

Abstract
The mechanisms by which neurotrophins regulate growth cone motility are unclear. We investigated the role of the p75 neurotrophin receptor (p75NTR) in mediating neurotrophin-induced increases in filopodial length. Our data demonstrate that neurotrophin binding to p75NTR is necessary and sufficient to regulate filopodial dynamics. Furthermore, retinal and dorsal root ganglion growth cones from p75 mutant mice are insensitive to neurotrophins but display enhanced filopodial lengths comparable with neurotrophin-treated wild-type growth cones. This suggests unoccupied p75NTR negatively regulates filopodia length. Furthermore, p75NTR regulates RhoA activity to mediate filopodial dynamics. Constitutively active RhoA blocks neurotrophin-induced increases in filopodial length, whereas inhibition of RhoA enhances filopodial lengths, similar to neurotrophin treatment. BDNF treatment of retinal neurons results in reduced RhoA activity. Furthermore, p75 mutant neurons display reduced levels of activated RhoA compared with wild-type counterparts, consistent with the enhanced filopodial lengths observed on mutant growth cones. These observations suggest that neurotrophins regulate filopodial dynamics by depressing the activation of RhoA that occurs through p75NTR signaling.
Keywords: filopodia, growth cone, p75, RhoA, neurotrophin, BDNF
Introduction
Axonal navigation depends on growth cone motility. Growth cone behaviors change in a region-specific manner as axons extend to their targets (Mason and Erskine, 2000), suggesting that guidance cues regulate growth cone motility. Therefore, to understand axon guidance, it is important to determine how guidance cues regulate growth cone motility. Filopodia are finger-like projections supported by a cytoskeleton of bundled actin filaments (Gallo and Letourneau, 2000). Filopodia detect extrinsic cues and transduce signals to guide growth cone migration (O'Connor et al., 1990; Kater and Rehder, 1995). Disruption of filopodia causes guidance defects in vivo (Bentley and Toroian-Raymond, 1986; Chien et al., 1993). Yet, the mechanisms underlying the regulation of filopodial dynamics are not well understood.
Neurotrophins (NTs), including nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), NT-3, and NT-4/5, regulate neuronal differentiation, axon extension, guidance, arborization, and synaptogenesis (Gundersen and Barrett, 1979; Cohen-Cory and Fraser, 1995; Gallo et al., 1997; Lom and Cohen-Cory, 1999; Ming et al., 1999; Yamashita et al., 1999; Alsina et al., 2001; Frost, 2001; Tucker et al., 2001). Neurotrophins bind two classes of receptors, the receptor tyrosine kinase family of Trk (A, B, and C) receptors and the p75 neurotrophin receptor (NTR). Trk receptors selectively bind neurotrophins, whereas p75NTR binds all neurotrophins with similar affinities (for review, see Hempstead, 2002; Chao, 2003). The signaling pathways activated by neurotrophins can regulate cytoskeletal dynamics, which in turn determine growth cone behaviors (Gallo and Letourneau, 1998, 2000). For example, chemotropic responses require coactivation of the phosphoinositide 3-kinase and phospholipase C-γ pathways through Trk receptors (Ming et al., 1999). p75NTR has been shown to mediate neurotrophin-induced increases in axon length (Brann et al., 1999; Yamashita et al., 1999; McQuillen et al., 2002). Recently, McQuillen et al. (2002) demonstrated that NT-3 binding to p75NTR in cortical subplate neurons increases neurite length and filopodial formation. However, the signaling pathways downstream of p75NTR that control growth cone functions, such as filopodial behavior, remain undefined.
The Rho family of GTPases regulate the actin cytoskeleton. RhoA is involved in regulating axon growth and growth cone morphology (Luo et al., 1997; Lehmann et al., 1999; Ruchhoeft et al., 1999; Bito et al., 2000; Kuhn et al., 2000). Recent evidence indicates that RhoA activity is regulated by the cytoplasmic domain of p75NTR (Yamashita et al., 1999; Yamashita and Tohyama, 2003). Yamashita et al. (1999) provided evidence that p75NTR can regulate axon extension by signaling through the RhoA pathway, such that the unbound state of p75NTR activates RhoA, whereas neurotrophin binding to p75NTR terminates this mode of RhoA activation. Growth cone behaviors were not investigated in previous studies, and it is unknown whether neurotrophin–p75NTR interactions regulate RhoA activity in growth cones.
To further analyze neurotrophin regulation of growth cone behaviors, we examined the role of p75NTR in regulating filopodial dynamics in peripheral and CNS neurons. We report that p75NTR is necessary and sufficient to mediate neurotrophin-induced increases in filopodial length. Additionally, we found that neurotrophin binding to p75NTR reduces RhoA activity. Our data demonstrate that neurotrophin-induced increases in filopodial length, mediated through p75NTR, are mimicked by inhibition of RhoA and are blocked by constitutively active RhoA. Significantly, BDNF reduces RhoA activity in individual growth cones. In addition, p75–/– growth cones exhibit a filopodial phenotype that is similar to wild-type growth cones treated with neurotrophins. p75–/– growth cones lack p75NTR expression and display reduced RhoA activity. These results establish that p75NTR regulates growth cone filopodia through the regulation of RhoA activity.
Materials and Methods
Reagents. BDNF and NT-3 were generously provided by Regeneron Pharmaceuticals (Tarrytown, NY). NGF and ciliary neurotrophic factor (CNTF) were purchased from R&D Systems (Minneapolis, MN), poly-d-lysine from ICN Pharmaceuticals (Costa Mesa, CA), laminin from Invitrogen (Carlsbad, CA), and K252a and lysophosphatidic acid (LPA) from BioMol (Plymouth Meeting, PA). All culture media were obtained from Invitrogen, and other reagents were obtained from Sigma (St. Louis, MO) unless specified otherwise.
Tissue culture. Dorsal root ganglion (DRG) and retina were dissected from embryonic day (E) 9 and E7 white Leghorn chicken embryos, respectively. Explants were cultured on glass coverslips coated with 25 μg/ml laminin. Retinal explants were cultured in F12 nutrient mixture medium supplemented with additives as described previously (Ernst et al., 2000). DRG explants were maintained in 16 pm NGF.
Ciliary ganglia were dissected from E10 white Leghorn chicken embryos. Explants were cultured on laminin-treated glass coverslips in F12 media containing 10% calf serum, 2 mm glutamine, and 10 ng/ml CNTF.
Mouse retina and DRG were dissected from E14.5 mice. Retinal explants were cultured on glass coverslips coated with 0.04 mg/ml poly-d-lysine and 25 μg/ml laminin. Mouse retinal explants were cultured in F12 medium as described above. Mouse DRG explants were cultured on laminin-coated glass coverslips in F12 medium, as described above, plus 1% calf serum and 40 pm NGF.
Genotyping of p75NTR mutant mice. Breeding pairs of mice heterozygous for a null mutation in the p75NTR gene (p75+/–) (Lee et al., 1992) were a gift from Dr. Frank Longo (University of North Carolina, Chapel Hill, NC). Heterozygous p75+/– mice were mated to obtain experimental animals. Littermates consisting of p75+/+ and p75–/– embryos were used. Genomic DNA obtained from embryonic mouse tissue was used to determine genotypes by PCR using primers as described previously (Yeo et al., 1997). The mouse colony was housed as per University of Minnesota Institutional Animal Care and Use Committee guidelines.
Protein loading. Chariot reagent (Active Motif, Carlsbad, CA) is a peptide carrier that delivers proteins into cells. Protein delivery was accomplished according to the manufacturer suggestions as described previously (Gallo et al., 2002). Briefly, Chariot was complexed with 1 μg of C3 transferase, constitutively active RhoA (C3 transferase, L63RhoA; Cytoskeleton, Denver, CO), or bovine serum albumin (Boehringer Mannheim, Gaithersburg, MD). After protein loading, cultures were experimentally treated or fixed with 0.25% glutaraldehyde.
Time-lapse video microscopy. Phase-contrast images were collected using an Olympus (Tokyo, Japan) XC-70 inverted microscope with a 60× objective equipped with a SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI). Images were collected every 6 sec for 30 min. Digital images were analyzed to measure extension and retraction lengths of filopodia using MetaVue (Universal Imaging, Downington, PA) imaging software.
Image acquisition and analysis. Fluorescent images were collected using an Olympus XC-70 inverted microscope. Growth cones were visualized with a 60× objective, and fluorescent images were obtained using a SPOT digital camera. Image analysis was accomplished using MetaVue imaging software. Individual filopodia were measured using the digital software. Only filopodia within 20 μm of the distal tip of the growth cone were counted and measured. Filopodial length and number were determined per growth cone.
Immunofluorescence. Actin filaments were stained with rhodamine-conjugated phalloidin (Molecular Probes, Eugene, OR). Cultures were fixed with 0.25% glutaraldehyde and quenched with 1 mg/ml sodium borohydride for 15 min. After rinsing with PBS, cells were incubated with 1 U of rhodamine-conjugated phalloidin for 45 min at room temperature.
Indirect immunofluorescence staining for p75NTR was performed using rabbit polyclonal antibodies to the extracellular or to the cytoplasmic domain of p75NTR. Antibodies against the extracellular domain of p75NTR were generously provided by Dr. Louis Reichardt (CHEX; University of California at San Francisco, San Francisco, CA) and obtained from Chemicon (AB1554; Temecula, CA). Antibodies against the cytoplasmic domain of p75NTR were obtained from Covance (PRB-602C; Berkeley, CA) and Promega (G3231; Madison, WI). Cultures were fixed with 2% paraformaldehyde and 5% sucrose for 15 min at 40°C. Cells were rinsed and blocked with 10% normal goat serum (NGS) (Jackson ImmunoResearch, West Grove, PA) for 1 hr at room temperature. Cells exposed to anti-cytoplasmic p75 were extracted with 0.1% Triton X-100. Cells were incubated with 1:800 (CHEX and AB1554) or 1:100 (Covance and Promega) p75 antibody (Ab) for 1 hr at room temperature followed by 1:800 Alexa568 goat anti-rabbit secondary (Molecular Probes) and 1 U of fluorescein-conjugated phalloidin (Molecular Probes) in block for 30 min. Negative controls involved exclusion of the primary Ab.
Western blot analysis. Brain lysates from E14.5 mouse embryos were probed for p75NTR using antibodies directed against the cytoplasmic domain (Covance or Promega) followed by incubation with HRP-conjugated goat anti-rabbit secondary Ab (Pierce, Rockford, IL). Membranes were developed using enhanced chemiluminescence (ECL) (Pierce). Membranes were stripped and reprobed with a monoclonal α-tubulin Ab (Sigma) and an HRP-conjugated goat anti-mouse secondary Ab.
RhoA activity assay. The RhoA activity assay was performed as described by Ren and Schwartz (2000). Briefly, 8 × 106 retinal cells were dissociated and cultured in 100 mm tissue culture dishes coated with 25 μg/ml laminin. The cultures were treated with 160 nm BDNF for various periods and then assayed for RhoA activity. For the mouse RhoA assay, cerebella were rapidly removed from postnatal day (P) 8 mice. Active RhoA was immunoprecipitated by incubating lysed cells for 45 min with the Rho-binding domain (RBD) of Rhotekin fused with glutathione S-transferase (GST) (GST-RBD; generously provided by Dr. Alan Hall, Medical Research Council, University College London, London, UK). The proteins were separated using SDS-PAGE, transferred to polyvinylidene difluoride membrane, and probed with a monoclonal RhoA Ab (Santa Cruz Biotechnology, Santa Cruz, CA) followed by incubation with an HRP-conjugated goat anti-mouse secondary (Pierce). Lysates were probed using a monoclonal α-tubulin Ab (Sigma) followed by incubation with an HRP-conjugated goat anti-mouse secondary. Membranes were developed using ECL. Activated RhoA was normalized against α-tubulin using densitometric analysis of the blots.
The in vitro RhoA activity assay on growth cones was modified from a previous description (Li et al., 2002). Briefly, cultures were fixed with 4% paraformaldehyde and 10% sucrose for 1 hr. Cultures were rinsed and incubated with 5% NGS plus 0.1% Triton X-100 for 1 hr. Cultures were subsequently exposed to 5 μg/ml GST-RBD in block for 4 hr followed by incubation with 1:200 polyclonal rabbit anti-glutathione S-transferase (Oncogene Research Products, Boston, MA) in 10% NGS overnight at 4°C. Cells were rinsed and incubated with 1:800 Alexa488 goat anti-rabbit secondary (Molecular Probes). Activated RhoA was quantified by measuring signal intensity in each growth cone minus background. Total protein was measured in growth cones by binding of fluoroscein-5-isothiocyanate (Molecular Probes).
Production of digital images. Digital images for micrographs and blots were processed and produced using Adobe Photoshop (Adobe Systems, San Jose, CA) software.
Results
Neurotrophins increase the length and number of filopodia on growth cones
To study the mechanisms by which neurotrophins control filopodial behavior, we investigated the effects of neurotrophins on CNS and peripheral nervous system growth cones. BDNF and NGF treatment of embryonic chick retinal and DRG growth cones, respectively, increased filopodial length in a dose-dependent manner (Fig. 1A,D). The addition of 160 nm BDNF to retinal neurons for 30 min resulted in a 28% increase in filopodial length (Fig. 1B,C) (mean, 10.0 vs 7.8 μm), whereas application of 20 nm NGF to DRG neurons for 30 min increased filopodial length by 27% over neurons maintained in 0.2 nm NGF (9.5 vs 7.5 μm). In addition, 160 nm BDNF produced a 60% increase in filopodial number on retinal growth cones (Fig. 1E). Lower concentrations of BDNF (4 and 40 nm) had significant effects on filopodial length but had no effect on filopodial number. Thus, neurotrophins regulate filopodial dynamics on growth cones.
Figure 1.

Neurotrophin treatment increases filopodial length on chick retinal and DRG growth cones. A, Treatment of chick retina or DRG cultures for 30 min with 160 nm BDNF or 20 nm NGF, respectively, caused a 28 and 27% increase in filopodial length. Data are presented as mean ± SEM from four independent experiments. B, C, Phalloidin staining of F-actin in examples of control BSA-treated (B) and BDNF-treated (C) chick RGC growth cones. Scale bar, 10 μm. D, BDNF increased filopodial length on RGC growth cones in a dose-dependent manner. Data are mean ± SEM from four independent experiments. E, BDNF increased filopodia number on RGC growth cones in a dose-dependent manner. Data are mean ± SEM from four independent experiments. *p < 0.001, **p < 0.005 indicate statistical significance relative to control; two-sample t test.
To further investigate neurotrophin effects on filopodial dynamics, extension and retraction rates of retinal growth cone filopodia were measured after BDNF treatment. BDNF caused a 34% increase in the extension rate after 3 min compared with controls (Fig. 2A,B) and maintained elevated rates of extension throughout a 30 min treatment. BDNF also increased the filopodial retraction rate by 26% after 6 min (Fig. 2C), but retraction rates subsided to control levels by 15 min. When the total time spent extending or retracting was analyzed, control growth cones spent 56% of the time extending and 44% retracting (Fig. 2D). Filopodia treated with BDNF spent 63% of the time extending and 37% retracting. These data provide evidence that BDNF enhanced filopodial length by increasing the extension rate coupled with an increase in time spent extending versus retracting.
Figure 2.

BDNF alters filopodial dynamics on retinal growth cones. A, Frames taken from time-lapse recordings show the enhanced filopodial extension after treatment with BDNF. Arrowheads indicate elongation between t = 0 and t = 12 sec. Scale bar, 1 μm. B, Treatment of retinal neurons with 160 nm BDNF increased the rate of filopodial extension within 3 min and remained elevated through a 30 min treatment. C, BDNF treatment caused an initial increase in filopodial retraction rate after 6 min but subsided by 15 min. D, Time spent extending or retracting was shifted by BDNF stimulation. Data are percentage ± SEM from four independent experiments. Numbers in parentheses indicate number of filopodia. *p < 0.001, **p < 0.02 indicate statistical difference relative to control; two-sample t test.
Neurotrophins regulate filopodial dynamics through p75NTR
We next determined whether neurotrophin-induced changes in filopodia dynamics were regulated by either Trk receptors or p75NTR. Axons emerging from E6 chick retinal explants are confirmed to be from retinal ganglion cells (RGCs) (McLoon and Barnes, 1989). E7 chick RGCs express TrkB, which mediates BDNF effects, but do not express TrkA, whereas TrkC expression is inconclusive (Frade et al., 1996; Hallbook et al., 1996; von Bartheld, 1998). We used immunofluorescence staining to determine whether p75NTR is expressed in our culture conditions. Growth cones from chick RGCs, DRGs, and ciliary neurons were labeled with a polyclonal Ab that recognizes an extracellular epitope of p75NTR (Fig. 3A–F). Expression of p75NTR was evident on all growth cone surfaces, including filopodia.
Figure 3.

BDNF-induced increases in filopodial length are mediated through p75NTR. A–F, p75NTR is expressed on filopodia (arrows) on growth cones of chick retinal (A), DRG (C), and ciliary neurons (E). Growth cones were counter stained with fluorescent phalloidin to show actin distribution (B, D, F). Scale bar, 10 μm. G, 160 nm BDNF, NGF, and NT-3 similarly increase filopodial length on RGC growth cones. H, Treatment of RGC growth cones with a Trk inhibitor, K252a, had no effect on BDNF-induced increases of filopodia length. I, Treatment of chick retinal cultures with a p75 Ab (CHEX) caused an increase in filopodial length that mimics BDNF treatment. Furthermore, pretreatment with CHEX blocked any effects of BDNF on filopodia length. *p < 0.001, significant difference relative to controls; two-sample t test. Data in G, H, and I are presented as percentage control ± SEM from four independent experiments.
We next asked whether p75NTR regulates filopodial dynamics in the absence of Trk activation. Because p75NTR binds all neurotrophins and TrkB is the only Trk receptor expressed on E7 chick RGC neurons, we asked whether NGF or NT-3 have similar effects as BDNF on RGC filopodial dynamics. Treatment with either 160 nm NGF or NT-3 increased filopodial length similar to BDNF (Fig. 3G) but had no effect on filopodia number (Table 1). That NGF, BDNF, and NT-3 all increase filopodial length on RGC growth cones suggests the involvement of p75NTR.
Table 1.
The role of BDNF in regulating filopodia number on retinal growth cones
|
Treatment |
Filopodia (n) |
|---|---|
| Chick | |
| No treatment | 6.3 ± 0.3 (102) |
| BDNF | 10.3 ± 0.3 (152)* |
| NGF | 6.3 ± 0.3 (74) |
| NT-3 | 6.2 ± 0.3 (62) |
| Control IgG | 6.2 ± 0.2 (80) |
| CHEX | 8.0 ± 0.3 (80)* |
| K252a | 6.76 ± 0.3 (80) |
| K252a plus BDNF | 8.66 ± 0.3 (80)* |
| Mouse | |
| p75+/+ no treatment | 7.1 ± 0.8 (85) |
| p75+/+ BDNF | 9.6 ± 1.2 (81) |
| p75-/- no treatment | 9.8 ± 0.5 (56)** |
| p75-/- BDNF |
10.5 ± 0.7 (69) |
Cultured retinal explants were treated with 160 nM BDNF, NGF, or NT-3 for 30 min or 1:100 CHEX for 4 hr. Cultures were pretreated with 1 μm K252a for 1 hr followed by addition of 160 nM BDNF for 30 min. Number of filopodia was determined for each growth cone. n denotes number of growth cones analyzed to determine filopodial number. *p<0.001 indicates statistical significance relative to controls; two-sample t test. **p<0.005 denotes statistical significance between p75-/- no treatment versus p75+/+ no treatment; two-sample t test.
To further determine whether Trk signaling is involved in neurotrophin regulation of filopodial length, we used a Trk inhibitor, K252a, to block Trk receptor activation. K252a (1 μm) has been shown previously to inhibit NGF-induced TrkA auto-phosphorylation in chick DRG and sympathetic neurons (Klinz and Heumann, 1995). In addition, we found that 1 μm K252a blocked neurotrophin-mediated neuritogenesis by chick DRGs in vitro (S. Gehler, unpublished data). Treatment of chick RGC growth cones with 1 μm K252a had no significant effect on filopodial length, whereas the addition of BDNF to K252a-treated cultures increased filopodial length by 31% compared with K252a alone (Fig. 3H). Similarly, 1 μm K252a had no effect on NGF-induced increases of filopodial length on E9 chick DRGs (Gehler, unpublished data). These results support our conclusion that neurotrophin-induced increases in filopodial length are independent of Trk activation.
We also tested the hypothesis that neurotrophin binding to p75NTR mediates neurotrophin-induced increases in filopodial length by investigating the effects of p75 antibodies on growth cone filopodia. Cultures were treated with a p75 Ab previously shown to block binding of neurotrophins to chicken p75NTR (von Bartheld et al., 1996). Treatment of chick RGC growth cones with the p75 Ab (CHEX) alone resulted in a 35% increase in filopodial length (Fig. 3I). The addition of BDNF to CHEX-treated cultures did not have an additional effect on filopodial length. Similarly, CHEX treatment mimicked the effects of NGF treatment on filopodial length of DRG growth cones (Gehler, unpublished data). These experiments were repeated using a different p75 Ab, AB1554, and we observed similar results (Gehler, unpublished data). These data suggest that the p75 Ab CHEX mimics neurotrophin treatment to increase filopodial length.
NGF-induced increases in filopodial length are mediated through p75NTR in the absence of TrkA expression
To further test whether p75NTR regulates filopodia dynamics in the absence of TrkA activation, we probed the effects of NGF on chick ciliary neurons, which express p75NTR but not TrkA (Allsopp et al., 1993; Hallbook et al., 1995; Yamashita et al., 1999). Treatment of ciliary neurons with 40 nm NGF increased filopodial length by 27% relative to controls (Fig. 4A). Similar increases in filopodial length were induced by BDNF and NT-3. Furthermore, treatment with CHEX or AB1554 elicited a 15–20% increase in filopodial length on ciliary neurons, supporting the idea that filopodial length is modulated through p75NTR (Fig. 4B). These data provide direct evidence that p75NTR signaling is sufficient to mediate neurotrophin-induced increases in filopodial length in the absence of Trk activation.
Figure 4.

NGF-induced increases in filopodia length are mediated through p75NTR in the absence of TrkA expression. A, NGF treatment of ciliary neurons, which express p75NTR but not TrkA, produced a dose-dependent increase in filopodial length. BDNF and NT-3 produced similar increases in filopodial length as NGF. *p < 0.001 indicates significant difference between NGF, BDNF, and NT-3 relative to control; two-sample t test. B, CHEX or AB1554 alone increased filopodial length on ciliary neurons. C, NGF stimulation of ciliary neurons enhanced the average rate of filopodial extension but did not affect the average rate of retraction. Data in A and B are presented as percentage control ± SEM from four independent experiments. *p < 0.001 indicates statistical difference relative to control; two-sample t test.
Similar to our studies of RGC growth cones, we investigated filopodial extension and retraction dynamics of ciliary growth cones. Videomicroscopy of filopodia dynamics revealed that the average extension rate of filopodia was enhanced by NGF treatment (Fig. 4C). The extension rate was increased by 30%, similar to neurotrophin-induced increases in filopodia length. NGF had no effect on the rate of filopodial retraction. These data demonstrate that neurotrophin binding to p75NTR increases filopodial length by enhancing filopodial extension rates.
Growth cones from p75–/– mice are insensitive to neurotrophin effects and exhibit increased filopodial length
If neurotrophin-induced effects on filopodial length are mediated through p75NTR, then filopodia on growth cones from p75 mutant mice (p75–/–) (Lee et al., 1992) may be insensitive to neurotrophin treatment. First, we examined p75NTR expression on growth cones from wild-type (p75+/+) and p75–/– mice using several antibodies against extracellular and cytoplasmic domains of p75NTR. p75+/+ retinal and DRG neurons express p75NTR throughout growth cone and filopodial surfaces (Fig. 5A,C). p75–/– DRG growth cones lacked staining for either extracellular or cytoplasmic epitopes of p75NTR (Fig. 5E,G). The absence of p75NTR expression by mutant neurons was confirmed by Western blot analysis of brain lysates from p75–/– embryos (Fig. 5I).
Figure 5.

p75–/– retinal and DRG growth cones do not express p75NTR. A–D, Indirect immunofluorescence for p75NTR on p75+/+ mouse retinal and DRG growth cones using anti-p75NTR (A, C) and fluorescein phalloidin (B, D). Arrows note the surface expression of p75NTR on individual filopodia. E–H, Indirect immunofluorescence using anti-p75NTR raised against the extracellular domain of p75NTR (ex-p75; E, F) or cytoplasmic domain of p75NTR (cyto-p75; G, H) on p75–/– DRG growth cones. Staining for the cytoplasmic domain produced similar results with two different antibodies recognizing different epitopes of the cytoplasmic domain. Fibroblasts were negative controls for p75NTR expression. Staining of p75–/– retinal growth cones displayed similar results as p75–/– DRG growth cones (Gehler, unpublished data). Scale bar, 10 μm. I, Immunoblot of brain lysates from p75–/– embryos. Lysates of NIH3T3 and PC12 cells were used as negative and positive controls, respectively. Western blot analysis of p75NTR expression was repeated three times using brain lysates from different embryos of each genotype.
Treatment of p75+/+ mouse retinal and DRG growth cones with BDNF and NGF, respectively, induced increased filopodial length, similar to our results with chick neurons (Fig. 6A–E, G–J). However, treatment of p75–/– retinal growth cones with 160 nm BDNF had no effect on filopodial length (Fig. 6F). Yet, untreated p75–/– retinal growth cones had longer filopodia, similar to those of p75+/+ growth cones that were treated with BDNF (Fig. 6F). Growth cones of p75–/– DRG neurons displayed similarly enhanced filopodial lengths when maintained in low NGF, and treatment with elevated NGF did not increase filopodial length (Fig. 6K). Because untreated p75–/– growth cones displayed enhanced filopodial lengths similar to neurotrophin-treated p75+/+ growth cones, we concluded that unoccupied p75NTR has a negative effect on filopodial length that is absent when p75NTR is not expressed.
Figure 6.

p75–/– growth cones display enhanced filopodial lengths in the absence of neurotrophin treatment and are insensitive to neurotrophin-induced increases in filopodial length. A–D, Phalloidin staining of p75+/+ and p75–/– retinal growth cones before and after treatment with 160 nm BDNF. Scale bar, 10 μm. E, BDNF and NGF treatment of p75+/+ mouse retinal and DRG neurons, respectively, caused a 28 and 23% increase in filopodial length. Data are presented as percentage control ± SEM from four independent experiments. F, 160 nm BDNF treatment had no effect on filopodial length on p75–/– retinal growth cones. However, untreated p75–/– growth cones displayed filopodial lengths comparable with BDNF-treated p75+/+ growth cones. Data are presented as percentage control ± SEM from seven independent experiments. G–J, Phalloidin staining of p75+/+ and p75–/– DRG growth cones raised in either low (0.2 nm) or high (8 nm) NGF. Scalebar, 10 μm. K, p75–/– DRG growth cones raised in low NGF displayed filopodial lengths comparable with p75+/+ growth cones treated with high NGF. However, treatment of p75–/– DRG growth cones with high NGF had no effect on filopodial length. Data are presented as percentage control ± SEM from at least four independent experiments. L, AB1554 mimicked NGF treatment when applied to p75+/+ DRG neurons, whereas p75–/– DRG neurons were insensitive to AB1554 treatment. Data are presented as percentage control ± SEM from two independent experiments. *p < 0.001 indicates statistically significant differences relative to control; two-sample t test.
Because neurotrophins do not increase filopodial length on p75–/– growth cones, we predicted that anti-p75NTR, AB1554, would also have no effect on filopodial length of p75–/– DRG growth cones. Similar to chick RGC and DRG cultures, AB1554 elicited a 22% increase in filopodial length on p75+/+ growth cones but had no effect on filopodial length of p75–/– growth cones (Fig. 6L).
Inactivation of RhoA mediates neurotrophin-induced increases in filopodial length
We next tested the hypothesis that the p75NTR-mediated effects of neurotrophins on filopodial length occur through a RhoA-dependent pathway. Yamashita et al. (1999) proposed that unoccupied p75NTR activates RhoA, and neurotrophin binding to p75NTR reduces p75NTR-mediated RhoA activation. We first determined whether RhoA activity regulates filopodial length. We loaded chick retinal neurons with either C3 exoenzyme, which ADP ribosylates and inhibits RhoA, or with constitutively active RhoA (L63RhoA) using a peptide-based protein-loading method (Gallo, 2003). Inhibition of RhoA activity with C3 increased filopodial length by 23% (Fig. 7A), similar to BDNF treatment, supporting the hypothesis that RhoA activity negatively regulates filopodial length. Furthermore, consistent with the hypothesis that neurotrophins affect filopodial length by reducing RhoA activity, BDNF had no additional effect on filopodial length of C3-treated growth cones (Fig. 7A). Loading growth cones with L63RhoA alone had no effect on filopodial length, but L63RhoA blocked the increased filopodial length that is induced by BDNF (Fig. 7A). These results suggest that BDNF-induced increases of filopodial length result from decreased RhoA activity.
Figure 7.

Inhibition of RhoA activity mimics BDNF treatment, and L63RhoA blocks BDNF-induced increases in filopodial length. A, C3 mimicked the effects of BDNF on filopodial length on chick RGC growth cones, whereas L63RhoA alone had no effect on filopodial length but blocked the effects of BDNF. Data are presented as percentage control ± SEM from a minimum of four independent experiments. B, C3 enhanced filopodial lengths of p75+/+ and p75–/– retinal growth cones, whereas L63RhoA reduced filopodial lengths of p75–/– growth cones to levels indistinguishable from p75+/+ growth cones. Data are presented as percentage control ± SEM from a minimum of three independent experiments. *p < 0.001 denotes significant difference relative to control; two-sample t test.
The enhanced filopodial length on p75–/– growth cones suggests that p75–/– neurons lack a negative influence that involves RhoA activity. To test this idea, we studied the effects of C3 and L63RhoA on filopodial length of p75–/– retinal growth cones. Treatment of p75+/+ and p75–/– retinal neurons with C3 produced a 45 and 52% increase in filopodial length, respectively (Fig. 7B). Conversely, although L63RhoA had no effect on filopodial length on p75+/+ retinal growth cones, L63RhoA reduced filopodial length of p75–/– growth cones to levels indistinguishable from p75+/+ growth cones. These observations indicate that the enhanced filopodial length of p75–/– growth cones can be blocked by elevating RhoA activity. These results suggest that unoccupied p75NTR activates RhoA and that p75NTR-mediated RhoA activation is absent in p75–/– growth cones.
RhoA activity is reduced after treatment with BDNF and in p75–/– neurons
To assess the effects of BDNF on RhoA activity, we performed a RhoA activity pull-down assay with E7 chick retinal neurons (Ren and Schwartz, 2000). Five minutes after BDNF treatment, RhoA activity levels were decrease compared with untreated cells (Fig. 8A). BDNF treatment for 30 min produced a 48% reduction in RhoA activity (Fig. 8B). These results are direct evidence that BDNF regulates RhoA activity in chick retinal neurons.
Figure 8.

BDNF reduces RhoA activity in cultured retinal neurons through a p75NTR-dependent mechanism. A, Representative example of the time-dependent reduction in RhoA activity after BDNF treatment. B, Thirty minute exposure to 160 nm BDNF caused a 48% reduction in activated RhoA normalized to tubulin. Results were obtained from a minimum of three independent experiments. C, Cerebella from P8 p75–/– mice displayed reduced RhoA activity compared with p75+/+ mice. D, p75–/– cerebella exhibited a 43% reduction in RhoA activity relative to p75+/+ controls. Results were obtained from a minimum of four mice for each genotype. **p < 0.05 denotes significant difference between treated cultures normalized to controls or p75–/– relative to p75+/+ control; two sample t test.
Based on our observations that p75–/– growth cones have longer filopodia, we hypothesized that p75–/– neurons lack p75NTR-mediated RhoA activation. Using the pull-down activity assay, we compared RhoA activity in lysates from p75+/+ and p75–/– cerebella. Mouse cerebella from P8 mice, which express high levels of p75NTR, were processed for RhoA activity. As predicted, cerebellar lysates from p75–/– mice displayed lower levels of activated RhoA compared with p75+/+ mice (Fig. 8C,D).
RhoA activity is reduced in individual growth cones
Neurotrophin-induced changes in RhoA activity have not been demonstrated in growth cones. To measure RhoA activity in individual growth cones, we modified the approach of Li et al. (2002), which used the GST-RBD recombinant protein and anti-glutathione S-transferase staining to assess endogenous Rho GTPase activity in situ. RhoA activity in individual growth cones was measured after BDNF treatment (Fig. 9A–I). Five or 10 min treatment with BDNF caused a 43 and 34% reduction, respectively, in RhoA activity and maintained this reduced RhoA activity after 30 min (Fig. 9I). These data correlate well with the rapid enhancement of filopodial extension after BDNF treatment (Fig. 2B). Controls for this technique included demonstration of reduced growth cone staining for GST-RBD binding after C3 treatment and increased staining after activation of RhoA with LPA. Exclusion of GST-RBD resulted in no staining. To determine whether differences in RhoA activity were attributable to changes in protein content, we quantified protein content in growth cones using fluoroscein-5-isothiocyanate (FITC), which binds amines and is a general protein marker. FITC staining of control and BDNF-treated growth cones produced similar intensity levels (Gehler, unpublished data), suggesting that differences in staining for RhoA activity are not attributable to changes in protein content.
Figure 9.
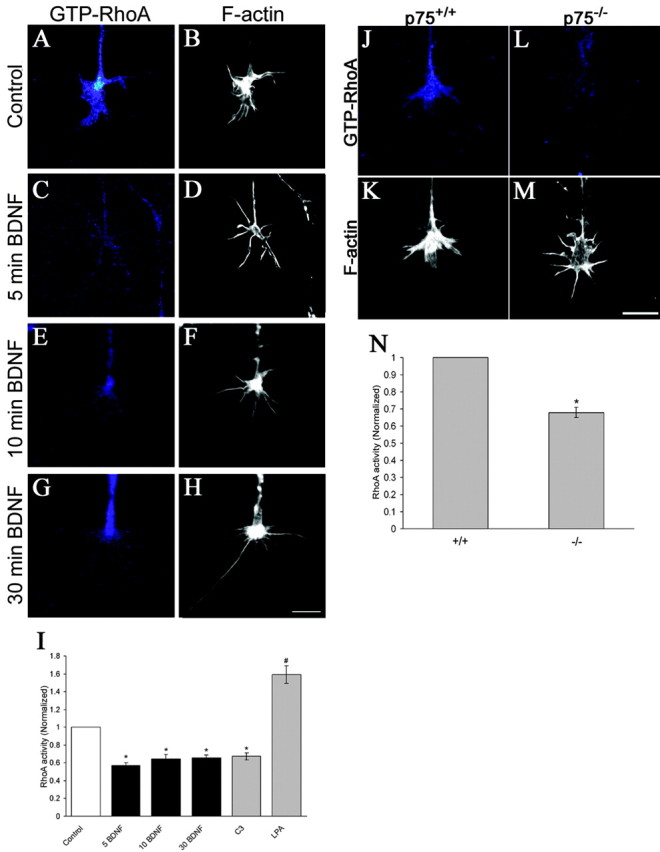
RhoA activity is reduced in growth cones after BDNF treatment. A–H, 160 nm BDNF caused a reduction of RhoA activity in individual RGC growth cones using the GST-RBD fusion protein plus anti-glutathione S-transferase (A, C, E, G) or rhodamine phalloidin (B, D, F, H). Scale bar, 10 μm. I, Quantification of RhoA activity within individual growth cones. There was a 43% reduction in staining for GST-RBD after treatment with BDNF for 5 min relative to control. GST-RBD staining was still reduced by 34% relative to control levels after 30 min exposure to BDNF. C3, a RhoA inhibitor, caused a 33% reduction in activated RhoA within individual growth cones. RhoA activity was elevated by 59% after treatment with LPA. Number of growth cones measured for each experimental manipulation was as follows: 5 min BDNF, 77; 10 min BDNF, 69; 30 min BDNF, 184; C3, 80; LPA, 74. J–M, p75–/– retinal growth cones displayed lower levels of endogenous RhoA activity relative to p75+/+ growth cones stained using GST-RBD (J, L) or rhodamine phalloidin (K, M). Scale bar, 10 μm. N, p75–/– retinal growth cones show a 32% reduction in RhoA activity relative to p75+/+ growth cones. Measurements were acquired from at least 165 growth cones obtained from a minimum of three embryos. *p < 0.001, #p < 0.001 denote significant difference between treated cultures normalized to controls or p75–/– relative to p75+/+ control; two sample t test.
We also measured RhoA activity in growth cones from p75+/+ and p75–/– neurons (Fig. 9J–N). GST-RBD staining of p75–/– retinal growth cones indicated a 32% reduction in RhoA activity relative to p75+/+ counterparts (Fig. 9N). These data suggest that reduced RhoA activity accounts for the longer filopodia on p75–/– growth cones.
BDNF regulates filopodia number on retinal growth cones
BDNF treatment of RGC growth cones also increased filopodial number in a dose-dependent manner (Fig. 1E). Treatment of chick or mouse retina with 160 nm BDNF enhanced filopodial number by 63 and 35%, respectively (Table 1). NGF and NT-3 had no effect on filopodial number on RGC growth cones even though they did increase filopodial length. Treatment of chick RGC neurons with CHEX alone increased filopodial number by 29% relative to controls. Regulation of filopodia number on RGCs is independent of Trk activity, because addition of BDNF to K252a-treated cultures increased filopodia number by ∼30%. Also, untreated p75–/– retinal growth cones exhibited a 38% increase of filopodia number, which is comparable with BDNF treatment of p75+/+ growth cones. Neurotrophin treatment of chick DRG and ciliary growth cones did not affect filopodial number (Gehler, unpublished data). These data suggest that BDNF binding to p75NTR regulates filopodial number in a cell-specific manner.
Discussion
The signaling mechanisms by which neurotrophins regulate growth cone motility are not fully understood. In this study, we investigated the role of p75NTR in regulating filopodial dynamics. Our data support the hypothesis that p75NTR is sufficient and necessary to mediate neurotrophin-induced increases in filopodial length. Furthermore, regulation of filopodial length by p75NTR involves modulation of RhoA activity. In addition, we show that BDNF stimulation can regulate filopodia number in a cell-specific manner. This suggests a signaling mechanism in which neurotrophin binding to p75NTR terminates an activity of RhoA that negatively regulates filopodial dynamics.
Filopodia are important sensors of guidance cues, and filopodial length influences the sampling area of growth cones. Filopodia are essential to growth cone turning, and manipulations of embryos that inhibit filopodia cause path-finding errors (Bentley and Toroian-Raymond, 1986; Chien et al., 1993; Zheng et al., 1996). BDNF treatment increases filopodial length by 28% in RGC growth cones, which would result in a 109% increase in the potential sampling volume of a growth cone. Thus, neurotrophin-induced increases in filopodial length may significantly increase the ability of growth cones to detect guidance cues or span substrates. Extrinsic regulation of filopodial length may also be significant during synaptogenesis, because both axonal arbors and dendrites extend exploratory filopodia.
Neurotrophins bind two classes of receptors with different binding selectivities and signaling pathways. Trk receptors selectively bind neurotrophins, whereas p75NTR binds all neurotrophins (Chao, 2003). Retinal ganglion cells in chick embryos express TrkB but not TrkA, whereas TrkC expression is not conclusively determined (von Bartheld, 1998). In view of evidence that chick RGCs express only TrkB, our finding that filopodial length is stimulated equally by three neurotrophins suggests that p75NTR mediates neurotrophin effects on filopodial length. Our evidence for the role of p75NTR in regulating filopodial dynamics included pharmacologically blocking Trk receptor activation, treatment with p75NTR antibodies, using chick ciliary neurons that do not express TrkA receptors, and using neurons from p75–/– mice. Our determinations of the concentration dependence of neurotrophin effects indicate that maximal increases of filopodial number and length require neurotrophin concentrations that exceed the published binding affinity of p75NTR (Chao, 2003). However, the published measurements involved transfected cells or neuronal cell bodies. Given the recently recognized diversity of ligands and binding associates of p75NTR (Dechant and Barde, 2003), the actual binding affinity of p75NTR on growth cone filopodia for neurotrophins may differ.
Neurotrophin binding to p75NTR is sufficient to increase filopodial length. Experiments using K252a to inhibit Trk signaling provided evidence for the sufficiency of p75NTR in increasing filopodial length. Ciliary neurons, which express p75NTR but not TrkA, also exhibited increased filopodial lengths and extension rates after NGF treatment, suggesting that neurotrophin binding to p75NTR alone regulates filopodial dynamics. Additional evidence that neurotrophin binding to p75NTR is sufficient to increase filopodial length came from studies using p75NTR antibodies. Treatment of chick RGCs, DRGs, and ciliary neurons with CHEX mimicked the effects of neurotrophins on filopodial length. These observations were confirmed using another p75 Ab (AB1554). We interpret these results as the Ab dimerizes the receptor and triggers p75NTR-mediated signaling to increase filopodial length. Previous studies report that p75 antibodies mimic the effects of neurotrophins in other p75NTR-mediated events (Brann et al., 1999; Freidin, 2001). Together, these results suggest that p75NTR regulates filopodial dynamics in the absence of Trk activation.
Our studies with p75 mutant mice showed that p75NTR is required for neurotrophins to modulate filopodial length. Treatment of p75–/– retinal and DRG neurons with neurotrophins failed to change filopodial length. Additionally, treatment of p75–/– DRG growth cones with AB1554 had no effect on filopodial length. Actually, growth cones of p75–/– neurons exhibited enhanced filopodial lengths, similar to p75+/+ neurons treated with elevated neurotrophins. These results suggest that unoccupied p75NTR exerts a negative effect on filopodial length that is absent in p75–/– neurons.
We tested the hypothesis that RhoA is a component of the pathway by which unoccupied p75NTR negatively regulates filopodial length. Consistent with this idea, inhibition of RhoA with C3 transferase mimicked the stimulatory effects of neurotrophins on filopodial length, and neurotrophins did not further increase filopodial length of C3-loaded growth cones. Conversely, introduction of constitutively active RhoA blocked BDNF-induced increase of filopodial length on chick RGC growth cones and also blocked the enhanced filopodial length of mouse p75–/– growth cones. Measuring RhoA activity using both a pull-down assay and a growth cone-staining assay showed decreased RhoA activity during a 30 min BDNF treatment. The time course of RhoA activity after BDNF treatment indicates that there is an inverse relationship between RhoA activity and filopodial extension rates. Consistent with the hypothesis, p75–/– neurons displayed reduced levels of activated RhoA compared with wild-type counterparts, using both cerebellar cell extracts and the growth cone-staining assay. This is consistent with the enhanced filopodial length of p75–/– neurons. These results indicate that RhoA activity, regulated by p75NTR signaling, modulates filopodial behavior.
The in vitro approach has allowed us to analyze regulation of filopodial length by neurotrophin signaling through p75NTR, independently of indirect or noncell autonomous functions of p75NTR that can complicate in vivo studies. For example, p75NTR receptors interact with trk receptors to modify the affinity and specificity of trk receptors (Huang and Reichardt, 2003), and it is suggested that p75NTR receptors expressed on Schwann cells bind neurotrophins and present them to growing axons (Taniuchi et al., 1986). In vivo axonal growth from sensory neurons of p75–/– mice is significantly diminished or delayed (Yamashita et al., 1999; Bentley and Lee, 2000; Gehler, unpublished data). However, we found that the mean neurite length extended from mouse E14.5 p75–/– DRG explants after 24 hr in vitro is indistinguishable from outgrowth of p75+/+ explants (Gehler, unpublished data). McQuillen et al. (2002) reported that growth cones of subplate neurons from p75–/– mice exhibit fewer filopodia in vivo, although we found that retinal growth cones from p75–/– neurons had more filopodia than wild-type neurons. The disparity between our study and that of McQuillen et al. (2002) may be attributable to the complexity of the in vivo situation. Alternatively, neurotrophin effects may be cell-type specific or may be limited to certain developmental stages. Consistent with this notion, we found that BDNF increased filopodia number on RGC neurons but not on DRG or ciliary neurons. The significance of reduced RhoA levels in neurons of p75–/– mice is unclear; however, Walsh et al. (1999) reported enhanced sprouting and growth of sympathetic axons in myelinated regions of p75–/– mice.
Collectively, these data suggest a model for p75NTR-mediated regulation of filopodial dynamics in both a ligand-dependent and ligand-independent state. In the unoccupied state, p75NTR negatively regulates filopodial dynamics through RhoA activation. After neurotrophin binding to p75NTR, negative regulation through RhoA would stop, producing enhanced filopodial lengths. Because p75–/– neuronal growth cones do not express p75NTR, this mode of activating RhoA activity is absent. As a result, filopodial lengths on p75–/– growth cones without neurotrophin stimulation resembles the enhanced filopodial length of p75+/+ growth cones treated with neurotrophins.
How is filopodial extension regulated downstream of RhoA? Filopodial extension is regulated by a balance of actin polymerization at filopodial tips and retrograde displacement of actin filaments toward the base of a filopodium (Mallavarapu and Mitchison, 1999). One link between actin polymerization and RhoA is possible through the RhoA effector mDia that regulates profilin, which in turn modulates the availability of actin monomers (Watanabe et al., 1997). However, contrary to our observations, downregulation of RhoA signaling through mDia would decrease filopodial extension. RhoA also acts via its downstream kinase Rho-associated kinase (ROCK) to regulate LIM-kinase (Maekawa et al., 1999), which regulates activity of actin-depolymerizing factor (ADF)/cofilin (Arber et al., 1998; Maekawa et al., 1999). ADF/cofilin regulates actin filament turnover, and ADF/cofilin overexpression increases filopodial number of rat cortical neurons (Meberg and Bamburg, 2000). Thus, RhoA could alter filopodial dynamics through ROCK-mediated regulation of ADF/cofilin. Additionally, ROCK regulates myosin by increasing phosphorylation of the regulatory light chain (Kimura et al., 1996). A decrease in RhoA activity would reduce actomyosin-based contractility. Although myosin II may not be involved in filopodial protrusion on growth cones (Diefenbach et al., 2002), myosin II may indirectly regulate filopodial length. There are examples of cross-talk between RhoA, Rac1, and Cdc42 in neurons (Kozma et al., 1997; van Leeuwen et al., 1997; Li et al., 2002; Yuan et al., 2003). For example, turning of Xenopus spinal neuron growth cones to attractive and repulsive cues involves filopodial asymmetry across a growth cone that is mediated by cross-talk between Cdc42 and RhoA signaling (Yuan et al., 2003). In light of these findings, it may be that neurotrophin signaling through p75NTR regulates filopodial dynamics through cross-talk between RhoA, Rac1, and Cdc42. Our results suggest that RhoA is necessary to regulate filopodial dynamics, but whether p75-mediated changes in RhoA affect Rac or Cdc42, which promote actin polymerization (Kuhn et al., 2000), or vice versa is not known. Future work should examine the regulation of these GT-Pases in the context of neurotrophin signaling to regulate growth cone motility and the filopodial dynamics.
In summary, signaling from p75NTR regulates filopodial dynamics. Neurotrophin binding to p75NTR reduces RhoA activity, which results in enhanced filopodial length. Growth cones that do not express p75NTR are insensitive to neurotrophin-induced increases in filopodial length. However, as a consequence of reduced RhoA activity, growth cones lacking p75NTR exhibit enhanced filopodial lengths that mimic neurotrophin treatment, suggesting that unbound p75NTR negatively regulates filopodial dynamics.
Footnotes
This research was supported by grants from the National Institutes of Health (NIH) (HD19950), National Science Foundation (IBN0080932), and Minnesota Medical Foundation. Scott Gehler is a trainee on NIH Eye Institute Vision Training Grant.
Correspondence should be addressed to Paul C. Letourneau, Department of Neuroscience, 6–145 Jackson Hall, University of Minnesota, Minneapolis, MN 55455. E-mail: letour@med.umn.edu.
DOI:10.1523/JNEUROSCI.0404-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244363-10$15.00/0
References
- Allsopp TE, Robinson M, Wyatt S, Davies AM (1993) Ectopic trkA expression mediates a NGF survival response in NGF-independent sensory neurons but not in parasympathetic neurons. J Cell Biol 123: 1555–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsina B, Vu T, Cohen-Cory S (2001) Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat Neurosci 4: 1093–1101. [DOI] [PubMed] [Google Scholar]
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P (1998) Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393: 805–809. [DOI] [PubMed] [Google Scholar]
- Bentley CA, Lee KF (2000) p75 is important for axon growth and schwann cell migration during development. J Neurosci 20: 7706–7715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentley D, Toroian-Raymond A (1986) Disoriented pathfinding by pioneer neurone growth cones deprived of filopodia by cytochalasin treatment. Nature 323: 712–715. [DOI] [PubMed] [Google Scholar]
- Bito H, Furuyashiki T, Ishihara H, Shibasaki Y, Ohashi K, Mizuno K, Maekawa M, Ishizaki T, Narumiya S (2000) A critical role for a Rho-associated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron 26: 431–441. [DOI] [PubMed] [Google Scholar]
- Brann AB, Scott R, Neuberger Y, Abulafia D, Boldin S, Fainzilber M, Futerman AH (1999) Ceramide signaling downstream of the p75 neurotrophin receptor mediates the effects of nerve growth factor on outgrowth of cultured hippocampal neurons. J Neurosci 19: 8199–8206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV (2003) Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci 4: 299–309. [DOI] [PubMed] [Google Scholar]
- Chien CB, Rosenthal DE, Harris WA, Holt CE (1993) Navigational errors made by growth cones without filopodia in the embryonic Xenopus brain. Neuron 11: 237–251. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S, Fraser SE (1995) Effects of brain-derived neurotrophic factor on optic axon branching and remodeling in vivo Nature 378: 192–196. [DOI] [PubMed] [Google Scholar]
- Dechant G, Barde YA (2003) The neurotrophin receptor p75NTR Novel functions and implications for diseases of the nervous system. Nat Neurosci 5: 1131–1136. [DOI] [PubMed] [Google Scholar]
- Diefenbach TJ, Latham VM, Yimlamai D, Liu CA, Herman IM, Jay DG (2002) Myosin 1c and myosin IIB serve opposing roles in lamellipodial dynamics of the neuronal growth cone. J Cell Biol 158: 1207–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst AF, Gallo G, Letourneau PC, McLoon SC (2000) Stabilization of growing retinal axons by the combined signaling of nitric oxide and brain-derived neurotrophic factor. J Neurosci 20: 1458–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frade JM, Rodriguez-Tebar A, Barde YA (1996) Induction of cell death by endogenous nerve growth factor through its p75 receptor. Nature 383: 166–168. [DOI] [PubMed] [Google Scholar]
- Freidin MM (2001) Antibody to the extracellular domain of the low affinity NGF receptor stimulates p75NGFR-mediated apoptosis in cultured sympathetic neurons. J Neurosci Res 64: 331–340. [DOI] [PubMed] [Google Scholar]
- Frost DO (2001) BDNF/trkB signaling in the developmental sculpting of visual connections. Prog Brain Res 134: 35–49. [DOI] [PubMed] [Google Scholar]
- Gallo G (2003) Making proteins into drugs: assisted delivery of proteins and peptides into living neurons. Methods Cell Biol 71: 325–338. [DOI] [PubMed] [Google Scholar]
- Gallo G, Letourneau PC (1998) Localized sources of neurotrophins initiate axon collateral sprouting. J Neurosci 18: 5403–5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G, Letourneau PC (2000) Neurotrophins and the dynamic regulation of the neuronal cytoskeleton. J Neurobiol 44: 159–173. [DOI] [PubMed] [Google Scholar]
- Gallo G, Lefcort FC, Letourneau PC (1997) The trkA receptor mediates growth cone turning toward a localized source of nerve growth factor. J Neurosci 17: 5445–5454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo G, Yee HF, Letourneau PC (2002) Actin turnover is required to prevent axon retraction driven by endogenous actomyosin contractility. J Cell Biol 158: 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen RW, Barrett JN (1979) Neuronal chemotaxis: chick dorsal-root axons turn toward high concentrations of nerve growth factor. Science 206: 1079–1080. [DOI] [PubMed] [Google Scholar]
- Hallbook F, Backstrom A, Kullander K, Kylberg A, Williams R, Ebendal T (1995) Neurotrophins and their receptors in chicken neuronal development. Int J Dev Biol 39: 855–868. [PubMed] [Google Scholar]
- Hallbook F, Backstrom A, Kullander K, Ebendal T, Carri NG (1996) Expression of neurotrophins and trk receptors in the avian retina. J Comp Neurol 364: 664–676. [DOI] [PubMed] [Google Scholar]
- Hempstead BL (2002) The many faces of p75NTR Curr Opin Neurobiol 12: 260–267. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF (2003) Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72: 609–642. [DOI] [PubMed] [Google Scholar]
- Kater SB, Rehder V (1995) The sensory-motor role of growth cone filopodia. Curr Opin Neurobiol 5: 68–74. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K (1996) Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase). Science 273: 245–248. [DOI] [PubMed] [Google Scholar]
- Klinz FJ, Heumann R (1995) Time-resolved signaling pathways for nerve growth factor diverge downstream of the p140trk receptor activation between chick sympathetic and dorsal root ganglion sensory neurons. J Neurochem 65: 1046–1053. [DOI] [PubMed] [Google Scholar]
- Kozma R, Sarner S, Ahmed S, Lim L (1997) Rho family GTPases and neuronal growth cone remodeling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol Cell Biol 17: 1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn TB, Meberg PJ, Brown MD, Bernstein BW, Minamide LS, Jensen JR, Okada K, Soda EA, Bamburg JR (2000) Regulating actin dynamics in neuronal growth cones by ADF/Cofilin and Rho family GTPases. J Neurobiol 44: 126–144. [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R (1992) Targeted mutation of the gene encoding the low affinity NGF receptor p75NTR leads to deficits in the peripheral sensory nervous system. Cell 69: 737–749. [DOI] [PubMed] [Google Scholar]
- Lehmann M, Fournier A, Selles-Navarro I, Dergham P, Sebok A, Leclerc N, Tigyi G, McKerracher L (1999) Inactivation of Rho signaling pathway promotes CNS axon regeneration. J Neurosci 19: 7537–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Aizenman CD, Cline HT (2002) Regulation of Rho GTPases by crosstalk and neuronal activity in vivo Neuron 33: 741–750. [DOI] [PubMed] [Google Scholar]
- Lom B, Cohen-Cory S (1999) Brain-derived neurotrophic factor differentially regulates retinal ganglion dendritic and axonal arborization in vivo J Neurosci 19: 9928–9938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, Jan LY, Jan YN (1997) Rho family small GTP-binding proteins in growth cone signalling. Curr Opin Neurobiol 7: 81–86. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K, Narumiya S (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM-kinase. Science 285: 895–898. [DOI] [PubMed] [Google Scholar]
- Mallavarapu A, Mitchison T (1999) Regulated actin cytoskeleton assembly at filopodium tips controls their extension and retraction. J Cell Biol 146: 1097–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason C, Erskine L (2000) Growth cone form, behavior, and interactions in vivo: retinal axon pathfinding as a model. J Neurobiol 44: 260–270. [DOI] [PubMed] [Google Scholar]
- McLoon SC, Barnes RB (1989) Early differentiation of retinal ganglion cells: an axonal protein expressed by premigratory and migrating retinal ganglion cells. J Neurosci 9: 1424–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillen PS, DeFreitas MF, Zada G, Shatz CJ (2002) A novel role for p75NTR in subplate growth cone complexity and visual thalamocortical innervation. J Neurosci 22: 3580–3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meberg PJ, Bamburg JR (2000) Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J Neurosci 20: 2459–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G, Song H, Berninger B, Inagaki N, Tessier-Lavigne M, Poo MM (1999) Phospholipase C-gamma and phosphoinositide 3-kinase mediate cytoplasmic signaling in nerve growth cone guidance. Neuron 23: 139–148. [DOI] [PubMed] [Google Scholar]
- O'Connor TP, Duerr JS, Bentley D (1990) Pioneer growth cone steering decisions mediated by single filopodial contracts in situ J Neurosci 10: 3935–3946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren XD, Schwartz MA (2000) Determination of GTP loading on Rho. Methods Enzymol 325: 264–272. [DOI] [PubMed] [Google Scholar]
- Ruchhoeft ML, Ohnuma S, McNeill L, Holt CE, Harris WA (1999) The neuronal architecture of Xenopus retinal ganglion cells is sculpted by Rho-family GTPases in vivo J Neurosci 19: 8454–8463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi M, Clark HB, Johnson EM (1986) Induction of nerve growth factor receptor in Schwann cells after axotomy. Proc Natl Acad Sci USA 83: 4094–4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker KL, Meyer M, Barde YA (2001) Neurotrophins are required for nerve growth during development. Nat Neurosci 4: 29–37. [DOI] [PubMed] [Google Scholar]
- van Leeuwen FN, Kain HET, van der Kammen RA, Michiels F, Kranenburg OW, Collard JG (1997) The guanine nucleotide exchange factor Tiam1 affects neuronal morphology; opposing roles for the small GTPases Rac and Rho. J Cell Biol 139: 797–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS (1998) Neurotrophins in the developing and regenerating visual system. Histol Histopathol 13: 437–459. [DOI] [PubMed] [Google Scholar]
- von Bartheld CS, Williams R, Lefcort F, Clary DO, Reichardt LF, Bothwell M (1996) Retrograde transport of neurotrophins from the eye to the brain in chick embryos: roles of the p75NTR and trkB receptors. J Neurosci 16: 2995–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh GS, Krol KM, Crutcher KA, Kawaja MD (1999) Enhanced neurotrophin-induced axon growth in myelinated portions of the CNS in mice lacking the p75 neurotrophin receptor. J Neurosci 19: 4155–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N, Madaule P, Reid T, Ishizaki T, Watanabe G, Kakizuka A, Saito Y, Nakao K, Jockusch BM, Narumiya S (1997) p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J 16: 3044–3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Tohyama M (2003) The p75 receptor acts as a displacement factor that releases Rho from Rho-GDI. Nat Neurosci 6: 461–467. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA (1999) Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron 24: 585–593. [DOI] [PubMed] [Google Scholar]
- Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM (1997) Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci 17: 7594–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan XB, Jin M, Xu X, Song YQ, Wu CP, Poo MM, Duan S (2003) Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat Cell Biol 5: 38–45. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Wan JJ, Poo MM (1996) Essential role of filopodia in chemotropic turning of nerve growth cones induced by a glutamate gradient. J Neurosci 16: 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]