

Abstract
Dopaminergic neurotransmission has been highly implicated in the reinforcing properties of many substances of abuse, including marijuana. Cannabinoids activate ventral tegmental area dopaminergic neurons, the main ascending projections of the mesocorticolimbic dopamine system, and change their spiking pattern by increasing the number of impulses in a burst and elevating the frequency of bursts. Although they also increase time-averaged striatal dopamine levels for extended periods of time, little is known about the temporal structure of this change. To elucidate this, fast-scan cyclic voltammetry was used to monitor extracellular dopamine in the nucleus accumbens of freely moving rats with subsecond timescale resolution. Intravenous administration of the central cannabinoid (CB1) receptor agonist, R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-(1-naphthalenyl) methanone mesylate, dose-dependently produced catalepsy, decreased locomotion, and reduced the amplitude of electrically evoked dopamine release while markedly increasing the frequency of detected (nonstimulated) dopamine concentration transients. The CB1 receptor antagonist [N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3-carboxamide] reversed and prevented all agonist-induced effects but did not show effects on dopamine release when injected alone. These data demonstrate that doses of a cannabinoid agonist known to increase burst firing produce ongoing fluctuations in extracellular dopamine on a previously unrecognized temporal scale in the nucleus accumbens.
Keywords: dopamine, accumbens, cannabinoids, drug abuse, cyclic voltammetry, neuromodulation
Introduction
Midbrain dopaminergic neurons normally fire at low frequencies of 5–10 Hz in freely moving rats, with periodic short bursts of high-frequency activity (∼20 Hz) that become more prevalent in response to salient stimuli (Hyland et al., 2002). Indeed, burst firing of dopamine (DA) neurons has been shown to be of functional significance in the context of sensory processing, specifically with regard to reward (Schultz, 1998). Increases in the burst rate of dopaminergic neurons produce greater peak extracellular dopamine concentrations at axon terminals (Gonon, 1988; Wightman et al., 1988; Overton and Clark, 1997). Recently, rapid surges in extracellular dopamine concentration or “transients” have been observed with fast-scan cyclic voltammetry at carbonfiber electrodes implanted in various terminal fields (Robinson et al., 2002) that would be consistent with bursting activity of dopaminergic neurons. The transients only last for a few seconds and can be time-locked to specific reward-related behaviors such as cocaine self-administration (Phillips et al., 2003) and operant responding for natural rewards (Roitman et al., 2004) during which they appear to promote reward-seeking.
Drugs of abuse share the ability to increase dopamine neurotransmission in the nucleus accumbens (NAc) (Di Chiara and Imperato, 1988; Nestler, 2002; Wise, 2002). Some abused drugs such as amphetamine and cocaine have direct actions on dopaminergic neurons, whereas others such as the main psychoactive component of marijuana, Δ9-tetrahydrocannabinol (Δ9-THC) (Mechoulam and Gaoni, 1967), exert their actions indirectly. Indeed, Δ9-THC exerts its reinforcing effects primarily via an action on central cannabinoid (CB1) receptors, the endogenous ligands of which are arachidonic acid derivatives such as anandamide and 2-arachidonyl-glycerol (Freund et al., 2003). This endogenous system interacts with many neurotransmitters in the brain, including GABA, glutamate, and acetylcholine; however, cannabinoid receptors are not found on dopaminergic neurons (Hernàndez et al., 2000). Nevertheless, extracellular dopamine in the NAc is elevated by Δ9-THC or synthetic CB1 agonists as shown by microdialysis studies (Chen et al., 1991; Tanda et al., 1997; Tanda and Goldberg, 2003).
A possible mechanism explaining this increase in NAc extracellular dopamine would be an indirect excitatory action of cannabinoids on ventral tegmental area (VTA) dopaminergic neurons, the main ascending dopaminergic projection to the NAc. Indeed, in the VTA-containing slice, CB1 agonists enhance the firing rate of dopaminergic neurons in a nondesensitizing manner (Cheer et al., 2000a) via a reduction of afferent GABAergic transmission (Szabo et al., 2002). This finding is consistent with the anatomical distribution of CB1 receptors and has added the VTA to a number of brain regions in which cannabinoids are known to exert their effects presynaptically (Schlicker and Kathmann, 2001). Cannabinoids given at doses that elevate dopamine levels in the NAc also excite dopaminergic neurons in the VTA through CB1 receptors in vivo (French et al., 1997; Gessa et al., 1998; Cheer at al., 2003). Furthermore, VTA dopamine neurons in anesthetized or paralyzed rats exhibit an elevated number of action potentials within bursts after cannabinoid application (French, 1997; Diana et al., 1998; Wu and French, 2000).
In this work we used fast-scan cyclic voltammetry in freely moving rats to measure the effects of systemic administration of a cannabinoid agonist at doses reported to increase phasic firing of VTA dopamine neurons to determine whether time-resolved dopamine release events that are functionally relevant can be observed after cannabinoid treatment. The effect of the agonist on extracellular dopamine was manifested as an increase in the frequency and amplitude of periodic dopamine transients, suggesting that changes in dopamine release dynamics follow a cannabinoid-induced enhancement of burst (phasic) activity in dopaminergic neurons.
Materials and Methods
Animals and surgery. Male Sprague Dawley rats (n = 21) implanted with a jugular vein catheter (250–350 gm; Charles River, Wilmington, MA) were housed individually in clear plastic cages with ad libitum access to food and water on an inverted 12 hr light/dark cycle. After 1 week, animals were anesthetized with xylazine hydrochloride (10 mg/kg, i.p.) and ketamine hydrochloride (100 mg/kg, i.p.) for implantation of electrodes for voltammetry as described previously (Robinson et al., 2002). Briefly, a guide cannula (BAS, West Lafayette, IN) aimed at the NAc core (1.3 mm anterior and 1.3 mm lateral relative to bregma) was permanently affixed to the skull, and a bipolar stimulating electrode (Plastics One, Roanoke, VA) was lowered to the medial forebrain bundle (MFB) (4.2 mm posterior, 1.4 mm lateral, and 8.6 mm ventral from the brain surface). A Ag/AgCl reference electrode was positioned in the contralateral hemisphere. Once stimulated dopamine release was voltammetrically detected in the ipsilateral NAc core with a carbon-fiber electrode, the stimulating electrode and guide cannula were permanently affixed to the skull with the aid of machine screws and cranioplastic cement. Rats were allowed to regain their presurgical weight (at least 48 hr postoperative) before freely moving experiments were initiated. All animal protocols and care were approved by the Institutional Animal Care and Use Committee of the University of North Carolina at Chapel Hill.
Fast-scan cyclic voltammetry. Cylindrical carbon-fiber microelectrodes with an exposed tip of 50–100 μm were prepared by pulling capillary glass (A-M Systems, Carlsborg, WA) around carbon fibers (T650, 6 μm diameter; Amoco Corporation, Greenville, SC). A fresh electrode was lowered each day to the core of the NAc (6–8 mm, ventral) with the aid of a microdrive that locked to the implanted guide cannula. A head-stage cable was connected to the electrode assembly, and animals were tethered to a rotating commutator situated in the top of the experimental chamber, which allowed for virtually unrestrained movement. The carbonfiber electrode was held at –0.4 V versus Ag/AgCl between scans and then driven to +1.3 V and back in a triangular manner at 400 V/sec every 100 msec. This waveform is similar to that documented previously that produces enhanced sensitivity for dopamine (Heien et al., 2003). The waveform was generated and current was monitored with an interface board (National Instruments, Austin, TX) in a Pentium IV computer running custom-designed LABVIEW (National Instruments) software. Medial forebrain stimulations consisted of 24 biphasic pulses (2 msec, 125 μA per phase) delivered at 60 Hz/min to elicit dopamine release.
Behavioral testing. Spontaneous locomotor activity was monitored using a DigiScan Animal Activity Monitor (AccuScan Instruments, Columbus, OH). A set of five infrared (IR) beams 3 cm above the floor of the box were used to determine locomotor activity. Ten readings of horizontal activity were obtained every minute for 10 min. Because animals were tested multiple times in 1 d and cannabinoids are known to suppress locomotion, each rat was exposed for 10 min to the locomotor activity box before testing to avoid a habituation confound. Catalepsy was evaluated with an 8 mm diameter metal bar suspended 10 cm above the floor of the cage by two wooden blocks 20 cm apart. Both forepaws were placed on the bar, and the time the animal kept them on the bar before both limbs returned to the ground was recorded (Wu et al., 2000). The average time from three identical trials in each rat was recorded. Trials were terminated after 180 sec.
Data analysis. Cyclic voltammograms (used for identification of detected species) were background subtracted from 10 cyclic voltammograms obtained before the stimulation to remove the capacitive and charging currents. To examine all of the detected voltammetric events during a recording period, two-dimensional plots were used with time as the abscissa, triangular wave voltage as the ordinate, and the background subtracted current encoded in false color. The temporal profile of voltammetrically detected dopamine changes was obtained by plotting the current at the peak oxidation of dopamine (+600 mV vs Ag/AgCl) as a function of time. To remove contributions from ionic changes that frequently accompany stimulated dopamine release (Venton et al., 2003), currents measured at a potential at which only ionic changes occurred were adjusted in amplitude and subtracted from current at the peak potential for dopamine oxidation (Phillips and Wightman, 2003). Similar data treatment was used for dopamine calibration (1 μm) experiments in a flow injection system used after freely moving experiments.
Cyclic voltammograms were collected into 1 min data files. An MFB stimulation to evoke dopamine release occurred at 30 sec within the collection interval. A background-subtracted cyclic voltammogram of the dopamine detected at the end of the stimulation in each animal was used as a template for identification of dopamine during other times in the recording interval. Typical noise levels were equivalent to 13 nm dopamine; only signals larger than this were considered for analysis. The entire file was evaluated for dopamine transients by taking a moving average of 10 scans and subtracting it from a cyclic voltammogram recorded 1 sec later. The resultant cyclic voltammogram was compared with the template voltammogram, and a correlation coefficient was obtained. The event was termed dopaminergic only if the correlation coefficient was ≥0.75. To obtain such a correlation coefficient, the cyclic voltammogram needed to have peaks that occurred at approximately +600 and –200 mV versus Ag/AgCl (oxidation and reduction potentials characteristic of dopamine). In addition, dopamine transients were required to meet these criteria for at least two consecutive scans.
Graphs were constructed from the data using Graph Pad Prism (Graph Pad Software, San Diego, CA). The amplitudes of stimulated dopamine release in files surrounding drug injections were analyzed with respect to pre-drug values with conventional ANOVA and Scheffe post hoc tests; the criterion of significance was set at p < 0.05. Drug-induced effects on spontaneous dopamine transients were determined by averaging the number of dopamine transients in 1 min files before and after the administration of drugs. Statistical analyses used STATISTICA (StatSoft, Tulsa, OK). Data are expressed as mean ± SEM.
Drugs. R(+)-[2,3-dihydro-5-methyl-3-[(morpholinyl)methyl]pyrrolo [1,2,3-DE]-1,4-benzoxazin-6-yl]-(1-naphtmalenyl)methanone mesylate (WIN55,212–2) (WIN) (Sigma, St. Louis, MO, USA) and [N-piperidino-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methylpyrazole-3-carboxamide] (SR141716A) (SR) (Research Triangle Institute–National Institute on Drug Abuse, Raleigh, NC) were freshly suspended in a 1:1:18 ratio of ethanol, emulphor (Alkamuls EL-620, Rhodia, Cranbury, NJ), and heparinized saline (0.9%) for intravenous injection. Initially, rats (n = 9) received the following dosing protocol (separated by at least 15 min for all dosing regimens): vehicle (200 μl), WIN (125 and 250 μg/kg), SR (300 μg/kg), and WIN again (250 μg/kg). To test the involvement of the endogenous cannabinoid system in the regulation of fast dopamine release, rats (n = 4) were given vehicle (200 μl) followed by SR (300 μg/kg) and WIN (250 μg/kg). The last experiment evaluated the dose-dependence of WIN-induced effects; animals (n = 8) were given intravenous bolus doses of vehicle (200 μl) followed by WIN at doses of 25, 50, and 75 μg/kg, yielding a cumulative dose of 150 μg/kg. Time-matched controls received saline injections.
Results
Electrically evoked and spontaneous dopamine release in freely moving rats
Electrical stimulation of the MFB with parameters that rats will readily self-administer (Garris et al., 1999) evokes dopamine release in the NAc of a freely moving rat that can be monitored by fast-scan cyclic voltammetry (Garris et al., 1997) (Fig. 1A). A representative recording obtained after adjusting the position of the carbon-fiber electrode for optimal response to dopamine is shown in Figure 1A. Detection of dopamine during and after the stimulation (Fig. 1A, delivered during the red bar above the color plot) is indicated by its oxidation current encoded as a green circle centered at +600 mV, accompanied by its reverse current at –200 mV encoded by the dark blue circle (Michael et al., 1999). A cyclic voltammogram extracted from the data at the end of the stimulation (first vertical line) is identical to that for dopamine in vitro (Fig. 1B). Longer-lasting changes in current with a peak at +225 mV (dark blue) are seen after uptake of dopamine. A cyclic voltammogram extracted at this later time (indicated by the second vertical line) has the characteristics of an alkaline pH shift, characterized previously in anesthetized animals (Fig. 1B) (Venton et al., 2003a). The current at +600 mV (Fig. 1A, x) can be corrected for these ionic changes (Fig. 1A, y) to reveal the time course of dopamine release and uptake.
Figure 1.

Spontaneous and electrically evoked dopamine release in the NAc monitored by cyclic voltammetry. A, The voltammetric current (encoded in color) is plotted against the applied potential (ordinate) and the acquisition time (abscissa). During electrical stimulation of the MFB (biphasic pulses, 0.4 sec duration, 60 Hz, ±120 μA; red bar), dopamine rapidly increased (green spot shows oxidative current). The dark blue area after the green spot is caused by a nondopaminergic (ionic) change. The time courses of the signals observed at different applied potentials (indicated by the dashed white lines) are shown to the right of the color plot: x, current at the peak oxidation potential (0.62 V) for dopamine; y, current at 0.21 V, a potential insufficient to oxidize dopamine but centered on the ionic change. Subtraction of y from x yields a signal that is attributable to dopamine changes; red boxes are times at which electrical stimulation began and ended; asterisk identifies the dopamine transient. B, Cyclic voltammograms extracted from this data set: Evoked DA, the cyclic voltammogram recorded at the end of the stimulation; DA transient, the cyclic voltammogram recorded at the time indicated by the asterisk on the color plot; r2, the correlation coefficient for the comparison of the two voltammograms. The cyclic voltammogram characteristic of the ionic change (at the time of the second vertical dashed bar on the color plot) is shown to the right.
Another dopamine concentration transient is also apparent in this record at the time indicated by the asterisk in Figure 1A. It precedes the electrical stimulation and was not associated with any overt stimulus. The correlation coefficient obtained by comparison of its cyclic voltammogram (Fig. 1B) with that obtained during the electrical stimulation was 0.77, indicating that it is caused by dopamine. This transient had a maximal amplitude of 32 nm, considerably less than the electrically evoked response (521 nm) in this example. Spontaneous dopamine transients such as this were observed in the NAc in nine of nine rats, with an average frequency of 1.4 ± 0.4 min-1.
The cannabinoid agonist WIN55,212–2 enhances dopamine transients but depresses electrically evoked release
Immediately after systemic administration of the cannabinimimetic WIN given at a dose of 125 μg/kg, a short-lived increase in ambulation (4 sec) followed by the onset of a cataleptic state characterized by enhanced muscle tone and rigidity was observed (Sañudo-Peña et al., 2000). Voltammetric responses during the cataleptic phase are shown in Figure 2. The data are from the same animal and recording site as in Figure 1. The number of dopamine transients in the recording interval was enhanced simultaneously with the onset of changes in locomotion (five transients after WIN compared with one before drug for this particular example over the 30 sec shown). The bottom panel shows the cyclic voltammograms obtained after MFB electrical stimulation (solid line) and a cyclic voltammogram corresponding to the WIN-induced dopamine transient (correlation coefficient of 0.93) that occurred immediately before the stimulation (dashed line).
Figure 2.
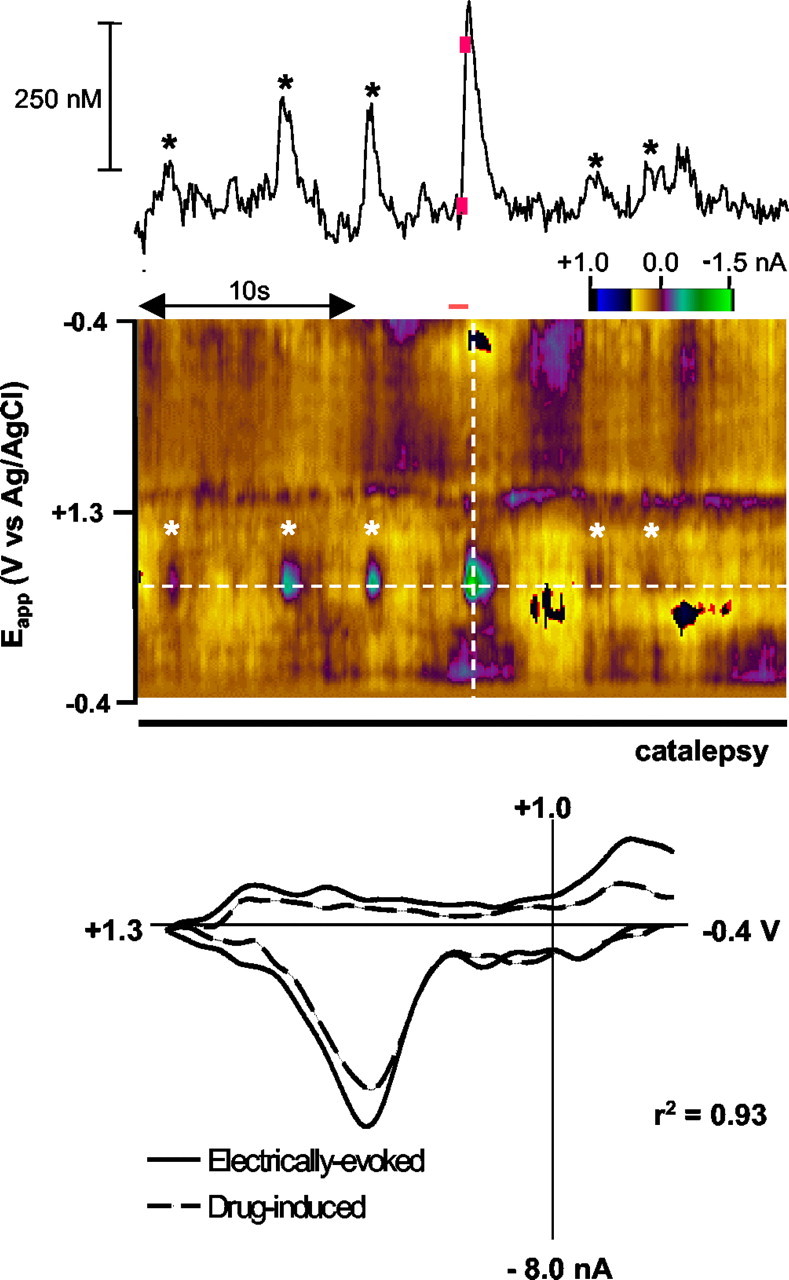
WIN55,212–2 increases the number of dopamine concentration transients in the NAc. Cyclic voltammograms recorded in the same animal and location as in Figure 1 after administration of WIN (125 μg/kg). The trace above the color plot is the current at the potential where dopamine is oxidized, corrected for ionic changes. Asterisks indicate times at which cyclic voltammograms reveal an increase in dopamine concentration. Electrical stimulation of the MFB (biphasic pulses, 0.4 sec duration, 60 Hz, ±120 μA) occurred at the time indicated by the red bar. The cyclic voltammogram corresponding to stimulated release (red bar) in the bottom panel (solid line) exhibits a correlation coefficient of 0.93 when superimposed on the WIN-induced transient (dashed line) occurring immediately before it.
Time course of cannabinoid-induced changes in phasic dopamine neurotransmission
After injection of WIN [WIN first dose (WIN1): 125 μg/kg] and the onset of catalepsy, two events occurred: the number of confirmed dopamine transients increased and remained elevated for at least 3 min after injection, and the evoked release of dopamine was greatly reduced immediately after administration of WIN (WIN1: 125 μg/kg). Before drug administration, electrical stimulations (indicated by black lines) repeated at 1 min intervals evoked a similar maximal dopamine concentration (Fig. 3). In the time between the stimulated responses, noise and some dopamine transients (indicated by the asterisks) are seen. The maximal concentration of stimulated dopamine release was diminished immediately after drug administration, but electrically evoked release of dopamine exhibited a tendency to return to pre-drug values 2 min after injection (Fig. 3).
Figure 3.

Time course of change in dopamine transients after WIN55,212–2. Dopamine concentrations extracted from cyclic voltammograms as in Figure 2 but recorded over a 6 min interval. The rat was administered WIN (125 μg/kg), which induced catalepsy at the time indicated. Dopamine release was evoked with one MFB stimulation per minute (biphasic pulses, 0.4 sec duration, 60 Hz, ±120 μA; black bars). Asterisks between the evoked responses indicate peaks that were identified as dopamine by cyclic voltammetry.
When examined across animals (n = 9), the maximal dopamine evoked by electrical stimulation was significantly decreased from 478 ± 92 nm (pre-drug) to 161 ± 54 nm (WIN1: 125 μg/kg) (F(10,425) = 3.59; p < 0.005). The maximal concentration of DA evoked by electrical stimulation is attributable to the balance between uptake and release (Wightman et al., 1988). Analysis of the disappearance of dopamine after stimulation revealed that the diminished response was not caused by an increase in the rate of uptake. Indeed, the rate of dopamine clearance was unchanged as shown in Figure 4, in which the uptake portions of the average dopamine responses obtained before and after drug administration (WIN1: 125 μg/kg; n = 9 for each condition) are overlaid. Statistical analysis of the slopes of dopamine clearance before and after WIN administration revealed no significant differences (F(60,1302) = 0.32; p = 0.95; interaction time × treatment). Therefore, we conclude that the decreased maximal dopamine evoked by electrical stimulation after WIN is caused by a decrease in the amount released.
Figure 4.

Lack of effect of WIN55,212–2 on dopamine uptake in freely moving rats. The descending portions of the evoked dopamine signals before and after WIN administration (125 μg/kg) are overlaid. Five evoked responses were averaged for each treatment. Data were time-shifted so that the initial dopamine concentration was 0.73 μm in each case (n = 3).
When evaluated in multiple animals, the increase in number of dopamine transients after WIN administration was found to be significant. Overall, the frequency of pre-drug dopamine transients before and after WIN (WIN1: 125 μg/kg) was enhanced from 1.4 ± 0.4 (pre-drug) to 3.87 ± 0.6 min-1 (F(10,503) = 11.99; p < 0.0001). The spontaneous frequency in the absence of drug is greater than we reported previously, but in that work a lower sensitivity approach was used and some transients may have been missed (Robinson et al., 2002). The amplitude distribution of dopamine transients before and after WIN showed that the cannabinoid agonist increased the amplitude of the dopamine transients (Fig. 5). Pre-drug spontaneous transients had a mean amplitude of 51.1 ± 9.2 nm, whereas after WIN (WIN1: 125 μg/kg) they increased to 98.7 ± 22.6 nm (p < 0.01; Student's t test), causing a rightward shift in the distribution. This effect was not observed after injection of vehicle in which the mean amplitude of transients was 47.1 ± 12.2 nm (p = 0.23; Student's t test).
Figure 5.

WIN55,212–2 increases the amplitude of dopamine concentration transients detected in the NAc. Histogram distributions of the amplitude of dopamine transients (WIN1: 125 μg/kg; n = 9). Bin size is 50 nm.
The actions of WIN55,212–2 are mediated through CB1 receptors
When given to rats rendered cataleptic by WIN [WIN second dose (WIN2): 250 μg/kg], SR (300 μg/kg), a CB1 receptor antagonist, reverted the catalepsy and animals regained their righting reflex and resumed exploratory behavior within 10 sec after administration. The antagonist also reversed both the WIN-induced increase in the rate of dopamine transients and the suppression of electrically evoked dopamine release. Administration of SR abolished high-amplitude transients, and mean transient amplitude in the presence of SR was shifted to 60.5 ± 13.1 nm, which was not significantly different from pre-drug (p = 0.09; Student's t test). A representative example from a different rat is shown in Figure 6, in which the example in the third panel was collected immediately after the injection of SR.
Figure 6.

Changes in dopamine neurotransmission after WIN55,212–2 are reversed by SR141716A. Left panel, Spontaneous dopamine transients (asterisk) and dopamine release evoked by electrical stimulation of the MFB (biphasic pulses, 0.4 sec duration, 60 Hz, ±120 μA; black bar). Center panel, Dopamine changes monitored in the 1 min interval after administration of WIN (125 μg/kg). Note that electrically evoked release is reduced by ∼50%. The right trace shows the reversal of these effects by SR (300 μg/kg).
Pooled data from multiple animals administered a series of doses of WIN and SR confirm the significance of these changes. Although administration of vehicle did not change the amplitude of stimulated release ([DA]max) (Fig. 7A) or the number of dopamine transients (Fig. 7B), administration of WIN (WIN1: 125 μg/kg) significantly suppressed [DA]max and increased the number of transients. A subsequent dose (WIN2: 250 μg/kg) did not alter either parameter further. SR reversed both effects of WIN and prevented further changes by a subsequent injection of WIN (WIN2: 250 μg/kg) (Fig. 7A,B).
Figure 7.

Mean responses of dopamine to a cannabinoid agonist and antagonist. A, The maximal dopamine concentration ([DA]max) elicited by electrical stimulation of the MFB is decreased after the first injection of WIN (125 μg/kg; WIN1) but not further decreased by a larger dose (250 μg/kg; WIN2). This effect is reversed and prevented by SR (300 μg/kg; SR; &p < 0.05 compared with WIN2), and a subsequent injection of WIN (250 μg/kg; WIN2) does not affect this (**p < 0.005 compared with vehicle; n = 9 rats). B, The frequency of NAc dopamine concentration transients (n = 9 rats) induced by an initial injection of WIN (125 μg/kg; WIN1) and a subsequent one (250 μg/kg; WIN2). SR (300 μg/kg; SR) reversed the effects of WIN and blocked further effects of WIN (250 μg/kg; WIN2). (***p < 0.001 compared with pre-drug). C, Administration of SR (300 μg/kg; SR) does not affect the frequency of dopamine transients (n = 4 rats) but prevents changes by WIN (250 μg/kg; WIN2). In each panel, vehicle consists of a 1:1:18 ethanol/emulphor/saline (0.9%) ratio. Bolus doses were each spaced by 15 min.
A separate group of rats (n = 4) was given SR after vehicle to test whether transient dopamine release was controlled tonically by endocannabinoids. The antagonist (300 μg/kg) prevented the appearance of catalepsy and blocked the enhancing effects of WIN (WIN2: 250 μg/kg) on the rate of dopamine transients, but had no effects when injected alone as shown in Figure 7C.
WIN55,212–2 dose-dependently modifies behavior and subsecond dopamine release
The dose-dependence of WIN on dopamine release and the onset of the well documented changes in movement that cannabinoids can induce was evaluated (n = 8) (Sañudo-Peña et al., 2000). The frequency of spontaneous dopamine transients increased in a dose-dependent manner (Fig. 8A) and reached statistical significance after the last intravenous infusion of WIN (75 μg/kg) of the dosing protocol (F(3,140) = 3.82; p < 0.05; cumulative dose of 150 μg/kg). Additionally, electrically stimulated dopamine release exhibited a trend to diminish, but this effect did not reach significance (F(3,152) = 1.71; p = 0.16) (Fig. 8B). To allow for drug clearance, the effects on locomotion were evaluated in the same rats 72 hr later. Rats were given the same dosing protocol at the same time of day. Locomotion was exquisitely sensitive to the treatment with WIN (black bars) because even the lowest dose (25 μg/kg) significantly lowered the number of IR beam breaks (134.4 ± 8.9) compared with vehicle (184.1 ± 11.6). Higher doses (50 and 75 μg/kg) further depressed locomotion (F(3,156) = 34.26; p < 0.001) (Fig. 8C). Catalepsy was also dose-dependently enhanced (Fig. 8D, black bars), and this effect reached significance compared with vehicle at the intermediate dose of 50 μg/kg and was further increased with the last dose of 75 μg/kg (F(3,44) = 9.25; p < 0.001). No significant effects on locomotion or catalepsy were observed when saline (Fig. 8C,D, white bars) was administered to drug-naive rats (n = 4) at similar intervals.
Figure 8.

Dose-dependent effects of WIN55,212–2 on dopamine release and behavior. For all panels vehicle = [1:1:18, ethanol/emulphor/saline (0.9%) ratio], 25 = 25 μg/kg, 50 = 50 μg/kg, and 75 = 75 μg/kg (n = 4 rats). Intravenous bolus doses were spaced by 15 min. A, Frequency of dopamine concentration transients in the NAc during a cumulative dosing regimen (*p < 0.05). B, The maximal dopamine concentration ([DA]max) elicited by electrical stimulation of the MFB. The response tends to decrease with the cumulative dosing regimen but was not significant at any dose. C, Locomotor activity after treatment with WIN. Black bars represent WIN-treated animals, whereas time-matched, saline controls are depicted with white bars. (***p < 0.001 compared with vehicle;&&&p < 0.001 compared with time-matched saline control;&&p < 0.005 compared with time-matched saline control). D, Administration of WIN dose-dependently increases catalepsy as measured using the bar test (*p < 0.05, ***p < 0.001 compared with vehicle;&&&p < 0.001 compared with time-matched saline control).
Discussion
This study addresses the role of cannabinoids on rapid dopamine neurotransmission. Although microdialysis studies have shown that cannabinoids increase dopamine over minutes (Chen et al., 1991; Tanda et al., 1997, 1999), the temporal structure of this change is not known. Here we confirm that the potent CB1 receptor agonist, WIN55,212–2, increases extracellular dopamine but reveal that this is manifested as an increase in the frequency and amplitude of rapid dopamine transients in the NAc. These effects are dose-dependent and reversed by the central cannabinoid receptor antagonist, SR141716A, indicating that they are receptor mediated.
The actions of WIN do not appear to involve altered dopamine uptake, as shown by unchanged clearance rates after stimulated release. Indeed, they are unlikely to be mediated in the NAc because cannabinoids applied directly to dopaminergic terminals in NAc brain slices do not affect evoked dopamine release (Szabo et al., 1999); however, cannabinoids do produce an increase in the overall firing rate of dopaminergic neurons in the VTA (French, 1997; French et al., 1997; Cheer et al., 2000a, 2003; Szabo et al., 2002). An augmentation in the burst-firing rate of dopaminergic neurons could increase the amplitude of transient dopamine release because, when neurons shift to a bursting pattern, the resulting action potentials cause greater extrasynaptic levels of transmitter compared with the same number of equally spaced action potentials at a lower frequency (Gonon and Buda, 1985; Gonon, 1988; Wightman et al., 1988). The failure of SR to alter the rate of dopamine transients concurs with electrophysiological findings that show no effect on the endogenous firing of VTA dopaminergic neurons (French, 1997; Cheer et al., 2003). These findings suggest that under basal physiological conditions, endogenous cannabinoid tone is not sufficient to modify neuronal activity in the VTA.
Cannabinoids are known to exert their effects presynaptically, via CB1 receptors located mainly on GABAergic and glutamatergic terminals (Schlicker and Kathmann, 2001; Wilson and Nicoll, 2002). Consistent with previous reports, WIN dose-dependently decreased spontaneous locomotion and produced marked cataleptic effects (Sañudo-Peña et al., 2000; Wiley and Martin, 2003). In this study, rats displayed vocalizations and aggressiveness toward handling when cataleptic (Sañudo-Peña et al., 2000). The dose–response experiment indicates that changes in locomotion precede the observed effects on dopamine release, which were detected when the dosing was sufficient to produce a loss of the righting reflex. Cataleptic doses of cannabinoids are also known to induce conditioned place aversion (Sañudo-Peña et al., 1997; Cheer et al., 2000b). In humans, catatonia, characterized by muscular rigidity accompanied by intense fear, is reported after withdrawal from benzodiazepines (Rosebush and Mazurek, 1995), which is associated with a reduction in GABA tone (Sañudo-Peña et al., 2000). These findings suggest that the cannabinoid-induced cataleptic state, accompanied by dysphoria, arises by diminishing GABAergic function. Presynaptic inhibition of GABA release is consistent with the current findings because dopaminergic neurons receive a strong GABAergic input in the VTA (Ikemoto and Panksepp, 1999; Szabo et al., 2002). In addition, a recent report by Melis et al. (2004) shows that WIN also inhibits excitatory synaptic transmission within the VTA. This finding may explain why cannabinoid agonists can elicit increases as well as decreases (albeit in a smaller population) in the firing rate of extracellularly recorded VTA neurons (Cheer et al., 2003), indicating that the generation of dopamine transients may require reciprocal actions on VTA neurons.
Dopamine transients in the NAc have been detected previously with cyclic voltammetry at carbon-fiber electrodes. They occur spontaneously when an animal is in solitude in the absence of any overt stimuli and without any obvious behavioral response. They become more frequent in response to salient stimuli such as presentation of a conspecific (Robinson et al., 2002). They also occur in response to cues that are associated with cocaine taking (Phillips et al., 2003) or with natural rewards (Roitman et al., 2004), during which they appear to promote reward-seeking. Interestingly, the doses of WIN that elicit dopamine transients have independently been shown to alter cocaine self-administration (Fattore et al., 1999), suggesting that cannabinoids exert their effects on goal-directed behavior by modifying dopamine release on a temporal scale relevant to the coding of discreet behavioral events. The neural basis for these concentration transients has yet to be unraveled, although they have a striking similarity in time course to dopamine release evoked by rapid trains of stimulus pulses that mimic burst firing. Moreover, the dopamine transients arise with regularity similar to that of low bursting activity rates of dopaminergic neurons in freely moving rats (Hyland et al., 2002). Because transients rapidly diminish in concentration as they diffuse from their release site (Venton et al., 2003b), this rapid form of chemical communication would restrict the spatial region for interactions of dopamine with its receptors.
The increase in the frequency and amplitude of dopamine transients in this study was accompanied by a decrease in the concentration of electrically evoked dopamine, demonstrating that the amount of dopamine released per impulse was diminished. Electrically evoked dopamine release was similarly depressed in freely moving rats after doses of ethanol that cause elevated extracellular dopamine as measured by microdialysis (Budygin et al., 2001). There, we proposed that evoked dopamine release was reduced because of increased spontaneous release resulting from the documented increased neuronal activity of dopaminergic neurons caused by ethanol (Gessa et al., 1985). Because of the increased sensitivity of the voltammetric technique used in this work (Heien et al., 2003), we were able to directly measure an increase in spontaneous dopamine transients after cannabinoid administration that may underlie the depression of evoked dopamine. Indeed, the reduction in stimulated dopamine release seen after both of these drugs is consistent with suppression of the amount of dopamine released per impulse after periods of sustained activity (Montague et al., 2004). Assuming that the transients are a result of burst firing, an increase in their amplitude, despite diminished release per impulse, would require a greater number of impulses to be present in each burst. Accordingly, electrophysiological studies in VTA dopaminergic neurons have shown that CB1 receptor agonists do produce such an increase in action potentials per burst (French, 1997; Gessa et al., 1998).
Overall, this study contributes to the growing body of evidence that dopamine transients are a major form of communication in the NAc by this neurotransmitter. Because dopamine release in limbic and cortical structures is considered to play an important role in the rewarding aspects of several drugs of abuse, including marijuana (Gardner, 2002; Tanda and Goldberg, 2003), the current findings provide further insight into the manner in which marijuana exerts its reinforcing effects.
Footnotes
This work was supported by Grant DA10900 from the National Institute on Drug Abuse. We thank the University of North Carolina Electronics Facility for the design and construction of the instrumentation used in this work. Thanks are also due to Dr. Donita Robinson and Garret Stuber for helpful comments regarding this manuscript.
Correspondence should be addressed to Prof. R. Mark Wightman, Department of Chemistry, CB 3290, Venable Hall, The University of North Carolina, Chapel Hill, NC 27599-3290. E-mail: mw@unc.edu.
DOI:10.1523/JNEUROSCI.0529-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244393-08$15.00/0
References
- Budygin EA, Phillips PEM, Robinson DL, Kennedy AP, Gainetdinov RR, Wightman RM (2001) Effect of acute ethanol on striatal dopamine neurotransmission in ambulatory rats. J Pharmacol Exp Ther 297: 27–34. [PubMed] [Google Scholar]
- Chen JP, Paredes W, Lowinson JH, Gardner EL (1991) Strain-specific facilitation of dopamine efflux by delta 9-tetrahydrocannabinol in the nucleus accumbens of rat: an in vivo microdialysis study. Neurosci Lett 129: 136–180. [DOI] [PubMed] [Google Scholar]
- Cheer JF, Marsden CA, Kendall DA, Mason R (2000a) Lack of response suppression follows repeated ventral tegmental cannabinoid administration: an in vitro electrophysiological study. Neuroscience 99: 661–667. [DOI] [PubMed] [Google Scholar]
- Cheer JF, Kendall DA, Marsden CA (2000b) Cannabinoid receptors and reward in the rat: a conditioned place preference study. Psychopharmacology (Berl) 151: 25–30. [DOI] [PubMed] [Google Scholar]
- Cheer JF, Kendall DA, Mason R, Marsden CA (2003) Differential cannabinoid-induced electrophysiological effects in rat ventral tegmentum. Neuropharmacology 44: 633–641. [DOI] [PubMed] [Google Scholar]
- Diana M, Melis M, Gessa GL (1998) Increase in meso-prefrontal dopaminergic activity after stimulation of CB1 receptors by cannabinoids. Eur J Neurosci 10: 2825–2830. [DOI] [PubMed] [Google Scholar]
- Di Chiara G, Imperato A (1988) Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA 85: 5274–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fattore L, Martellotta MC, Cossu G, Mascia MS, Fratta W (1999) CB1 cannabinoid receptor agonist WIN 55,212–2 decreases intravenous cocaine self-administration in rats. Behav Brain Res 104: 141–146. [DOI] [PubMed] [Google Scholar]
- French ED (1997) delta9-Tetrahydrocannabinol excites rat VTA dopamine neurons through activation of cannabinoid CB1 but not opioid receptors. Neurosci Lett 226: 159–162. [DOI] [PubMed] [Google Scholar]
- French ED, Dillon K, Wu X (1997) Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. NeuroReport 8: 649–652. [DOI] [PubMed] [Google Scholar]
- Freund TF, Katona I, Piomelli D (2003) Role of endogenous cannabinoids in synaptic signaling. Physiol Rev 83: 1017–1066. [DOI] [PubMed] [Google Scholar]
- Gardner EL (2002) Addictive potential of cannabinoids: the underlying neurobiology. Chem Phys Lipids 121: 267–290. [DOI] [PubMed] [Google Scholar]
- Garris PA, Christensen JRC, Rebec GV, Wightman RM (1997) Real-time measurement of electrically evoked extracellular dopamine in the striatum of freely moving rats. J Neurochem 68: 152–161. [DOI] [PubMed] [Google Scholar]
- Garris PA, Kilpatrick M, Bunin MA, Michael D, Walker QD, Wightman RM (1999) Dissociation of dopamine release in the nucleus accumbens from intracranial self-stimulation. Nature 398: 67–69. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Muntoni F, Collu M, Vargiu L, Mereu G (1985) Low doses of ethanol activate dopaminergic neurons in the ventral tegmental area. Brain Res 348: 201–203. [DOI] [PubMed] [Google Scholar]
- Gessa GL, Melis M, Muntoni AL, Diana M (1998) Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. Eur J Pharmacol 341: 39–44. [DOI] [PubMed] [Google Scholar]
- Gonon FG (1988) Nonlinear relationship between impulse flow and dopamine released by rat midbrain dopaminergic neurons as studied by in vivo electrochemistry. Neuroscience 24: 19–28. [DOI] [PubMed] [Google Scholar]
- Gonon FG, Buda MJ (1985) Regulation of dopamine release by impulse flow and by autoreceptors as studied by in vivo voltammetry in the rat striatum. Neuroscience 14: 765–774. [DOI] [PubMed] [Google Scholar]
- Heien MLAV, Phillips PEM, Stuber GD, Seipel AT, Wightman RM (2003) Overoxidation of carbon-fiber microelectrodes enhances dopamine adsorption and increases sensitivity. Analyst 128: 1413–1419. [DOI] [PubMed] [Google Scholar]
- Hernàndez M, Berrendero F, Suàrez I, García-Gil L, Cebeira M, Mackie K, Ramos JA, Fernàndez-Ruiz J (2000) Cannabinoid CB1 receptors colocalize with tyrosine hydroxylase in cultured fetal mesencephalic neurons and their activation increases the levels of this enzyme. Brain Res 857: 57–65. [DOI] [PubMed] [Google Scholar]
- Hyland BI, Reynolds JNJ, Hay J, Perk CG, Miller R (2002) Firing modes of midbrain dopamine cells in the freely moving rat. Neuroscience 114: 475–492. [DOI] [PubMed] [Google Scholar]
- Ikemoto S, Panksepp J (1999) The role of nucleus accumbens dopamine in motivated behavior: a unifying interpretation with special reference to reward-seeking. Brain Res Brain Res Rev 31: 6–41. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Gaoni Y (1967) The absolute configuration of delta-9-tetrahydrocannabinol, the major active constituent of hashish. Tetrahedron Lett 12: 1109–1111. [DOI] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL (2004) Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci 24: 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael DJ, Joseph JD, Kilpatrick MR, Travis ER, Wightman RM (1999) Improving data acquisition for fast-scan cyclic voltammetry. Anal Chem 71: 3941–3947. [DOI] [PubMed] [Google Scholar]
- Montague PR, McClure SM, Baldwin PR, Phillips PEM, Budygin EA, Stuber GD, Kilpatrick MR, Wightman RM (2004) Dynamic gain control of dopamine delivery in freely moving animals. J Neurosci 24: 1754–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ (2002) From neurobiology to treatment: progress against addiction. Nat Neurosci 5: 1076–1079. [DOI] [PubMed] [Google Scholar]
- Overton PG, Clark D (1997) Burst firing in midbrain dopaminergic neurons. Brain Res Brain Res Rev 3: 312–334. [DOI] [PubMed] [Google Scholar]
- Phillips PEM, Wightman RM (2003) Critical guidelines for validation of the selectivity of in-vivo chemical microsensors. Trends Anal Chem 22: 509–514. [Google Scholar]
- Phillips PEM, Stuber GD, Heien MLAV, Wightman RM, Carelli RM (2003) Subsecond dopamine release promotes cocaine seeking. Nature 22: 614–618. [DOI] [PubMed] [Google Scholar]
- Robinson DL, Heien MLAV, Wightman RM (2002) Frequency of dopamine concentration transients increases in dorsal and ventral striatum of male rats during introduction of conspecifics. J Neurosci 2: 10477–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roitman MF, Stuber GD, Phillips PEM, Wightman RM, Carelli RM (2004) Dopamine operates as a subsecond modulator of food seeking. J Neurosci 24: 1265–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosebush PI, Mazurek MF (1995) Complicating factors in the analysis of acute drug-induced akathisia. Arch Gen Psychiatry 52: 878–880. [DOI] [PubMed] [Google Scholar]
- Sañudo-Peña MC, Tsou K, Delay ER, Hohman AG, Force M, Walker JM (1997) Endogenous cannabinoids as an aversive or counter-rewarding system in the rat. Neurosci Lett 223: 125–128. [DOI] [PubMed] [Google Scholar]
- Sañudo-Peña MC, Romero J, Seale GE, Fernandez-Ruiz JJ, Walker JM (2000) Activational role of cannabinoids on movement. Eur J Pharmacol 391: 269–274. [DOI] [PubMed] [Google Scholar]
- Schlicker E, Kathmann M (2001) Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol Sci 22: 565–572. [DOI] [PubMed] [Google Scholar]
- Schultz W (1998) Predictive reward signal of dopamine neurons. J Neurophysiol 1: 1–27. [DOI] [PubMed] [Google Scholar]
- Szabo B, Muller T, Koch H (1999) Effects of cannabinoids on dopamine release in the corpus striatum and the nucleus accumbens in vitro. J Neurochem 73: 1084–1089. [DOI] [PubMed] [Google Scholar]
- Szabo B, Siemes S, Wallmichrath I (2002) Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. Eur J Neurosci 15: 2057–2061. [DOI] [PubMed] [Google Scholar]
- Tanda G, Pontieri FE, Di Chiara G (1997) Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 276: 2048–2050. [DOI] [PubMed] [Google Scholar]
- Tanda G, Loddo P, Di Chiara G (1999) Dependence of mesolimbic dopamine transmission on delta9-tetrahydrocannabinol. Eur J Pharmacol 376: 23–26. [DOI] [PubMed] [Google Scholar]
- Tanda G, Goldberg SR (2003) Cannabinoids: reward, dependence, and underlying neurochemical mechanisms—a review of recent preclinical data. Psychopharmacology (Berl) 169: 115–134. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Michael DJ, Wightman RM (2003a) Correlation of local changes in extracellular oxygen and pH that accompany dopaminergic terminal activity in the rat caudate-putamen. J Neurochem 84: 373–381. [DOI] [PubMed] [Google Scholar]
- Venton BJ, Zhang H, Garris PA, Phillips PEM, Sulzer D, Wightman RM (2003b) Real-time decoding of dopamine concentration changes in the caudateputamen during tonic and phasic firing. J Neurochem 87: 1284–1295. [DOI] [PubMed] [Google Scholar]
- Wightman RM, Amatore C, Engstrom RC, Hale PD, Kristensen EW, Kuhr WG, May LJ (1988) Real-time characterization of dopamine overflow and uptake in the rat striatum. Neuroscience 25: 513–523. [DOI] [PubMed] [Google Scholar]
- Wiley JL, Martin BR (2003) Cannabinoid pharmacological properties common to other centrally acting drugs. Eur J Pharmacol 471: 185–193. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA (2002) Endocannabinoid signaling in the brain. Science 296: 678–682. [DOI] [PubMed] [Google Scholar]
- Wise RA (2002) Brain reward circuitry: insights from unsensed incentives. Neuron 36: 229–240. [DOI] [PubMed] [Google Scholar]
- Wu X, French ED (2000) Effects of chronic delta9-tetrahydrocannabinol on rat midbrain dopamine neurons: an electrophysiological assessment. Neuropharmacology 39: 391–398. [DOI] [PubMed] [Google Scholar]