

Abstract
Plasma membrane neurotransmitter transporters determine in part the concentration, time course, and diffusion of extracellular transmitter. Much has been learned about how substrate translocation through the transporter occurs; however, the precise way in which transporter structure maps onto transporter function has not yet been fully elucidated. Here, biochemical and electrophysiological approaches were used to test the hypothesis that intracellular domains of the rat brain GABA transporter (GAT1) contribute to the transport process. Injection of a peptide corresponding to the presumed fourth intracellular loop of the transporter (IL4) into oocytes expressing GAT1 greatly reduced both forward and reverse transport and reduced the transport rate in a dose-dependent manner. Coinjection of the IL4 peptide with a peptide corresponding to the N-terminal cytoplasmic tail of GAT1 reversed the IL4-mediated inhibition; this reversal, and direct binding between these two domains, was prevented by mutagenesis of charged residues in either the IL4 or N-terminal domains. Furthermore, syntaxin 1A, a soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein that inhibits GAT1 transport rates via interactions with the N-terminal tail of GAT1 was unable to regulate the GAT1 IL4 mutant. Together, these data suggest a model in which the GAT1 IL4 domain serves as a barrier for transport, and this barrier can be regulated through intra-molecular and inter-molecular interactions.
Keywords: transport, GABA, protein, uptake, synapse, peptide
Introduction
GABA is the major inhibitory neurotransmitter in the CNS, and its action is regulated in part by GABA transporters. At particular synapses, these transporters reduce the time course and amplitude of inhibitory currents (Dingledine and Korn, 1985; Isaacson et al., 1993) and prevent synaptic spillover (Ichinose and Lukasiewicz, 2002; Overstreet and Westbrook, 2003). Previous studies identified functional parameters of GABA uptake, including ion dependencies, transport rates, and pharmacology (Radian et al., 1986; Borden, 1996). The cloning of the first GABA transporter (GAT1) (Guastella et al., 1990) began the process of mapping function onto structure. GAT1 likely has 12 transmembrane domains with intracellular N and C termini. The functional role of the C-terminal tail is unclear, because its removal has little effect on substrate translocation (Mabjeesh and Kanner, 1992). GABA binding is affected by mutations in the third transmembrane domain (Bismith et al., 1997) and in extracellular loops (Tamura et al., 1995). Ion binding maps onto residues at or near the first transmembrane domain (Pantanowitz et al., 1993; Mager et al., 1996). However, transporter conformations that correspond to states of substrate binding and permeation remain to be determined.
GAT1 binds the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein syntaxin 1A (Deken et al., 2000), and many transporters are regulated in this way (Geerlings et al., 2000; Haase et al., 2001; Sung et al., 2003). For GAT1, syntaxin 1A decreases GABA transport rates fourfold (Deken et al., 2000). Because syntaxin 1A binds to the N-terminal tail of GAT1 (N-Tail), it suggests that intracellular transporter domains regulate substrate transport. This hypothesis is supported by studies involving mutagenesis of the N-terminal tail of GAT1. Mutations at position R44 greatly reduce unidirectional flux by slowing reorientation of GAT1 to its outward facing conformation (Bennett et al., 2000). Mutagenesis of charged residues at the syntaxin 1A binding site in GAT1 mimic the reduction in transport rates seen with syntaxin 1A binding (Deken et al., 2000).
However, the mechanism by which the N-terminal tail of GAT1 regulates substrate flux remains unclear. One possibility is that the tail interacts with the permeation path through intra-molecular interactions. For ion channels, such interactions among intracellular domains are responsible for regulating substrate permeation in many different channel families. A classic example is the inactivation ball of Shaker K+ channels (Hoshi et al., 1990), in which amino acids in the N-terminal tail bind near, and occlude, the channel pore. In cystic fibrosis transmembrane regulator (CFTR) Cl- channels, syntaxin 1A binds the N-terminal cytoplasmic tail of CFTR, and this interaction influences tail binding to the regulatory domain of CFTR (Naren et al., 1999). Thus, a common mechanism for ion channel and transporter regulation may be through a series of intra-molecular and inter-molecular interactions. Here, we examine intracellular interactions that govern activity of the rat brain GABA transporter GAT1.
Materials and Methods
Cell culture. Xenopus oocyte culture has been described previously (Quick and Lester, 1994). Oocytes were maintained at 18°C in medium containing ND96 (96 mm NaCl, 2 mm KCl, 1 mm MgCl2, 5 mm HEPES, pH 7.4), 1.8 mm CaCl2,50 μg/ml gentamicin, and 5% horse serum. RNA was synthesized in vitro using Message Machine (Ambion, Austin, TX). To produce approximately equal expression levels, oocytes were injected with 1 ng of wild-type (WT) GAT1 cRNA or 10 ng of mutant GAT1 cRNA. Syntaxin 1A cRNA was injected at a 1:1 molar ratio with each GAT1 construct.
Constructs. For the fusion protein generation, cDNAs encoding the particular domains were amplified by PCR and subcloned into pGEX (Amersham Biosciences, Arlington Heights, IL). These domains were expressed as glutathione S-transferase (GST) fusion proteins in E. coli, purified on glutathione agarose, eluted in 20 mm glutathione, and dialyzed against PBS. GST was removed using thrombin. Synthetic peptides were made using a PerSeptive Biosystems (Foster City, CA) 9050 peptide synthesizer. Mutant constructs were made using Altered Sites (Promega, Madison, WI); specific sites were replaced with alanine residues. All mutants were verified by DNA sequencing. The following GAT1 and syntaxin 1A constructs were used. cRNAs: WT-GAT1; aa 1-599; GAT1 triple mutant at positions D41, D43, and D45 (3D-GAT1); GAT1 double mutant at positions G39 and L41 (2M-GAT1); GAT1 quadruple mutant at positions R414, R417, R419, and R420 (IL4ΔR-GAT1); syntaxin 1A; wild-type syntaxin 1A; aa 1-288. Peptides: N-Tail; aa 1-54; N-Tail with a 9 amino acid hemagglutinin sequence (HA-N-Tail); IL putative intracellular loops of GAT1; IL1, aa 100-124; IL2, aa 230-242; IL3, aa 308-328; fourth intracellular domain (IL4), aa 394-422; IL5, aa 477-497; IL4 quadruple mutant at positions R414, R417, R419, and R420 (IL4ΔR); IL4 triple mutant at positions L415, L416, N418 (IL4C); randomly scrambled IL4 sequence (IL4-Scram); the H3 domain of syntaxin 1A (Syn1AH3), aa 188-266.
GABA flux assays and statistical analysis. Standard [3H]GABA uptake and efflux assays were performed as described previously (Wang et al., 2003). Statistical analyses were performed using SPSS (Chicago, IL). Two-sample comparisons were made using t tests; multiple comparisons were made using one-way ANOVAs followed by Tukey's honestly significant difference test. Asterisks indicate results significantly different from control (p < 0.05).
Biochemical assays. For quantitative solution binding assays, fusion protein constructs were cleaved from GST, dialyzed in buffer (0.2% Triton X-100 in PBS), and mixed with the appropriate GST fusion proteins in 0.2 ml volume for 3 hr at 4°C. GST fusion proteins were then precipitated with excess glutathione agarose, washed extensively in buffer, and immunoblotted using the appropriate antibody (Ab) (Chemicon, Temecula, CA). Pull-down assays were performed as described previously (Naren et al., 1999). Briefly, soluble GST constructs were added to Triton X-100 lysates (1% Triton X-100 in PBS with protease inhibitors). The samples were diluted in PBS to bring the Triton X-100 concentration to 0.2% and mixed for 12 hr at 4°C. Bound proteins were precipitated with excess glutathione agarose, washed extensively, and immunoblotted. Biotinylation experiments were performed as described previously (Deken et al., 2000).
Electrophysiology. Currents were measured at room temperature using a GeneClamp 500 amplifier (Axon Instruments, Foster City, CA). Electrodes were filled with 3 m KCl and had resistances of 1-3 MΩ. The oocyte was voltage clamped at -80 mV and superfused continuously with ND96. Measurements of charge movements were performed as described previously (Parent et al., 1992; Mager et al., 1993; Deken et al., 2000). Each oocyte was tested in the presence and absence of the GAT1-specific antagonist SKF89976A to isolate (by subtraction) the charge movements associated with the presence of GAT1 in the plasma membrane. Transient currents generated by voltage jumps were integrated to yield the amount of charge movement in and out of the membrane field of the oocyte. Transporter number was calculated from the equation N = Qmax/qzδ, where N is the number of transporters per oocyte, Qmax is the total charge movement, q is the elementary charge, and zδ is the distance that all charges move within the membrane field. We assumed that the value of zδ for the GAT1 transporter is 1.0 (Mager et al., 1993). Turnover rates were determined by measuring the number of charges translocated per second (calculated from GABA-induced peak currents) and dividing by the number of functional transporters (calculated from GAT1-specific charge movements) in the same oocyte.
Results
To test whether intracellular domains of GAT1 regulate transport, we acutely injected fusion proteins corresponding to various intracellular loops of the transporter into oocytes expressing GAT1 and then subjected these oocytes to transport assays (Fig. 1A). Injection of a fusion protein (to ∼1 μm final concentration) corresponding to the putative fourth intracellular domain of GAT1 resulted in an 80% reduction in GABA flux and efflux. Injection of fusion proteins corresponding to the other intracellular loops had no effect. The IL4 fusion protein (data not shown) or a synthetic IL4 peptide inhibited GABA flux in a concentration-dependent manner with a half-maximal effective concentration of ∼200 nm; a synthetic, scrambled IL4 peptide had no effect on GABA transport (Fig. 1B). Mixing IL4 with any or all of the other intracellular loop fusion proteins had no additional effect (data not shown).
Figure 1.
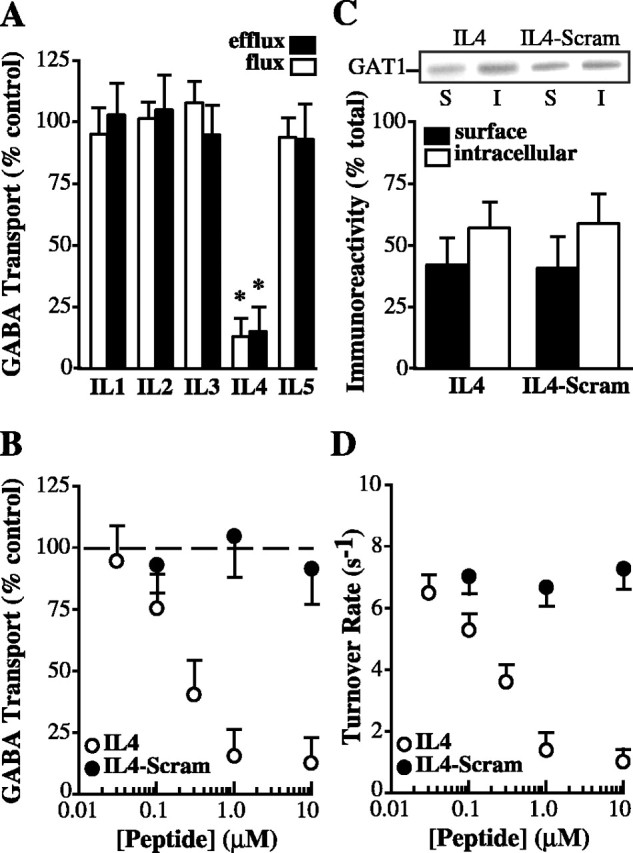
GAT1 IL4 domain inhibits GABA transport rates. A, IL4 fusion protein inhibits GABA flux and efflux. Oocytes expressing GAT1 were injected 15 min before assay with fusion proteins corresponding to each of five GAT1 intracellular domains (IL1-IL5; ∼1 μm final concentration) and were subjected to [3H]GABA flux (open bars) and efflux (filled bars) assays. Data are plotted relative to nonpeptide-injected oocytes and are from three experiments (4-7 oocytes per condition per experiment). B, Synthetic IL4 peptide inhibition of GABA uptake is concentration dependent. GAT1-expressing oocytes were acutely injected with various concentrations of IL4 peptide (open circles) or scrambled IL4 peptide (filled circles) and subjected to [3H]GABA flux assays. Data are plotted relative to nonpeptide-injected oocytes and are from three oocyte batches (5-9 oocytes per data point). C, IL4 peptide does not alter GAT1 surface expression. GAT1-expressing oocytes were acutely injected with 1 μm final concentration of IL4 or scrambled IL4 peptide and subjected to biotinylation. Immunoblot shows GAT1 immunoreactivity for avidin-bound (S, surface; filled bars) and nonbound (I, intracellular; open bars) fractions. Data from two experiments (20 oocytes per condition per experiment) are quantified relative to total GAT1 expression. D, IL4 peptide reduces GAT1 turnover rate in a concentration-dependent manner. Oocytes were treated as in B and evaluated using two-electrode voltage clamp. Data are from 6-13 oocytes per data point.
Given that GAT1 trafficking is regulated by several mechanisms, one possible reason for the IL4-mediated decrease in GABA transport was a change in GAT1 surface expression. To test this, GAT1-expressing oocytes were acutely injected with IL4 peptide or scrambled IL4 peptide and subjected to surface biotinylation assays (Fig. 1C). Approximately 50% of total cellular GAT1 was found on the plasma membrane in either condition, suggesting that changes in GAT1 expression were not responsible for the IL4-mediated inhibition. Therefore, these data suggested that the IL4 domain was altering the GAT1 turnover rate (i.e., the rate at which GABA moves through GAT1). To test this idea more directly, turnover rates were measured (as described in Materials and Methods). In GAT1-expressing oocytes acutely injected with the scrambled IL4 peptide, the turnover rate was ∼7 sec-1 at 22°C and saturating GABA concentrations, similar to previous estimates (Deken et al., 2000). Acute injection of IL4 peptide reduced the turnover rate in a dose-dependent manner, and the half-maximal effective concentration and the maximal inhibition were comparable with that seen in the radiolabeled flux assays. These data suggest that the IL4 domain inhibits GAT1 turnover rates.
Previously, we showed that the N-terminal tail of GAT1 is a positive regulator of GAT1 turnover rates: a GAT1 construct lacking the N-terminal tail shows rates of ∼2 sec-1, and acute injection of a fusion protein corresponding to the tail (N-Tail) rescues wild-type rates (Deken et al., 2000). Thus, we reasoned that N-Tail might reverse IL4-mediated transport inhibition (Fig. 2A). In oocytes expressing wild-type GAT1, acute injection of N-Tail fusion protein had no effect on GAT1 turnover rates. However, coinjection of N-Tail and IL4 fusion proteins reversed the inhibition seen in GAT1-expressing oocytes injected with IL4 fusion protein alone (Fig. 2A). The magnitude of the reversal was concentration dependent (Fig. 2B).
Figure 2.
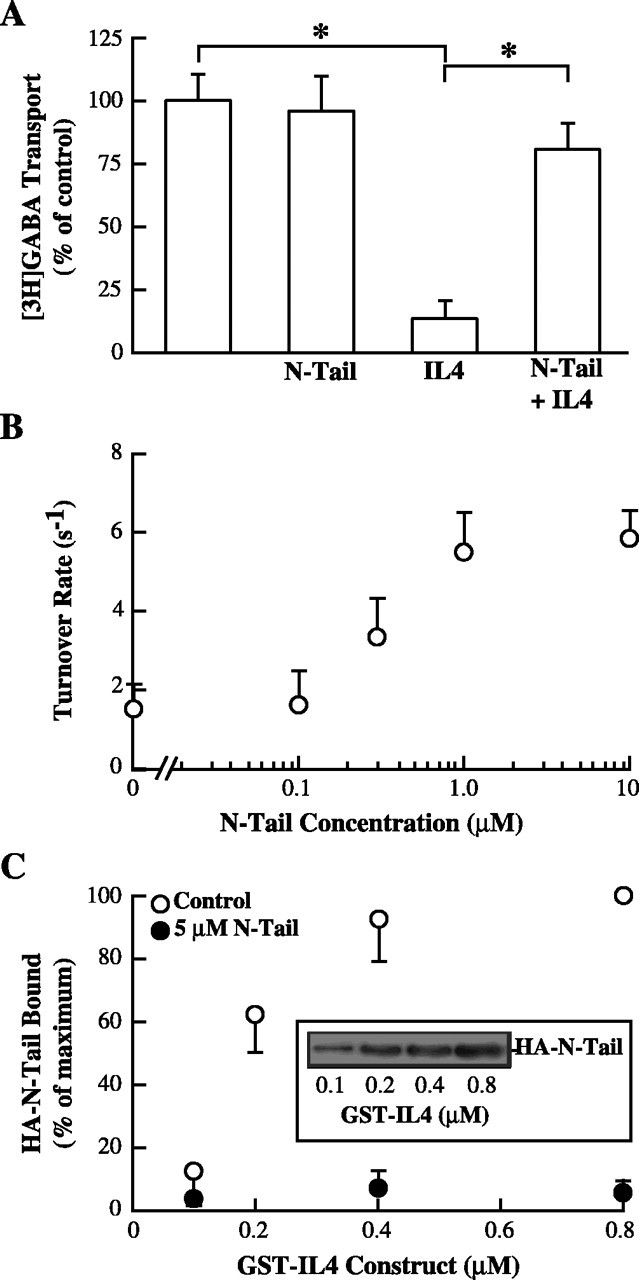
N-terminal tail and IL4 domain of GAT1 functionally interact. A, Coinjection of N-Tail peptide reverses IL4 peptide inhibition of GABA uptake. Oocytes expressing GAT1 were left untreated or injected 15 min before assay with IL4 fusion protein, N-Tail fusion protein, or both (∼1 μm) and subjected to [3H]GABA flux assays. Data are from three experiments (4-7 oocytes per condition per experiment). Asterisks indicate significant differences (p < 0.05) between the two indicated groups. B, N-Tail fusion protein reverses IL4-mediated inhibition of GAT1 turnover rates. GAT1-expressing oocytes were acutely injected with 1 μm IL4 fusion protein alone or in combination with increasing concentrations of N-Tail fusion protein and then subjected to two-electrode voltage clamp. Data are from 5-11 oocytes per data point. C, N-terminal tail and IL4 domain directly interact. Recombinant GAT1 HA-N-Tail cytosolic domain was bound to increasing concentrations of GST-IL4 and immunoblotted with HA-specific Ab. Data from three experiments are plotted relative to binding with 0.8 μm GST-IL4 fusion protein in the absence (open circles) or presence (filled circles) of excess (5 μm) N-Tail fusion protein.
To determine whether there was a direct association between the N-terminal and IL4 domains, we performed in vitro binding assays (Fig. 2C). Recombinant IL4 (GST-IL4) was added to recombinant HA-tagged N-Tail (GST-HA-N-Tail) in which the GST was cleaved free by thrombin. These complexes were precipitated with glutathione agarose and immunoblotted using an HA-specific Ab. Association between the N-Tail of GAT1 and the IL4 domain was saturable with half-maximal binding of ∼150 nm. Binding was competitively inhibited by adding excess N-Tail fusion protein (without the HA tag) to the assay. No binding was evident when GST alone was used, and the addition of fusion proteins corresponding to other GAT1 intracellular loops failed to alter the binding of HA-N-Tail to GST-IL4 (data not shown).
We next sought to determine the specific residues that might participate in this interaction. We showed previously that a mutant GAT1 construct in which three aspartic acid residues were removed from the N-terminal tail (3D-GAT1) exhibited decreased turnover rates (Deken et al., 2000); this decrease is consistent with an inability of this construct to bind to the IL4 domain. To examine this possibility, we performed pull-down assays using immobilized GST-IL4 in the presence of lysates from oocytes expressing either 3D-GAT1 or a control GAT1 construct in which two residues near the aspartic acid residues were mutated (Fig. 3A). Recombinant GST-IL4 was unable to pull down the GAT1 construct lacking N-terminal aspartic acid residues.
Figure 3.
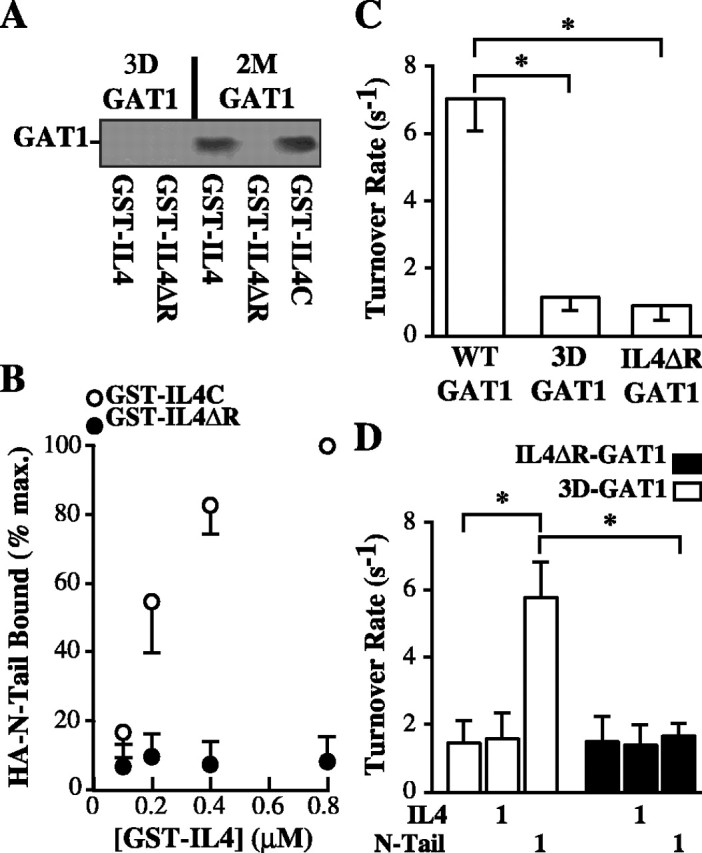
Charged amino acid residues in the N-terminal and IL4 domains are important for the functional interaction. A, Mutations in the N-terminal or IL4 domains eliminate the physical interaction. Cell lysates from oocytes expressing a GAT1 construct lacking three aspartic acid residues in the N-terminal tail (3D-GAT1) or a control mutant (2M-GAT1) were subjected to pull-down assays using GST constructs containing wild-type IL4 domain (GST-IL4), IL4 domain lacking three arginine residues (GST-IL4ΔR), or a control IL4 domain mutant (GST-IL4C). Bound proteins were subjected to immunoblotting using GAT1 Ab. Data are representative of three experiments. B, Mutation of arginine residues in the IL4 domain eliminates N-terminal tail binding. Recombinant GAT1 HA-N-Tail cytosolic domain was bound to increasing concentrations of GST-IL4C (open circles) or GST-IL4ΔR (filled circles), precipitated, and immunoblotted with an HA-specific Ab. Data are from three experiments plotted relative to binding with 0.8 μm GST-IL4C fusion protein. C, N-terminal and IL4 mutant GAT1 constructs show decreased turnover rates. Oocytes expressing wild-type GAT1, 3D-GAT1, or IL4ΔR-GAT1 were subjected to two-electrode voltage clamp. Data are from 8-10 oocytes per condition. D, N-Tail fusion protein does not rescue the turnover rate of IL4ΔR-GAT1. Oocytes expressing 3D-GAT1 (open bars) or IL4ΔR-GAT1 (filled bars) were left untreated or acutely injected with 1 μm IL4 or N-Tail fusion protein and then subjected to two-electrode voltage clamp. Data are from 6-11 oocytes per condition. Asterisks indicate significant differences (p < 0.05) between the two indicated groups.
Next, we reasoned that these acidic residues in the N-terminal tail might interact with four basic arginine residues in the IL4 domain. Pull-down assays (Fig. 3A) showed that recombinant GST-IL4 in which these arginine residues were mutated (GSTIL4ΔR) was unable to precipitate wild-type GAT1, whereas a GST-IL4 construct harboring mutations in nearby uncharged residues (GST-IL4C) was able to precipitate wild-type GAT1. Direct in vitro binding assays showed that the GST-IL4C construct bound the GAT1 N-terminal tail comparably with wild-type IL4 domain; however, N-Tail binding to GST-IL4ΔR was almost eliminated (Fig. 3B).
The above data suggested a model in which IL4 domain inhibits transport rates, and that this inhibition can be overcome by interactions between the N-terminal and IL4 domains. One prediction of this model is that GAT1 constructs in which this inter-domain interaction is prevented should show reduced transport rates. We examined GAT1 turnover rates in oocytes expressing 3D-GAT1 and a GAT1 mutant in which the arginine residues in the IL4 domain were replaced (IL4ΔR-GAT1). Compared with wild-type GAT1, both constructs showed reduced turnover rates (Fig. 3C). In addition, whereas wild-type turnover rates could be rescued in the 3D-GAT1 mutant by acute injection of N-Tail fusion protein, acute injection of N-Tail fusion protein could not rescue turnover rates in the IL4ΔR-GAT1 construct (Fig. 3D). Acute injection of wild-type IL4 fusion protein had no additional effect on transport rates of the IL4ΔR-GAT1 construct, suggesting that the arginine residues are important for N-terminal regulation but not for inhibition of transport rates per se.
The SNARE protein syntaxin 1A regulates GAT1 turnover rates through direct interactions between the H3 domain of syntaxin 1A and aspartic acid residues in the N-terminal tail (Deken et al., 2000). Those data and the present results suggest that regulation may occur through competition between syntaxin 1A and the IL4 domain for N-terminal tail binding. To test this hypothesis, turnover rates were examined in oocytes expressing wild-type syntaxin 1A with wild-type or mutant GAT1 constructs (Fig. 4A). Coexpression of syntaxin 1A reduced wild-type GAT1 turnover rates; this inhibition was reversed by acute injection of Syn1AH3 fusion protein, most likely because it out-competes full-length syntaxin 1A binding to the N-terminal tail. Syn1AH3 fusion protein was unable to reverse syntaxin 1A-mediated inhibition of the 3D-GAT1 construct, consistent with elimination of syntaxin 1A binding (Deken et al., 2000). Acute injection of Syn1AH3 fusion protein was also unable to rescue turnover rates in the IL4ΔR-GAT1 construct, consistent with the idea that syntaxin 1A inhibits GAT1 function by preventing the N-terminal tail of GAT1 from interacting with the IL4 domain.
Figure 4.
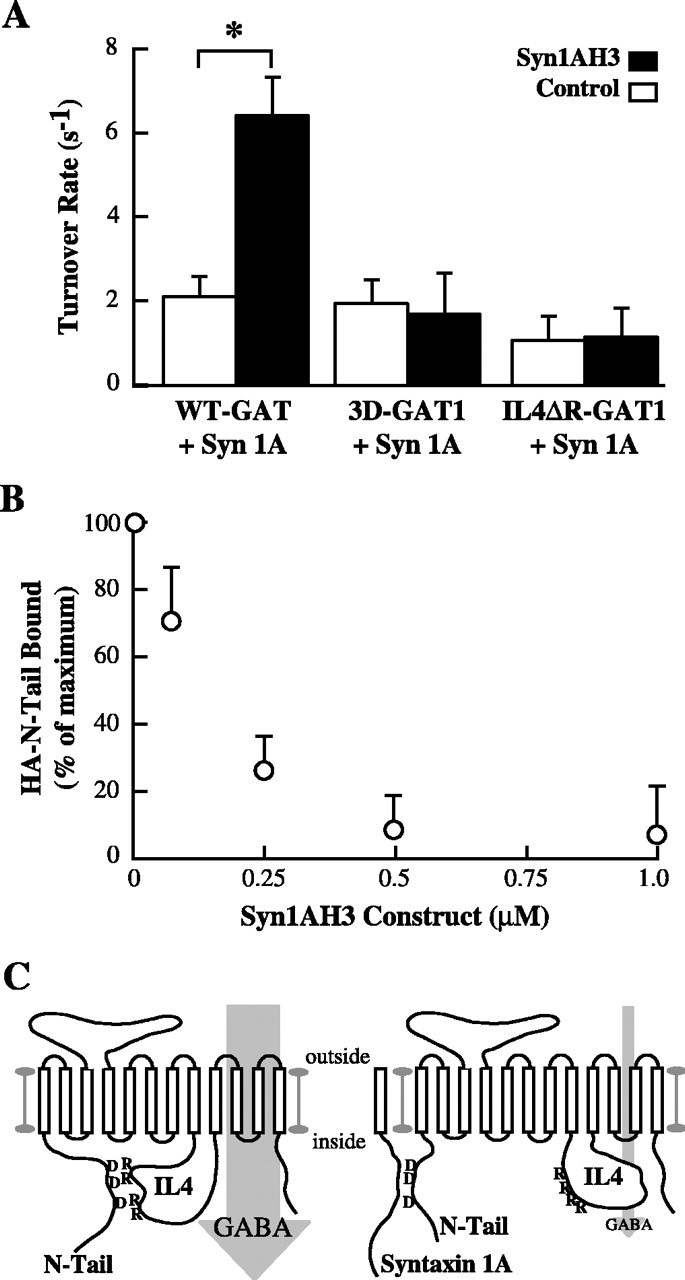
Syntaxin 1A functionally regulates the interaction between the N-terminal tail and the IL4 domain. A, Syntaxin 1A H3 domain reverses full-length syntaxin 1A inhibition of wild-type GAT1 but not N-terminal or IL4 GAT1 mutants. Oocytes coexpressing syntaxin 1A with wild-type GAT1, 3D-GAT1, or IL4ΔR-GAT1 were subjected to two-electrode voltage clamp before (open bars) and after (filled bars) acute injection with 1 μm Syn1AH3 fusion protein. Data are from 5-9 oocytes per condition. Asterisks indicate significant differences (p < 0.05) between the two indicated groups. B, Syn1AH3 fusion protein inhibits the binding of the N-terminal tail to the IL4 domain. Recombinant HA-N-Tail cytosolic domain of GAT1 was bound to equal amounts of GST-IL4 domain in the presence of increasing concentrations of Syn1AH3 fusion protein and immunoblotted using HA-specific Ab. Data are from three separate experiments plotted relative to binding in the absence of Syn1AH3 fusion protein. C, A cartoon illustrating inter-domain interactions that govern GABA flux through GAT1. D, Aspartic acid residues in the N-terminal tail; R, arginine residues in the IL4 domain.
To test this hypothesis directly, competitive in vitro binding assays were performed among the H3 domain of syntaxin 1A, the IL4 domain, and the N-terminal tail (Fig. 4B). Recombinant IL4 (GST-IL4) was added to recombinant GST-HA-N-Tail (in which the GST was cleaved free by thrombin) in the absence or presence of increasing concentrations of Syn1AH3 fusion protein. These complexes were precipitated with glutathione agarose and immunoblotted using an HA-specific Ab. Direct binding of the N-terminal tail to the IL4 domain was inhibited by the addition of Syn1AH3 fusion protein with a half-maximal effective concentration of ∼200 nm. These data are consistent with the idea that inhibition in GST-IL4 binding is attributable to competition by Syn1AH3 for the same binding site in the N-terminal tail.
Discussion
The present data show that a chain of cytoplasmic inter-domain associations within GAT1, and between GAT1 and its interacting partners, act to regulate substrate flux (Fig. 4C). The fourth intracellular loop of GAT1 acts as a negative regulator of transport rates, and we speculate that it does so by forming a barrier near the substrate permeation path, similar to inactivation or regulatory domains of other carriers and channels. The N-terminal tail of GAT1 serves as a positive regulator of substrate flux, because it directly binds to the fourth intracellular loop and prevents the loop from binding at its transport inhibition site. The interaction between these two GAT1 domains likely occurs via acidic and basic residues in each domain because mutagenesis of these residues prevents inter-domain binding and eliminates the N-terminal reversal of the inhibition. GAT1 binding partners, such as syntaxin 1A, then exert their effects on transport by regulating the availability of the N-terminal tail of GAT1 to interact with the fourth intracellular loop. A similar model has been proposed for syntaxin 1A regulation of cystic fibrosis transmembrane regulator Cl- channels via N-terminal and R domain interactions (Naren et al., 1999).
The present data raise some interesting questions. First, the arginine residues in the fourth intracellular loop appear necessary for N-terminal tail regulation of transport; however, they are not necessary for transport inhibition by the IL4 domain. How the loop mediates inhibition has yet to be determined. Second, cytosolic syntaxin 1A peptides are able to reverse full-length syntaxin 1A-mediated inhibition, but they are insufficient to regulate GAT1 in the absence of full-length syntaxin 1A (Deken et al., 2000). This is surprising, given that cytosolic syntaxin 1A peptide binding should prevent N-terminal tail aspartic acid residues of GAT1 from binding to arginine residues in the IL4 domain. Such a result suggests that the cytosolic syntaxin 1A peptides are exerting their effects independent of their binding to the aspartic acid residues in the N-terminal tail. Third, GAT1 likely functions as a multimer (Scholze et al., 2002). This raises questions as to whether one IL4 domain is sufficient for inhibition, whether IL4 domains act independently or cooperatively, and whether associations between the N-terminal tail and the IL4 domain occur through homomeric interactions, heteromeric interactions, or both. Fourth, the translocation step in which the IL4 domain decreases transport has yet to be determined. We have shown previously that syntaxin 1A inhibition occurs after substrate binding but which involves flux, efflux, and homoexchange (Wang et al., 2003). We speculate that the IL4 domain may be important in conformational steps related to bidirectional substrate translocation. Fifth, the physiological relevance of this regulation is unknown. Although the time scale of a single turnover may be too slow to affect fast synaptic transmission directly, altering the translocation step could influence ambient extracellular GABA levels or spillover to neighboring synapses (Ichinose and Lukasiewicz, 2002; Overstreet and Westbrook, 2003) by determining whether bound substrates either unbind or are translocated. Regardless, regulating flux rates will likely be important at slow GABAB receptor-mediated synapses and in modulating GABA efflux, a process known to have both physiological and pathophysiological consequences (Richerson and Wu, 2003).
Footnotes
This work was supported by National Institutes of Health Grant MH61468. We thank P. Saxena for oocyte preparation.
Correspondence should be addressed to Michael W. Quick, Department of Biological Sciences, HNB 228, University of Southern California, 3641 Watt Way, Los Angeles, CA 90089-2520. E-mail: mquick@usc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244082-06$15.00/0
References
- Bennett ER, Su H, Kanner BI (2000) Mutation of arginine 44 of GAT-1, a (Na(+) + Cl(-))-coupled gamma-aminobutyric acid transporter from rat brain, impairs net flux but not exchange. J Biol Chem 275: 34106-34113. [DOI] [PubMed] [Google Scholar]
- Bismith Y, Kavanaugh MP, Kanner BI (1997) Tyrosine 140 of the gamma-aminobutyric acid transporter GAT-1 plays a critical role in neurotransmitter recognition. J Biol Chem 272: 16096-16102. [DOI] [PubMed] [Google Scholar]
- Borden LA (1996) GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem Int 29: 335-356. [DOI] [PubMed] [Google Scholar]
- Deken SL, Beckman M, Boos L, Quick MW (2000) Transport rates of GABA transporters: regulation by the N-terminal domain and syntaxin 1A. Nat Neurosci 3: 998-1003. [DOI] [PubMed] [Google Scholar]
- Dingledine R, Korn SJ (1985) Gamma-aminobutyric acid uptake and the termination of inhibitory synaptic potentials in the rat hippocampal slice. J Physiol (Lond) 366: 387-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geerlings A, Lopez-Corcuera B, Aragon C (2000) Characterization of the interactions between the glycine transporters GLYT1 and GLYT2 and the SNARE protein syntaxin 1A. FEBS Lett 470: 51-54. [DOI] [PubMed] [Google Scholar]
- Guastella J, Nelson N, Nelson H, Czyzyk L, Keynan S, Miedel MC, Davidson N, Lester HA, Kanner BI (1990) Cloning and expression of a rat brain GABA transporter. Science 249: 1303-1306. [DOI] [PubMed] [Google Scholar]
- Haase J, Killian AM, Magnani F, Williams C (2001) Regulation of the serotonin transporter by interacting proteins. Biochem Soc Trans 29: 722-728. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW (1990) Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science 250: 533-538. [DOI] [PubMed] [Google Scholar]
- Ichinose T, Lukasiewicz PD (2002) GABA transporters regulate inhibition in the retina by limiting GABA(C) receptor activation. J Neurosci 22: 3285-3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Solis JM, Nicoll RA (1993) Local and diffuse synaptic actions of GABA in the hippocampus. Neuron 10: 165-175. [DOI] [PubMed] [Google Scholar]
- Mabjeesh NJ, Kanner BI (1992) Neither amino nor carboxyl termini are required for function of the sodium- and chloride-coupled gamma-aminobutyric acid transporter from rat brain. J Biol Chem 267: 2563-2568. [PubMed] [Google Scholar]
- Mager S, Naeve J, Quick MW, Labarca C, Davidson N, Lester HA (1993) Steady states, charge movements, and rates for a cloned GABA transporter expressed in Xenopus oocytes. Neuron 10: 177-188. [DOI] [PubMed] [Google Scholar]
- Mager S, Kleinberger-Doron N, Keshet GI, Davidson N, Kanner BI, Lester HA (1996) Ion binding and permeation at the GABA transporter GAT1. J Neurosci 16: 5405-5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naren AP, Cormet-Boyaka E, Fu J, Villain M, Blalock JE, Quick MW, Kirk KL (1999) CFTR chloride channel regulation by an interdomain interaction. Science 286: 544-548. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Westbrook GL (2003) Synapse density regulates independence at unitary inhibitory synapses. J Neurosci 23: 2618-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantanowitz S, Bendahan A, Kanner BI (1993) Only one of the charged amino acids located in the transmembrane alpha-helices of the gamma-aminobutyric acid transporter (subtype A) is essential for its activity. J Biol Chem 268: 3222-3225. [PubMed] [Google Scholar]
- Parent L, Supplisson S, Loo DD, Wright EM (1992) Electrogenic properties of the cloned Na+/glucose cotransporter: I. Voltage-clamp studies. J Membr Biol 125: 49-62. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester HA (1994) Expression of excitability proteins in oocytes, in methods in neuroscience, ion channels of excitable cells (Conn PM, ed), pp 261-279. San Diego: Academic.
- Radian R, Bendahan A, Kanner BI (1986) Purification and identification of the functional sodium- and chloride-coupled gamma-aminobutyric acid transport glycoprotein from rat brain. J Biol Chem 261: 15437-15441. [PubMed] [Google Scholar]
- Richerson GB, Wu Y (2003) Dynamic equilibrium of neurotransmitter transporters: not just for reuptake anymore. J Neurophysiol 90: 1363-1374. [DOI] [PubMed] [Google Scholar]
- Scholze P, Freissmuth M, Sitte HH (2002) Mutations within an intramembrane leucine heptad repeat disrupt oligomer formation of the rat GABA transporter 1. J Biol Chem 277: 43682-43690. [DOI] [PubMed] [Google Scholar]
- Sung U, Apparsundaram S, Galli A, Kahlig KM, Savchenko V, Schroeter S, Quick MW, Blakely RD (2003) A regulated interaction of syntaxin 1A with the antidepressant-sensitive norepinephrine transporter established catecholamine clearance capacity. J Neurosci 23: 1697-1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura S, Nelson H, Tamura A, Nelson N (1995) Short external loops as potential substrate binding site of gamma-aminobutyric acid transporters. J Biol Chem 270: 28712-28715. [DOI] [PubMed] [Google Scholar]
- Wang D, Deken SL, Whitworth TL, Quick MW (2003) Syntaxin 1A inhibits both GABA flux and efflux through the rat brain GABA transporter GAT1. Molec Pharm 64: 905-913. [DOI] [PubMed] [Google Scholar]