

Abstract
The early inflammatory response to spinal cord injury (SCI) causes significant secondary damage. Strategies that nonselectively suppress inflammation have not improved outcomes after SCI, perhaps because inflammation has both adverse and beneficial effects after SCI. We have shown that the selective, time-limited action of a monoclonal antibody (mAb) to the CD11d subunit of the CD11d/CD18 integrin, delivered intravenously during the first 48 hr after SCI in rats, markedly decreases the infiltration of neutrophils and delays the entry of hematogenous monocyte-macrophages into the injured cord. We hypothesized that this targeted strategy would lead to neuroprotection and improved neurological outcomes. In this study the development of chronic pain was detected in rats by assessing mechanical allodynia on the trunk and hindpaws 2 weeks to 3 months after a clinically relevant clip-compression SCI at the twelfth thoracic segment. The anti-CD11d mAb treatment reduced this pain by half. Motor performance also improved as rats were able to plantar-place their hindpaws and use them for weight support instead of sweeping movements only. Improved cardiovascular outcome was shown after SCI at the fourth thoracic segment by significant decreases in autonomic dysreflexia. Locomotor performance was also improved. These functional changes correlated with significantly greater amounts and increased organization of myelin and neurofilament near the lesion. The improved neurological recovery after the specific reduction of early inflammation after SCI demonstrates that this selective strategy increases tissue at the injury site and improves its functional capacity. This early neuroprotective treatment would be an ideal foundation for building later cell-based therapies.
Keywords: spinal cord injury, integrin, inflammation, pain, autonomic, locomotor, myelin, neurofilament
Introduction
Secondary damage after a spinal cord injury (SCI) is caused, in part, by ischemia, cellular and tissue edema, and oxidative damage. These changes often relate to early inflammation and lead to myelin degradation as well as intensified necrosis and apoptosis of neurons and glia that all contribute to increases in lesion size (Taoka and Okajima, 1998; Bethea and Dietrich, 2002). Neutrophils and hematogenous macrophages invade the spinal cord, peaking at 12 hr and 5-7 d after SCI, respectively (Popovich et al., 1997; Taoka et al., 1997; Leskovar et al., 2000), and releasing pro-inflammatory cytokines and reactive oxygen and nitrogen species (Taoka and Okajima, 1998; Popovich et al., 1999). These cytotoxic substances further damage the white and gray matter and contribute to scar formation (Jones et al., 2002).
The progression of secondary damage deteriorates neurological function and renders the spinal cord less amenable to repair strategies such as cell replacement or therapeutic gene transfer. Preventing this progression is of key importance. No practical way to control the early inflammatory response has yet been devised, despite our understanding of its destructive role in SCI. We postulated that a successful anti-inflammatory treatment must selectively disrupt the early, destructive, leukocyte-mediated actions in the injured cord, while leaving an opportunity for later regenerative interventions and wound-healing responses. After SCI, intraspinal leukocyte infiltration first requires leukocyte tethering by selectins on the surface of endothelial cells (Bevilacqua, 1993). This is followed by the interaction of endothelial cell-adhesion molecules with integrins on the leukocyte surface (Neish et al., 1995; Shanley et al., 1998), facilitating leukocyte extravasation through the blood-CNS barrier. To prevent this interaction, we used a mAb to the CD11d subunit of the CD11d/CD18 integrin (Grayson et al., 1999; Van der Vieren et al., 1999). This mAb treatment substantially decreases the numbers of neutrophils and macrophages at the lesion at 2-3 d after SCI (Mabon et al., 2000; Saville et al., 2002). In the present study, we delivered the anti-CD11d mAb intravenously in three consecutive doses at 2, 24, and 48 hr after a clinically relevant clip-compression SCI (Rivlin and Tator, 1977) and assessed the neurological outcomes and effects on the spinal cord lesion. The disabilities faced by those with SCI include development of incapacitating chronic pain (Siddall et al., 1995; Yezierski, 1996) and loss of control of cardiovascular, bowel, urinary bladder, and sexual function (Mathias and Frankel, 1992; Karlsson, 1999). Therefore, our study assessed some of these functions, as well as motor outcomes, to obtain a balanced view of the neurological outcome. In one series of experiments, we induced an injury at the twelfth thoracic segment (T12) that causes incomplete sensorimotor loss and consistently leads to mechanical allodynia, a type of neuropathic pain in which non-noxious stimuli generate avoidance responses (Bruce et al., 2002). In a second series, we induced an injury at the fourth thoracic segment (T4) that, in addition to major motor and sensory impairment, greatly affects autonomic function, leading to episodic hypertension termed autonomic dysreflexia (Weaver et al., 2001).
Materials and Methods
Spinal cord injury and treatment paradigm
All animal procedures were done following the Canadian Guide to Care and Use of Experimental Animals. Male rats were used to study mechanical allodynia and housed in pairs. Female rats were used to study autonomic dysreflexia and housed individually. Forty-one female and sixteen male Wistar rats (Harlan Bioproducts, Indianapolis, IN), were anesthetized as described previously (Weaver et al., 2001). The T4 or T12 spinal cord segment was exposed by a dorsal laminectomy and injured, without disrupting the dura, by 60 sec of clip compression (Weaver et al., 2001). A 50 gm calibrated clip was used at T4 and a 35 gm clip was used at T12, producing models of autonomic dysreflexia and mechanical allodynia, respectively (Weaver et al., 2001; Bruce et al., 2002). Postoperative care was provided as described previously (Weaver et al., 2001). Rats were blindly assigned to one of two groups. One of the following treatments was administered intravenously via the tail vein in three consecutive doses at 2, 24, and 48 hr after SCI. The control groups received normal saline (2 week study) or an isotype-matched irrelevant antibody (Ab) (1B7, 1 mg/kg, gift of the ICOS Corporation, Bothell, WA; 6 and 12 week studies), and a second group received the anti-CD11d mAb (1.0 mg/kg; gift of the ICOS Corporation). All aspects of the testing and data analysis were done using a blinded experimental design.
Behavioral and physiological testing Locomotor function.
Locomotor recovery of the animals was assessed by two independent observers using the 21 point Basso, Beattie, and Bresnahan (BBB) open field locomotor score (Basso et al., 1995) from 1 d to 12 weeks after SCI. Testing was done twice per week, and scores were averaged to generate a weekly score. Scores of left and right legs were averaged. In addition, motor testing after injury at T12 included the inclined plane test (Rivlin and Tator, 1977), in which the animal is placed on a rubber mat secured to a flat board that is then tilted from the horizontal plane in 5° increments until the animal loses its grip. The maximal sustainable angle that the rat can maintain for 3 sec in a head-up position is the score for that animal at that time. Rats that recovered sufficient locomotor skill were also tested using a more difficult grid-walking test in which they are placed on a 30 cm long runway with grid openings of 4 × 4 cm, and the number of foot-falls through the grid are counted after videotaping (Kunkel-Bagden et al., 1993). Rats must be able to accomplish weight supporting hindlimb plantar placement and some degree of coordinated stepping before using this test. Otherwise they simply drag their hindquarters across the runway.
Mechanical allodynia. Rats were tested for the presence of mechanical allodynia in the dorsal trunk and the hindpaws before and 2-12 weeks after SCI at T12. Mechanical allodynia is one type of neuropathic pain resulting in an increased sensitivity to innocuous stimuli. In this state, pain is produced by mechanical stimuli that are not painful to an uninjured animal or person (Christensen et al., 1996). Using a modified Semmes-Weinstein monofilament (Stoelting Co., Wood Dale, IL) calibrated to generate a force of 15 mN, rats were tested for their response to innocuous mechanical stimulation. Mechanical allodynia was assessed once per week on the dorsal trunk immediately rostral to the lesion level in dermatomes corresponding to the T9-T11 spinal segments. Mechanical allodynia occurring below the SCI level was assessed every second week by stimulation to the hindpaws, as described elsewhere (Bruce et al., 2002). A testing session consisted of 10 stimulations with the filament lasting 3 sec, each separated by a 5 sec interval. Avoidance responses, defined as flinching, escape, paw withdrawal, and/or licking, vocalization, or abnormal aggressive behaviors, indicated that the rat perceived the stimulus as noxious. The number of these responses to 10 stimuli was quantified.
Autonomic dysreflexia. On the thirteenth or approximately fortieth day after SCI at T4, a femoral or carotid artery was cannulated under halothane anesthesia. Autonomic dysreflexia, assessed 6-48 hr later, is an increase in arterial pressure and reflex decrease in heart rate, induced by a 1 min distension of the colon with a balloon-tipped catheter that is inflated with 2.0 ml of air (Weaver et al., 2001). This generates a colon distension similar in size to a large fecal bolus and is only slightly noxious to a rat with an intact spinal cord (Marsh and Weaver, 2004). The average change during the minute of colon stimulation was used for statistical comparisons. Cardiovascular data acquisition and analysis were performed using Powerlab software (AD Instruments, Mountain View, CA).
Lesion assessment
At 2, 6, or 12 weeks after SCI, the rats were perfused with 4% formaldehyde, and the spinal cord was removed and cryostat-sectioned transversely at 20 μm and serially thaw-mounted on alternate slides. One set of sections was processed for luxol fast blue (2 week study) or solochrome cyanin (6 and 12 week study) staining to identify tightly packed myelin (Page, 1965; Weaver et al., 2001). A second, adjacent, set of sections from the 6 and 12 week studies was immunoprocessed for neurofilament 200 using an anti-NF200 mouse mAb (Chemicon, Temecula, CA) and standard procedures including the diaminobenzidine reaction to visualize the reaction product (Bruce et al., 2002). Sections reacted without the primary Ab revealed no nonspecific staining. A third set of sections from the 6 and 12 week studies was stained with hematoxylin and eosin for analysis of morphological structure at the lesion site.
Using a blinded assessment, 10 hematoxylin and eosin-stained sections per lesion site were rated on a scale of 1-3 for necrosis, fibrovascular tissue, and inflammatory cells. These 10 sections sampled approximately a 1 mm length of cord. Necrosis was identified as gross areas of cellular breakdown, debris, and tissue fragmentation. The inflammatory infiltrates included cells with morphology typical of macrophages and lymphocytes. Two different individuals scored these lesions, and their scores were averaged. For analysis of myelin and neurofilament staining, digitized images of every eighth section on a slide were collected, and the stained area was quantified using a calibrated analysis program of ImagePro Plus software (Media Cybernetics, Silver Spring, MD). The area measurements were normalized for analysis because of the distortion of the overall cord area by a large cystic cavity at and near the lesion in some rats. We first established that the cord cross-sectional area within a length 3 mm rostral and caudal to the T4 or T12 dorsal roots is uniform in the normal intact spinal cord. Areas of staining were then expressed in each rat as a percentage of the total cross-sectional area of the relatively intact thoracic cord sampled 3-4 mm rostral to the epicenter of the injury. The averaged area of five sections was calculated and plotted as a sample at 0.4 mm lengths along the cord. The smallest average area was considered the epicenter of the lesion. In addition, the normalized areas of neurofilament in gray and white matter at 2 mm rostral and caudal to the lesion epicenter were determined to assess changes in each specific region with treatment. At 2 mm from the lesion, the gray and white matter could readily be defined by morphological characteristics, and the area of interest used in this subanalysis encompassed exclusively gray or white matter. All analyses were done blinded to the treatment the rat had received.
Statistical analysis
Mean values are expressed ± SE. Results were subjected to parametric statistical analysis using one- or two-way ANOVA. Fischer's protected t test was used to determine differences between mean values (Snedecor and Cochran, 1989). In addition, a simple regression analysis was used to assess the effect of anti-CD11d mAb treatment on the slopes of the area of myelin and neurofilament with distance from the lesion epicenter. This was used to assess the steepness of the increment in area with distance. For all analyses, significance was accepted at p < 0.05.
Results
Anti-CD11d mAb improves locomotor function
After the T12 injury, locomotor improvement was rapid in both treated and control rats. In the control rats, the BBB scores stabilized at about 2 weeks, reaching a maximum score of 7.8 ± 0.2 points (Fig. 1a). In contrast, the BBB scores of the treated rats continued to improve after this time and stabilized at ∼5 weeks, reaching a score of 9.7 ± 0.5 points. The significantly higher scores of the treated rats starting at 4 weeks are notable because the treated rats were able to execute weight-supporting stepping, whereas the control rats could only sweep their hind legs. In the inclined plane test, the treated rats were able to maintain their position on a tilted platform at a significantly higher angle of incline than the control rats (Fig. 1c). The control rats held their position at an average angle of 39° and the treated rats at 44° overall in the 8 week testing period. In our laboratory, intact rats can maintain their position at angles up to 65 ± 1.5° (n = 8). Finally, at 6-12 weeks after T12 SCI, the rats were tested by a grid-walking task on a 30-cm-long grid. This difficult motor task requires significant precision of hindpaw placement, greatly challenging animals with the severity of impairment that we studied. None of the seven control rats were able to perform this task. From 8-12 weeks after SCI, 3 of the 9 treated rats accurately placed their hindpaws on the bars of the grid with an average score of 5 ± 1 footfalls per trial.
Figure 1.

Anti-CD11d mAb treatment improves open field locomotor scores (BBB) (a) and inclined plane test scores (c) for 12 weeks after SCI at T12 and for 6 weeks after SCI at T4 (b). BBB scores of 1-7 indicate increasing movement of the three hindlimb joints, 8 indicates sweeping of the hind limbs with no weight support, and 10 indicates development of more complex motor control, including consistent weight-supported plantar stepping. The inclined plane test (Rivlin and Tator, 1977) determines the ability of the rat to hold its position on an inclined plane with forelimbs and hindlimbs. □, Control rats (a, c, n = 6); ▪, mAb-treated rats (a, c, n = 9). b, Control 1 and 2 weeks, n = 20; 3-6 weeks, n = 6; mAb 1 and 2 weeks, n = 19; 3-6 weeks, n = 7. Decreased number at 3 weeks in c is caused by autonomic testing and perfusion of rats at 2 weeks after SCI. *p < 0.05 compared with control rats.
The SCI at T4 caused more severe paralysis with a less rapid course of recovery (Fig. 1b). At the second, fifth, and sixth weeks after SCI the treated rats had higher scores BBB than the control rats. Treated rats reached a plateau of performance at 5 weeks with a score of 8.4 ± 0.3, indicating that they could make sweeping movements with their hindlegs, whereas the control rats reached an average score of 7.4 ± 0.3 and could not sweep their hind legs.
Anti-CD11d mAb reduces mechanical allodynia at and below the SCI level
Mechanical allodynia was assessed on the dorsal trunk and hindpaws before and at 2-12 weeks after SCI at T12 using a 15 mN modified Semmes Weinstein filament. Before SCI, rats rarely exhibited avoidance responses to stimulation with the filament (Fig. 2a). After SCI, the control rats showed increasing numbers of avoidance responses to the 10 stimuli applied to the dorsal trunk (immediately rostral to the injury), consistent with the development of mechanical allodynia (Fig. 2a) (Bruce et al., 2002). Significant increases in the number of avoidance responses to trunk stimulation commenced at 4 weeks after SCI and subsequently remained at a plateau level of ∼4.1 ± 0.8 avoidance responses to 10 stimulations until the tenth week. Avoidance responses were further increased at 12 weeks after SCI (5.3 ± 0.6), possibly indicating a continuation of the development of mechanical allodynia. The anti-CD11d mAb treatment significantly decreased the frequency with which these avoidance responses occurred, reducing the mechanical allodynia by half at 4 weeks after SCI and lasting for the duration of the study (Fig. 2a). Similarly, the anti-CD11d mAb treatment resulted in a significantly decreased number of avoidance responses to innocuous hindpaw stimulation at 6, 8, and 10 weeks when compared with control treated rats (Fig. 2b). The control rats reached a score of 2.8 ± 0.3 responses, and the treated rats responded approximately half as often (1.3 ± 0.2 responses). The testing of the hind paws was terminated at 10 weeks after injury, perhaps because of the abnormal gait of the rats, the pads on their hindfeet became thickened, causing inconsistency in responses.
Figure 2.

Anti-CD11d mAb treatment decreases mechanical allodynia for 12 weeks after SCI at T12. Avoidance responses were elicited by stimulating the trunk (a) and the hindpaw (b) in controlrats (□, a, n = 7; b, n = 6) and in mAb-treated rats (▪, a, n = 8; b, n = 7).Scores from 2 weeks of testing for allodynia on the trunk were averaged at each time point plotted. *p < 0.05 compared with control rats.
Anti-CD11d mAb reduces autonomic dysreflexia
To assess autonomic dysreflexia, we measured the increases in mean arterial pressure caused by balloon distension of the colon. Dysreflexia increases in magnitude with time after SCI in rats, reaching a plateau at 4-5 weeks (Marsh and Weaver, 2004). At 2 weeks after SCI, this visceral stimulation increased arterial pressure and decreased heart rate for the duration of the stimulus (Fig. 3a). The anti-CD11d mAb treatment markedly reduced these increases in arterial pressure (Fig. 3b). Mean increases during the 1 min stimulation period were reduced significantly from 30 ± 3 mmHg in the control rats to 21 ± 2 mmHg in the treated rats (Fig. 3c). Thirteen of fourteen control rats had an average increase in arterial pressure >20 mmHg, whereas significantly fewer rats treated with the anti-CD11d mAb (3 of 10) had responses of this magnitude (p < 0.05). Because the normal reflex response to this stimulus in an intact rat is an ∼15 mmHg increase in arterial pressure (Maiorov et al., 1997), this treatment affected much of the reflex increment attributable to SCI. The average decreases in heart rate during the 1 min colon distension in control rats (64 ± 10 beats/min) tended to be greater than in treated rats (46 ± 9 beats/min; p = 0.09). At 2 weeks after SCI, resting arterial pressures of control and treated rats were 100 ± 2 and 104 ± 3, respectively and did not differ from each other. Likewise, resting heart rate (503 ± 9 beats/min) in control rats was not different from that in treated rats (495 ± 9 beats/min).
Figure 3.

Anti-CD11d mAb treatment reduces the magnitude of autonomic dysreflexia. Changes in pulsatile arterial pressure (AP), mean arterial pressure (MAP), and heart rate (HR) are shown during a 2 ml balloon distension of the colon in control (a, d) and mAb-treated rats (b, e) 2 weeks (top row) and 6 weeks (bottom row) after SCI at T4. The large increase in pressure evoked at 6 weeks was often associated with arrhythmias (d). Average change in MAP was assessed at 2 (c) and 6 (f) weeks after SCI in control rats (□, c, n = 14; f, n = 6) and mAb-treated rats (▪, c, n = 10; f, n = 7). The dotted lines on the bar graphs show the approximate change in pressure that would be elicited by such stimulation in a rat with an intact spinal cord (Maiorov et al., 1997). bpm, beats per minute; *p < 0.05 compared with control rats.
At 6 weeks after SCI, the increases in arterial pressure during dysreflexia were greater than at 2 weeks by ∼10 and ∼7 mmHg in the control and treated rats, respectively. Heart rate responses to colon distension in the control rats more often included arrhythmias (Fig. 3d). Arterial pressure responses of the treated rats remained smaller than those of the control rats at this time (Fig. 3e). Again, the mean changes in arterial pressure of the control rats (40 ± 6 mmHg) were significantly greater than those of the treated rats (28 ± 2 mmHg) (Fig. 3f). Four of six control rats had increases in arterial pressure >30 mmHg, whereas only 1 of 7 treated rats had an increase this large (p < 0.05). Heart rate decreases during colon distension were inconsistent in control (30 ± 33 beats/min) and treated rats (22 ± 10 beats/min) and did not differ between the groups. At 6 weeks after SCI the resting mean arterial pressures of control and treated rats were 111 ± 2 and 111 ± 3 mmHg, respectively. Likewise, the resting heart rates in control (501 ± 10 beats/min) and treated (520 ± 17 beats/min) rats were not different from each other.
Characteristics of the lesion site including inflammatory cells
The tissue at the lesion site of all rats at 6 and 12 weeks after SCI was characterized by areas of necrotic debris, bundles of fibrovascular tissue, and a chronic state of inflammation (Fig. 4a-d). Foamy macrophages and clusters of small, intensely stained cells with morphology suggestive of lymphocytes were the characteristic inflammatory cells within the lesion (Fig. 4e,f). The abundance of inflammatory cells appeared greater at 6 than 12 weeks after SCI. At these times only the occasional, single, neutrophil was found in these lesion sites. Areas within the lesion contained small and large arterioles and bundles of fibrous tissue (Fig. 4g,h). As shown on Figure 4 and Table 1, most of these morphological characteristics did not differ between the anti-CD11d mAb-treated and control rats. The density of fibrovascular bundles and the relative abundance of inflammatory cells in the treated and control lesion sites and adjacent neuropil were similar in both groups of rats. In contrast, the extent of necrotic debris within the lesion was significantly less in the treated rats: the mean rating score for necrosis was significantly lower at the T12 lesion of mAb-treated rats than in the control rats and tended to be lower at T4 in treated rats as well (Fig. 4c,d, Table 1).
Figure 4.
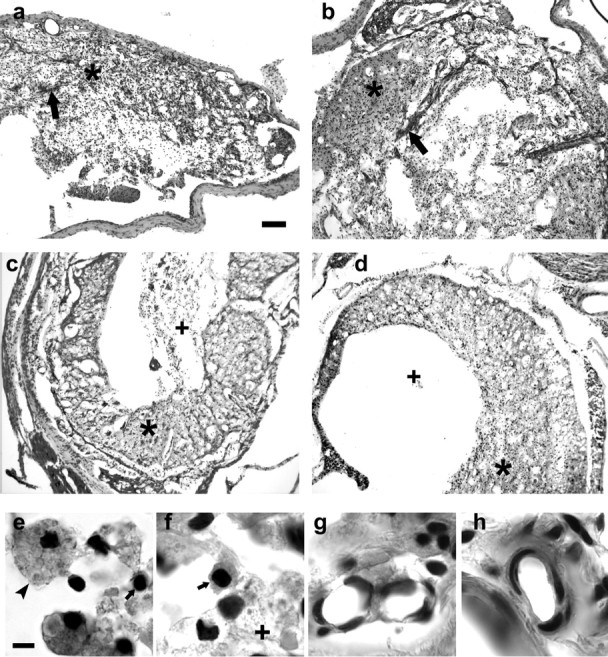
Morphological and cellular characteristics of the lesion site at T4 (6 weeks) and T12 (12 weeks) in control and mAb-treated rats. Low-power photomicrographs of sections stained with hematoxylin and eosin show the general appearance of the lesion at T4 in control (a) and mAb-treated (b) rats and at T12 in control (c) and mAb-treated rats (d). Asterisks on panels indicate areas containing inflammatory cells. Large arrows on a and b indicate bands of fibrovascular tissue. Plus signs (+) indicate areas of necrotic debris. e and f illustrate examples of foamy macrophages (arrowhead) and lymphocytes (small arrow) present in these lesions. An area of necrotic debris is indicated by the + in f. g and h show examples of arterioles within bundles of fibrous tissue. Scale bars: (in a) a-d, 100 μm; (in e) e-h,= 5.0 μm.
Table 1.
Morphological and cellular characteristics of lesion site
|
|
Necrosis |
Fibrovascular tissue |
Inflammatory cells |
|---|---|---|---|
| T4-6 weeks (n = 5) | |||
| Control | 2.7 ± 0.2 | 2.0 ± 0.2 | 2.4 ± 0.1 |
| Anti-CD11d | 2.2 ± 0.3 | 2.0 ± 0.3 | 2.7 ± 0.1 |
| T12-12 weeks (n = 6) | |||
| Control | 2.8 ± 0.1 | 1.5 ± 0.1 | 2.6 ± 0.2 |
| Anti-CD11d |
2.4 ± 0.2*
|
1.6 ± 0.2 |
2.4 ± 0.3 |
Relative quantity of necrotic debris, fibrovascular tissue, and inflammatory cells were ranked 1-3, with a score of 3 indicating the greatest amount. *Significantly different from control.
Compact myelin is greater after anti-CD11d mAb
After the T4 or T12 SCI, the staining for compact myelin revealed the overall integrity of the remaining white matter. At and near the lesion epicenter, myelin was detected in small patches that constituted a small percentage of the normalized cross-sectional area of the cord in both control and treated rats (Fig. 5a-d). Samples of spinal cord sections at 6 weeks after SCI at T4 illustrate myelin in the lesion epicenter that appears as small areas of dark staining (Fig. 5a). At 2.0 mm caudal to the T4 injury, this myelin staining encompassed much of the white matter and, because the treated rats had substantially more intact white matter in this area, more myelin was visible in these rats than in the control rats (Fig. 5a). This treatment effect was also apparent at the T12 lesion, although injury in this area led to a larger, longer area of damage (photomicrographs not shown).
Figure 5.

Anti-CD11d mAb treatment increases myelin in the injured cord after SCI. Solochrome cyanin-stained sections taken at 6 weeks after T4 SCI are shown at the lesion epicenter and at 2.0 mm caudal to the epicenter in control and mAb-treated rats (a). Arrows on photomicrographs at the epicenter indicate patches of intact dark blue compact myelin. Note the more intact neuropil at 2 mm caudal to the injury site in the rat treated with anti-CD11d mAb, reflected by more abundant compact myelin and a smaller cavitation than in the control rat. Scale bar, 200 μm (applies to all photomicrographs). Treatment effects on normalized areas of compact myelin are illustrated after SCI at T4 (b, 2 weeks; c, 6 weeks) and at T12 (d, 12 weeks). Myelin was stained with solochrome cyanin in c and d and with luxol fast blue in b. □, Control rats (b, n = 10; c, n = 5; d, n = 5). ▪, mAb-treated rats (b, n = 8; c, n = 7; d, n = 6). *p < 0.05 compared with control rats. +Shortest distance where area is larger than area at epicenter, p < 0.05.
We determined areas of staining in the injured spinal cords by quantitative morphometry. In all rats the area of myelin was smallest at the epicenter of the lesion and increased incrementally with distance from the epicenter (Fig. 5b-d). Areas of myelin in the treated animals were significantly greater than in those in control rats at most cord levels up to, and beyond, 2.0 mm from the T4 injury site at 2 and 6 weeks after the injury (Fig. 5b,c). This treatment effect was present at all cord levels examined. At the lesion epicenter, a small amount of myelin was present in the treated rats at 2 weeks after SCI but was no longer detected by 6 weeks. We defined a core of the lesion as all areas that were not significantly different from the epicenter. The boundaries of this lesion core were the shortest distance rostral and caudal from the epicenter where the areas were significantly different from the area at the epicenter. At 2 weeks, the width of this core at T4 was 2.0 mm in control rats and only 1.2 mm in treated rats (Fig. 5b). At 6 weeks after SCI, this difference was no longer apparent; cores in treated and control rats were 1.6 mm (Fig. 5c). However, the increment in myelin area with distance from the epicenter was greater in the anti-CD11d mAb-treated rats both rostral and caudal to the epicenter at 6 weeks after SCI (Table 2). These measurements reveal that the overall volume of myelin present in the treated rats was greater than that in the control rats.
Table 2.
Effect of anti-CD11d mAb treatment on slope of increase in area of myelin and neurofilament with distance from epicentre
|
Myelin staina |
T4-2 weeks |
T4-6 weeks |
T12-12 weeks |
|---|---|---|---|
| Rostral | |||
| Control | 0.151b | 0.207 | 0.061 |
| Anti-CD11d | 0.160 | 0.252* | 0.112* |
| Caudal | |||
| Control | 0.161 | 0.179 | 0.044 |
| Anti-CD11d | 0.194 | 0.273* | 0.108* |
| Neurofilamenta | |||
| Rostral | |||
| Control | 0.033 | 0.025 | |
| Anti-CD11d | 0.054* | 0.048* | |
| Caudal | |||
| Control | 0.032 | 0.017 | |
| Anti-CD11d |
|
0.056*
|
0.049*
|
bSlope of line Y = A + BX, where Y = normalized area of stain (%), and X = distance from lesion epicenter (in millimeters). *Slope is significantly different from control, determined by ANOVA for homogeneity of regression line slopes. All lines demonstrated significant correlation (range, 0.96-0.57; average, 0.82), tested by a regression ANOVA; p<0.05.
The anti-CD11d mAb treatment also increased the areas of myelin at the T12 injury site at 12 weeks after SCI (Fig. 5d). The areas of myelin were significantly greater in the treated group at most cord levels up to 3.2 mm from the lesion epicenter. The core of the lesion extended 3.2 mm in the control rats and 2.4 mm in the treated rats (Fig. 5d). The increment in myelin area with distance from the T12 lesion epicenter was significantly greater in the anti-CD11d mAb treated rats than in the control rats (Table 2). Again, the data reflect a greater volume of myelin in the treated than in the control rats.
Neurofilament is greater after anti-CD11d mAb
Serial sections adjacent to those stained for myelin were processed for neurofilament 200-immunoreactive protein to determine if the regions containing larger areas of myelin also had more area containing intact axons. Moreover, the staining also permitted an assessment of gray matter neurofilament. As shown in cord sections at 6 weeks after T4 SCI in control and treated rats, bundles of neurofilament at the lesion epicenter were arrayed irregularly in the remaining white and gray matter, and only isolated small areas of the neuropil appeared intact with punctate staining of axon bundles in cross section (Fig. 6a). Sections sampled 2.0 mm from the lesion epicenter showed more intact white and gray matter, especially in rats treated with the anti-CD11d mAb. These treated rats had more regions with regularly dispersed axon bundles than the control rats, and the high-power photomicrographs revealed more axons in cross section.
Figure 6.

Anti-CD11d mAb treatment increases neurofilament in the injured cord after SCI. Neurofilament-stained sections taken at 6 weeks after T4 SCI are illustrated at low and higher (inset) power at the lesion epicenter and at 2.0 mm caudal to the epicenter in control and mAb-treated rats (a). These sections are within 40 μm of those stained for myelin illustrated in Figure 5a. Within the lesion, neurofilament fibers were mostly arrayed in disorganized bundles in control and mAb-treated rats. At 2.0 mm caudal to the lesion, the neurofilament was more intact throughout the gray and white matter, and the greater integrity of the neuropil after anti-CD11d mAb treatment is again reflected by the smaller cavitation in the section. White matter axon bundles were aligned more normally at 2 mm from the lesion site, and greater numbers were visualized in cross-section in the treated rats (arrowheads in insets). *Indicates area from which high-power inset was taken. Scale bar is 200 μm for the low-power photomicrographs and 10 μm for the insets. Treatment effects on normalized areas of neurofilament are illustrated after SCI at T4 (b,6 weeks) and T12 (c,12 weeks). □, Control rats (b, c, n = 5). ▪, mAb-treated rats (b, n = 7; c, n = 6). *p < 0.05 compared with control rats. +Shortest distance where area is larger than area at epicenter, p < 0.05.
The quantitative assessment of areas of neurofilament revealed the smallest areas at the epicenter of the lesion and incremental increases in this staining with distance from the epicenter (Fig. 6b,c). The normalized areas of neurofilament near the T4 injury were significantly greater in the treated rats at 1 and 2 mm from the epicenter at 6 weeks after SCI (Fig. 6b). The core of the lesion with respect to neurofilament (areas not different from the epicenter) extended 4.0 mm in the control rats and only 2.0 mm in the treated rats. The increment in area of neurofilament with distance from the epicenter, both rostrally and caudally, was greater in the treated rats than in control rats (Table 2). These data demonstrate a greater volume of neurofilament in the treated rats.
Normalized areas of neurofilament near the T12 lesion were significantly greater in the treated than control rats at 12 weeks after SCI (Fig. 6c), even at the epicenter of the injury. The core of the lesion extended >4.0 mm in the control rats and only 2.0 mm in the treated rats (Fig. 6c). The increment in neurofilament area with distance from the lesion epicenter was significantly greater in the treated rats both rostral and caudal to the epicenter (Table 2). Again, the data reflect a greater volume of neurofilament in the treated than in the control rats.
As shown in Figure 6, neurofilament immunoreactivity was present in both white matter and gray matter in our experiments. This permitted an additional analysis of the treatment effects on normalized areas of neurofilament in the white matter versus gray matter. The total normalized areas of neurofilament compartmentalized for white and gray matter are shown in Figure 7. At the T4 injury, the CD11d mAb treatment led to significant increases in the areas of both white matter and gray matter both rostral and caudal to the injury (Fig. 7a). At 2 mm caudal to this injury the area of neurofilament within the gray matter was small but, in treated rats, this area was twice the size of that in the controls. At the T12 injury, the increased area of neurofilament caused by the treatment could be attributed to increases in white matter rostral to the injury and to both white and gray matter caudal to the injury site (Fig. 7b).
Figure 7.

Anti-CD11d mAb-treatment increases neurofilament in white matter and gray matter at 2 mm rostral and caudal to the lesion site. The normalized total areas of neurofilament sampled at 2 mm from the T4 and T12 lesions were compartmentalized according to their locations in white or gray matter. Treatment effects on the white matter and gray matter neurofilament are illustrated after SCI at T4 (a, 6 weeks) and T12 (b, 12 weeks). □, Control rats (a, b, n = 5). ▪, mAb-treated rats (a, n = 7; b, n = 6). *p < 0.05 compared with control rats.
Discussion
A brief intervention that has been shown to block the inflammatory cell infiltration (Mabon et al., 2000; Saville et al., 2002) led to significant preservation of the integrity of the injured spinal cord and long-lasting improvement of sensory, autonomic, and motor function. This improvement was particularly notable because this anti-CD11d mAb treatment is limited in duration and highly selective (Saville et al., 2002). Part of the success of this strategy may relate to the time-dependent balance between the early destructive actions of infiltrating pro-inflammatory neutrophils and macrophages, and the subsequent beneficial actions of macrophages and T-cells to support wound healing (Blight, 1985; Tator and Fehlings, 1991; Young, 1993; Streit et al., 1998; Taoka and Okajima, 1998). Studies in our laboratories have shown the importance of early delivery of this mAb treatment. Delay until 24 hr after SCI does not reduce the intraspinal influx of neutrophils and monocyte-macrophages (Saville et al., 2002) or decrease the myeloperoxidase activity and lipid peroxidation at 72 hr after SCI (F. Bao, G. Dekaban, and L. Weaver, unpublished observations). We also have evidence that the early dosing schedule of this treatment does not interfere with the later accumulation of intraspinal macrophages necessary for wound repair. The density of macrophages at the injury site of treated rats is similar to that in control rats at 7 d after the SCI (L. Saville, C. Pospisil, L. Mawhinney, F. Bao, C. Simedrea, A. Peters, P. O'Connell, L. Weaver, and G. Dekaban, unpublished observations). Moreover, the present study demonstrated that the chronic state of inflammation did not differ between treated and control rats. Our selective approach tempered only the early actions of inflammatory cells and had a significant impact on functional recovery that was still marked at 12 weeks after SCI.
The treatment improved the integrity of the neuropil and the state of myelination at all times of assessment. The positive effects of the anti-CD11d mAb intervention on spinal cord myelin could have been caused by sparing of myelin as well as improved remyelination in the 6 and 12 week groups. The loss of myelin at the lesion epicenter with time is likely caused by degeneration and removal by phagocytes. Treatment effects that increased the area of neurofilament adjacent to the T4 and T12 injury sites were associated with a higher incidence of normally aligned axons in the white matter, suggesting that these axons had survived the injury. At T12, even the lesion epicenter had more neurofilament and less gross necrosis in the treated rats. The compartmentalized analysis of neurofilament adjacent to the lesion epicenter demonstrated both gray and white matter sparing.
The anti-CD11d mAb treatment greatly affected pain induced both rostral and caudal to the injury site. Chronic pain occurs in 48-96% of cord-injured people (Siddall et al., 1995; Yezierski, 1996), seriously impacting on the quality of life and causing further incapacity and depression. Chronic pain is not easily treated, perhaps because it is generated by multiple mechanisms (Yezierski, 1996; Christensen and Hulsebosch, 1997; Woolf and Mannion, 1999). Clinically, mechanical allodynia is particularly disturbing because normal contact with a peripheral stimulus (and therefore pain) is inevitable. Part of the etiology of allodynia is the loss of normal continuous restraint of pain transmission by descending inhibitory pathways (Fields et al., 1991). Disruption of this modulation by SCI creates an opportunity for exaggerated transmission of pain sensation, including mechanical allodynia. Studies in cord-injured rats have identified several drugs that have short-lived effects on, but do not relieve, chronic allodynia (Xu et al., 1992; Hains et al., 2002). In contrast, our anti-inflammatory treatment had substantial long-lasting effects that limited the mechanical allodynia. The decreased chronic pain after the mAb treatment was likely accomplished by preserving descending connections to spinal neurons in pain pathways rostral and caudal to the T12 injury site as well as preserving the spinal neurons. The improved motor function induced by the mAb treatment after the T12 lesion may have been greatly attributable to sparing of propriospinal neurons adjacent to the injury that contribute to spinal motor rhythm generators (Barbeau et al., 1999).
The anti-CD11d treatment also reduced autonomic dysreflexia by ∼50%. This dysfunction in cardiovascular control occurs in 50-90% of people with tetraplegia or high paraplegia (Mathias and Frankel, 1992; Karlsson, 1999). After SCI, life-threatening increases in arterial pressure can develop in response to sensory input entering the spinal cord below the level of the lesion. This episodic hypertension can result in debilitating headaches, seizures, strokes, and even death. After severe SCI, the loss of supraspinal inhibitory influences (Dembowsky et al., 1980) permits unchecked activity of spinal reflexes to produce autonomic dysreflexia. These exaggerated reflexes begin within weeks of SCI and are caused by any stimulation of the skin, muscle, or organs (Mathias and Frankel, 1992). The neuroprotection afforded by the anti-CD11d mAb strategy increased the neurofilament rostral and caudal to the injury site at T4, and these effects were directed to white and gray matter. By 6 weeks after this injury the protective effect on myelin and neurofilament were detected adjacent to but not within the lesion epicenter itself. We have previously shown that a very small preservation of white and gray matter is adequate to control the excitatory spinal reflexes that cause autonomic dysreflexia, eliminating or greatly decreasing the condition (Weaver et al., 2001). In contrast, at the T4 injury, the neuroprotection afforded by this treatment caused only a small increase in motor function. The higher location of this injury leaves many more segments of spinal cord lacking control from the brain than does the T12 injury, and this greater deficit appears to generate a more intractable loss of motor function.
The anti-CD11d mAb intervention is a very selective neuroprotective strategy because it uses a specific blockade of the association between the CD18/Cd11d integrin and VCAM-1. This action of the mAb is well documented in vitro (Van der Vieren et al., 1999) and greatly affects the extravasation of circulating neutrophils and macrophages into the injured spinal cord (Mabon et al., 2000). The anti-CD11d mAb intervention promoted more neurological recovery than the complete, long-lasting depletion of hematogenous macrophages used by others (Popovich et al., 1999), perhaps because the blockade was transient. For example, remyelination and regeneration may be accomplished more effectively if macrophages are permitted to phagocytose myelin debris (Rapalino et al., 1998; Kotter et al., 2001; Popovich and Hickey, 2001). The beneficial effects of the anti-CD11d mAb intervention also may be explained by an action to temper the activated state of the neutrophils and macrophages during the early phases of SCI (Shanley et al., 1998). We have also shown that our anti-CD11d mAb treatment decreases myeloperoxidase activity, lipid peroxidation, protein nitration, and inducible nitric oxide expression in the injured cord by 30-60% within the first 3 d after clip-compression SCI (Bao et al., 2004)
This anti-CD11d strategy for neuroprotection contrasts with other acute therapies that have failed to offer a practical way to control the early inflammatory response. Drugs such as methylprednisolone have had inconsistent effects on recovery from SCI both in human and animal studies (Hurlbert, 2000; Rabchevsky et al., 2002). Although this steroid has anti-oxidant and anti-inflammatory actions (Grevel, 1992; Hall, 1992; Rabchevsky et al., 2002), it can have destructive effects on neurons and glia, mediated by several mechanisms (Sapolsky, 1996). Furthermore, we have evidence that, at 1 week after SCI, this steroid prolongs the presence of neutrophils in the injured spinal cord and decreases the number of macrophages (L. Saville, C. Pospisil, L. Mawhinney, F. Bao, C. Simedrea, A. Peters, P. O'Connell, L. Weaver and G. Dekaban, unpublished observations).
Several earlier studies have shown that blocking the influx of leukocytes into the injured cord can be advantageous. Blocking antibodies to P-selectin or intercellular adhesion molecule-1 (ICAM-1) limit the intraspinal influx of neutrophils, reducing oxidative damage and improving motor function (Hamada et al., 1996; Taoka et al., 1997). These studies clearly demonstrated that neutrophils augment secondary damage after SCI. Studies in ICAM-1 and P-selectin knockout mice also show improved motor recovery for 2 weeks after SCI, confirming a negative role of leukocyte tethering and adhesion after SCI (Farooque et al., 1999). The advantage of our method is the powerful early effect to prevent neutrophil infiltration, coupled with a delay, rather than a complete eradication, of macrophage infiltration.
In summary, the novelty of our anti-integrin approach is the targeting of the CD11d/CD18 integrin, because transient blockade of its function reduces and delays the first encounter between the injured CNS and the systemic immune system, affecting both neutrophils and macrophages. If this selective early blocking strategy were to be considered as an initial treatment for SCI, it could be coupled with other neuroprotective treatments such as the antibiotic minocycline (Wells et al., 2003), induction of protective immunity (Hauben et al., 2000), and regeneration-promoting strategies (McDonald et al., 1999; Coumans et al., 2001; Kwon et al., 2002). If the ultimate neurological outcome of SCI depends on initial gray and white matter sparing, early neuroprotection is a mandatory foundation that should precede other methods of repair.
Footnotes
This research was supported by the Canadian Institutes of Health Research and the Ontario Neurotrauma Foundation(ONF).D.G. and M.O. hold studentships from ONF. We are also indebted to Carmen Simedrea, Leyanna Saville, Jennifer Fleming, and Paul Gris for assistance with the studies. Dr. David Ramsay of the Department of Pathology, London Health Sciences Centre assisted us greatly in the analysis of the chronically injured spinal cords. We thank the ICOS Corporation for partial support of this research and for providing the anti-CD11d mAb used in the study. We appreciate the critiques of the manuscript provided by Drs. Arthur Brown and Canio Polosa.
Correspondence should be addressed to Dr. Lynne C. Weaver, Spinal Cord Injury Laboratory, BioTherapeutics Research Group, Robarts Research Institute, 100 Perth Drive, P.O. Box 5015, London, Ontario N6A 5K8, Canada. E-mail: lcweaver@robarts.ca.
D. R. Marsh's present address: Department of Anatomy and Neurobiology, Dalhousie University, Halifax, Nova Scotia B3H 1X5, Canada.
Copyright © 2004 Society for Neuroscience 0270-6474/04/244043-09$15.00/0
References
- Bao F, Chen Y, Dekaban G, Weaver LC (2004) Early anti-inflammatory treatment reduces lipid peroxidation and protein nitration after spinal cord injury in rats. J Neurochem 88: 1335-1344. [DOI] [PubMed] [Google Scholar]
- Barbeau H, Mccrea DA, O'Donovan MJ, Rossignol S, Grill WM, Lemay MA (1999) Tapping into spinal circuits to restore motor function. Brain Res Brain Res Rev 30: 27-51. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC (1995) A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma 12: 1-21. [DOI] [PubMed] [Google Scholar]
- Bethea JR, Dietrich WD (2002) Targeting the host inflammatory response in traumatic spinal cord injury. Curr Opin Neurol 15: 355-360. [DOI] [PubMed] [Google Scholar]
- Bevilacqua MP (1993) Endothelial-leukocyte adhesion molecules. Annu Rev Immunol 11: 767-804. [DOI] [PubMed] [Google Scholar]
- Blight AR (1985) Delayed demyelination and macrophage invasion: a candidate for “secondary” cell damage in spinal cord injury. CNS Trauma 2: 299-315. [DOI] [PubMed] [Google Scholar]
- Bruce JC, Oatway MA, Weaver LC (2002) Chronic pain after clip compression injury of the rat spinal cord. Exp Neurol 178: 33-48. [DOI] [PubMed] [Google Scholar]
- Christensen MD, Hulsebosch CE (1997) Chronic central pain after spinal cord injury. J Neurotrauma 14: 517-537. [DOI] [PubMed] [Google Scholar]
- Christensen MD, Everhart AW, Pickelman JT, Hulsebosch CE (1996) Mechanical and thermal allodynia in chronic central pain following spinal cord injury. Pain 68: 97-107. [DOI] [PubMed] [Google Scholar]
- Coumans JV, Lin TT, Dai HN, MacArthur L, McAtee M, Nash C, Bregman BS (2001) Axonal regeneration and functional recovery after complete spinal cord transection in rats by delayed treatment with transplants and neurotrophins. J Neurosci 21: 9334-9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dembowsky K, Czachurski J, Amendt K, Seller H (1980) Tonic descending inhibition of the spinal somato-sympathetic reflex from the lower brain stem. J Auton Nerv Syst 2: 157-182. [DOI] [PubMed] [Google Scholar]
- Farooque M, Isaksson J, Olsson Y (1999) Improved recovery after spinal cord trauma in ICAM-1 and P-selectin knockout mice. NeuroReport 10: 131-134. [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P (1991) Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci 14: 219-245. [DOI] [PubMed] [Google Scholar]
- Grayson MH, Van der Vieren M, Sterbinsky SA, Gallantin WM, Hoffman P, Staunton D, Bochner BS (1999) alphadbeta2 integrin is a ligand for vascular cell adhesion molecule-1. Int Arch Allergy Immunol 118: 263-264. [DOI] [PubMed] [Google Scholar]
- Grevel J (1992) Optimisation of immunosuppressive therapy using pharmacokinetic principles. Clin Pharmacokinet 23: 380-390. [DOI] [PubMed] [Google Scholar]
- Hains BC, Yucra JA, Eaton MJ, Hulsebosch CE (2002) Intralesion transplantation of serotonergic precursors enhances locomotor recovery but has no effect on development of chronic central pain following hemisection injury in rats. Neurosci Lett 324: 222-226. [DOI] [PubMed] [Google Scholar]
- Hall ED (1992) The neuroprotective pharmacology of methylprednisolone. J Neurol 76: 13-22. [DOI] [PubMed] [Google Scholar]
- Hamada Y, Ikata T, Katoh S, Kauchi K, Wa M, Wai Y, Kuzawa K (1996) Involvement of an intercellular adhesion molecule 1-dependent pathway in the pathogenesis of secondary changes after spinal cord injury in rats. J Neurochem 66: 1525-1531. [DOI] [PubMed] [Google Scholar]
- Hauben E, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Akselrod S, Neeman M, Cohen IR, Schwartz M (2000) Autoimmune T cells as potential neuroprotective therapy for spinal cord injury. Lancet 355: 286-287. [DOI] [PubMed] [Google Scholar]
- Hurlbert RJ (2000) Methylprednisolone for acute spinal cord injury: an inappropriate standard of care. J Neurosurg 93: 1-7. [DOI] [PubMed] [Google Scholar]
- Jones LL, Yamaguchi Y, Stallcup WB, Tuszynski MH (2002) NG2 is a major chondroitin sulfate proteoglycan produced after spinal cord injury and is expressed by macrophages and oligodendrocyte progenitors. J Neurosci 22: 2792-2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson A-K (1999) Autonomic dysreflexia. Spinal Cord 37: 383-391. [DOI] [PubMed] [Google Scholar]
- Kotter MR, Setzu A, Sim FJ, Van Rooijen N, Franklin RJ (2001) Macrophage depletion impairs oligodendrocyte remyelination following lysolecithin-induced demyelination. Glia 35: 204-212. [DOI] [PubMed] [Google Scholar]
- Kunkel-Bagden E, Dai HN, Bregman BS (1993) Methods to assess the development and recovery of locomotor function after spinal cord injury in rats. Exp Neurol 119: 153-164. [DOI] [PubMed] [Google Scholar]
- Kwon BK, Liu J, Messerer C, Kobayashi NR, McGraw J, Oschipok L, Tetzlaff W (2002) Survival and regeneration of rubrospinal neurons 1 year after spinal cord injury. Proc Natl Acad Sci USA 99: 3246-3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leskovar A, Moriarty LJ, Turek JJ, Schoenlein IA, Borgens RB (2000) The macrophage in acute neural injury: changes in cell numbers over time and levels of cytokine production in mammalian central and peripheral nervous systems. J Exp Biol 203 Pt 12: 1783-1795. [DOI] [PubMed] [Google Scholar]
- Mabon PJ, Weaver LC, Dekaban GA (2000) Inhibition of monocyte/macrophage migration to a spinal cord injury site by an antibody to the integrin alphaD: a potential new anti-inflammatory treatment. Exp Neurol 166: 52-64. [DOI] [PubMed] [Google Scholar]
- Maiorov DN, Krenz NR, Krassioukov AV, Weaver LC (1997) Role of spinal NMDA and AMPA receptors in episodic hypertension in conscious spinal rats. Am J Physiol 273: H1266-H1274. [DOI] [PubMed] [Google Scholar]
- Marsh DR, Weaver LC (2004) Autonomic dysreflexia, induced by noxious or innocuous stimulation, does not depend on changes in dorsal horn substance P. J Neurotrauma, in press. [DOI] [PubMed]
- Mathias CJ, Frankel HL (1992) Autonomic disturbances in spinal cord lesions. In: Autonomic failure. A textbook of clinical disorders of the autonomic nervous system (Mathias CJ, Bannister R, eds), pp 494-513. Oxford: Oxford UP.
- McDonald JW, Liu XZ, Qu Y, Liu S, Mickey SK, Turetsky D, Gottlieb DI, Choi DW (1999) Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med 5: 1410-1412. [DOI] [PubMed] [Google Scholar]
- Neish AS, Read MA, Thanos D, Pine R, Maniatis T, Collins T (1995) Endothelial interferon regulatory factor 1 cooperates with NF-kappa B as a transcriptional activator of vascular cell adhesion molecule 1. Mol Cell Biol 15: 2558-2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page KM (1965) A stain for myelin using solochrome cyanin. J Med Lab Tech 22: 224. [PubMed] [Google Scholar]
- Popovich PG, Hickey WF (2001) Bone marrow chimeric rats reveal the unique distribution of resident and recruited macrophages in the contused rat spinal cord. J Neuropathol Exp Neurol 60: 676-685. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Wei P, Stokes BT (1997) Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol 377: 443-464. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van RN, Stokes BT (1999) Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 158: 351-365. [DOI] [PubMed] [Google Scholar]
- Rabchevsky AG, Fugaccia I, Sullivan PG, Blades DA, Scheff SW (2002) Efficacy of methylprednisolone therapy for the injured rat spinal cord. J Neurosci Res 68: 7-18. [DOI] [PubMed] [Google Scholar]
- Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, Hadani M, Schwartz M (1998) Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med 4: 814-821. [DOI] [PubMed] [Google Scholar]
- Rivlin AS, Tator CH (1977) Objective clinical assessment of motor function after experimental spinal cord injury in the rat. J Neurosurg 47: 577-581. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM (1996) Stress, glucocorticoids, and damage to the nervous system: the current state of confusion. Stress 1: 1-19. [DOI] [PubMed] [Google Scholar]
- Saville LR, Simedria FC, Weaver LC, Dekaban GA (2002) Treatment of spinal cord injury by using a antibody to the leukocyte integrin alpha D/beta 2 effectively prevents the early infiltration of phagocytes and monocytes/macrophages into the lesion. J Neurotrauma 19: 1352. [Google Scholar]
- Shanley TP, Warner RL, Crouch LD, Dietsch GN, Clark DL, O'Brien MM, Gallatin WM, Ward PA (1998) Requirements for αd in IgG immune complex-induced rat lung injury. J Immunol 160: 1014-1020. [PubMed] [Google Scholar]
- Siddall PJ, Taylory D, Cousins MJ (1995) Pain associated with spinal cord injury. Curr Opin Neurol 8: 447-450. [DOI] [PubMed] [Google Scholar]
- Snedecor G, Cochran WG (1989) Statistical methods. Iowa: Iowa State UP.
- Streit WJ, Semple-Rowland SL, Hurley SD, Miller RC, Popovich PG, Stokes BT (1998) Cytokine mRNA profiles in contused spinal cord and axotomized facial nucleus suggest a beneficial role for inflammation and gliosis. Exp Neurol 152: 74-87. [DOI] [PubMed] [Google Scholar]
- Taoka Y, Okajima K (1998) Spinal cord injury in the rat. Prog Neurobiol 56: 341-358. [DOI] [PubMed] [Google Scholar]
- Taoka Y, Okajima K, Uchiba M, Murakami K, Kushimoto S, Johno M, Naruo M, Okabe H, Takatsuki K (1997) Role of neutrophils in spinal cord injury in the rat. Neuroscience 79: 1177-1182. [DOI] [PubMed] [Google Scholar]
- Tator CH, Fehlings MG (1991) Rev of the secondary injury theory of acute spinal cord trauma with emphasis on vascular mechanisms. J Neurosurg 75: 15-26. [DOI] [PubMed] [Google Scholar]
- Van der Vieren M, Crowe DT, Hoekstra D, Vazeux R, Hoffman PA, Grayson MH, Bochner BS, Gallatin WM, Staunton DE (1999) The leukocyte integrin αDβ2 binds VCAM-1: evidence for a binding interface between I domain and VCAM-1. J Immunol 163: 1984-1990. [PubMed] [Google Scholar]
- Weaver LC, Verghese P, Bruce JC, Fehlings MG, Krenz NR, Marsh DR (2001) Autonomic dysreflexia and primary afferent sprouting after clip-compression injury of the rat spinal cord. J Neurotrauma 18: 1107-1119. [DOI] [PubMed] [Google Scholar]
- Wells JE, Hurlbert RJ, Fehlings MG, Yong VW (2003) Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain 126: 1628-1637. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ (1999) Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet 353: 1959-1964. [DOI] [PubMed] [Google Scholar]
- Xu X-J, Hao JX, Aldskogius H, Seiger Å, Wiesenfeld-Hallin Z (1992) Chronic pain-related syndrome in rats after ischemic spinal cord lesion: a possible animal model for pain in patients with spinal cord injury. Pain 48: 279-290. [DOI] [PubMed] [Google Scholar]
- Yezierski RP (1996) Pain following spinal cord injury: the clinical problem and experimental studies. Pain 68: 185-194. [DOI] [PubMed] [Google Scholar]
- Young W (1993) Secondary injury mechanisms in acute spinal cord injury. J Emerg Med 11: 13-22. [PubMed] [Google Scholar]