

Abstract
Ischemic injury to the CNS results in loss of ionic homeostasis and the development of neuronal death. An increase in intracellular Ca2+ is well established, but there are few studies of changes in intracellular Cl– ([Cl–]i) after ischemia. We used an in vitro model of cerebral ischemia (oxygen–glucose deprivation) to examine changes in [Cl–]i and GABAA receptor-mediated responses in hippocampal slices from adult rats. Changes in [Cl–]i were measured in area CA1 pyramidal neurons using optical imaging of 6-methoxy-N-ethylquinolinium chloride, a Cl–-sensitive fluorescent indicator. Oxygen–glucose deprivation induced an immediate rise in [Cl–]i, which recovered within 20 min. A second and more prolonged rise in [Cl–]i occurred within the next hour, during which postsynaptic field potentials failed to recover. The sustained increase in [Cl–]i was not blocked by GABAA receptor antagonists. However, oxygen–glucose deprivation caused a progressive downregulation of the K+–Cl– cotransporter (KCC2), which may have contributed to the Cl– accumulation. The rise in [Cl–]i was accompanied by an inability of the GABAA agonist muscimol to cause Cl– influx. In vivo, diazepam is neuroprotective when given early after ischemia, although the mechanism by which this occurs is not well understood. Here, we added diazepam early after oxygen–glucose deprivation and prevented the downregulation of KCC2 and the accumulation of [Cl–]i. Consequently, both GABAA responses and synaptic transmission within the hippocampus were restored. Thus, after oxygen–glucose deprivation, diazepam may decrease neuronal excitability, thereby reducing the energy demands of the neuron. This may prevent the activation of downstream cell death mechanisms and restore Cl– homeostasis and neuronal function.
Keywords: ischemia, GABAA receptor, MEQ, KCC2, Cl–-ATPase, benzodiazepines
Introduction
Transient cerebral ischemia induces a cascade of cellular events that eventually produces neuronal death, especially in vulnerable areas such as the hippocampus. One of the first consequences of oxygen and glucose deprivation during cerebral ischemia is membrane depolarization, the rapid depletion of cellular ATP, and the accumulation of extracellular excitatory amino acids. During these events, there is a progressive increase in extracellular K+ and an increase in intracellular Na+, Cl–, and Ca2+, accompanied by suppression of neuronal activity and synaptic transmission (for review, see Martin et al., 1994; Kristian and Siesjo, 1998; Lipton, 1999).
The accumulation of Na+ and Ca2+ during ischemia or oxygen–glucose deprivation (OGD), an in vitro model for transient global ischemia, has been investigated extensively. Recently, more attention has been focused on changes in intracellular Cl– and its role in ischemia-induced neuronal death. Previously, we demonstrated in hippocampal slices from immature rats that OGD induced an immediate increase in intracellular Cl– (Inglefield and Schwartz-Bloom, 1998a). Similarly, a short anoxic episode increased Cl– in rat hypoglossal neurons (Jiang et al., 1992). In both studies, the changes in intracellular Cl– were reversible, and there was complete recovery of electrophysiological parameters measured during reoxygenation. In contrast, if anoxia is prolonged or if adult hippocampal slices are subjected to OGD under conditions that model cerebral ischemia, in vivo, the loss of synaptic transmission may become irreversible (Zhu and Krnjević, 1999). Furthermore, the ionic imbalance in Na+, K+, Ca2+, and Cl– worsens during reoxygenation, causing irreversible damage to area CA1 neurons (Taylor et al., 1999; LoPachin et al., 2001).
One of the major consequences of a rise in intracellular Cl– is that GABAA responses, which are normally hyperpolarizing, can become depolarizing (Thompson and Gahwiler, 1989; Kaila, 1994). A loss of GABAA-mediated inhibition may contribute to neuronal hyperexcitability observed in the hippocampus during the first 12 hr of reperfusion (Urban et al., 1989; Gao et al., 1999), leading to neuronal damage. However, enhancement of GABA neurotransmission with benzodiazepines after transient cerebral ischemia prevents neuronal death in the adult hippocampus (Schwartz-Bloom et al., 1998; for review, see Schwartz-Bloom and Sah, 2001). The narrow therapeutic window for benzodiazepines suggests that there is a critical time after ischemia when enhancement of GABA neurotransmission is beneficial to neurons.
Previously, we have used the adult hippocampal slice preparation to study the effects of diazepam on biochemical consequences of OGD. When hippocampal slices were exposed to diazepam after OGD, there was complete recovery of ATP, which is required for neuronal viability (Galeffi et al., 2000). In this study, we investigated changes in intracellular Cl– with a Cl–-sensitive indicator 6-methoxy-N-ethylquinolinium chloride (MEQ) in living brain slices (Inglefield and Schwartz-Bloom, 1997) during OGD and up to 2 hr after the onset of reoxygenation in the adult hippocampus. Second, we determined whether GABAA receptormediated Cl– influx is compromised during reoxygenation. Finally, we determined whether diazepam can restore any OGD-induced changes in intracellular Cl–, GABAA receptor activity, and synaptic activity in area CA1 hippocampal neurons, independent of sedation, hypothermia, or change in blood flow.
Materials and Methods
Hippocampal slice preparation and OGD. Adult male Sprague Dawley rats (250–300 gm) were used in these experiments. The rats were decapitated, and each brain was removed from the skull rapidly and placed in ice-cold physiological buffer oxygenated with a 95% O2/5% CO2 mixture for 1 min. The solution consisted of (in mm) 119 NaCl, 2.5 KCl, 1.0 Na H2PO4, 1.3 MgCl2, 1.5 CaCl2, 26 NaHCO3, and 11 glucose, pH 7.4. When low Cl– (10 mm) solution was used, Cl– was replaced with either sulfate or gluconate. Coronal slices (300 μm) were prepared from the hippocampal region of the brain with a vibratome. The slices were placed in an incubation chamber and submerged ∼1 mm in buffer that was supplemented with 1 mm kynurenic acid and 400 μm ascorbic acid, for 60–90 min at 32°C. After incubation, the slices were transferred to a chamber that contained oxygenated buffer (without kynurenic and ascorbic acid) and kept at 36°C before additional experimental manipulation.
OGD was produced by exposing the slices to a d-glucose-deficient buffer equilibrated with a 95% N2/5% CO2 gas mixture for 7 min at 36°C. This period of OGD was chosen because it produced mild histological damage without rapid necrosis (see Results and Fig. 1) and it caused incomplete recovery of ATP (Galeffi et al., 2000). In some experiments, OGD was performed after slices were loaded with 6-methoxy-N-ethyl-1,2-dihydroquinoline (dihydro-MEQ; see below); in other experiments, OGD was performed before slices were loaded with dihydro-MEQ, as noted in the figure legends. After OGD, slices were superfused or placed in an oxygenated buffer containing glucose (“reoxygenation”) for up to 2.5 hr, as noted. In certain experiments, diazepam (5 μm) was included in the reoxygenation buffer immediately after OGD; it was removed 1 hr later, before any optical imaging. Diazepam at 5 μm is a maximally effective concentration at restoring ATP after OGD in adult brain slices (Galeffi et al., 2000).
Figure 1.
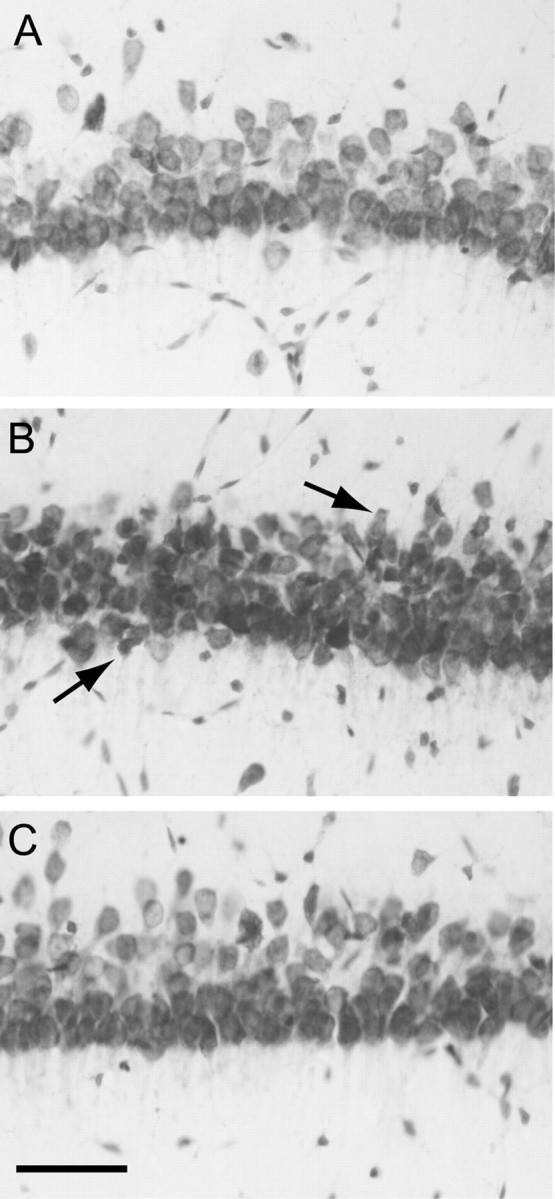
Morphology of area CA1b pyramidal neurons in adult hippocampal slices subjected to OGD (7 min) and reoxygenated for 3 hr. Sections (30 μm) were prepared from hippocampal slices and stained with cresyl violet as described in Materials and Methods. A, control. B, OGD. Note the mild shrinkage of neurons, indicated by arrows (C), OGD plus diazepam (5 μm, added after OGD). Scale bar, 50 μm.
Determination of area CA1 field responses. For the electrophysiology experiments, hippocampal slices were transferred to a chamber superfused with oxygenated standard buffer (36°C). After 1 hr of superfusion, the Schaffer collateral–commissural pathway was stimulated with a bipolar electrode situated in the stratum radiatum at the CA2–CA3 border. The stimulation consisted of rectangular stimulus pulses of 0.1 msec duration at 30 sec intervals at 0.04 Hz (single stimulus, 0.1–1.5 mA). Extracellular potentials were recorded every 30 sec with glass micropipettes filled with 200 mm NaCl (4–8 MΩ) positioned in the medial portion of the stratum pyramidale of area CA1. Slices were incubated for 30–40 min to obtain stable population spike amplitude. OGD was induced by switching from oxygenated buffer to glucose-free buffer equilibrated with 95% N2/5% CO2. After 7 min of OGD, the slices were reoxygenated with standard buffer for 60 min. In some experiments, the slices were exposed to 5 μm diazepam for either 15 min before OGD or immediately after OGD. In both cases, diazepam was removed from the superfusion buffer 30 min after OGD. Throughout the experiment, electrophysiological recordings were amplified (100×), digitized at 5 kHz, and stored for offline analysis of population spike amplitude with pClamp (Axon Instruments, Union City, CA). The percentage of population spike recovery was calculated as the population spike amplitude 1 hr after OGD divided by the population spike amplitude before OGD times 100.
Preparation of dihydro-MEQ. MEQ (Molecular Probes, Eugene, OR) was reduced to its cell-permeable derivative dihydro-MEQ before bath-loading slices, as described previously by our laboratory (Inglefield and Schwartz-Bloom, 1997; Sah et al., 2002). Briefly, reduction of MEQ to dihydro-MEQ was performed by the gradual addition of sodium borohydride (12% in 0.1 mm NaOH; 32 μm) to an aqueous solution of MEQ (16 μm in 0.1 mm NaOH) under nitrogen for 30 min (note: we have determined that NaOH is required to completely reduce the MEQ currently available from Molecular Probes). Formation of the dihydro derivative was indicated by the appearance of a reddish-yellow oil. The oil was extracted twice with ethyl acetate (0.5 ml). The organic extracts were combined and dried with anhydrous MgSO4, and the ethyl acetate was evaporated under N2 in a glass microvial. Reduced dye was stored at –80°C under N2, and it was used the same day for optimal loading of brain slices.
Measurement of MEQ, calcein acetoxymethyl ester, and Calcium Green-1 acetoxymethyl ester fluorescence. Adult slices were bath-loaded with dihydro-MEQ using a method modified from that used for neonatal and juvenile rats (Inglefield and Schwartz-Bloom, 1997). For the adult slices, it was necessary to use a two-step loading approach for dihydro-MEQ to improve cell penetration (Sah et al., 2002). During the first step, the slices were incubated in oxygenated standard buffer (at 32°C) that contained 4 mm dihydro-MEQ resuspended in ethyl acetate (0.5%) for 2 min. During the second step, the slices were incubated in oxygenated buffer (32°C) containing 400 μm dihydro-MEQ resuspended in DMSO (0.05%) for 30 min. After incubation with dihydro-MEQ, the slices were washed in oxygenated buffer for 15 min before being transferred to the imaging chamber.
The use of UV laser scanning confocal microscopy to measure Cl–-sensitive changes in MEQ fluorescence in hippocampal slices has been described previously by our laboratory (Inglefield and Schwartz-Bloom, 1997). After loading with MEQ, slices were submerged in a chamber that was superfused with oxygenated buffer (flow rate, ∼2 ml/min). The temperature in the imaging chamber was maintained at 32°C for all the experiments, except as noted. For all experiments, slices were allowed to equilibrate in the chamber for at least 5–10 min before the baseline-recording period. The imaging chamber was positioned on the stage of an upright Optiphot microscope (Nikon, Melville, NY). The laser scanning confocal microscope (Noran Odyssey; Noran Instruments, Middleton, WI) was equipped with an argon ion UV laser (50 mW output, Enterprise 653; Coherent, Carlsbad, CA) and a digital imaging system (MetaMorph; Universal Imaging, Downingtown, PA) for image and data acquisition. MEQ-loaded neurons of the hippocampus were imaged in area CA1 by excitation with the 364 nm line of the UV laser. Fluorescent light was transmitted through a UV water-immersible objective (40× numerical aperture; Olympus, Melville, NY). Emission (Emax = 440 nm) was imaged with use of a 400 nm barrier filter. Photomultipliers detected the fluorescent signal through a confocal slit at 1× electronic zoom. The video frame rates (16 images per second) of the Odyssey confocal microscope (Noran Instruments) allowed rapid, full image (512 × 480 pixels) acquisition. To minimize photobleaching of the dye, repeated long-term illumination by the UV laser was avoided. Images were 8-bit (256 intensity levels) and saved for offline analysis. Before any experimental manipulation, a minimum of three images over a 5 min period was recorded and then averaged to determine the baseline fluorescence.
To determine changes in cell volume after OGD, slices were bath-loaded with calcein acetoxymethyl ester (AM; Molecular Probes) (Crowe et al., 1995; Inglefield and Schwartz-Bloom, 1998a). This indicator is insensitive to changes in intracellular ions. Slices were incubated with 5 μm calcein AM at 36°C for 30 min, and subsequently, they were washed for 10 min. The intracellular calcein fluorescence was detected by exciting at 488 nm and using a 515 nm barrier filter. A change in fluorescence intensity was expressed as (ΔF/F) × 100, as described for MEQ. A decrease in calcein fluorescence indicates an increase in cell volume associated with cell swelling.
Changes in intracellular Ca2+ after OGD were measured using Calcium Green-1 AM (Molecular Probes). We used this indicator because it can be bath-loaded into adult hippocampal slices and provide fluorescent images in individual cell bodies. To achieve good penetration into the adult slice, a two-step loading procedure was used (Smetters et al., 1999). Slices were incubated with 2 mm Calcium Green-1 AM for 2 min, followed by 10 μm for 45 min at 32°C. Slices were transferred to an imaging chamber (36°C) before OGD. Calcium Green-1 fluorescence was detected with the 488 nm excitation line and a 515 nm barrier filter. Binding of Ca2+ produces an increase in Calcium Green-1 fluorescence and reflects an increase in intracellular Ca2+; absolute concentrations of Ca2+ are not calculated because the indicator is non-ratioable (Zhang and Lipton, 1999). A change in fluorescence intensity was expressed as (ΔF/F) × 100 using values corrected for photobleaching using a curve-fitting program and linear regression (Prism; Graph Pad, San Diego, CA).
Data analysis for MEQ fluorescence imaging. Because MEQ fluorescence is quenched collisionally by Cl–, increases in intracellular Cl– correspond directly with a decrease in fluorescence. MEQ is not a ratiometric indicator, therefore absolute intracellular concentrations of Cl– were not measured. (However, changes in MEQ fluorescence can be calibrated to estimate changes in intracellular Cl– using the Stern–Volmer equation; see below.) The change in intracellular Cl– was expressed as the percentage change in MEQ fluorescence of single cells above the baseline, for a given frame, calculated by the equation (ΔF/F) × 100. Because there was a slow decrease of MEQ fluorescence in control slices (15.5 ± 1.6% over 10 min), the percentage change in MEQ fluorescence for any experimental condition was compared with the percentage change in control slices. At least three rats were used to collect data for all experiments.
Only those cells that had <10% change in MEQ fluorescence during the initial baseline recording period (5 min) and an initial fluorescence intensity between 90 and 200 optical density units were used for data analysis. Data were subjected to two-way ANOVAs, and when the interactions were significant, groups of means were compared using Tukey's post hoc analysis. A probability of <0.05 was considered statistically significant.
Intracellular calibration of MEQ fluorescence. Changes in MEQ fluorescence can be calibrated to estimate changes in intracellular [Cl–] using the Stern–Volmer relationship, F0/FCl = 1 + Kq[Cl–], where F0 is the total quenchable signal, FCl is the fluorescence in the presence of a given Cl– concentration, and Kq is the Stern–Volmer quench constant (in m–1) (Verkman, 1990). We have calibrated changes in MEQ fluorescence with intracellular Cl– under the experimental conditions of the present study (adult hippocampal slices at 32°C). A detailed description of the methods is provided by Inglefield and Schwartz-Bloom (1997). In this study, the Stern–Volmer constant (the slope, Kq in m–1) was calculated to be 44.3 ± 1.12 in m–1. The K–1 q, or 22 mm, is the Cl– concentration that quenches the fluorescence by 50%. The equation can be used to estimate the change in intracellular Cl– over a defined recording period.
Measurement of NKCC-1 and KCC2 protein expression. Hippocampi subjected to OGD (in the absence or presence of diazepam) were dissected from acute slices in ice-cold isolation buffer (in mm: 250 sucrose, 10 Tris, 10 HEPES, and 1 EDTA, pH 7.2 at 24°C). Three hippocampi were combined and homogenized with a glass Teflon homogenizer in ice-cold isolation buffer containing a protease inhibitor mixture (1:200; Sigma, St. Louis, MO). Tissue samples were prepared as in Yan et al. (2001). Tissue was lysed with 30 sec of sonication, and cellular debris was removed by centrifugation at 420 × g for 5 min at 4°C. The resulting supernatant was collected and frozen at –80°C for later use. For immunoblotting, the protein content of samples was determined using the Protein Assay kit (Bio-Rad, Hercules, CA), and samples were denatured in SDS reducing buffer (Lammli's buffer) for 30 min at 37°C before gel electrophoresis. Equal amounts of protein samples were loaded onto 4–12% Tris-glycine gradient gels (Invitrogen, Carlsbad, CA). Proteins were separated by SDS-PAGE and transferred to polyvinyl difluoride membrane (Millipore, Billerica, MA). After two rinses with water, membranes to be blotted for NKCC-1 were incubated in blocking solution (7.5% milk in TBS with 0.1% Tween 20) overnight at 4°C and incubated with T4 mouse monoclonal antibody for NKCC-1 (1:1000; Hybridoma Bank, University of Iowa, Iowa City, IA) and mouse anti-βIII tubulin antibody (1:3000; Promega, Madison, WI) for 2 hr at 24°C. For KCC2 immunoblotting, membranes were incubated in blocking solution (as above) for 1 hr at 24°C, followed by an incubation with rabbit polyclonal antibody against KCC2 (1:2000; Upstate Technologies, Waltham, MA) and mouse monoclonal antibody for βIII tubulin (1:3000) overnight at 4°C. Membranes were exposed to HRP-linked secondaries (anti-mouse antibody for NKCC-1/βIII tubulin immunoblots, anti-mouse and antirabbit antibodies for KCC-2/βIII tubulin immunoblots; Amersham Biosciences, Piscataway, NJ) for 1 hr at 24°C. Protein bands were detected by ECL on film. Films were scanned, and data were digitized using NIH Image and expressed as calibrated optical density units times area (normalized).
Measurement of Cl–-ATPase activity. Cl–-ATPase activity was measured in membrane fractions obtained from hippocampus homogenates or in membrane fractions prepared from hippocampal slices exposed to OGD (7 min at 37°C), as described by Kitagawa et al. (2001). Hippocampal slices subjected to OGD 2 hr earlier were homogenized with ice-cold buffer containing 0.25 m sucrose, 1 mm EDTA-Tris, pH 7.4, 12.5 mm MES, pH 7.4, and protease inhibitors. The homogenate was centrifuged (1000 × g for 10 min), and the supernatant was centrifuged first at 10,000 × g for 15 min and then at 100,000 × g for 20 min. The resulting pellet was suspended in 5 mm EDTA-Tris, pH 7.4, and centrifuged again (100,000 × g for 20 min). The final pellet was suspended in 5 mm EDTA-Tris, pH 7.4, and used as the plasma membrane-rich fraction. ATPase activity was determined by fluorimetric measurement of inorganic phosphate released, using the Pi Per Phosphate Assay kit (Molecular Probes). Fourteen to 20 μg of protein were assayed for 15 min at 36°C in 200 μl reaction buffer containing (in mm) 100 Tris-MES, pH 7.4, 1 EDTA-Tris, 100 NaCl, 10 KCl, 6 Mg acetate, and 6 ATP-Tris, pH 7.4, in the presence or the absence of 1 mm ouabain (to inhibit the Na+/K+-ATPase) or 0.3 mm ethacrynic acid (Sigma) (to inhibit the Cl–-ATPase). Trichloroacetic acid (10%) was added to terminate the reaction. The activity in the presence of 1 mm ouabain or 0.3 mm ethacrynic acid was subtracted from the total ATPase activity to determine the activities of Na+/K+-ATPase and Cl–-ATPase, respectively.
General histology and cytochrome c immunohistochemistry. In parallel to the imaging and the electrophysiology experiments, slices were collected 2 hr after reoxygenation–glucose and fixed in phosphate-buffered 4% paraformaldehyde, pH 7.4, overnight. The fixed slices were embedded in gelatin/albumin as described by Okazaki et al. (1995). Immediately after embedding, the slices were cut into 30 μm sections with a vibrotome and either stained with cresyl violet for histological observation or subjected to cytochrome c immunohistochemistry. For the cytochrome c immunohistochemistry, the sections were rinsed in PBS and incubated in two stages in solutions containing 0.2% Triton-X, 2% BSA, and 10% NGS (Vector Laboratories, Burlingame, CA) for 30 min each. The sections were incubated with an anti-cytochrome c monoclonal primary antibody (PharMingen, San Diego, CA) at a dilution of 1:500 overnight at 4°C. The slices were washed in PBS and incubated with a biotinylated secondary anti-mouse antibody (Vector Laboratories) at a dilution of 1:500 for 2 hr at room temperature. Visualization of cytochrome c-immunopositive cells was achieved with an avidin–biotin reagent (ABC kit; Vector Laboratories), followed by DAB staining.
Results
Typically, hippocampal slices used in imaging studies are prepared from neonatal or juvenile rats. Neurons in slices from neonatal or juvenile rats are loaded easily with membrane-permeable fluorescent dyes, and they are more resistant to slicing-induced trauma. Here, we used a set of conditions (see Materials and Methods) to prepare hippocampal slices from adult rats with a well preserved cellular morphology of the CA1 region for at least 6 hr after slicing (Fig. 1A), allowing us to perform both electrophysiology and optical imaging experiments.
Effect of OGD on area CA1 morphology and evoked CA1 field responses
When hippocampal slices were subjected to OGD for 7 min, they displayed consistent abnormalities in area CA1 neuronal shape 2–3 hr into reoxygenation. Some neurons appeared shrunken or pyknotic, but massive cell necrosis was not observed (Fig. 1B). When diazepam (5 μm) was added to the reoxygenation buffer after OGD, the morphology of area CA1 neurons was similar to that in control slices 3 hr after reoxygenation (Fig. 1C). Because the relatively mild morphological changes attributable to OGD are not quantifiable, we examined adjacent slices for the presence of cytosolic cytochrome c immunoreactivity, an early marker of cell damage. Two hours after OGD, the number of cytochrome c-immunopositive cells in area CA1 was significantly increased (Fig. 2), similar to results after cerebral ischemia in vivo (Sugawara et al., 1999). Diazepam, included in the reoxygenation buffer, completely prevented the OGD-induced increase in cytochrome c-immunopositive cells.
Figure 2.

Cytochrome c immunostaining in hippocampal area CA1 2 hr after reoxygenation. Sections (30 μm) were prepared from hippocampal slices and subjected to cytochrome c immunohistochemistry as described in Materials and Methods. A, Control slice. B, Slice subjected to OGD. SP, Stratum pyramidale. C, Quantitiation of immunopositive cells in area CA1 of the hippocampus 2 hr after OGD. Diazepam (5 μm) was added immediately after OGD. Data are the means ± SEM number of immunopositive cells counted in area CA1. *p < 0.01 versus control; ANOVA, followed by Tukey's multiple comparison test (n = 6–10 slices). Scale bar, 25 μm.
This OGD model was used to mimic changes that occur after transient global cerebral ischemia in vivo; ischemia triggers irreversible functional damage to the hippocampus, yet morphological signs of cell death are delayed in area CA1. As a parameter for assessing permanent functional damage, we measured the recovery of synaptic transmission after OGD in area CA1 stratum pyramidale. We recorded orthodromic population spikes in area CA1 stratum pyramidale, induced by stimulation of the Schaffer collateral–commissural pathway. After obtaining a stable population spike amplitude, OGD was performed for 7 min (see Materials and Methods). During OGD, the population spike amplitude decreased rapidly, and within 1.5–2 min, it was essentially eliminated (Fig. 3A). The presynaptic fiber volley was also eliminated by 6 min, indicating that energy-dependent ion gradients had collapsed and the membranes depolarized completely. The presynaptic fiber volley recovered within 10 min of reoxygenation in buffer containing glucose (reflecting conduction of action potentials along the Schaffer collaterals). However, the postsynaptic population spike failed to recover by 60 min of reoxygenation in eight of nine slices (Fig. 3). In one slice, 30% recovery was observed. Interestingly, the population spikes in four of nine slices recovered temporarily (∼25% of baseline) during the initial 20 min after OGD (data not shown), possibly reflecting an attempt to recover the membrane potential.
Figure 3.

Effect of OGD on the evoked population spike in area CA1 pyramidal cells. A, Representative individual recordings before, during, and 60 min after 7 min OGD in a hippocampal slice from an adult rat. Postsynaptic population spikes were recorded from area CA1 pyramidal cells after stimulation of the Schaffer collaterals. During OGD, the postsynaptic response was abolished. The failure of the postsynaptic population spike to recover indicates irreversible damage. The presynaptic fiber volley is indicated with an asterisk. B, Data summary. The baseline values consisted of the average of 10 consecutive recordings before OGD, and the 60 min values consisted of the average of five consecutive recordings. The percentage recovery was calculated as the 60 min values/baseline values × 100. Data are the mean ± SEM of nine slices. *p < 0.001 versus baseline; ANOVA and Tukey's multiple comparison test.
Effect of OGD on MEQ fluorescence
Using a two-step bath-loading procedure for MEQ (see Materials and Methods), adult hippocampal slices exhibited numerous fluorescent neurons with uniform dye distribution in area CA1 stratum pyramidale (Fig. 4A). To assess changes in intracellular Cl– during and immediately after OGD, hippocampal slices were loaded with MEQ and subjected to OGD for 7 min. OGD decreased MEQ fluorescence (within ∼5 min) in pyramidal neurons in area CA1, indicating an increase in intracellular Cl– (Fig. 4). The increase in intracellular Cl– was maximal 10 min after the onset of OGD, consistent with the nonspecific increase in membrane permeability that occurs as membranes become severely depolarized. Subsequently, the intracellular Cl– recovered toward control levels by ∼25 min into reoxygenation–glucose. However, the recovery was not permanent. When MEQ-loaded slices were imaged at different times during reoxygenation, a secondary rise in intracellular Cl– was evident. By 1 hr after reoxygenation, there was a significant decrease in MEQ fluorescence compared with control slices (ΔF/F = 44.11 ± 2.7% vs 14.7 ± 1.5%, OGD vs control) (Fig. 5A). Over the next 1.5 hr, the decrease in MEQ fluorescence persisted. To rule out the possibility that the second wave of decreased MEQ fluorescence resulted from dye leakage out of injured cells or cell swelling during reoxygenation, hippocampal slices were perfused with a buffer containing 10 mm Cl–. In the presence of the low Cl– buffer, the decrease in MEQ fluorescence 1 hr after reoxygenation was prevented completely (Fig. 5B). The same results were found at 2 hr after reoxygenation (data not shown), suggesting that the decrease in MEQ fluorescence 1 and 2 hr after OGD was attributable to an accumulation in intracellular Cl–. We also measured changes in calcein fluorescence, which is sensitive to changes in cell volume (Crowe et al., 1995; Inglefield and Schwartz-Bloom, 1998a). The calcein ΔF/F at 2 hr after OGD did not differ from control slices (Fig. 5C), indicating that cell swelling was not a factor in the decrease in MEQ fluorescence. At 2 hr of reoxygenation, the OGD-induced change in intracellular Cl– over the 10 min recording period could be estimated to be 5.7 mm, according to the Stern–Volmer relationship. Therefore, during the second wave (∼90 min) of a decrease in MEQ fluorescence, up to 54 mm Cl– may have accumulated in the imaged neurons.
Figure 4.

Effect of OGD on MEQ fluorescence (intracellular Cl–) in are a CA1b pyramidal cells. A, B, Confocal video images of MEQ fluorescence in adult hippocampal neurons in area CA1b stratum pyramidale. Special conditions were required to bath-load adult slices successfully with dihydro-MEQ (see Materials and Methods). MEQ fluorescence before (A) and after (B) OGD, at the onset of reoxygenation. C, Continuous recording of the change in MEQ fluorescence within individual CA1 pyramidal neurons before, during, and immediately after OGD. The temperature was maintained at 36°C. The bar indicates the 7 min OGD period. The original ΔF/F values were corrected for baseline by subtracting extrapolated values from the baseline drift of each cell (before OGD) to give the ΔF/F values. D, The peak change in MEQ fluorescence 10 min after the onset of OGD was compared with the fluorescence change in control slices not subjected to OGD over a 10 min recording period. Values are expressed as the percentage decrease in fluorescence during 10 min from the baseline [(ΔF/F) × 100]. Data are the means ± SEM of three to seven cells per condition, respectively. *p < 0.01 versus control; unpaired Student's t test.
Figure 5.

OGD causes a secondary rise in intracellular Cl– that is independent of cell swelling. A, Slices were subjected to OGD, loaded with dihydro-MEQ, and imaged for 10 min at various times during reoxygenation. Values are expressed as the percentage decrease in fluorescence during 10 min from baseline [(ΔF/F) × 100]. Data are the means ± SEM of 29, 9, 6, and 21 cells per condition, respectively. *p < 0.001, **p < 0.01 versus control. B, Slices were superfused with a low-Cl–-containing buffer before imaging at 1 hr after OGD. Data are the means ± SEM of 29, 11, 9, and 4 cells percondition, respectively. *p < 0.001 versus control. C, Slices were imaged at 2 hr after OGD for changes in calcein fluorescence under identical conditions as for MEQ. Values are expressed as the percentage decrease in calcein fluorescence during 10 min from baseline [(ΔF/F) × 100]. Data are the means ± SEM of 14 and 16 cells per condition, respectively. p > 0.05 versus control, NS. All statistical procedures included ANOVA and Tukey's multiple comparison test.
To determine a possible mechanism(s) for the secondary rise in intracellular Cl–, we superfused the GABAA receptor antagonist bicuculline and GABA-gated Cl– channel blocker picrotoxin before imaging slices 2 hr after OGD. Both antagonists failed to reduce significantly the increase in intracellular Cl–, indicating that the accumulation of Cl– was not attributable to an increase in GABAA receptor activity at this time (Fig. 6).
Figure 6.

GABAA antagonists do not prevent Cl– accumulation after OGD. Bicuculline (30 μm) or picrotoxin (100 μm) was added to the superfusate during the imaging, 2 hr after reoxygenation. Values are expressed as the percentage decrease in MEQ fluorescence during 10 min from the baseline [(ΔF/F) × 100]. Data are the means ± SEM of 29, 19, 8, and 11 cells per condition, respectively. *p < 0.001, **p < 0.05 versus control; ANOVA and Tukey's multiple comparison test.
Effect of OGD on NKCC-1 and KCC2 protein levels
To address other possible mechanisms for the sustained increase in intracellular Cl–, we focused on the major Cl– transporter systems known to be present in neurons. The Na+–K+–2Cl– cotransporter (NKCC-1) facilitates Na+, K+, and Cl– influx into neurons and glial cells. The K+ and Cl– cotransporter (KCC2) is neuronal specific and normally transports K+ and Cl– out of the neurons to maintain a low intracellular Cl–. To determine whether these cotransporters might have a role in the OGD-induced increase in intracellular Cl–, we investigated the effect of OGD on NKCC-1 and KCC2 protein levels. [A more direct approach using Cl– imaging and the Cl– transport inhibitors bumetanide and furosemide is confounded by the interference of these inhibitors with UV-activated indicators (i.e., MEQ). This precluded us from examining their direct effects on OGD-induced Cl– accumulation.] NKCC-1 protein levels in the hippocampus were unchanged up to 2 hr after reoxygenation (Fig. 7A). Interestingly, KCC2 protein levels were significantly lower 1 hr (by 30%) after OGD and had declined even further (by 70%) 2 hr after reoxygenation (Fig. 7B). Thus, the downregulation of this important Cl– extrusion mechanism (KCC2) may contribute to Cl– accumulation after OGD.
Figure 7.
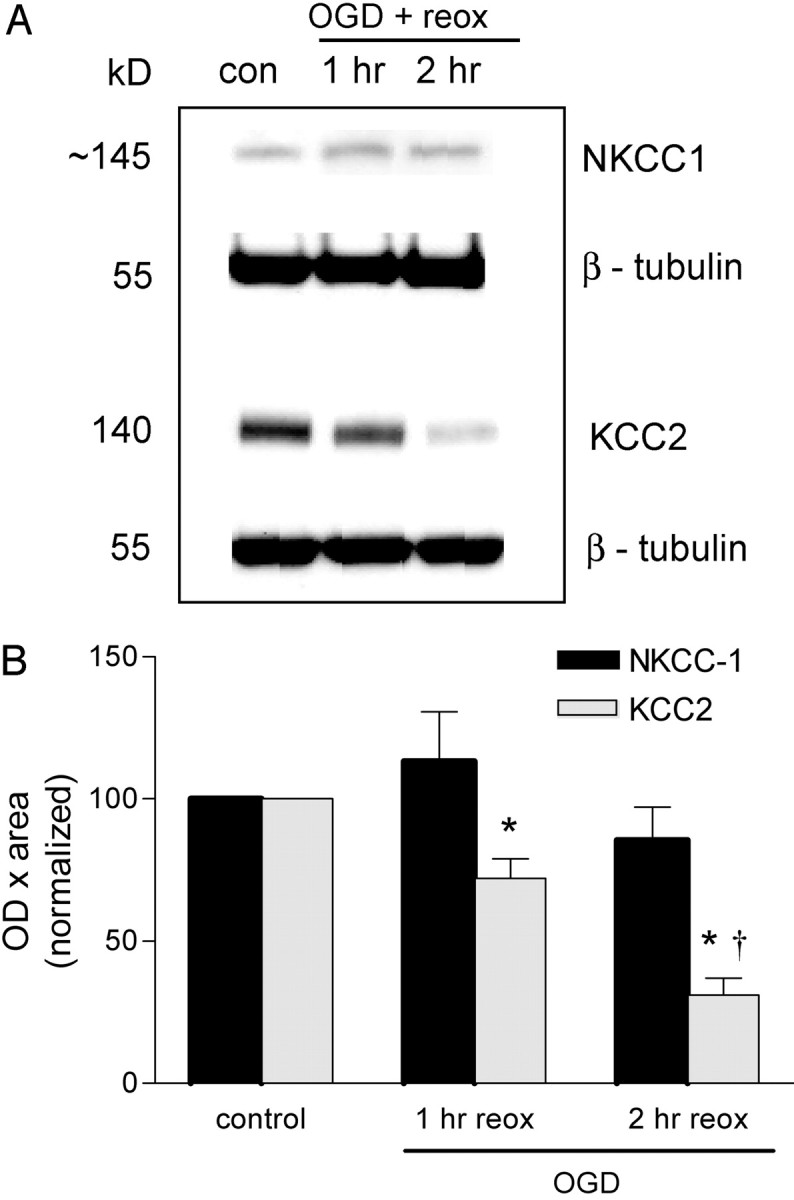
Effect of OGD on NKCC-1 and KCC2 cotransporter expression in the hippocampus. The hippocampus was dissected from hippocampal slices subjected to OGD and reoxygenated (plus glucose) for 1–2 hr. Samples were prepared for immunoblotting as described in Materials and Methods. A, Representative immunoblots for NKCC1 and KCC2 expression at 1 and 2 hr after OGD. B, Quantitated data from all experiments. Data are the means ± SEM of OD × area digitized normalized to 100. *p < 0.05 versus control; †p < 0.001 versus OGD plus 1 hr of reoxygenation (reox); ANOVA and Tukey's multiple comparison test (n = 4–10 slices for NKCC1 and n = 6–18 slices for KCC2).
Effect of OGD on Cl–-ATPase activity
Another mechanism to help maintain the Cl– transmembrane gradient is the Cl–-ATPase, a plasma membrane ion pump that extrudes Cl– from neurons (Inagaki et al., 1985; Shiroya et al., 1989). Because there is incomplete recovery of ATP on reoxygenation (Galeffi et al., 2000), we determined whether the accumulation of intracellular Cl– after OGD was attributable to a reduction in Cl–-ATPase activity. Hippocampal slices were subjected to 7 min OGD and after 2 hr of reoxygenation–glucose, Cl–-ATPase activity was measured. Surprisingly, in the slices subjected to OGD, there was a 52% increase in Cl–-ATPase activity compared with control slices (Table 1). A significant increase (20%) in Na+/K+-ATPase activity also occurred 2 hr after reoxygenation.
Table 1.
Effect of OGD on ATPase activity in hippocampal slices
|
Treatment |
ATPase activity (Pi μmol/mg protein/hr) |
||
|---|---|---|---|
|
|
Cl- ATPase |
Na+/K+-ATPase |
|
| Control | 7.49 ± 1.59 | 44.24 ± 6.67 | |
| OGD + 2 hr reoxygenation |
20.02 ± 3.77* |
55.33 ± 5.61* |
|
Data are the means ± SEM of six experiments. *p < 0.05 versus control; paired t test.
Effect of OGD on GABAA receptor activity
An accumulation of intracellular Cl– reduces the inward Cl– gradient, and this should attenuate the influx of Cl– in response to GABAA receptor activation. We tested this possibility with use of the GABAA receptor agonist muscimol. In control slices, muscimol (50 μm) exposure reduced MEQ fluorescence significantly, indicating an increase in intracellular Cl– (Table 2). In slices subjected to OGD, muscimol was added to the superfusate 2 hr after reoxygenation. At this time, muscimol not only failed to decrease MEQ fluorescence in the slices subjected to OGD, but it slightly enhanced MEQ fluorescence. The reversed direction of the change in MEQ fluorescence indicates possible efflux of Cl– on opening of the GABA-gated Cl– channels by muscimol (but see Discussion for additional explanations).
Table 2.
Effect of OGD and diazepam on the ability of muscimol to increase intracellular Cl– in area CA1 pyramidal neurons
|
Condition |
Muscimol response (% of control ΔF/Fa) |
|---|---|
| Standard | |
| Controlb | 68 ± 8.6 |
| OGD + 2 hr reoxygenation | -7.3 ± 5.9* |
| Diazepam | |
| Controlb | 42 ± 11.8 |
| OGD + 2 hr reoxygenation |
56 ± 11.1 |
Slices were superfused with muscimol (50 μm) for 10 min at 2 hr after OGD. In these experiments, slices were loaded with dihydro-MEQ 1 hr after OGD. In the diazepam condition, slices were superfused with diazepam (5 μm) for 1 hr during reoxygenation and imaged 1 hr later (no diazepam present). The muscimol response was calculated by expressing the (ΔF/Fmuscimol overΔF/Fcontrol) - 1 × 100. Data are the means ± SEM of 16-31 cells per condition. *p < 0.05 versus corresponding control; ANOVA and Tukey's multiple comparison test.
This control refers to the absence of muscimol.
This control refers to the absence of OGD.
Effect of diazepam on OGD-induced changes in intracellular Cl– and GABAA receptor activity
Benzodiazepines such as diazepam are neuroprotective when administered after an ischemic insult in vitro and in vivo (for review, see Schwartz-Bloom and Sah, 2001). Therefore, we determined whether the OGD-induced increase in intracellular Cl– occurring during reoxygenation could be prevented by diazepam. Diazepam (5 μm) was added to the reoxygenation buffer immediately after OGD. One hour later, diazepam was removed, and the slices were bath-loaded with dihydro-MEQ. Two hour after reoxygenation, slices were subjected to optical imaging as described above (no diazepam was present). At this time, the second rise in intracellular Cl– was prevented completely by the previous addition of diazepam (MEQ ΔF/F = 10.3 ± 1.1 vs 6.9 ± 1.1, for control vs OGD, respectively; NS, ANOVA and Tukey's multiple comparison test). In addition, diazepam restored the ability of muscimol to induce Cl– influx in area CA1 2 hr after OGD (Table 2).
Effect of diazepam on OGD-induced KCC2 downregulation
Because the rise in intracellular Cl– was prevented by diazepam, we determined whether the downregulation of KCC2 could also be sensitive to the presence of diazepam. Both the rise in intracellular Cl– and the downregulation of KCC2 occurred within the first hour after OGD. Therefore, we exposed slices to diazepam (5 μm) immediately after OGD. Diazepam prevented the downregulation of KCC2 protein levels significantly 2 hour after OGD (Fig. 8). By maintaining normal levels of KCC2 during reoxygenation, accumulation of intracellular Cl– may have been prevented.
Figure 8.

Diazepam attenuates OGD-induced downregulation of KCC2 protein expression. A, Immunodetection of KKC2 protein expression in hippocampal slices. Slices were exposed to diazepam (dz) during reoxygenation and collected 2 hr after OGD. B, Quantification of the immunoblots by densitometry, as described in Materials and Methods. Data are the means ± SEM of 8, 14, and 20 slices per condition, expressed as normalized OD × area. *p < 0.001 versus control; †p < 0.01 versus OGD; ANOVA and Tukey's multiple comparison test.
Effect of diazepam on OGD-induced rise in intracellular Ca2+
Using Calcium Green-1, we found that OGD induced an early rise in intracellular Ca2+ in individual area CA1 hippocampal cells. The rise in intracellular Ca2+ is transient because the Ca2+ becomes buffered or sequestered intracellularly (Fig. 9). The rise in intracellular Ca2+ is a key factor in OGD-induced neuronal damage and irreversible synaptic failure; preventing the rise limits the extent of membrane dysfunction (Rader and Lanthorn, 1989; Yamamoto et al., 1997). Therefore, we superfused the slices with diazepam (5 μm) during OGD, which reduced the rise in intracellular Ca2+ by 62% (Fig. 9).
Figure 9.

Effect of diazepam on OGD-induced changes in intracellular Ca2+. Slices were loaded with Calcium Green-1 AM and subjected to OGD for 7 min. A, Video image of area CA1 pyramidal cells shows Calcium Green-1 fluorescence during baseline recording. B, An increase in fluorescence is indicated after OGD. C, Continuous recording of Calcium Green-1 fluorescence before, during, and after OGD in representative slices. In these experiments, diazepam (5 μm) was added to the superfusion buffer at the onset of OGD to affect the immediate rise in intracellular Ca2+. D, The peak increase in intracellular Ca2+ occurred ∼10 min after the onset of OGD. Values are expressed as the percentage increase in fluorescence intensity [(ΔF/F) × 100] of Calcium Green-1 above baseline. Data are the means ± SEM of six and five cells per condition, respectively. *p < 0.01 versus OGD; unpaired Student's t test.
Effect of diazepam on evoked synaptic activity after OGD
Maintenance of intracellular Cl– homeostasis is crucial for preserving synaptic transmission. Loss of synaptic transmission occurs after OGD, and its recovery is a useful end point to assess the efficacy of neuroprotective agents. Two types of experiments were performed with diazepam. In the first set of experiments, diazepam (5 μm) was added 15 min before OGD and removed 30 min later. Before OGD, diazepam produced a small decrease (7%) in the amplitude of the population spike (p < 0.05, Student's t test), consistent with its ability to enhance GABA inhibition (Fig. 10A,B). During OGD, the population spikes were suppressed within 1–1.5 min, as observed in the absence of diazepam. In the diazepam-treated slices, the evoked population spikes started to recover within 8–12 min after OGD, and they recovered completely by 60 min of reoxygenation. In the non-diazepam-treated slices, no recovery was evident at the corresponding time point. In the second set of experiments, we determined whether diazepam added immediately after OGD (for 30 min) could also improve the recovery of the population spikes. When diazepam was added after OGD, the evoked population spikes started to recover within 10–12 min after OGD (Fig. 10C,D). By 60 min of reoxygenation, the population spikes recovered ∼77% (p > 0.05 vs baseline).
Figure 10.
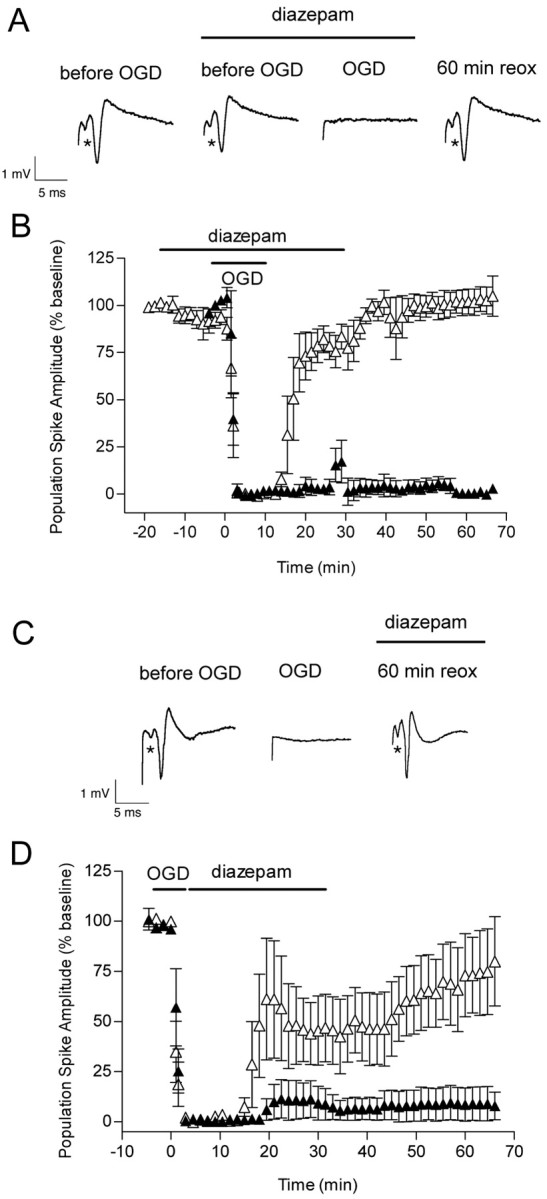
Effect of OGD and diazepam (applied before and after OGD) on the evoked population spike in area CA1 pyramidal cells. A, Diazepam (5 μm) was added 15 min before OGD, and it was removed from the slice chamber 30 min after OGD. Representative traces are shown before, during, and 60 min after OGD in the presence of diazepam. The first trace was recorded before the addition of diazepam. B, The time course is shown for changes in the population spike amplitude recorded from pyramidal CA1 neurons in the absence (▴) and in the presence (▵) of diazepam in control and OGD slices. Note the small decrease in population spike amplitude (p < 0.05) starting at –12 min because of the presence of diazepam. To reduce crowding of data points, each data point is the average of three consecutive recordings that were made every 30 sec. Data are the means ± SEM of four slices per condition. C, Diazepam (5 μm) was added immediately after OGD, and it was removed from the slice chamber 30 min later. Individual recordings are shown for a control hippocampal slice before, during, and 60 min after 7 min OGD plus diazepam. D, The time course is shown for changes in the population spike amplitude recorded from pyramidal CA1 neurons in the absence (▴) and in the presence (▵) of diazepam in control and OGD slices. Each data point is the average of three consecutive recordings that were made every 30 sec. Data are the means ± SEM of five slices per condition. The recovery of the population spike amplitude by 60 min of reoxygenation was not different from baseline (p > 0.05; ANOVA and Tukey's multiple comparison test).
Discussion
In this study, we have used optical imaging techniques to demonstrate that OGD increases intracellular Cl– in two phases in area CA1b neurons from the adult hippocampus. The secondary increase in intracellular Cl– during reoxygenation could be attributable, in part, to dysfunctional or downregulated Cl– extrusion mechanisms. We also found that diazepam was able to (1) prevent the accumulation of intracellular Cl–, (2) restore GABA-mediated Cl– influx, (3) prevent the downregulation of KCC2, and (4) prevent the rise in intracellular Ca2+, thereby restoring synaptic transmission within area CA1 of the hippocampus.
OGD effects on intracellular Cl– and synaptic transmission
Under the conditions used here, neurons were probably severely depolarized during OGD because the cells did not respond to synaptic stimulation and the presynaptic volley had completely disappeared (Zhu and Krnjević, 1999; Obeidat et al., 2000). At the same time, OGD induced an immediate rise in intracellular Cl–. Similarly, others have shown that when neurons lose their membrane potential and undergo spreading depression-like depolarization, extracellular Cl– decreases and intracellular Cl– increases (Phillips and Nicholson 1979; Jiang et al.,1992), along with a general increase in membrane permeability (Hansen 1985; Tanaka et al., 1997). This can occur during hypoxia or OGD (Somjen et al. 1992; Taylor and Weber, 1993; Obeidat et al., 2000). The source of the increase in intracellular Cl– during OGD cannot be established definitively because as the membrane potential approaches zero, an inwardly directed electrochemical potential is imposed on Cl–, driving Cl– into the cell through whatever conductance is available. Because extracellular K+ rises simultaneously, it is possible that NKCC1 becomes activated and KCC2 reverses to contribute to the rise in intracellular Cl– (Payne et al., 2003).
If OGD persists for more than a few minutes after the onset of anoxic depolarization, synaptic transmission will remain compromised, causing irreversible damage to neurons (Balestrino et al. 1989; Rader and Lanthorn, 1989). Taylor et al. (1999) showed that both synaptic transmission and a rise in intracellular Cl– (assessed with x-ray electron analysis) failed to recover by 30 min after OGD in the adult hippocampal slice. However, Wang et al. (1999) reported that the ionic distribution had returned to pre-hypoxia levels on reoxygenation, although 70% of the neurons remained depolarized and unresponsive to synaptic stimulation. In the present study, Cl– levels returned toward baseline by ∼20 min after reoxygenation; this recovery was most likely associated with the attempt of the neurons to restore their membrane potential (Rader and Lanthorn, 1989) and ionic distribution during the early phase of reoxygenation. This recovery was followed by a secondary rise in intracellular Cl– sometime within the next hour, when area CA1 field potentials failed to recover.
The secondary rise in intracellular Cl– appears to be unrelated to activation of GABA-gated Cl– channels. The GABAA receptor antagonists bicuculline and picrotoxin did not alter the Cl– influx 2 hr into reoxygenation. We ruled out the possibility that the decrease in MEQ fluorescence after OGD was because of an increase in dye leakage or to cell swelling. The decrease in MEQ fluorescence was prevented in a low Cl– buffer, and there was no change in calcein fluorescence (i.e., cell volume) at 2 hr of reoxygenation. There are other possible explanations for the secondary rise in intracellular Cl–. First, because there is synaptic failure during reoxygenation, neurons most likely are still depolarized, driving Cl– into the cell. Second, Cl– cotransporters or dysfunctional Cl– extrusion mechanisms may have contributed to the rise in intracellular Cl–. Although NKCC-1 levels did not change during reoxygenation, NKCC-1 can be activated by elevated extracellular K+ and glutamate, bringing Na+, K+, and Cl– into the neuron (Su et al., 2000; Schomberg et al., 2001). Interference of Cl– cotransporter inhibitors with MEQ precluded us from testing this idea directly. However, Beck et al. (2003) found that Cl– cotransporter inhibitors do inhibit Cl– influx and cell death after glutamate-mediated excitotoxicity and OGD in cultured cortical neurons. In addition, inhibition of NKCC1 is neuroprotective after anoxia or OGD (Müller, 2000; Pond et al., 2004) and in rats after focal ischemia (Yan et al., 2001). In the case of Cl– extrusion mechanisms, the KCC2 and the Cl–-ATPase help to maintain the Cl– gradient in adult neurons (Inagaki et al., 1985; Shiroya et al., 1989; Rivera et al., 1999); they extrude Cl– in response to GABAA receptor-activated Cl– channel opening. KCC2 is regulated in a variety of ways after different types of brain insults. Under conditions that mimic ischemia/OGD, KCC2 can reverse in response to small increases in extracellular K+, bringing K+ and Cl– into the neuron (Payne, 1997; DeFazio et al., 2000). In addition, several investigators have found that KCC2 is downregulated after various types of neuronal injury (for review, see Payne et al., 2003). Low levels of KCC2 are associated with a raised intracellular Cl–, producing depolarizing GABA responses (Rivera et al., 1999). Here, under conditions that promote Cl– influx after OGD, the substantial decrease in KCC2 levels may compromise the ability of hippocampal neurons to restore the Cl– gradient.
There is considerable evidence that ATPase activity is compromised during and after cerebral ischemia (for review, see Lees, 1991). However, we found that both Cl–-ATPase and Na+/K+-ATPase activities were increased during reoxygenation. It is very likely that increased ATPase activity is triggered by the increased requirement for Cl– and Na+ extrusion from an accumulating intracellular pool. Our results support the findings of others who show that the Na+/K+-ATPase is increased after cerebral ischemia, when intracellular Na+ is elevated (Lees, 1991). In the present study, the rate of Cl– accumulation (perhaps because of KCC2 downregulation) may have exceeded the ability of an activated Cl–-ATPase to extrude Cl–.
OGD and GABAA receptor activity
Two hours into reoxygenation, during the secondary rise in intracellular Cl–, the GABA agonist muscimol failed to decrease MEQ fluorescence. It is unlikely that the decrease in MEQ fluorescence was already maximal after OGD, thereby preventing any additional decrease in MEQ fluorescence by muscimol; other compounds such as NMDA produce even greater decreases in MEQ fluorescence than reported here (Inglefield and Schwartz-Bloom, 1998b). Other investigators have reported that GABAA-mediated inhibition is suppressed immediately after anoxia in the hippocampal slice, because of a loss of excitatory input onto GABA-ergic interneurons (Krnjević et al., 1991; Khazipov et al., 1993). In contrast, Katchman et al. (1994) proposed that the loss of GABAA-mediated inhibition immediately after anoxia was attributable to a decrease in extracellular Cl–.
Several mechanisms may underlie the failure of muscimol to induce Cl– influx via the GABAA receptor observed 2 hr after OGD. First, it is possible that the accumulation of intracellular Cl– during reoxygenation moves the equilibrium potential for Cl– (ECl) closer to the resting membrane potential; this would reduce or eliminate a net Cl– influx after GABAA receptor activation by muscimol. With the accumulation of intracellular Cl–, it is likely that muscimol may have induced Cl– efflux through the GABAA receptor. However, changes in GABAA receptor sensitivity by Ca2+ and reactive oxygen species may also decrease the ability of muscimol to induce Cl– influx (for review, see Schwartz-Bloom and Sah, 2001) after OGD.
OGD and the effect of diazepam
Several investigators have demonstrated that benzodiazepines and barbiturates can restore synaptic transmission and membrane potential after anoxia (Abramowicz et al., 1991; Kass et al., 1992; Wang et al., 1999). In agreement with these studies, we have shown that diazepam can reduce the rise in intracellular Ca2+ and prevents the irreversible loss of synaptic transmission in hippocampal slices after OGD. Moreover, diazepam prevents the downregulation of KCC2 and the accumulation of intracellular Cl– during reoxygenation. Interestingly, the downregulation of KCC2 may be regulated indirectly by Ca2+-induced signaling events (for review, see Payne et al., 2003).
Based on our findings, we suggest that diazepam can reduce neuronal excitability if it is given early during reoxygenation. [In vivo, if diazepam administration is delayed >2 hr after ischemia, its neuroprotective ability is lost (Schwartz-Bloom et al., 1998)]. At this time, diazepam could still promote neuronal inhibition even as intracellular Cl– starts to rise. When the membrane depolarization places the resting membrane potential above the positive shift in the ECl, GABAA receptor activation will still cause neuronal hyperpolarization (DeFazio et al., 2000). Second, even under conditions when ECl does become more positive relative to the resting membrane potential, diazepam could shunt depolarizing currents evoked by excitatory input via a decrease in membrane resistance (Staley and Mody, 1992). In addition, diazepam could enhance an inward GABA current to provide a major extrusion path for Cl–. By reducing neuronal excitability early after OGD, diazepam reduces the energy demands of the cell. This may prevent several downstream signaling events that lead to cell death, including the downregulation of KCC2 (Rivera et al., 2002), which is important in maintaining Cl– homeostasis and GABA inhibition (Payne et al., 2003).
Footnotes
This work was supported by National Institutes of Health Grant NS28791 (R.D.S.-B.) and American Heart Association Grant 0020264U (F.G.). We thank Dr. George Somjen for critical reading of this manuscript.
Correspondence should be addressed to Dr. Rochelle D. Schwartz-Bloom, Department of Pharmacology and Cancer Biology, Duke University Medical Center, Box 3813, Durham, NC 27710. E-mail: schwa001@duke.edu.
R.Sah's present address: Pharmacology Research Center, Children's Hospital Medical Center, ML 7025, Cincinnati, OH 45229.
DOI:10.1523/JNEUROSCI.0755-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244478-11$15.00/0
References
- Abramowicz EA, Kass IS, Chambers G, Cottrell E (1991) Midazolam improves electrophysiologic recovery after anoxia and reduces changes in ATP levels and calcium influx during anoxia in the rat hippocampal slice. Anesthesiology 74: 1121–1123. [DOI] [PubMed] [Google Scholar]
- Balestrino M, Aitken PG, Somjen GG (1989) Spreading depression-like depolarization in CA1 and fascia dentata of hippocampal slices: relationship to selective vulnerability. Brain Res 497: 102–107. [DOI] [PubMed] [Google Scholar]
- Beck J, Lenart B, Kintner DB, Sun D (2003) Na–K–Cl cotransporter contributes to glutamate-mediated excitotoxicity. J Neurosci 23: 5061–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe WE, Altamirano J, Huerto L, Alvarez-Leefmans FG (1995) Volume changes in single NIE-115 neuroblastoma cells measured with a fluorescent probe. Neuroscience 69: 283–296. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ (2000) Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J Neurosci 20: 8069–8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeffi F, Sinnar S, Schwartz-Bloom RD (2000) Diazepam promotes ATP recovery and prevents cytochrome c release in hippocampal slices after in vitro ischemia. J Neurochem 75: 1242–1249. [DOI] [PubMed] [Google Scholar]
- Gao TM, Pulsinelli WA, Xu ZC (1999) Changes in membrane properties of CA1 pyramidal neurons after transient forebrain ischemia in vivo Neuroscience 90: 771–780. [DOI] [PubMed] [Google Scholar]
- Hansen AJ (1985) Effect of anoxia on ion distribution in the brain. Physiol Rev 65: 101–148. [DOI] [PubMed] [Google Scholar]
- Inagaki C, Tanaka T, Hara M, Ishiko J (1985) Novel microsomal anion-sensitive Mg2+-ATPase activity in rat brain. Biochem Pharmacol 34: 1705–1712. [DOI] [PubMed] [Google Scholar]
- Inglefield JR, Schwartz-Bloom RD (1997) Confocal imaging of intracellular chloride in living brain slices: measurement of GABAA receptor activity. J Neurosci Methods 75: 127–135. [DOI] [PubMed] [Google Scholar]
- Inglefield JR, Schwartz-Bloom RD (1998a) Optical imaging of hippocampal neurons with a chloride-sensitive dye: early effects of in vitro ischemia. J Neurochem 70: 2500–2509. [DOI] [PubMed] [Google Scholar]
- Inglefield JR, Schwartz-Bloom RD (1998b) Activation of excitatory amino acid receptors in the rat hippocampal slice increases intracellular Cl– and cell volume. J Neurochem 71: 1396–1404. [DOI] [PubMed] [Google Scholar]
- Jiang C, Agulian S, Haddad GG (1992) Cl– and Na+ homeostasis during anoxia in rat hypoglossal neurons: intracellular and extracellular in vitro studies. J Physiol (Lond) 448: 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K (1994) Ionic basis of GABAA receptor channel function in the nervous system. Prog Neurobiol 42: 489–537. [DOI] [PubMed] [Google Scholar]
- Kass IS, Abramowicz EA, Cottrell J, Chambers G (1992) The barbiturate thiopental reduces ATP levels during anoxia but improves electrophysiological recovery and ionic homeostasis in the rat hippocampal slice. Neuroscience 49: 537–543. [DOI] [PubMed] [Google Scholar]
- Katchman AN, Vicini S, Hershkowitz N (1994) Mechanism of early anoxia-induced suppression of the GABAA-mediated inhibitory postynaptic current. J Neurophysiol 71: 1128–1138. [DOI] [PubMed] [Google Scholar]
- Khazipov R, Bregestovski P, Ben-Ari Y (1993) Hippocampal inhibitory interneurons are functionally disconnected from excitatory inputs by anoxia. J Neurophysiol 70: 2251–2259. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Yagyu K, Yamamoto A, Hattori N, Omori K, Zeng XT, Inagaki C (2001) Molecular cloning and characterization of the Cl– pump-associated 55-Kda protein in rat brain. Biochem Biophys Res Commun 289: 363–371. [DOI] [PubMed] [Google Scholar]
- Kristian T, Siesjo BK (1998) Calcium in ischemic cell death. Stroke 29: 705–718. [DOI] [PubMed] [Google Scholar]
- Krnjević K, Xu YZ, Zhang L (1991) Anoxic block of GABAergic IPSPs. Neurochem Res 16: 279–284. [DOI] [PubMed] [Google Scholar]
- Lees GJ (1991) Inhibition of sodium-potassium-ATP-ase: a potentially ubiquitous mechanism contributing to central nervous system neuropathology. Brain Res Rev 16: 283–300. [DOI] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1432–1538. [DOI] [PubMed] [Google Scholar]
- LoPachin RM, Gaughan CL, Lehning J, Weber ML, Taylor CP (2001) Effects of ion channel blockade on the distribution of Na, K, Ca and other elements in oxygen-glucose deprived CA1 hippocampal neurons. Neuroscience 103: 971–983. [DOI] [PubMed] [Google Scholar]
- Martin RL, Lloyd HGH, Cowan AI (1994) The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends Neurosci 1 7: 251–257. [DOI] [PubMed] [Google Scholar]
- Müller M (2000) Effects of chloride transport inhibition and chloride substitution on neuron function and on hypoxic spreading-depression-like depolarization in rat hippocampal slices. Neuroscience 97: 33–45. [DOI] [PubMed] [Google Scholar]
- Obeidat AS, Jarvis CR, Andrew RD (2000) Glutamate does not mediate acute neuronal damage after spreading depression induced by O2/glucose deprivation in the hippocampal slice. J Cereb Blood Flow Metab 20: 412–422. [DOI] [PubMed] [Google Scholar]
- Okazaki MM, Evenson DA, Nadler VJ (1995) Hippocampal mossy fiber sprouting and synapse formation after status epilepticus in rat: visualization after retrograde transport with biocytin. J Comp Neurol 352: 515–534. [DOI] [PubMed] [Google Scholar]
- Payne JA (1997) Functional characterization of neuronal-specific K–Cl– cotransporter: implication for [K+]o regulation. Am J Physiol 42: C1516–C1525. [DOI] [PubMed] [Google Scholar]
- Payne JA, Rivera C, Voipio J, Kaila K (2003) Cation-chloride co-transporter in neuronal communication, development and trauma. Trends Neurosci 26: 199–206. [DOI] [PubMed] [Google Scholar]
- Phillips JM, Nicholson C (1979) Anion permeability in spreading depression investigated with ion-sensitive microelectrodes. Brain Res 173: 567–571. [DOI] [PubMed] [Google Scholar]
- Pond BB, Galeffi F, Ahrens R, Schwartz-Bloom RD (2004) Chloride transport inhibitors influence recovery from oxygen-glucose deprivation-induced cellular injury in adult hippocampus. Neuropharmacology, in press. [DOI] [PubMed]
- Rader RK, Lanthorn TH (1989) Experimental ischemia induces a persistent depolarization blocked by decreased calcium and NMDA antagonists. Neurosci Lett 99: 125–130. [DOI] [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999) The K+/Cl–co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251–255. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M (2002) BDNF-induced TrkB activation down-regulates the K+–Cl– cotransporter KCC2 and impairs neuronal Cl– extrusion. J Cell Biol 159: 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R, Galeffi F, Ahrens R, Jordan G, Schwartz-Bloom RD (2002) Modulation of the GABAA-gated chloride channel by reactive oxygen species. J Neurochem 80: 383–391. [DOI] [PubMed] [Google Scholar]
- Schomberg SL, Su G, Haworth RA, Sun D (2001) Stimulation of Na–K–2Cl cotransporter in neurons by activation of non-NMDA ionotropic receptor and group-I mGluRs. J Neurophys 85: 2563–2575. [DOI] [PubMed] [Google Scholar]
- Schwartz-Bloom RD, Sah R (2001) γ-Aminobutyric acidA neurotransmission and cerebral ischemia. J Neurochem 77: 353–371. [DOI] [PubMed] [Google Scholar]
- Schwartz-Bloom RD, McDonough KJ, Chase PJ, Chadwick LE, Inglefield JR, Levin ED (1998) Long-term neuroprotection by benzodiazepine full versus partial agonists after transient cerebral ischemia in the gerbil. J Cereb Blood Flow Metab 18: 548–558. [DOI] [PubMed] [Google Scholar]
- Shiroya T, Fukunaga R, Akashi K, Shimada N, Takagi Y, Nishino T, Hara M, Inagaki C (1989) An ATP-driven Cl– pump in the brain. J Biol Chem 264: 17416–17421. [PubMed] [Google Scholar]
- Smetters D, Majewska A, Yuste R (1999) Detecting action potentials in neuronal populations with calcium imaging. Methods 18: 215–221. [DOI] [PubMed] [Google Scholar]
- Somjen GG, Aitken PG, Czéh GL, Herreras O, Jing J, Young JN (1992) Mechanisms of spreading depression: a review of recent finding and a hypothesis. Can J Physiol Pharmacol 70: S248–S258. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Mody I (1992) Shunting of excitatory input to dentate gyrus granule cells by a depolarizing GABAA receptor-mediated postsynaptic conductance. J Neurophysiol 68: 197–212. [DOI] [PubMed] [Google Scholar]
- Su G, Haworth RA, Dempsey RJ, Sun D (2000) Regulation of Na+–K+–Cl– cotransporter in primary astrocytes by dibutyryl cAMP and high [K+]o Am J Physiol 279: C1710–C1721. [DOI] [PubMed] [Google Scholar]
- Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH (1999) Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19: RC39(1–6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H (1997) Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol 78: 891–902. [DOI] [PubMed] [Google Scholar]
- Taylor CG, Weber ML (1993) Effect of temperature on synaptic function after reduced oxygen and glucose in hippocampal slices. Neuroscience 52: 555–562. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Weber ML, Gaughan CL, Lehning EJ, LoPachin RM (1999) Oxygen/glucose deprivation in hippocampal slices: altered intraneuronal elemental composition predicts structural and functional damage. J Neurosci 19: 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SM, Gahwiler BH (1989) Activity-dependent disinhibition. II. Effects of extracellular potassium, furosemide, and membrane potential on ECl– in hippocampal CA3 neurons. J Neurophysiol 61: 512–523. [DOI] [PubMed] [Google Scholar]
- Urban L, Neill KH, Crain BJ, Nadler JV, Somjen GG (1989) Postischemic synaptic physiology in area CA1 of the gerbil hippocampus studied in vitro J Neurosci 9: 3966–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkman AS (1990) Development and biological applications of chloride-sensitive fluorescent indicators. Am J Physiol 259: C375–C388. [DOI] [PubMed] [Google Scholar]
- Wang T, Raley-Susman KM, Wang J, Chambers G, Cottrell JE, Kass IS (1999) Thiopental attenuates hypoxic changes of electrophysiology, biochemistry, and morphology in rat hippocampal slice CA1 pyramidal cells. Stroke 30: 2400–2407. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Tanaka E, Shoji Y, Kudo Y, Inokuchi H, Higashi H (1997) Factors that reverse the persistent depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol 78: 903–911. [DOI] [PubMed] [Google Scholar]
- Yan Y, Dempsey RJ, Sun D (2001) Na+–K+–Cl– cotransporter in rat focal cerebral ischemia. J Cereb Blood Flow Metab 21: 711–721. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Lipton P (1999) Cytosolic Ca2+ changes during in vitro ischemia in rat hippocampal slices: major roles for glutamate and Na+-dependent Ca2+ release from mitochondria. J Neurosci 19: 3307–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JP, Krnjević K (1999) Persistent block of CA1 synaptic function by prolonged hypoxia. Neuroscience 90: 759–770. [DOI] [PubMed] [Google Scholar]