

Abstract
The tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a lipid and protein phosphatase. We report here that PTEN physically associates with the NR1 and NR2B subunits of NMDA receptors (NMDARs) in rat hippocampus. Downregulating the protein expression of PTEN inhibits the function of extrasynaptic NMDARs and decreases NMDAR surface expression, suggesting a crucial role for endogenous PTEN in the modulation of NMDAR-mediated neuronal function. Reducing PTEN expression also enhances Akt/Bad phosphorylation in hippocampal neurons. Importantly, suppressing lipid and protein phosphatase activity of PTEN, respectively, activates Akt and inhibits extrasynaptic NMDAR activity and thereby protects against ischemic neuronal death in vitro and in vivo. Thus, our study reveals a dual neuroprotective mechanism by which Akt/Bad and extrasynaptic NMDARs are regulated via downregulation of two distinct PTEN phosphatase activities and present the possibility of PTEN as a potential therapeutic target for stroke treatment.
Keywords: phosphatase, NMDA receptor, cerebral ischemia, neuroprotection, stroke, synapse
Introduction
The tumor suppressor PTEN (phosphatase and tensin homolog deleted on chromosome 10) is a lipid phosphatase that can negatively regulate the serine/threonine kinase Akt to exert its tumor suppressor function (Maehama and Dixon 1998; Stambolic et al., 1998). PTEN is also a protein phosphatase and has been shown to dephosphorylate phosphotyrosine- and phosphoserine/threonine-containing substrates (Myers et al., 1997). Recent studies demonstrate that the protein phosphatase activity of PTEN can regulate cell migration, spreading, and growth (Tamura et al., 1999). However, little is known about the role of PTEN in the CNS (Backman et al., 2001; Groszer et al., 2001; Kwon et al., 2001).
The NMDA receptor (NMDAR) plays a key role in regulating synaptic plasticity, neuronal development, and excitotoxicity in the CNS (Choi, 1995; Dingledine et al., 1999). Among many extracellular and intracellular processes that can modulate NMDARs, phosphorylation and dephosphorylation by protein kinases and phosphatases are especially important, because they can critically regulate trafficking, surface expression, and the channel properties of NMDARs (Dingledine et al., 1999; Swope et al., 1999; Carroll and Zukin, 2002).
Glutamate-dependent excitotoxicity is thought to result primarily from the excessive release of glutamate that overactivates NMDARs (Choi, 1995). Recent studies provide evidence suggesting that calcium flux through extrasynaptic NMDARs plays a critical role in cell death pathways (Hardingham et al., 2002; Vanhoutte and Bading, 2003). Thus, understanding the cellular and molecular mechanisms regulating extrasynaptic NMDARs may offer the possibility of developing effective and clinically useful drugs for preventing neuronal death.
Although recent studies suggest that the downregulation of PTEN may play a neuroprotective role in CNS insults (Gary and Mattson, 2002; Lee et al., 2004), the underlying mechanisms remain unclear. The present study was undertaken to examine the regulation of NMDARs and the cell survival-promoting kinase Akt by the protein and lipid phosphatase activities of PTEN in hippocampal neurons and to determine whether such regulation could mediate the neuroprotective effects of PTEN downregulation in ischemic neuronal injury.
Materials and Methods
Transient global ischemia in rat. The modified four-vessel occlusion model was described previously (Hsu et al., 1998). Briefly, adult male Wistar rats weighing 200-300 gm were anesthetized with 1.5% halothane, and the vertebral arteries were electrocauterized on day one. Twenty-four hours later, the animal was reanesthetized, and both common carotid arteries were clamped with aneurysm clips for 15 min. Animals were included for subsequent experiments only if they displayed an isoelectric EEG during the entire occlusion period, displayed a dilated pupil and a lack of corneal light reflex, and recovered EEG activity within 30 min of reperfusion. Body temperature, as measured with a rectal thermometer, was maintained between 36.5 and 38°C throughout the procedure with a heating lamp. Sham animals received identical surgical exposure and handling without vessel occlusion.
Hippocampal neuronal cultures and oxygen-glucose deprivation treatment. The hippocampal neuronal cultures were prepared from Wistar rats at gestation day 18 using a modified published protocol (Brewer et al., 1993). Briefly, dissociated neurons were suspended in plating medium (Neurobasal medium, 2% B-27 supplement, 0.5% FBS, 0.5 μm l-glutamine, and 25 μm glutamic acid) and plated on poly-d-lysine-coated glass coverslips set in 35 mm Petri dishes at ∼100,000 cells per coverslip. After 3 d in culture, three-quarters of the plating medium was removed and replaced with maintenance medium (Neurobasal medium, B-27 supplement, and 0.5 μm l-glutamine). Thereafter, maintenance medium was changed in the same manner every 3-4 d. The cultures were used for experiments at 12-15 d after plating. The oxygen-glucose deprivation (OGD) experiments were performed using a specialized, humidified chamber (Plas-Labs, Lansing, MI) kept at 37°C, which contained an anaerobic gas mixture (85% N2, 10% H2, and 5% CO2) (Goldberg and Choi, 1993; Ying et al., 1997). To initiate OGD, culture medium was replaced with deoxygenated, glucose-free extracellular solution (in mm: 140 NaCl, 5.4 KCl, 1.3 CaCl2, 1.0 MgCl2, and 10 HEPES). After 1 hr challenge, cultures were removed from the anaerobic chamber, and the OGD solution in the cultures was replaced with maintenance medium. Cells were then allowed to recover for varying lengths of time in a humidified incubator with 95% O2 and 5% CO2 at 37°C. To block NR2B-containing NMDA receptors at extrasynaptic sites, 10 μm ifenprodil (Tocris Cookson, Ballwin, MO) was added in the maintenance medium after OGD.
Coimmunoprecipitation, Western blotting, and in vitro binding assays. Coimmunoprecipitation, Western blotting, and in vitro binding experiments were performed as described previously (Wan et al., 1997a; Liu et al., 2000; Lee et al., 2002). Monoclonal antibody (mAb) against PTEN (Chemicon, Temecula, CA), polyclonal antibody (pAb) against PTEN (Cell Signaling Technology, Beverly, MA), pAb against NR1 (Chemicon), pAb against NR2A (Novus Biologicals, Littleton, CO), pAb against NR2B (Novus Biologicals), pAb against glutamate receptor 1 (GluR1) (Oncogene, San Diego, CA), mAb against actin (Chemicon), or mAb against glutathione S-transferase (GST) (Chemicon) were used. Primary antibodies were labeled with horseradish peroxidase-conjugated secondary antibodies, and bands were imaged using an ECL detection system (Amersham Biosciences, Baie D'urfe, Quebec, Canada).
NR1-1aCT, NR2ACT, and NR2BCT cDNA-encoding fragments were amplified by PCR from full-length cDNA clones. All 5′ and 3′ oligonucleotides incorporated BamHI and EcoRI sites, respectively, to facilitate subcloning into pGEX4T-2. Initiation methionine residues and stop codons were also incorporated when appropriate (Lee et al., 2002). GST-fusion proteins were prepared from bacterial lysates as described by the manufacturer (Amersham Biosciences). To confirm appropriate splice fusion and the absence of spurious PCR-generated nucleotide errors, all constructs were resequenced. GST-NR1-1aCT, GST-NR2ACT, NR2BCT, or GST (10 μg) alone was incubated with [35S]PTEN probe for 4 hr at room temperate. The beads were then washed six times with tissue homogenizing buffer and eluted with 10 mm glutathione. Eluates were separated by SDS-PAGE and visualized by autoradiography (Lee et al., 2002).
PTEN small interfering RNA expression vectors. An expression vector that directs the synthesis of small interfering RNAs (siRNAs) was constructed as described previously (Brummelkamp et al., 2002). A 19-nucleotide PTEN target sequence (5′-AGATGAGAGACGGCGGCGG-3′) corresponding to nucleotides 206-224 of PTEN mRNA (Steck et al., 1997) was selected for constructing siRNApten-green fluorescent protein (GFP) vector that mediates the expression of PTEN siRNAs (Brummelkamp et al., 2002). A scrambled PTEN target sequence (5′-CCCTCTATCGCGCCCTGCT-3′) was designed to construct the control SsiRNApten-GFP (scrambled siRNApten-GFP) vector (Brummelkamp et al., 2002). Plasmid transfections were done using LipofectAMINE 2000 (Invitrogen, Burlington, Ontario, Canada) as described previously (Wan et al., 1997b).
Immunocytochemical staining. Immunocytochemical staining was performed as described previously (Wan et al., 1997b; Snyder et al., 2001). mAb against NeuN (neuronal-specific nuclear protein) (Chemicon), mAb against PTEN (Chemicon), pAb against PTEN (Cell Signaling Technology), pAb against phosphorylated Akt on Ser-473 (New England Biolabs, Beverly, MA), and pAb against phosphorylated Bad on Ser-136 (New England Biolabs) were used for staining.
Surface NMDARs were examined using specific antibodies against the N-terminal extracellular domain of NR1 subunits (Chemicon) under nonpermeant conditions (Wan et al., 1997b; Snyder et al., 2001). The same NR1 antibody Ab was used to examine the total NR1 expression under permeant conditions (Wan et al., 1997b). The secondary Ab Alexa Fluor 350 (blue fluorescence) and Alexa Fluor 594 (red fluorescence) were purchased from Molecular Probes (Eugene, OR).
Fluorescence-labeled receptors in the cultured neurons were examined using a Zeiss (Oberkochen, Germany) LSM 510 META confocal microscope, and the intensity of fluorescence staining was measured as described previously (Wan et al., 1997b; Beattie et al., 2000; Passafaro et al., 2001; Snyder et al., 2001). All morphometric measurements were done using Image Pro Plus analysis software (Image Processing Solutions, Silver Spring, MD).
Electrophysiology. For recordings of NMDAR-mediated whole-cell currents (Wang et al., 1996), the cultures were bathed in an extracellular recording solution (140 mm NaCl, 5.4 mm KCl, 25 mm HEPES, 1.3 mm CaCl2,33mm glucose, 0.001 mm glycine, and 0.001 mm tetrodotoxin, pH 7.35, 310-320 mOsm osmolarity). To remove the blockade of NMDA channels by Mg2+, Mg2+ was not added to the extracellular solution. Pipettes (2-5MΩ) were filled with a standard intracellular solution (140 mm CsCl, 10 mm HEPES, and 10 mm BAPTA, pH 7.25, 300-315 mOsm osmolarity). NMDAR-mediated whole-cell currents were evoked at regular intervals by pressure application (150 kPa, 100 msec) of l-aspartate (250 μm) from a micropipette with its tip located 20-30 μm from the cell under study. The holding potential was -60 mV. The recording of miniature EPSCs (mEPSCs) was described previously (Wang et al., 1996; Wan et al., 1997b). The mEPSCs were recorded immediately after formation of the whole-cell patch configuration and continuously monitored for 20-30 min thereafter. The mEPSCs were filtered at 2 kHz and analyzed with Mini Analysis program (Synaptosoft, Leonia, NY).
All single-channel recordings were performed in the cell-attached configuration. NMDA (10 μm) and glycine (3 μm) were supplemented in standard extracellular solution filled in the recording pipette (15-25 MΩ) to evoke NMDAR-mediated single-channel currents (Wang et al., 1996). The holding potential of the patch was +70 mV. Current recordings were filtered at 10 kHz (-3 dB) and stored on tape. Recordings were replayed from tape, filtered at 3 kHz (-3 dB, eight-pole Bessel), and digitized at 20 kHz sampling rate for off-line analysis (Colquhoun and Sigworth, 1995). Channel openings and closings were determined off-line by using a 50% crossing threshold. The channel open probability (Po) was calculated by dividing the mean open time by the sum of mean open time and mean shut time. Mean open time and mean shut time were calculated by integration of the mixture of exponential components fitted to open-time and shut-time distributions, respectively.
The method for examining the number of functional channels in the membrane (N) was described previously (Jahr, 1992; Rosenmund et al., 1995; Lan et al., 2001). The N was analyzed by taking advantage of the essentially irreversible block of NMDA-elicited currents by the open-channel blocker MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate]. We recorded NMDA whole-cell currents that were elicited by 1 μm NMDA and 50 μm glycine in the continuous presence of MK-801 (5 μm) from neurons expressing SsiRNApten-GFP and siRNApten-GFP. The N can be calculated from the cumulative charge transfer (Q), an integral of NMDA-elicited current during the time required for complete block by MK-801, as follows: N = Q/[γ(V - Erev)tbl], where tbl is the time constant for MK-801 block [tbl = 1/kbl(MK-801)], and kbl = 2.5 × 107 · m-1 · sec-1 (Jahr, 1992). As indicated in our single-channel recordings, PTEN downregulation does not alter single-channel conductance (γ) or mean open time and should not affect kbl.
Propidium iodide labeling. To determine the neuronal cell death, culture medium was replaced by extracellular solution containing propidium iodide (PI) (Molecular Probes) at a final concentration of 50 μg/ml. After 10 min incubation in an ambient gas incubator at 37°C, cultures were washed with extracellular solution and then fixed with 4% paraformaldehyde. Neuronal death (percentage) was determined by calculating the number of PI labeling in neurons expressing GFP over total number of the green neurons.
In vivo siRNApten expression. Adult male Wistar rats were anesthetized with 2% halothane and placed in a David Kopf Instruments (Tujunga, CA) stereotaxic apparatus. SsiRNApten, siRNApten, and vehicle were injected into the CA1 pyramidal cell layer of rats at the following coordinates: 3.5 mm posterior to bregma, 1.6 mm lateral from the midline, and at a depth of 3.0 mm from the skull surface. A volume of 1 μl of concentrated vector (1 × 106 particles/μl) was injected into one side of the hippocampus at a rate of 0.05 μl/min using the protocol established recently by Wei et al. (2003). For ischemia experiments, the transient global ischemic model was established 2 d after plasmid injection. At 48 hr after global ischemia, rats were perfused with 4% paraformaldehyde as described previously (Zhang et al., 2002). Coronal sections (40-μm-thick) through the dorsal hippocampus were cut on a sliding microtome and then processed for Fluoro-Jade B histochemical staining (Hopkins et al., 2000), which consistently reveals the dying or degenerating neurons (Hopkins et al., 2000; Zhang et al., 2002). Quantitative assessment was conducted by visually counting Fluoro-Jade B-stained neurons using a 20× objective lens. Average cell numbers were obtained by averaging counts over one consistent field at the middle point of the dorsal hippocampus where vectors and vehicle were injected and over three sections (80 μm apart) in each brain.
Statistics. All population data were expressed as mean ± SE. The Student's paired t test or the ANOVA test was used when appropriate to examine the statistical significance of the differences between groups of data. Significance was placed at p < 0.05.
Results
PTEN forms a complex with NMDARs
To investigate the possible regulation of NMDARs by the phosphatase PTEN, we examined whether PTEN can associate with NMDARs using rat hippocampal homogenates. We found that the NMDAR NR1 and NR2B subunits were coimmunoprecipitated with PTEN by anti-PTEN Ab (Fig. 1A,C). Conversely, immunoprecipitating with either an anti-NR1 or NR2B Ab coprecipitated PTEN in the hippocampal tissue (Fig. 1B,D). In contrast, PTEN did not coimmunoprecipitate with the NR2A subunits and the AMPA receptor GluR1 subunit (Fig. 1A,C,E). These results suggest that PTEN may physically interact with NR1/NR2B complexes of NMDA receptors within the hippocampal neurons. Because NR1/NR2B receptors are mainly distributed in extrasynaptic membranes (Li et al., 1998, 2002; Stocca and Vicini, 1998; Rumbaugh and Vicini, 1999; Tovar and Westbrook, 1999), PTEN may play a role in regulating the function of extrasynaptic NMDARs.
Figure 1.

Physical association between PTEN and NMDARs in rat hippocampus. A, C, Coimmunoprecipitation of hippocampal NR1 and NR2B subunits, but not GluR1 subunit of AMPA receptors, by PTEN Ab. B, D, E, Coimmunoprecipitation of hippocampal PTEN by NR1 and NR2B Ab but not by NR2A Ab. F, In vitro binding assay showing the direct binding of [35S]PTEN to GST-NR1-1aCT but not NR2A-CT and NR2B-CT. GST was used as a control. IP, Immunoprecipitation.
Direct protein-protein interaction between PTEN and NMDARs
To determine whether PTEN could directly couple to NMDARs, in vitro binding assays were performed. Because the carboxyl tail (CT) of NR1/NR2 subunits contains potential binding sites for various proteins important for signaling (Scannevin and Huganir, 2000; Barria and Malinow, 2002; Sheng and Kim, 2002), GST-fusion proteins encoding the CTs of NR1-1a (GST-NR1-1aCT:E834-S938), NR2A (GST-NR2ACT:D1350-V1464), and NR2B (GST-NR2BCT: P1365-V1482) were prepared and probed with in vitro translated [35S]methionine-labeled PTEN (Lee et al., 2002). As depicted in Figure 1F, the [35S]PTEN probe bound with GST-NR1-1aCT but failed to bind to GST-NR2ACT and GST-NR2BCT. Thus, it appears that the CT of the NR1 subunit contains a domain that mediates the direct interaction between PTEN and NMDARs.
PTEN regulates the function and surface expression of NMDARs
To determine whether the physical association between PTEN and NMDAR subunits in the hippocampus had a functional implication, we recorded NMDAR-mediated whole-cell currents in cultured hippocampal neurons in which PTEN protein expression was suppressed by using an siRNA expression system (Brummelkamp et al., 2002). As illustrated in Figure 2, B and C, expression of siRNApten-GFP, but not the SsiRNApten-GFP as a control, markedly suppressed PTEN expression in cultured hippocampal neurons, indicating a specific knockdown of PTEN by siRNApten-GFP introduction. Whole-cell patch-clamp recordings were obtained from pairs of nearby transfected and nontransfected neurons under visual guidance using fluorescence and transmitted light illumination. We found that neurons expressing siRNApten-GFP exhibited significantly lower peak amplitudes of NMDA currents than the untransfected neighboring control cells (Fig. 3A,B). In contrast, there was no significant difference in peak amplitudes of NMDA currents between the neurons expressing SsiRNApten-GFP and the untransfected neighboring control cells (Fig. 3A,B). PTEN deficiency-induced reduction did not cause alterations of the I-V curve or the reversal potential (Fig. 3C), suggesting that reduction of the currents by PTEN knockdown is not attributable to an alteration of driving force. Thus, these results provide first evidence indicating that PTEN functions as an upstream regulator to modulate NMDAR function.
Figure 2.

Suppression of PTEN protein expression by siRNA in cultured hippocampal neurons. A, Representative images of siRNApten-GFP expression in cultured hippocampal neurons 24 hr after transfection. Scale bar, 40 μm. B, An example of PTEN knockdown by siRNApten-GFP but not by SsiRNApten-GFP introduction in cultured hippocampal neurons 24 hr after transfection. Scale bar, 20 μm. C, Time course of siRNA-induced decrease of PTEN expression in cultured hippocampal neurons (n = 12 for each group; *p < 0.05). Data are normalized to values from nontransfected neurons.
Figure 3.

Down regulating PTEN inhibits extrasynaptic NMDAR function. A, An example of decreased NMDAR-mediated whole-cell currents in neurons expressing siRNApten-GFP (bottom) but not in neurons expressing SsiRNApten-GFP (top). B, Graph showing peak NMDA currents inhibited by PTEN downregulation (Non-GFP, n = 28; SsiRNApten-GFP, n = 23; siRNApten-GFP, n = 25; *p < 0.05). C, I-V relationship in neurons expressing non-GFP and siRNApten-GFP. D, Quantification analyses indicate that PTEN knockdown has no significant effects on NMDAR-mediated mEPSC amplitudes (left, n = 8 for each group) and frequency (right, n = 8 for each group). Data are normalized to non-GFP values. E, Representative traces of NMDA-activated channels recorded in cell-attached patches from neurons expressing SsiRNApten-GFP (left) and siRNApten-GFP (right). F, Effects of PTEN knockdown on NMDA channel open probability (Po), mean open time (to), and channel conductance (γ). Data are normalized to values from neurons expressing SsiRNApten-GFP as control (n = 6; *p < 0.05).
Because the recorded whole-cell currents were mainly mediated through extrasynaptic NMDARs, we further examined the synaptic responses of NMDARs to PTEN downregulation. NMDAR-mediated mEPSCs in PTEN-deficient neurons were recorded. Our data showed that downregulation of PTEN had no significant effects on both amplitude and frequency of NMDAR-mediated synaptic currents (Fig. 3D). These results are consistent with our observations that PTEN forms complexes with NR1/NR2B that are primarily located at extrasynaptic sites (Li et al., 1998, 2002; Stocca and Vicini, 1998; Rumbaugh and Vicini, 1999; Tovar and Westbrook, 1999) and thereby suggest that the phosphatase PTEN is a positive regulator of extrasynaptic NMDARs, and downregulation of endogenous PTEN expression by siRNApten-GFP is sufficient to inhibit NMDAR function at the extrasynaptic sites of hippocampal neurons.
To examine whether the decline in the extrasynaptic NMDA peak current could be attributable to a change in NMDA single-channel properties, we performed cell-attached single-channel recordings in neurons expressing SsiRNApten-GFP and siRNApten-GFP to analyze NMDA channel conductance (γ), mean open time (to), open probability (Po), and reversal potential (Erev). We found that PTEN downregulation decreased Po (n = 6 patches; p < 0.05) but did not alter to, γ, and Erev [Po, to, and γ were (7.6 ± 0.8) × 10-3, 3.02 ± 0.71 msec, and 43.3 ± 5.6 pS in neurons expressing SsiRNApten-GFP; (5.4 ± 0.6) × 10-3, 2.84 ± 0.53 msec, and 44.6 ± 4.7 pS in neurons expressing siRNApten-GFP]. These results suggest that reduced NMDA single-channel activity by the downregulation of endogenous PTEN may contribute to the decrease of NMDA peak currents in neurons expressing siRNApten-GFP.
Although the decline in the NMDA peak current could be attributable to a reduction in single-channel open probability, it could be also attributable to a decreased expression of NMDARs on the cell surface (Snyder et al., 2001; Barria and Malinow, 2002; Carroll and Zukin, 2002). As shown in Figure 4, A and B, whereas protein expression of PTEN was reduced by siRNApten-GFP, the surface expression of NR1 subunits in the PTEN-deficient neurons was also significantly reduced. Because the expression of siRNApten-GFP did not alter the total NR1 levels in the cultured hippocampal neurons (Fig. 4C), the decreased surface expression of NR1 could be the result of a decreased delivery and/or increased internalization of NMDARs. Thus, PTEN knockdown-induced inhibition of NMDA currents may be mediated by an altered trafficking of NMDARs in the hippocampal neurons.
Figure 4.
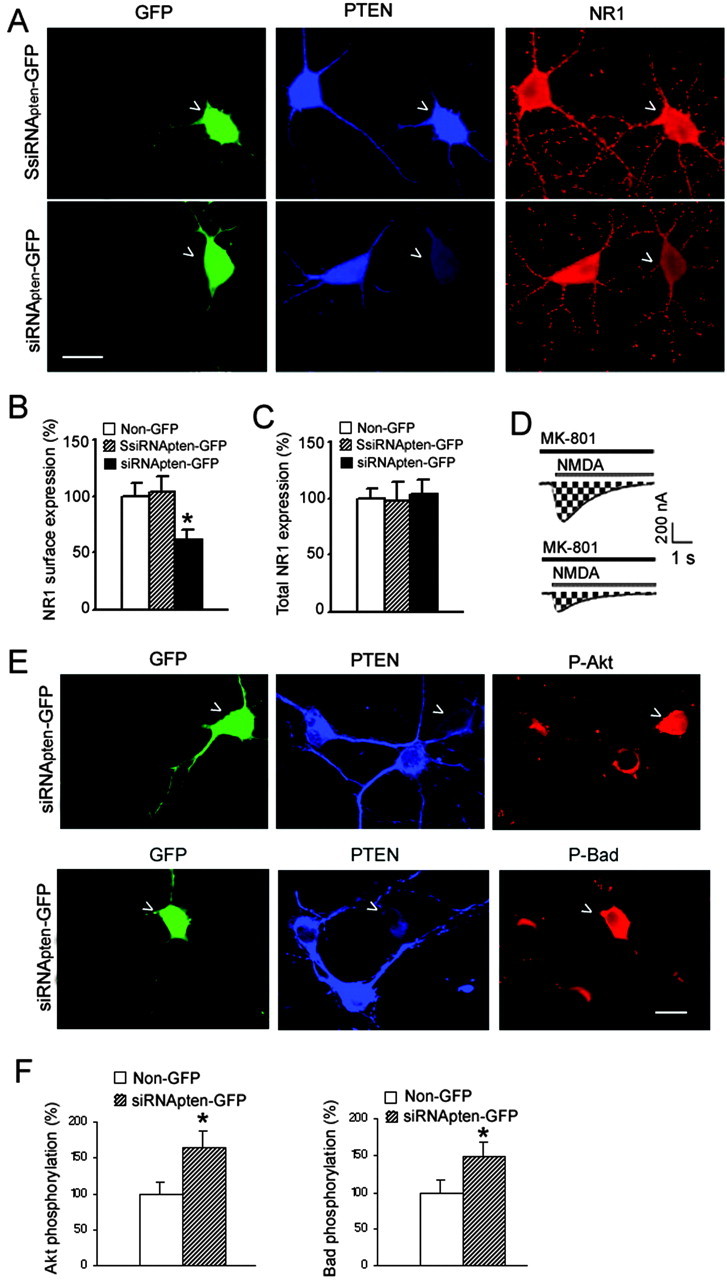
PTEN knockdown reduces surface expression of NMDARs and increases Akt/Bad phosphorylation. A, An example showing decreased surface expression of NMDARs in PTEN-deficient hippocampal neurons. Scalebar, 30 μm. B, Summarized data showing surface expression of NMDARs (Non-GFP, n = 57; SsiRNApten-GFP, n = 69; siRNApten-GFP, n = 65; *p < 0.05). C, Graph showing that suppressing PTEN did not alter total NR1 expression in hippocampal neurons (Non-GFP, n = 22; SsiRNApten-GFP, n = 26; siRNApten-GFP, n = 19). D, Downregulation of PTEN reduced the number of functional NMDA channels (N) at the membrane surface. Currents were elicited by application of NMDA (1 μm NMDA with 50 μm glycine) in the continuous presence of the open-channel blocker MK-801 (5 μm) in the neurons expressing SsiRNApten-GFP (top) and the neurons expressing siRNApten-GFP(bottom) at a holding potential of -60 mV. The NMDA inward current increased to a peak value and then decayed exponentially because of MK-801 block of NMDA channels as they opened. The cumulative charge transfer (Q), which is the total current flow during the time interval for complete block by MK-801, was obtained by integration of (Figure legend continues.) (Figure legend continued.) the current trace over time (area indicated by shading). E, Examples showing that both Akt and Bad phosphorylation are increased in neurons expressing siRNApten-GFP. Scale bar, 20 μm. F, Graph plotting the increased phosphorylation levels of Akt and Bad in PTEN-deficient hippocampal neurons (left: Non-GFP, n = 39; siRNApten-GFP, n = 51; *p < 0.05; right: Non-GFP, n = 45; siRNApten-GFP, n = 55; *p < 0.05). SsiRNApten-GFP transfection has no significant effects on both Akt and Bad phosphorylation (data not shown).
To further determine whether the observed decrease of surface NR1 expression represented a reduced number of functional NMDA channels, we performed whole-cell patch-clamp recordings of NMDAR currents to examine the cumulative charge transfer (Q) in the presence of NMDA and the open-channel blocker MK-801 (Fig. 4D). The number of functional channels (N) can be calculated from Q as described in Materials and Methods (Jahr, 1992; Rosenmund et al., 1995; Lan et al., 2001). Our results showed that PTEN downregulation in neurons expressing siRNApten-GFP decreased N by 23.6% compared with neurons expressing SsiRNApten-GFP (n = 6; p < 0.05), suggesting that the reduction of functional NMDA channels on membrane surface by PTEN downregulation may contribute to the decline of NMDA peak currents in the hippocampal neurons.
Downregulation of PTEN enhances Akt and Bad phosphorylation
Deletion of PTEN in mouse brain has been shown to elevate the levels of phosphorylated Akt (the activated form of the kinase) in neuronal cells (Groszer et al., 2001; Kwon et al., 2001). Consistent with these findings, our study demonstrates that, in cultured rat hippocampal neurons, PTEN deficiency mediated by siRNApten-GFP expression increased Akt phosphorylation (Fig. 4E), suggesting the negative regulation of Akt by PTEN in the rat hippocampus.
Akt phosphorylates several down-stream key proteins to exert its survival-promoting effect. Bad is a proapoptotic member of the bcl-2 gene family that is a direct target of Akt. Phosphorylation of Bad by Akt at Ser-136 inactivates its ability to promote cell death (Yang et al., 1995). As illustrated in Figure 4F, our study shows that specific PTEN knockdown, although increasing Akt phosphorylation, results in elevated phosphorylation levels of Bad in hippocampal neurons. These results suggest that PTEN downregulation can activate Akt-dependent downstream signaling pathways and thereby promote neuronal survival within hippocampal neurons.
Lipid and protein phosphatase activities of PTEN, respectively, regulate Akt and NMDARs
To examine whether the phosphatase activity of PTEN is involved in the regulation of Akt/Bad signaling and NMDAR activity, two different dominant-negative forms of phosphatase-inactivated PTEN mutants, C124A and G129E, were used to inhibit endogenous lipid and protein phosphatase activities of PTEN (Myers et al., 1998; Tamura et al., 1998; Weng et al., 2001). The C124A mutation causes a loss of both lipid and protein phosphatase activities, and the mutant G129E results only in loss of lipid phosphatase activity with retention of protein phosphatase activity (Myers et al., 1998; Tamura et al., 1998; Weng et al., 2001). The phosphorylation levels of Akt and the surface expression of NMDARs in neurons expressing GFP plasmids containing C124A or G129E mutants were first examined (Tamura et al., 1998). As expected, Akt phosphorylation was increased in neurons expressing both C124A-GFP and G129E-GFP (Fig. 5A). However, the surface expression of NR1 was decreased in neurons expressing C124A-GFP but increased in neurons expressing G129E-GFP (Fig. 5A,B). There was no significant difference in surface expression of NMDA receptors between the neurons expressing GFP and the untransfected neighboring control cells (Fig. 5B). Subsequently, we found that neurons expressing C124A-GFP exhibited significantly lower peak amplitudes of NMDA currents than the untransfected neighboring control cells (Fig. 5C,D). In contrast, neurons expressing G129E-GFP exhibited significantly increased peak amplitudes of NMDA currents than the untransfected neighboring control cells (Fig. 5C,D). There was no significant difference in peak amplitudes of NMDA currents between the neurons expressing GFP and the untransfected neighboring control cells (Fig. 5D). Together, these results suggest that Akt phosphorylation and NMDAR activities are predominately regulated by the lipid and protein phosphatase activities, respectively.
Figure 5.

Akt and NMDARs are, respectively, regulated by PTEN lipid and protein phosphatase activities. A, Representative images showing that Akt phosphorylation is increased in neurons transfected with either C124A-GFP or G129E-GFP. However, transfection of C124A-GFP decreases NR1 surface expression, and expression of G129E-GFP increases NR1 surface expression. Scale bar, 20 μm. B, Summarized data showing surface NR1 expression (Non-GFP, n = 52; GFP, n = 48; C124A-GFP, n = 46; G129E-GFP, n = 61; *p < 0.05). C, Examples of responses of NMDA currents to GFP, C124A-GFP, or G129E-GFP expression. D, Graph showing peak NMDA currents inhibited by C124A-GFP transfection but increased by G129E-GFP expression (Non-GFP, n = 21; GFP, n = 27; C124A-GFP, n = 31; G129E-GFP, n = 35; *p < 0.05).
Suppressing PTEN expression protects against ischemic neuronal death
The responses of Akt and extrasynaptic NMDARs to PTEN knockdown in our observations suggest that PTEN downregulation may be involved in the regulation of neuronal survival processes. To determine whether PTEN downregulation was neuroprotective, PI staining was performed to assess neuronal death in hippocampal neurons expressing siRNApten at 48 hr after 1 hr OGD. Of note, neurons transfected with siRNApten-GFP vectors had significantly less neuronal death than those transfected with SsiRNApten-GFP vectors (Fig. 6A), supporting a neuroprotective role of PTEN downregulation in ischemic neuronal injury.
Figure 6.

Neuroprotection mediated by PTEN down regulation. A, Left, Examples of PI staining. Scale bar, 25 μm. Right, Summarized data from PI staining (GFP, n = 216; SsiRNApten-GFP, n = 189; siRNA-GFP, n = 207; *p < 0.05). B, Top, An example of reduced PTEN expression in the CA1 pyramidal cell layer 7 d after microelectroporation. Bottom, Time course of PTEN expression after microelectroporation of PTEN siRNA-GFP (n = 6 for each group; *p < 0.05). Data are normalized to values from nontransfected neurons. C, Left, Examples of Fluoro-Jade B staining of degenerating neurons in the CA1 pyramidal cell layer. Scale bar, 60 μm. Right, Summarized data from Fluoro-Jade B staining (*p < 0.05, difference from control; **p < 0.05, difference from siRNApten expression; data shown are means ± SE of 4 determinations). D, DN-Akt blocks the neuroprotection induced by PTEN downregulation (SsiRNApten-GFP, n = 125; siRNApten-GFP, n = 151; siRNApten-GFP + DN-Akt, n = 116. *p < 0.05, difference from SsiRNApten-GFP; #p < 0.05, difference from siRNApten-GFP). E, PI labeling showing that ifenprodil treatment significantly reduces OGD-induced neuronal death in neurons expressing GFP (left: control, n = 183; ifenprodil treatment, n = 162; *p < 0.05) but does not have an effect on neurons coexpressing C124A-GFP and DN-Akt (right: control, n = 151; ifenprodil treatment, n = 178).
Importantly, our study has provided direct evidence that suppressing PTEN expression in vivo protects against delayed hippocampal CA1 neuronal death in the transient global ischemic model (Fig. 6 B, C). As shown in Figure 6C, massive degenerating neurons labeled with Fluoro-Jade B are present in the CA1 pyramidal cell layer 48 hr after global ischemia in vehicle-injected animals, whereas the in vivo administration of siRNApten into the CA1 pyramidal cell layer 3 d before global ischemia prominently reduces the number of neurons stained with Fluoro-Jade B around the injection site, as indicated by arrowheads. As expected, the control injection of SsiRNApten-GFP did not have neuroprotective effect (Fig. 6C). These findings strongly suggest that PTEN downregulation is neuroprotective in cerebral ischemia.
Enhanced Akt signaling and inhibited activities of extrasynaptic NMDARs mediate the neuroprotection
To gain direct evidence that elevated Akt activity contributed to the neuroprotection, we cotransfected dominant-negative Akt constructs (DN-Akt) (Stambolic et al., 1998; Wang et al., 1999) with siRNApten-GFP to inhibit PTEN deficiency-induced Akt activation. When compared with SsiRNApten-GFP or siRNApten-GFP, the expression of DN-Akt with siRNApten-GFP significantly blocked siRNApten-GFP-induced decrease of neuronal death at 48 hr after 1 hr OGD (Fig. 6D), suggesting that the neuroprotective effects mediated by PTEN downregulation require Akt activation.
We also tested whether the neuroprotection could be attributable to the inhibition of extrasynaptic NMDARs. We cotransfected DN-Akt with C124A-GFP to inhibit C124A-induced Akt activation, and the effect of downregulated protein phosphatase activity mediated by C124A introduction could be observed. The PI staining data showed that addition of ifenprodil, a selective inhibitor of NR2B-containing NMDARs (Williams 1993; Tovar and Westbrook, 1999; Williams, 2001; Hardingham et al., 2002), resulted in a significant reduction of cell death in neurons expressing GFP (Fig. 6E, left) but failed to reduce cell death in neurons expressing both C124A-GFP and DN-Akt (Fig. 6E, right). Because ifenprodil can efficiently block extrasynaptic NMDAR-mediated neuronal death (Hardingham et al., 2002), these data suggest that ifenprodil-induced neuroprotection shares a common final pathway, namely the inhibited extrasynaptic NMDAR-dependent signaling, with the protective effect mediated by downregulation of PTEN protein phosphatase activity. Together with the observed effects of PTEN on NMDARs, these results suggest that downregulation of protein phosphatase activity of PTEN may exert its neuroprotective effects through inhibiting single-channel activity and/or surface expression of extrasynaptic NMDARs after ischemic neuronal injury.
Discussion
Modulation of NMDARs by kinases and phosphatases is a crucial cellular process that regulates diverse neuronal function (Swope et al., 1999; Carroll and Zukin, 2002). However, little is known about the role of extrasynaptic NMDARs in this regulatory process. We demonstrated that specific downregulation of the protein phosphatase activity of PTEN results in decrease of both single-channel activity and surface expression of extrasynaptic NMDARs. Thus, we identified the protein phosphatase activity of PTEN as a crucial upstream signal to regulate extrasynaptic NMDARs. Importantly, our work has provided direct evidence that suppressing the protein phosphatase activity of PTEN, through downregulating extrasynaptic NMDARs, protects against ischemic neuronal death. Thus, downregulating protein phosphatase activity of PTEN may represent a novel pharmacological approach for stroke treatment by which extrasynaptic NMDAR-mediated neuronal death can be prevented without significantly interfering with NMDAR-mediated synaptic function (Hardingham et al., 2002; Vanhoutte and Bading, 2003).
Although additional investigations are required to reveal the precise mechanisms underlying the PTEN regulation of NMDARs, the present studies have demonstrated that PTEN is physically associated with NR1 and NR2B subunits. Interestingly, our data indicate that PTEN can bind directly with the carboxyl tail of NR1 but not NR2 subunits, implying that direct protein-protein interaction may occur between PTEN and NR1 subunits. The NR1 subunit has four splice variants that differ in their C-terminal domain with specific relevance to phosphorylation modifications. To gain direct implications on PTEN regulation of NMDARs, it would be interesting to know in the future whether PTEN can interact with each of the NR1 splice variants. Because NR2 subunits play a critical role in controlling the number and composition of NMDARs at synapses (Barria and Malinow, 2002), the indirect modulation of NR2B subunits by PTEN may contribute to the altered surface NMDAR expression at the extrasynaptic sites. Additional characterization of cellular and molecular mechanisms mediating PTEN regulation of extrasynaptic NMDARs and identification of intracellular signaling coupled to the interaction between PTEN and extrasynaptic NMDARs may explain the neuroprotective effects of PTEN downregulation.
Akt is a crucial mediator of neuronal survival (Dudek et al., 1997; Yuan and Yankner, 2000). As an upstream regulator of Akt, the phosphatase PTEN may play a role in the regulation of survival signaling during the neuronal injury process. Supporting this idea, our study has provided direct evidence that downregulating the lipid phosphatase activity of PTEN results in increased phosphorylation levels of both Akt and its downstream target Bad in hippocampal neurons, which leads to decreased hippocampal neuronal death after ischemic injury. Although activation of the Akt-dependent pathway by factors such as neurotrophins has been suggested for the treatment of stroke, the efficacy may be impaired by the antagonizing effect of PTEN, because endogenous PTEN within neurons continuously reduces the levels of phosphatidylinositol 3, 4, 5-trisphosphate, the positive regulator of Akt activity (Stambolic et al., 1998). On the basis of our findings, we propose a synergistic approach for the treatment of stroke in which combining Akt signaling activators with PTEN downregulation may be more effective in preventing ischemic neuronal death.
Footnotes
This work was supported by grants to Q.W. from the Heart and Stroke Foundation of Ontario, Christopher Reeve Paralysis Foundation, and Canadian Institutes of Health Research (CIHR). Q.W. is a CIHR New Investigator. M.G.F. holds the Krembil Chair in Neural Repair and Regeneration. We thank R. Agami and K. M. Yamada for pSUPER vector and PTEN plasmids. We also thank P. Yang and P. Z. Sun for technical assistance.
Correspondence should be addressed to Dr. Qi Wan, Toronto Western Research Institute and Department of Physiology, University of Toronto, 399 Bathurst Street, McLaughling Wing, Room 14-409, Toronto, Ontario, Canada M5T 2S8. E-mail: qi.wan@utoronto.ca.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244052-09$15.00/0
References
- Backman SA, Stambolic V, Suzuki A, Haight J, Elia A, Pretorius J, Tsao MS, Shannon P, Bolon B, Ivy GO, Mak TW (2001) Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet 29: 396-403. [DOI] [PubMed] [Google Scholar]
- Barria A, Malinow R (2002) Subunit-specific NMDA receptor trafficking to synapses. Neuron 35: 345-353. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Carroll RC, Yu X, Morishita W, Yasuda H, von Zastrow M, Malenka RC (2000) Regulation of AMPA receptor endocytosis by a signaling mechanism shared with LTD. Nat Neurosci 3: 1291-1300. [DOI] [PubMed] [Google Scholar]
- Brewer GJ, Torricelli JR, Evege EK, Price PJ (1993) Optimized survival of hippocampal neurons in B27-supplemented Neurobasal, a new serum-free medium combination. J Neurosci Res 35: 567-576. [DOI] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550-553. [DOI] [PubMed] [Google Scholar]
- Carroll RC, Zukin RS (2002) NMDA-receptor trafficking and targeting: implications for synaptic transmission and plasticity. Trends Neurosci 25: 571-577. [DOI] [PubMed] [Google Scholar]
- Choi DW (1995) Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci 18: 58-60. [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ (1995) Fitting and statistical analysis of single-channel records. In: Single-channel recording, Ed 2 (Sakmann B, Neher E, eds), pp 483-587. New York: Plenum.
- Dingledine R, Borges K, Bowie D, Traynelis SF (1999) The glutamate receptor ion channels. Pharmacol Rev 51: 7-61. [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275: 661-665. [DOI] [PubMed] [Google Scholar]
- Gary DS, Mattson MP (2002) PTEN regulates Akt kinase activity in hippocampal neurons and increases their sensitivity to glutamate and apoptosis. Neuromolecular Med 2: 261-269. [DOI] [PubMed] [Google Scholar]
- Goldberg MP, Choi DW (1993) Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci 13: 3510-3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Lesche R, Trumpp A, Zack JA, Kornblum HI, Liu X, Wu H (2001) Negative regulation of neural stem/progenitor cell proliferation by the Pten tumor suppressor gene in vivo. Science 294: 2186-2189. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5: 405-414. [DOI] [PubMed] [Google Scholar]
- Hopkins KJ, Wang G, Schmued LC (2000) Temporal progression of kainic acid induced neuronal and myelin degeneration in the rat forebrain. Brain Res 864: 69-80. [DOI] [PubMed] [Google Scholar]
- Hsu JC, Zhang Y, Takagi N, Gurd JW, Wallace MC, Zhang L, Eubanks JH (1998) Decreased expression and functionality of NMDA receptor complexes persist in the CA1, but not in the dentate gyrus after transient cerebral ischemia. J Cereb Blood Flow Metab 18: 768-775. [DOI] [PubMed] [Google Scholar]
- Jahr CE (1992) High probability opening of NMDA receptor channels by l-glutamate. Science 255: 470-472. [DOI] [PubMed] [Google Scholar]
- Kwon CH, Zhu X, Zhang J, Knoop LL, Tharp R, Smeyne RJ, Eberhart CG, Burger PC, Baker SJ (2001) Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet 29: 404-411. [DOI] [PubMed] [Google Scholar]
- Lan JY, Skeberdis VA, Jover T, Grooms SY, Lin Y, Araneda RC, Zheng X, Bennett MV, Zukin RS (2001) Protein kinase C modulates NMDA receptor trafficking and gating. Nat Neurosci 4: 382-390. [DOI] [PubMed] [Google Scholar]
- Lee FJ, Xue S, Pei L, Vukusic B, Chery N, Wang Y, Wang YT, Niznik HB, Yu XM, Liu F (2002) Dual regulation of NMDA receptor functions by direct protein-protein interactions with the dopamine D1 receptor. Cell 111: 219-230. [DOI] [PubMed] [Google Scholar]
- Lee JH, Kim KY, Lee YK, Park SY, Kim CD, Lee WS, Rhim BY, Hong KW (2004) Cilostazol prevents focal cerebral ischemic injury by enhancing CK2 phosphorylation and suppression of PTEN phosphorylation in rats. J Pharmacol Exp Ther 308: 896-903. [DOI] [PubMed] [Google Scholar]
- Li B, Chen N, Luo T, Otsu Y, Murphy TH, Raymond LA (2002) Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat Neurosci 5: 833-834. [DOI] [PubMed] [Google Scholar]
- Li JH, Wang YH, Wolfe BB, Krueger KE, Corsi L, Stocca G, Vicini S (1998) Developmental changes in localization of NMDA receptor subunits in primary cultures of cortical neurons. Eur J Neurosci 10: 1704-1715. [DOI] [PubMed] [Google Scholar]
- Liu F, Wan Q, Pristupa ZB, Yu XM, Wang YT, Niznik HB (2000) Direct protein-protein coupling enables cross-talk between dopamine D5 and gamma-aminobutyric acid A receptors. Nature 403: 274-280. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem 273: 13375-13378. [DOI] [PubMed] [Google Scholar]
- Myers MP, Stolarov JP, Eng C, Li J, Wang SI, Wigler MH, Parsons R, Tonks NK (1997) P-TEN, the tumor suppressor from human chromosome 10q23, is a dual-specificity phosphatase. Proc Natl Acad Sci USA 94: 9052-9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MP, Pass I, Batty IH, Van der Kaay J, Stolarov JP, Hemmings BA, Wigler MH, Downes CP, Tonks NK (1998) The lipid phosphatase activity of PTEN is critical for its tumor supressor function. Proc Natl Acad Sci USA 95: 13513-13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Piech V, Sheng M (2001) Subunit-specific temporal and spatial patterns of AMPA receptor exocytosis in hippocampal neurons. Nat Neurosci 4: 917-926. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Feltz A, Westbrook GL (1995) Synaptic NMDA receptor channels have a low open probability. J Neurosci 15: 2788-2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumbaugh G, Vicini S (1999) Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J Neurosci 19: 10603-10610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannevin RH, Huganir RL (2000) Postsynaptic organization and regulation of excitatory synapses. Nat Rev Neurosci 1: 133-141. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ (2002) Postsynaptic signaling and plasticity mechanisms. Science 298: 776-780. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF (2001) Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci 4: 1079-1085. [DOI] [PubMed] [Google Scholar]
- Stambolic V, Suzuki A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, Ruland J, Penninger JM, Siderovski DP, Mak TW (1998) Negative regulation of PKB/Akt-dependent cell survival by the tumor suppressor PTEN. Cell 95: 29-39. [DOI] [PubMed] [Google Scholar]
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV (1997) Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers. Nat Genet 15: 356-362. [DOI] [PubMed] [Google Scholar]
- Stocca G, Vicini S (1998) Increased contribution of NR2A subunit to synaptic NMDA receptors in developing rat cortical neurons. J Physiol (Lond) 507: 13-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Raymond LA, Huganir RL (1999) Regulation of ligandgated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res 33: 49-78. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada KM (1998) Inhibition of cell migration, spreading, and focal adhesions by tumor suppressor PTEN. Science 280: 1614-1617. [DOI] [PubMed] [Google Scholar]
- Tamura M, Gu J, Takino T, Yamada KM (1999) Tumor suppressor PTEN inhibition of cell invasion, migration, and growth: differential involvement of focal adhesion kinase and p130Cas. Cancer Res 59: 442-449. [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro J Neurosci 19: 4180-4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanhoutte P, Bading H (2003) Opposing roles of synaptic and extrasynaptic NMDA receptors in neuronal calcium signalling and BDNF gene regulation. Curr Opin Neurobiol 13: 366-371. [DOI] [PubMed] [Google Scholar]
- Wan Q, Man HY, Braunton J, Wang W, Salter MW, Becker L, Wang YT (1997a) Modulation of GABAA receptor function by tyrosine phosphorylation of beta subunits. J Neurosci 17: 5062-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT (1997b) Recruitment of functional GABA(A) receptors to postsynaptic domains by insulin. Nature 388: 686-690. [DOI] [PubMed] [Google Scholar]
- Wang Q, Somwar R, Bilan PJ, Liu Z, Jin J, Woodgett JR, Klip A (1999) Protein kinase B/Akt participates in GLUT4 translocation by insulin in L6 myoblasts. Mol Cell Biol 19: 4008-4018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YT, Yu XM, Salter MW (1996) Ca2+-independent reduction of N-methyl-d-aspartate channel activity by protein tyrosine phosphatase. Proc Natl Acad Sci USA 93: 1721-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei F, Xia XM, Tang J, Ao H, Ko S, Liauw J, Qiu CS, Zhuo M (2003) Calmodulin regulates synaptic plasticity in the anterior cingulate cortex and behavioral responses: a microelectroporation study in adult rodents. J Neurosci 23: 8402-8409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng LP, Brown JL, Eng C (2001) PTEN coordinates G(1) arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum Mol Genet 10: 599-604. [DOI] [PubMed] [Google Scholar]
- Williams K (1993) Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol 44: 851-859. [PubMed] [Google Scholar]
- Williams K (2001) Ifenprodil, a novel NMDA receptor antagonist: site and mechanism of action. Curr Drug Targets 2: 285-298. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ (1995) Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80: 285-291. [DOI] [PubMed] [Google Scholar]
- Ying HS, Weishaupt JH, Grabb M, Canzoniero LMT, Sensi SL, Sheline CT, Monyer H, Choi DW (1997) Sublethal oxygen-glucose deprivation alters hippocampal neuronal AMPA receptor expression and vulnerability to kainate-induced death. J Neurosci 17: 9536-9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Yankner BA (2000) Apoptosis in the nervous system. Nature 407: 802-809. [DOI] [PubMed] [Google Scholar]
- Zhang X, Cui SS, Wallace AE, Hannesson DK, Schmued LC, Saucier DM, Honer WG, Corcoran ME (2002) Relations between brain pathology and temporal lobe epilepsy. J Neurosci 22: 6052-6061. [DOI] [PMC free article] [PubMed] [Google Scholar]