNAP function results in elevated SNARE complex levels and reduced neurotransmitter release are consistent with the proposed biochemical role of SNAP in SNARE complex disassembly and secretion. Although the effect of reduced SNAP dosage on SNARE complex abundance in Drosophila conflicts with previous work in yeast (Sogaard et al., 1994), our results are consistent with those observed in Drosophila and yeast NSF mutants (Sogaard et al., 1994; Littleton et al., 1998; Tolar and Pallanck, 1998; Mohtashami et al., 2001). Thus, our results support previous biochemical studies indicating that these proteins collaborate to disassemble SNARE complexes but do not support studies in yeast suggesting that SNAP functions to stabilize SNARE complexes. The finding that SNARE complex abundance is decreased in yeast SNAP mutants (Sogaard et al., 1994) may indicate that yeast SNARE complexes are unstable in the absence of SNAP or may reflect a difference in constitutive and regulated secretory mechanisms. Although we do not observe decreased SNARE complex abundance on overexpression of SNAP, as is seen on overexpression of Drosophila NSF (Golby et al., 2001), this finding may simply indicate that NSF is limiting under normal circumstances.
Although our studies support a positive role for SNAP in SNARE complex disassembly and regulated secretion, there are several important differences between our results and previous work on NSF function in Drosophila. Electrophysiological and ultrastructural analyses of dNSF1 mutants have shown that these mutants display an activity-dependent reduction in neurotransmitter release and an accumulation of docked synaptic vesicles at sites of neurotransmitter release (Ordway et al., 1994). Although SNAPM4 mutants also display reduced neurotransmitter release, the magnitude of this reduction is quite mild, and this defect is not accompanied by an activity-dependent component, nor is there an alteration in the distribution of synaptic vesicles in the nerve terminal. SNAPM4 hemizygotes also display increased mEJP amplitude, whereas dNSF1 mutants do not appear to display an altered mEJP amplitude (R. Ordway, personal communication).
One potential explanation for these discordant results relates to the nature of the SNAP and dNSF1 alleles analyzed. The dNSF1 mutations analyzed in previous work are conditional, temperature-sensitive alleles that appear to completely lack dNSF1 function at elevated temperatures (Kawasaki et al., 1998), whereas the SNAPM4 mutation appears to be a weak hypomorph. The SNAPM4 mutation may not be severe enough to result in a substantial reduction in neurotransmitter release or in detectable ultrastructural alterations. Furthermore, the significantly larger muscle input resistance observed in the SNAPM4 hemizygotes (Fig. 3E) raises the possibility that the increased mEJP amplitude derives from postsynaptic compensation in response to a presynaptic deficit in neurotransmitter release during development. Such a phenomenon would not be expected in conditional dNSF1 mutants, given that these mutants exhibit normal, or nearly normal, neurotransmitter release kinetics at permissive temperatures. The lack of an activity-dependent component of the SNAPM4 electrophysiological phenotype is not easily reconciled with previous work on NSF function in Drosophila; further work, possibly involving additional NSF and SNAP alleles may resolve this conflict.
Another feature that complicates a direct comparison of Drosophila SNAP mutants with Drosophila dNSF1 mutants relates to the possible existence of factors with functions redundant to SNAP and dNSF1. Although temperature-sensitive dNSF1 mutants manifest a striking electrophysiological defect at restrictive temperatures in adults (Kawasaki et al., 1998), electrophysiological studies of these same alleles at the larval stage of development failed to detect phenotypic differences from controls (Mohtashami et al., 2001). Although these results may reflect differences in the properties of the synaptic preparations used in these studies, another distinct possibility is that the dNSF1 and dNSF2 genes play redundant roles at the larval neuromuscular junction (Golby et al., 2001). A similar explanation may underlie the lack of electrophysiological phenotypes in embryos hemizygous for the SNAPG8 and SNAPP2 mutations and the mild electrophysiological phenotypes displayed by SNAPM4 mutants. In addition to the SNAP gene analyzed in this work, there appear to be at least two additional SNAP-like genes in the Drosophila genome (CG3988 and CG6208) that might play redundant roles with SNAP. The CG3988 and CG6208 genes have not previously been subjected to genetic or molecular analysis, so further work will be required to explore the possible redundancy of these genes with SNAP and to investigate other models for the lack of electrophysiological phenotypes in SNAP mutant embryos and the mild electrophysiological deficits in SNAPM4 mutants.
Perhaps the most unexpected result of our work relates to the toxicity associated with ectopic expression of SNAP. Several lines of evidence suggest that this toxicity derives from a defect in vesicle trafficking. First, flies that ectopically express SNAP in the nervous system display phenotypes that are consistent with defective neuropeptide secretion (McNabb et al., 1997). Second, the SNAP overexpression phenotypes observed in flies are modified by altering the abundance of syntaxin, dNSF1, and dNSF2, all of which function intimately with SNAP in the secretory pathway. Finally, ectopic expression of SNAP in Drosophila imaginal disks has been shown to result in the accumulation of Golgi membranes (Dunne et al., 2002), demonstrating that excess SNAP is capable of disrupting constitutive secretion. Although our results suggest that ectopic expression of SNAP can result in a perturbation of the secretory pathway, whether SNAP overexpression facilitates or inhibits secretion in a given tissue will likely depend on the relative abundance of SNAP, NSF, SNAREs, and possibly other secretory factors in that tissue.
A study of the yeast SNAP protein sec17p has shown that elevated levels of sec17p inhibit homotypic vacuolar fusion by driving the assembly of cis-SNARE complexes, thereby precluding the formation of trans-SNARE complexes required for vesicle fusion (Wang et al., 2000). This inhibitory effect of sec17p on vacuole fusion can be overcome by addition of excess sec18p, the yeast NSF counterpart. Our findings suggest that a similar mechanism may underlie the SNAP overexpression phenotypes in Drosophila. Although increased Drosophila SNAP abundance does not induce increased SNARE complex abundance, as is seen in the yeast system, reducing the abundance of the neuronal SNARE syntaxin1A enhances the phenotype resulting from ectopic expression of SNAP. Thus, our results are consistent with the model that SNAP overexpression reduces the availability of this SNARE. Furthermore, the Drosophila SNAP overexpression phenotypes are at least partially suppressed by co-overexpression of dNSF1 or dNSF2. Nevertheless, there are additional models that could explain our findings, and further work will be required to definitively explain the SNAP overexpression phenotypes.
In summary, our findings that loss of SNAP function results in reduced neurotransmitter release and elevated SNARE complex abundance strongly support current models proposing that NSF and SNAP promote neurotransmitter release by collaborating to disassemble SNARE complexes. However, our results also demonstrate that overexpression of SNAP is toxic, likely as a result of secretory pathway inhibition. Although the mechanistic basis for the inhibitory effects of SNAP on secretion requires further work, our finding that the SNAP overexpression phenotypes can be modified by altering the abundance of other secretory factors suggests a genetic means for identifying novel components of the vesicle trafficking-neurotransmitter release apparatus. Further studies involving the reagents described here should provide additional insight into the molecular mechanisms of vesicle trafficking.
Footnotes
This work was supported by National Science Foundation Grant 9734125 (L.P.). We thank the Berkeley Drosophila Genome Project and Bloomington Drosophila Stock Center for sequence data and fly stocks used in this work and the Electron Microscopy Shared Resource Laboratory of the Fred Hutchinson Cancer Research Center for assistance with scanning and transmission electron microscopic analysis. We also thank Mark Fortini, Nina Lukinova, and Edward Giniger for fly stocks, Hugo Bellen and Troy Littleton for providing antisera, and Jesse Goldmark, Carrie Jones, and Robert Kreber for help with technical aspects of this work. We thank all members of the Pallanck laboratory for critical comments on this manuscript. Finally, we thank Steve Stowers for helping with electrophysiological studies on embryos and Barry Ganetzky and Bing Zhang for sponsoring some of this work in their laboratories.
Correspondence should be addressed to Leo Pallanck, Department of Genome Sciences, University of Washington, 1705 Northeast Pacific Street, Health Sciences J-113, Seattle, WA 98195. E-mail: pallanck@gs.washington.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/243964-10$15.00/0
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS (2002) Wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila Neuron 33: 545-558. [DOI] [PubMed] [Google Scholar]
- Barnard RJ, Morgan A, Burgoyne RD (1997) Stimulation of NSF ATPase activity by alpha-SNAP is required for SNARE complex disassembly and exocytosis. J Cell Biol 139: 875-883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen HJ, Budnik V (2000) The neuromuscular junction. In: Drosophila protocols (Sullivan W, Hawley RS, Ashburner M, eds), pp 175-199. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Budnik V, Zhong Y, Wu CF (1990) Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci 10: 3754-3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg MG, Sarthy PV, Koliantz G, Pak WL (1993) Genetic and molecular identification of a Drosophila histidine decarboxylase gene required in photoreceptor transmitter synthesis. EMBO J 12: 911-919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clary DO, Rothman JE (1990) Purification of three related peripheral membrane proteins needed for vesicular transport. J Biol Chem 265: 10109-10117. [PubMed] [Google Scholar]
- Dunne JC, Kondylis V, Rabouille C (2002) Ecdysone triggers the expression of Golgi genes in Drosophila imaginal discs via broad-complex. Dev Biol 245: 172-186. [DOI] [PubMed] [Google Scholar]
- Feeney CJ, Karunanithi S, Pearce J, Govind CK, Atwood HL (1998) Motor nerve terminals on abdominal muscles in larval flesh flies, Sarcophaga bullata: comparisons with Drosophila J Comp Neurol 402: 197-209. [PubMed] [Google Scholar]
- Fujita SC, Zipursky SL, Benzer S, Ferrus A, Shotwell SL (1982) Monoclonal antibodies against the Drosophila nervous system. Proc Natl Acad Sci USA 79: 7929-7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golby JA, Tolar LA, Pallanck L (2001) Partitioning of N-ethylmaleimide-sensitive fusion (NSF) protein function in Drosophila melanogaster: dNSF1 is required in the nervous system, and dNSF2 is required in mesoderm. Genetics 158: 265-278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham ME, Burgoyne RD (2000) Comparison of cysteine string protein (Csp) and mutant alpha-SNAP overexpression reveals a role for csp in late steps of membrane fusion in dense-core granule exocytosis in adrenal chromaffin cells. J Neurosci 20: 1281-1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigliatti TA (1998) Mutagenesis. In: Drosophila: a practical approach, Ed 2 (Roberts BD, ed), pp 55-64. New York: Oxford UP.
- He P, Southard RC, Chen D, Whiteheart SW, Cooper RL (1999) Role of alpha-SNAP in promoting efficient neurotransmission at the crayfish neuromuscular junction. J Neurophysiol 82: 3406-3416. [DOI] [PubMed] [Google Scholar]
- Hummel T, Krukkert K, Roos J, Davis G, Klambt C (2000) Drosophila Futsch/22C10 is a MAP1B-like protein required for dendritic and axonal development. Neuron 26: 357-370. [DOI] [PubMed] [Google Scholar]
- Jahn R, Sudhof TC (1999) Membrane fusion and exocytosis. Annu Rev Biochem 68: 863-911. [DOI] [PubMed] [Google Scholar]
- Kawasaki F, Mattiuz AM, Ordway RW (1998) Synaptic physiology and ultrastructure in comatose mutants define an in vivo role for NSF in neurotransmitter release. J Neurosci 18: 10241-10249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenchin VA, Martin TF (2000) Priming in exocytosis: attaining fusion-competence after vesicle docking. Biochimie 82: 399-407. [DOI] [PubMed] [Google Scholar]
- Lin RC, Scheller RH (2000) Mechanisms of synaptic vesicle exocytosis. Annu Rev Cell Dev Biol 16: 19-49. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Bellen HJ, Perin MS (1993) Expression of synaptotagmin in Drosophila reveals transport and localization of synaptic vesicles to the synapse. Development 118: 1077-1088. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B (1998) Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron 21: 401-413. [DOI] [PubMed] [Google Scholar]
- Lnenicka GA, Keshishian H (2000) Identified motor terminals in Drosophila larvae show distinct differences in morphology and physiology. J Neurobiol 43: 186-197. [PubMed] [Google Scholar]
- Lukinova NI, Roussakova VV, Fortini ME (1999) Genetic characterization of cytological region 77A-D harboring the presenilin gene of Drosophila melanogaster Genetics 153: 1789-1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Sambrook J, Fritsch EF (1989) In situ hybridization of bacteriophage lambda plaques. In: Molecular cloning: a laboratory manual, Ed 2 (Nolan C, ed), pp 2.108-102.121. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- McLachlan EM, Martin AR (1981) Non-linear summation of end-plate potentials in the frog and mouse. J Physiol (Lond) 311: 307-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNabb SL, Baker JD, Agapite J, Steller H, Riddiford LM, Truman JW (1997) Disruption of a behavioral sequence by targeted death of peptidergic neurons in Drosophila Neuron 19: 813-823. [DOI] [PubMed] [Google Scholar]
- Mohtashami M, Stewart BA, Boulianne GL, Trimble WS (2001) Analysis of the mutant Drosophila N-ethylmaleimide sensitive fusion-1 protein in comatose reveals molecular correlates of the behavioural paralysis. J Neurochem 77: 1407-1417. [DOI] [PubMed] [Google Scholar]
- Nie Z, Ranjan R, Wenniger JJ, Hong SN, Bronk P, Zinsmaier KE (1999) Overexpression of cysteine-string proteins in Drosophila reveals interactions with syntaxin. J Neurosci 19: 10270-10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway RW, Pallanck L, Ganetzky B (1994) Neurally expressed Drosophila genes encoding homologs of the NSF and SNAP secretory proteins. Proc Natl Acad Sci USA 91: 5715-5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pak WL, Grossfield J, White NV (1969) Nonphototactic mutants in a study of vision of Drosophila Nature 222: 351-354. [DOI] [PubMed] [Google Scholar]
- Pardue M (2000) In situ hybridization to polytene chromosomes. In: Drosophila protocols (Sullivan W, Hawley RS, Ashburner M, eds), pp 119-129. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Pennetta G, Hiesinger P, Fabian-Fine R, Meinertzhagen I, Bellen H (2002) Drosophila VAP-33A directs bouton formation at neuromuscular junctions in a dosage-dependent manner. Neuron 35: 291-306. [DOI] [PubMed] [Google Scholar]
- Robinow S, White K (1988) The locus elav of Drosophila melanogaster is expressed in neurons at all developmental stages. Dev Biol 126: 294-303. [DOI] [PubMed] [Google Scholar]
- Snustad DP, Simmons MJ (2000) Transcription and RNA processing. In: Principles of genetics, Ed 2 (Harras D, ed), pp 292-325. New York: Wiley.
- Sogaard M, Tani K, Ye RR, Geromanos S, Tempst P, Kirchhausen T, Rothman JE, Sollner T (1994) A rab protein is required for the assembly of SNARE complexes in the docking of transport vesicles. Cell 78: 937-948. [DOI] [PubMed] [Google Scholar]
- Sollner TH (2003) Regulated exocytosis and SNARE function (Rev). Mol Membr Biol 20: 209-220. [DOI] [PubMed] [Google Scholar]
- Spradling AC, Rubin GM (1982) Transposition of cloned P elements into Drosophila germ line chromosomes. Science 218: 341-347. [DOI] [PubMed] [Google Scholar]
- Stewart BA, Atwood HL, Renger JJ, Wang J, Wu CF (1994) Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J Comp Physiol [A] 175: 179-191. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Schwarz TL (1999) A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics 152: 1631-1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar LA, Pallanck L (1998) NSF function in neurotransmitter release involves rearrangement of the SNARE complex downstream of synaptic vesicle docking. J Neurosci 18: 10250-10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstreken P, Kjaerulff O, Lloyd TE, Atkinson R, Zhou Y, Meinertzhagen IA, Bellen HJ (2002) Endophilin mutations block clathrin-mediated endocytosis but not neurotransmitter release. Cell 109: 101-112. [DOI] [PubMed] [Google Scholar]
- Wang L, Ungermann C, Wickner W (2000) The docking of primed vacuoles can be reversibly arrested by excess Sec17p (alpha-SNAP). J Biol Chem 275: 22862-22867. [DOI] [PubMed] [Google Scholar]
- Wickner W, Haas A (2000) Yeast homotypic vacuole fusion: a window on organelle trafficking mechanisms. Annu Rev Biochem 69: 247-275. [DOI] [PubMed] [Google Scholar]