

Abstract
Peripheral nerve injury is known to upregulate the rapidly repriming Nav1.3 sodium channel within first-order spinal sensory neurons. In this study, we hypothesized that (1) after peripheral nerve injury, second-order dorsal horn neurons abnormally express Nav1.3, which (2) contributes to the responsiveness of these dorsal horn neurons and to pain-related behaviors. To test these hypotheses, adult rats underwent chronic constriction injury (CCI) of the sciatic nerve. Ten days after CCI, allodynia and hyperalgesia were evident. In situ hybridization, quantitative reverse transcription-PCR, and immunocytochemical analysis revealed upregulation of Nav1.3 in dorsal horn nociceptive neurons but not in astrocytes or microglia, and unit recordings demonstrated hyperresponsiveness of dorsal horn sensory neurons. Intrathecal antisense oligodeoxynucleotides targeting Nav1.3 decreased the expression of Nav1.3 mRNA and protein, reduced the hyperresponsiveness of dorsal horn neurons, and attenuated pain-related behaviors after CCI, all of which returned after cessation of antisense delivery. These results demonstrate for the first time that sodium channel expression is altered within higher-order spinal sensory neurons after peripheral nerve injury and suggest a link between misexpression of the Nav1.3 sodium channel and central mechanisms that contribute to neuropathic pain after peripheral nerve injury.
Keywords: ion channel, sensitization, neuropathy, dorsal horn, wide dynamic range, pain
Introduction
Injury of peripheral nerves can result in the development of a chronic neuropathic pain syndrome associated with hyperresponsiveness of sensory neurons within the dorsal root ganglion (DRG) (Wall and Devor, 1983; Study and Kral, 1996) and the spinal cord dorsal horn (Woolf, 1983; Sotgiu et al., 1992; Laird and Bennett, 1993).
Voltage-gated sodium channels underlie action potential generation and play a critical role in electrical signaling between neurons. The TTX-sensitive (TTX-S) Nav1.3 sodium channel is normally expressed at relatively high levels in the rat embryonic nervous system but is barely detectable in the adult DRG or spinal cord (Beckh et al., 1989; Waxman et al., 1994; Felts et al., 1997; Hains et al., 2002). However, expression of Nav1.3 is upregulated within DRG neurons after their axotomy by sciatic nerve transection (Waxman et al., 1994; Black et al., 1999) and after chronic constriction injury (CCI) of the sciatic nerve in adult rats (Dib-Hajj et al., 1999). This is functionally important, because Nav1.3 produces a rapidly repriming TTX-S current that permits neuronal firing at higher-than-normal frequencies within DRG neurons (Cummins and Waxman, 1997; Cummins et al., 2001). To date, however, the question of whether peripheral nerve injury triggers changes in sodium channel expression within higher-order sensory neurons has not been examined.
In this study, we hypothesized that upregulated expression of Nav1.3 occurs in second-order dorsal horn sensory neurons after peripheral nerve injury and that Nav1.3 contributes to hyperresponsiveness of these neurons and to behavioral allodynia and hyperalgesia. To test this hypothesis, we examined Nav1.3 expression in dorsal horn neurons and, using a targeted antisense (AS) oligodeoxynucleotide (ODN) knockdown strategy, examined the functional contribution of Nav1.3 to the hyperresponsiveness of dorsal horn neurons and pain-related behaviors after CCI.
Materials and Methods
Animal care. Experiments were performed in accordance with National Institutes of Health guidelines for the care and use of laboratory animals; all animal protocols were approved by the Yale University Institutional Animal Use Committee. Adult male Sprague Dawley rats (200–225 gm) were used for this study. Animals were housed under a 12 hr light/dark cycle in a pathogen-free area with ad libitum access to water and food.
Chronic constriction injury. Rats (n = 63) were deeply anesthetized with ketamine/xylazine (80/5 mg/kg, i.p.), and the left sciatic nerve was exposed at the mid-thigh level by blunt dissection of the biceps femoris. For CCI (n = 48), four chromic gut (4-0) ligatures were tied loosely around the nerve ∼1 mm apart, proximal to its trifurcation, as described by Bennett and Xie (1988). For sham surgery (n = 15), the sciatic nerve was isolated but not ligated. After CCI or sham surgery, the overlying muscles and skin were closed in layers with 4-0 silk sutures and staples, respectively, and the animal recovered on a 30°C heating pad. Postoperative treatments included saline (2.0 cc, s.c.) for rehydration and enro-floxacin (0.3 cc; 22.7 mg/ml, s.c.). After surgery, animals were maintained under the same conditions and fed ad libitum.
ODN synthesis and delivery. Seven days after CCI, animals (n = 50) were anesthetized with ketamine/xylazine (80/5 mg/kg, i.p.), and a sterile premeasured 32 gauge intrathecal catheter (ReCathCo, Allison Park, PA) was introduced through a slit in the atlanto-occipital membrane and threaded to the lumbar enlargement for antisense administration, as described in detail previously (Hains et al., 2003c). Four days after catheter placement (day 11 after CCI), intrathecal administration of an AS ODN sequence corresponding to the translation initiation site of Nav1.3 (5′-CAGTGCCTGGGCCATCTTTTC-3′) (CCI plus 1.3 AS; n = 20) or its mismatch (MM) (5′-CGATCGCGTGCGCTATCTTCT-3′) (CCI plus 1.3 MM; n = 13) was initiated. A search of the GenBank nucleotide sequence database for short conserved sequences using BLAST algorithms and the Nav1.3 AS ODN sequence as a query returned only matches from rat, mouse, and human Nav1.3. However, manual alignment of the sequence from all of the Nav1 channel sequences showed that the AS ODN sequence of Nav1.3 is identical at 17/21 to Nav1.1 and Nav1.2 and identical at 14/21 to Nav1.6. However, the identical residues were separated by mismatches, such that the longest contiguous stretch of the sequence was 7 for Nav1.1 and Nav1.6, and 11 for Nav1.2. Thus, it is not likely that the Nav1.3 AS can form a stable duplex with channel sequences other than Nav1.3. For 4 d, 45 μg/5 μl twice daily of either AS or MM in artificial CSF (aCSF; in mm: 1.3 CaCl2–2 H2O, 2.6 KCl, 0.9 MgCl, 21.0 NaHCO3, 2.5 Na2HPO4–7 H2O, and 125.0 NaCl; prepared in sterile H2O) was injected followed by a 10 μl aCSF flush. On day 4, Cy3-tagged AS or MM was delivered in the same manner to a subset of these animals. This confirmed uptake of AS by neurons within laminas I–V. In a subset of CCI plus 1.3 AS animals (n = 8), AS injections were stopped after 4 d (on day 14 after CCI), and outcome measures continued for another 3 d. A separate group of injured animals (CCI; n = 15) underwent intrathecal injection of aCSF only.
In situ hybridization. Ten days after CCI or sham surgery, tissue was collected from the ipsilateral and contralateral DRG (n = 6 animals/group) and spinal cord lumbar enlargement (n = 6 animals/group) (L3–L5) of rats after perfusion with 4% paraformaldehyde PBS and fixation in 30% sucrose. Twelve micrometer transverse cryosections (n = 4 sections/animal) from each treatment group were processed for detection of Nav1.3 mRNA as described previously (Black et al., 1996), with incubation in 4% paraformaldehyde increased to 12 min and permeabilization with proteinase K reduced to 6 min. Digoxigenin-labeled antisense and sense riboprobes for Nav1.3 (nucleotides 6335–6813; GenBank accession number Y00766) were synthesized as described previously. Sense riboprobes yielded no signal on in situ hybridization (data not shown).
Quantitative reverse transcription-PCR. Ten days after CCI or sham surgery, fresh tissue was collected from the ipsilateral and contralateral DRG and L3–L5 of the ipsilateral and contralateral sides of the spinal cord (n = 5–8 animals/group) and flash frozen. Total tissue RNA was extracted using RNeasy minicolumns (Qiagen, Valencia, CA), and purified RNA was treated with RNase-free DNase I and repurified using an RNeasy mini-column. First-strand cDNA was reverse-transcribed using 5 μl of purified total RNA, 1 mm random hexamer primer (Roche, Indianapolis, IN), 40 U of SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA), and 40 U of RNase inhibitor (Roche). The buffer consisted of (in mm) 50 Tris-HCl, pH 8.3, 75 KCl, 3 MgCl2, 10 DTT, and 5 deoxyNTP. The reaction proceeded at 37°C for 90 min, continued at 42°C for 30 min, and was terminated by heating to 95°C for 5 min. A parallel reaction omitting reverse transcriptase was performed to demonstrate the absence of contaminating genomic DNA.
Expression of Nav1.3 mRNA was quantified using the relative standard curve method of PCR using methods described previously (Hains et al., 2003c). 18S ribosomal RNA (rRNA) was used as an endogenous control to normalize expression levels. Nav1.3 cDNA was obtained from human embryonic kidney 293 cells stably transfected with an Nav1.3 construct (Cummins et al., 2001). Standards and unknowns were amplified in quadruplets. Primer and probe sequences for the Nav1.3 target were designed using Primer Express software (Applied Biosystems, Foster City, CA) according to the specifications of the TaqMan protocol. Sequences for the forward primer, reverse primer, and probe are as follows: 5′-AGGACAATGTCCAGAAGGGTAC-3′, 5′-AGTAGTCCTGAGTCATGAGTCGAAAC-3′, and 5′-TGGGACGAAACCCCAACTACGGCTACAC-3′, respectively. The amplicon comprises nucleotides 1442–1556 of the Nav1.3 gene. Standard curves for sodium channel primer–probe sets and 18S rRNA endogenous control (rRNA) were constructed from the respective mean critical threshold (CT) value, and the equation describing the curve was derived using Sequence Detection System software, version 1.6.3 (Applied Biosystems).
Immunocytochemistry. Tissue was collected in the same manner as for in situ hybridization for both ipsilateral and contralateral DRG and lumbar spinal cord (n = 6 animals/group) 10 d after sham or CCI surgery. For detection of Nav1.3 protein, cryosections (n = 4 sections/animal) were mounted, and slides were incubated at room temperature in the following: (1) blocking solution (PBS containing 5% NGS, 2% BSA, 0.1% Triton X-100, and 0.02% sodium azide) for 30 min, (2) subtype-specific primary antibody raised in rabbit against Nav1.3 (16153) overnight in blocking solution, (3) PBS, six times for 5 min each, (4) goat anti-rabbit IgG–Cy3 (1:2000; Amersham Biosciences, Piscataway, NJ) in blocking solution for 2 hr, and (5) PBS, six times for 5 min each. Characterization of the specificity of the 16153 Nav1.3 antibody has been performed in previous studies (Hains et al., 2002, 2003c). To further categorize cells that express Nav1.3, we used the guinea pig anti-substance-P receptor neurokinin-1 receptor (NK1R) (1:250; Affiniti Research Products Ltd., Exeter, UK), mouse anti-GFAP (1:250; Chemicon, Temecula, CA), and mouse anti-OX-42 CD11b/c (1:250; BD Biosciences, San Jose, CA) primary antibodies. Secondary antibodies included goat anti-rabbit Cy3 (1:2000; Amersham Biosciences), goat anti-mouse Alexa 488 (1:1000; Molecular Probes, Eugene, OR), and goat anti-guinea pig Cy2 (1:250; Rockland Immunochemicals, Gilbertsville, PA). Control experiments were performed without primary or secondary antibodies, yielding only background levels of signal.
Quantitative image analysis. Images were captured with a Nikon (Melville, NY) Eclipse E800 light microscope equipped with epifluorescence and Nomarski optics, using a SPOT RT camera (Diagnostic Instruments Inc., Sterling Heights, MI). Quantitative analysis was performed by a blinded observer using IPLab Spectrum software (Scanalytics, Fair-fax, VA), and the number of positively labeled neurons was counted for each dorsal horn. Cells were sampled only if the nucleus was visible within the plane of section and if cell profiles exhibited distinctly delineated borders. Background levels of signal were subtracted, and control and experimental conditions were evaluated in an identical manner.
Electrophysiological procedures. Ten days after CCI or sham surgery, animals (n = 4/group; each yielding 7–12 units ipsilateral or contralateral to the injury site per group) underwent extracellular single-unit recording as described by Hains et al. (2003a,b). Rats were anesthetized with halothane by tracheal intubation (1.1%; 2–2.5 cc tidal volume; 60–70 strokes/min). Rectal temperature was maintained at 37°C. Units were isolated from L3–L5 medially near the dorsal root entry zone up to a depth of 1000 μm. Recordings were made with a low-impedance 5 MΩ tungsten-insulated microelectrode (A-M Systems, Carlsborg, WA) after a cell was identified and its receptive field was mapped. Three routine mechanical stimuli were applied: (1) brush (BR) stimulation of the skin with a cotton applicator, (2) pressure (PR), by attaching a large arterial clip with a weak grip to a fold of the skin (144 gm/mm2), and (3) pinch (PI), by applying a small arterial clip with a strong grip to a fold of skin (583 gm/mm2). Multireceptive (MR) neurons were identified by their relative magnitude of responsiveness to all stimuli, criteria that were used in all groups to ensure recording of MR neurons. Because phenotype shifts can occur after CCI, such that more neurons could assume a MR functional classification, our search paradigm ensured that in all groups we sampled MR neurons. Stimulation was applied with the experimenter blinded to the output of the cell during stimulation. Background activity was recorded for 20 sec, and stimuli were applied serially for 20 sec, separated by another 20 sec of spontaneous activity without stimulation. Care was taken to ensure that the responses were maximal, that each stimulus was applied to the primary receptive field of the cell, and that isolated units remained intact for the duration of each experiment using Spike2 template-matching routines. Neurons were considered to be hyperresponsive if evoked frequencies were >150% of control levels.
Electrical signals were amplified and filtered at 300–3000 Hz (DAM80; World Precision Instruments, Sarasota, FL), processed by a data-collection system (CED Micro1401; Cambridge Electronic Design, Cambridge, UK), and stored on a computer (Pentium 4 personal computer; Dell, Austin, TX) to construct peristimulus time histograms or wavemark files. The stored digital record of individual unit activity was retrieved and analyzed off-line with Spike2 software, version 3.13 (Cambridge Electronic Design).
Behavioral testing. Behavioral analysis (n = 8–10 animals/group) was performed daily from day 10 to day 17 after CCI. After acclimation to the testing area (30 min), mechanical sensory thresholds were determined by paw withdrawal to application of a series of von Frey filaments (Stoelting, Wood Dale, IL) to the glabrous surface of the paw. After application of calibrated von Frey filaments (0.4–26 gm, verified by calibration against a force transducer) with enough force to cause buckling of the filament, a modification of the “up–down” method of Dixon (1980) was used to determine the value at which paw withdrawal occurred 50% of the time (Chaplan et al., 1994), which was interpreted to be the mechanical nociceptive threshold.
After acclimation to the test chamber, thermal hyperalgesia was assessed by measuring the latency of paw withdrawal in response to a radiant heat source (Dirig et al., 1997). Animals were placed in Plexiglas boxes on an elevated glass plate, under which a radiant heat source (4.7 A) was applied to the glabrous surface of the paw through the glass plate. The heat source was turned off automatically by a photocell when the limb was lifted, allowing the measurement of paw-withdrawal latency. Three minutes were allowed between each trial, and four trials were averaged for each limb.
Statistical analysis. All statistical tests were performed at the α level of significance of 0.05 by two-tailed analyses using parametric tests. Data were tested for significance using one-way ANOVA, followed by Bonferroni post hoc analysis. Tests of factors including pairwise comparisons were applied with either the paired Student's t test or the two-sample Student's t test. Data management and statistical analyses were performed using SAS (1992) statistical procedures with SigmaStat (version 1.0; Jandel Scientific, Corte Madera, CA) and graphed as mean ± SD using Jandel SigmaPlot (version 7.0).
Results
In situ hybridization
In situ Nav1.3 mRNA hybridization studies were performed on lumbar spinal cord sections 10 d after CCI (Fig. 1). Sections from intact animals (Fig. 1A) and from the contralateral side (data not shown) showed very little hybridization signal for Nav1.3. After CCI, Nav1.3 hybridization signal was clearly present on the ipsilateral side within small (5–10 μm) neurons in laminas I–II, in addition to larger (20–40 μm) neurons in laminas III–IV (Fig. 1B). Neurons expressing Nav1.3 were localized in the medial aspect of the dorsal horn, which is known to be the predominant projection territory of somatosensory afferents with receptive fields on the hindlimbs/paws. Quantification by densitometry (Fig. 1C) showed the ipsilateral increase in Nav1.3 signal to be statistically significant (p < 0.05). No Nav1.3 signal was observed in afferent fibers or in dendritic and/or axonal arborizations, and no signal was detected within white-matter tracts. Nav1.3 hybridization signal was not observed in the DRG contralateral to the CCI (Fig. 1E), but signal was present on the ipsilateral side (Fig. 1F) in small- and large-diameter DRG neurons.
Figure 1.
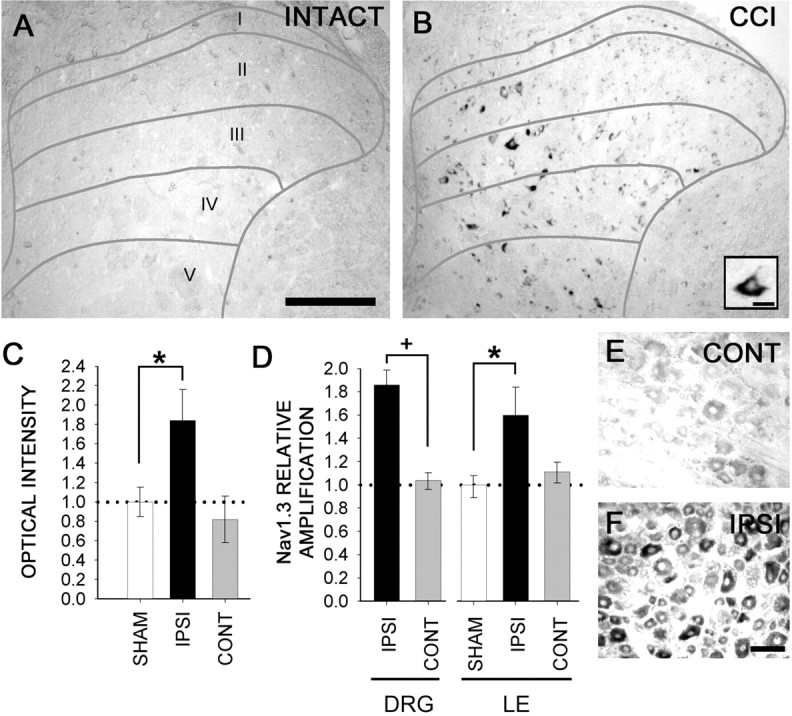
A, In situ hybridization for Nav1.3 transcripts revealed no signal in the dorsal horn gray matter of sham-operated animals. However, 10 d after CCI of the sciatic nerve, Nav1.3 signal is detectable in cells exhibiting neuronal profiles within the superficial and deep dorsal horn, mostly in the medial region where afferent fibers terminate, on the ipsilateral side (B). Under high magnification, cells (B, inset) demonstrate multipolar neuronal morphologies and cytoplasmic staining. No labeling was observed in primary afferent fibers or white matter after CCI. C, Signal was significantly (*p < 0.05) increased in neurons expressing Nav1.3 after CCI on the ipsilateral side. D, Quantitative RT-PCR amplification showed a significant (*+p < 0.05) increase in ipsilateral DRG (183% compared with the contralateral DRG) and ipsilateral dorsal horn [158% compared with the contralateral side of the lumber (LE) cord] Nav1.3 mRNA in CCI animals. Dotted lines indicate control levels in C and D. No changes were observed in the dorsal horn contralateral to CCI. Upregulation of Nav1.3 mRNA was observed in the ipsilateral DRG (F) but not in the contralateral DRG (E). Scale bars: (in A) A, B, 300 μm; B, inset, 10 μm; (in F) E, F, 50 μm. CONT, Contralateral; IPSI, ipsilateral.
The change in Nav1.3 mRNA, measured by quantitative reverse transcription (RT)-PCR amplification from the DRG and lumbar enlargement of sham-operated and CCI groups, is shown in Figure 1D. Quantitative RT-PCR results showed a statistically significant increase in the ipsilateral DRG to ∼183% of the contralateral side after CCI. In lumbar dorsal horn ipsilateral to CCI mRNA was increased to ∼158% of sham-operated Nav1.3 mRNA levels. No significant changes were observed on the side contralateral to the CCI in either the DRG or lumbar enlargement.
Immunocytochemistry
Immunocytochemistry with an isoform-specific antibody raised against Nav1.3 revealed very little expression of Nav1.3 in the lumbar dorsal horn of the contralateral spinal cord after CCI (Fig. 2A) and in sham-operated intact animals (data not shown). Ten days after CCI, however, expression of Nav1.3 was clearly increased in the ipsilateral dorsal horn (Fig. 2B). Nav1.3 signal was observed in dorsal horn cells exhibiting multipolar neuronal profiles in laminas I–IV, and the distribution was increased in neurons located medially in these laminas.
Figure 2.
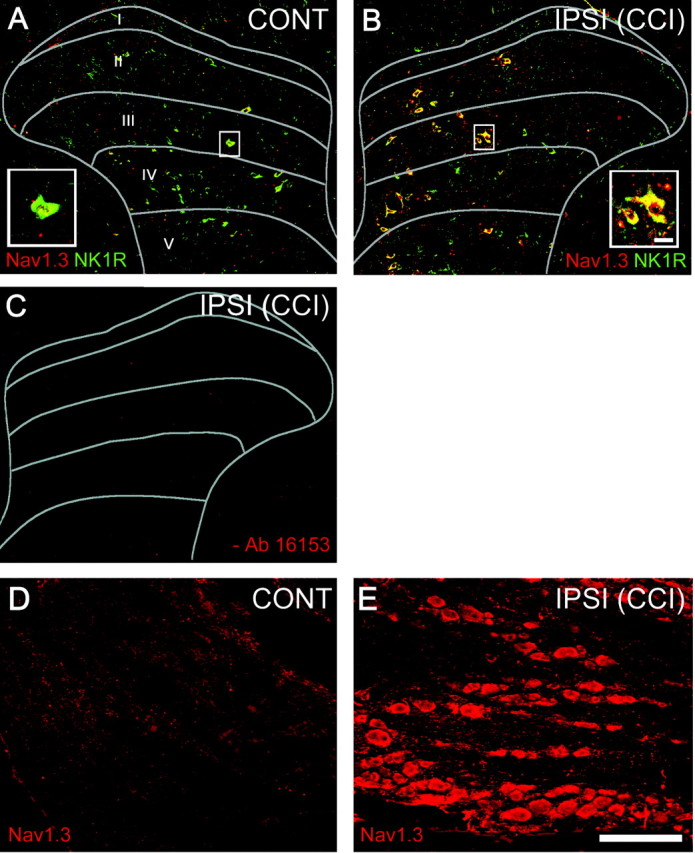
Immunocytochemical localization of Nav1.3 (red channel) and substance-P receptor (NK1R, green channel) protein within the ipsilateral and contralateral dorsal horn 10 d after CCI. A, On the contralateral side, very few Nav1.3 immunopositive profiles were detectable, whereas NK1R signal was present in cells exhibiting neuronal morphologies throughout all dorsal horn laminas. Inset, High-magnification of double-labeled neuron. B, On the side ipsilateral to CCI, Nav1.3 and NK1R positively labeled cell profiles were observed throughout laminas I–V. Merged images show colocalization of Nav1.3 and NK1R (yellow) within ipsilateral dorsal horn after CCI (B). C, In reactions for which primary antibody (16153) was omitted, no Nav1.3 signal was detectable. Increased signal for Nav1.3 was observed in the ipsilateral DRG (E) compared with the contralateral side of CCI animals (D). Nav1.3 AS reduced Nav1.3 but not NK1R signal in the ipsilateral dorsal horn after CCI (data not shown) (Table 1). Scale bar, 300 μm. CONT, Contralateral; IPSI, ipsilateral.
Staining for NK1R, which provides a marker for nociceptive neurons, demonstrated a distribution of NK1R-positive dorsal horn neurons consistent with previous reports (Ishigooka et al., 2001), with signal detected in dorsal horn laminas I–VI. After CCI, colocalization with Nav1.3 revealed that ipsilateral dorsal horn neurons expressing Nav1.3 were also positive for NK1R (Fig. 2 B). In immunocytochemistry reactions in which primary antibody (16153, 3-1) was omitted (Fig. 2C), no Nav1.3 signal was detectable.
Increased signal for Nav1.3 was observed in the ipsilateral DRG (Fig. 2E), compared with the contralateral side of CCI animals (Fig. 2D) and sham-operated animals (data not shown).
Double-labeling experiments revealed that, in the ipsilateral dorsal horn after CCI, Nav1.3 protein did not colocalize with OX-42, a marker for microglia (Fig. 3A–C), or with GFAP, a marker for astrocytes (Fig. 3D–F).
Figure 3.
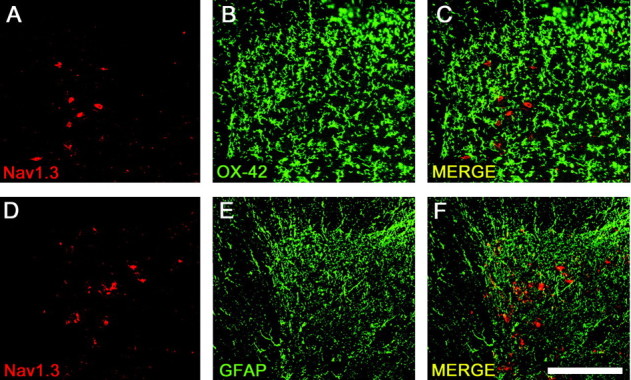
Colocalization experiments demonstrate that after CCI, upregulation of Nav1.3 did not take place within glial cells. Nav1.3 (A) was not found to colocalize with OX-42 (B), a marker for microglia (C, MERGE). Nav1.3 (D) was not found to colocalize with GFAP (E), a marker for astrocytes (F, MERGE). Scale bar, 300 μm.
Extracellular unit recordings
Units were sampled from dorsal horn laminas I–V (24–988 μm). Representative peristimulus time histograms (spikes/1 sec bin) are shown in Figure 4 for MR neurons in sham-operated control animals (Fig. 4 A) and the ipsilateral side of CCI groups 10 d after injury (Fig. 4 B). When compared with controls, CCI animals showed increased evoked activity to all peripheral stimuli (brush, pressure, pinch) on the ipsilateral side. After CCI, spontaneous background activity was elevated (Fig. 4 B). Of 33 MR neurons sampled in CCI animals, 78% (26 of 33) were hyperresponsive.
Figure 4.

Representative peristimulus time histograms (spikes/1 sec bin) of dorsal horn multireceptive neurons recorded extracellularly from L3–L5 in response to BR, PR, and PI stimulation to mapped receptive fields on the hindpaw. Unit waveforms are also shown. Records are shown for sham-operated animals (A) and for animals 10 d after CCI (B). Compared with controls, injured animals demonstrated increased spontaneous activity and evoked hyperresponsiveness to all peripheral stimuli.
Quantification of mean ± SD evoked discharge rates of MR neurons, over background, from sham-operated and CCI groups is shown in see Figure 7C. Responses were typical for MR neurons, demonstrating high evoked activity to all stimuli. Compared with sham-operated animals that demonstrated evoked response rates between 5 and 15 Hz, CCI animals demonstrated significantly increased response rates of up to 20–60 Hz.
Figure 7.

After 4 d of intrathecal administration, beginning 11 d after CCI, Nav1.3 MM had no effect on spontaneous or evoked activity (A), but Nav1.3 AS reduced both spontaneous activity and evoked responses to BR, PR, and PI stimulation after CCI (B). Unit waveforms are also shown. C, Quantification of mean ± SD spontaneous and evoked discharge rates of neurons sampled from sham-operated CCI plus 1.3 MM and CCI plus 1.3 AS groups demonstrated significantly decreased evoked activity to all peripheral stimuli after injury in the AS group (*p < 0.05; CCI plus 1.3 MM vs CCI plus 1.3 AS).
Antisense knockdown of Nav1.3
Because our results showed upregulated Nav1.3 expression, hyperresponsiveness of dorsal horn sensory neurons, and allodynia and hyperalgesia, we then asked whether targeted ODN knockdown of Nav1.3 after CCI would reduce dorsal horn neuronal hyperresponsiveness and ameliorate pain-related behaviors. In these experiments we intrathecally administered Nav1.3 AS, as well as its mismatch sequence, beginning on day 11 after injury.
A single injection (5 μl) of Cy3-labeled AS diffused to a depth of 800–900 μm in spinal parenchyma and was taken up by cells exhibiting a neuronal morphology as deep as lamina V (Fig. 5A). In contrast to its robust penetration into the spinal cord, Cy3-labeled AS was unable to penetrate DRG neurons (Fig. 5B), a result similar to our previous findings (Hains et al., 2003c). Neither Nav1.3 MM (Fig. 5C) nor AS (Fig. 5D) administration was effective in knocking down Nav1.3 mRNA expression in ipsilateral DRG neurons after CCI.
Figure 5.

A single injection (5 μl) of Cy3-labeled AS ODN diffused to a depth of 800–900 μm [through the dorsolateral funiculus (DLF) and into laminas I–V] in spinal parenchyma and was taken up by cells exhibiting a neuronal morphology (A). Cy3-labeled AS was unable to penetrate into the DRG neurons after intrathecal injection (B). Neither MM (C) nor AS (D) administration was effective in reducing Nav1.3 mRNA expression in ipsilateral DRG neurons after CCI. Scale bars: (in A) A, B, 300μm; (in D) C, D, 50 μm. p, Proximal; d, distal to intrathecal space.
To show that administration of Nav1.3 AS into the spinal cord in fact resulted in knockdown of Nav1.3, we assessed its effect on Nav1.3 mRNA and immunoreactivity. In situ hybridization demonstrated that Nav1.3 MM administration after CCI had no effect on Nav1.3 mRNA signal (Fig. 6A), whereas administration of Nav1.3 AS for 4 d resulted in significantly decreased Nav1.3 signal in the ipsilateral dorsal horn (Fig. 6B). When measured by RT-PCR 4 d after Nav1.3 AS administration, levels of Nav1.3 mRNA were significantly decreased in the ipsilateral dorsal horn, falling to ∼115% of intact levels (Fig. 6C), whereas after MM administration, Nav1.3 mRNA levels remained elevated at 160% of intact levels.
Figure 6.

After 4 d of intrathecal administration of Nav1.3 AS or MM oligodeoxynucleotides, beginning 11 d after CCI, Nav1.3 in situ hybridization signal intensity and distribution were unchanged in the MM group (A), whereas in AS-receiving animals, Nav1.3 signal was markedly reduced (B). C, Quantitative RT-PCR amplification showed a significant (*p < 0.05) decrease in ipsilateral dorsal horn Nav1.3 mRNA in CCI animals receiving Nav1.3 AS compared with animals receiving MM. The dotted line indicates mRNA levels in sham-operated animals. Scale bar, 300 μm. CONT, Contralateral; IPSI, ipsilateral.
Immunostaining for Nav1.3 revealed a loss of Nav1.3 immunoreactivity in AS-treated CCI compared with untreated or MM CCI. Quantification (Table 1) revealed that AS but not MM significantly reduced the number of NK1R-positive neurons that express Nav1.3.
Table 1.
Number of NK1R neuronal profiles and Nav 1.3 neuronal profiles per section in superficial laminas of dorsal horn at L3-L5
|
Group |
Contralateral to CCI |
|
|
Ipsilateral to CCI |
|
|
|
|---|---|---|---|---|---|---|---|
|
|
NK1R (a) |
Nav 1.3 (b) |
Merge b/a × 100% |
NK1R (c) |
Nav 1.3 (d) |
Merge d/c × 100% |
|
| Sham | 15.14 ± 3.22 | 1.13 ± 1.09 | 7.46 ± 1.36% | 20.29 ± 4.72 | 2.50 ± 1.41 | 19.90 ± 10.37% | |
| CCI | 17.86 ± 4.12 | 1.22 ± 1.15 | 6.83 ± 1.07% | 33.38 ± 5.15 | 22.00 ± 3.74 | 66.48 ± 10.68% | (-49.23%) |
| CCI plus Nav 1.3 MM | 14.28 ± 5.94 | 0.99 ± 1.03 | 6.93 ± 1.49% | 30.89 ± 5.80 | 24.59 ± 3.05 | 81.63 ± 16.76% | (-49.23%) |
| CCI plus Nav 1.3 AS |
15.43 ± 2.64 |
1.06 ± 0.94 |
6.87 ± 2.54% |
25.57 ± 5.46 |
8.07 ± 2.99*
|
33.63 ± 13.57%*
|
(-49.23%) |
Data represent mean ± SD values (n = 4 sections/6 animals/group).
p < 0.05, significant difference between the CCI and CCI plus 1.3 AS groups.
Unit recordings showed that knockdown of Nav1.3 resulted in decreased hyperresponsiveness of dorsal horn neurons. Four days after administration of Nav1.3 AS (Fig. 7B), but not after administration of MM (Fig. 7A), hyperresponsiveness of MR neurons was reduced in the ipsilateral dorsal horn of CCI rats. Quantification revealed that Nav1.3 AS treatment significantly reduced evoked hyperresponsiveness of dorsal horn neurons (Fig. 7C).
Behavioral testing
By 10 d after CCI, animals developed mechanical allodynia, as demonstrated by decreases in paw-withdrawal thresholds to von Frey stimulation (Fig. 8A). Mean ± SD mechanical thresholds were significantly decreased on the side ipsilateral to the CCI (3.3 ± 1.5 gm) compared with the contralateral side (23.0 ± 2.0 gm), with a mean ipsilateral versus a contralateral difference score of 19.7 ± 1.7 gm for all CCI animals. Thermal behavioral testing revealed that by 10 d after CCI, animals developed thermal hyperalgesia, as indicated by decreases in paw-withdrawal latency (Fig. 8B). In the injured animals, the mean difference score between ipsilateral (withdrawal latency, 6.21 ± 1.19 sec) and contralateral (withdrawal latency, 9.74 ± 1.09 sec) limbs was 3.53 ± 1.14 sec at this time point.
Figure 8.

Pain-related behaviors after CCI and effects of administration of Nav1.3 MM or AS, for 4 d, beginning 11 d after CCI. A, Mean ± SD von Frey (VF) filament threshold values, displayed as the difference between ipsilateral and contralateral paw thresholds, show that by 10 d after CCI, paw-withdrawal thresholds were significantly decreased on the ipsilateral side compared with the contralateral side and sham-operated controls. Intrathecal administration (indicated by gray shading) of Nav1.3 MM had no effect on mechanical thresholds, whereas Nav1.3 AS resulted in a significant (*p < 0.05) reduction in mechanical allodynia. After cessation of AS administration, mechanical thresholds returned to levels consistent with CCI. B, Mean ± SD paw-withdrawal latency (in seconds) to radiant thermal stimuli after CCI demonstrates decreased latencies and development of thermal hyperalgesia ipsilaterally by 10 d after CCI. In sham-operated animals, no differences in paw-withdrawal latency were observed. Administration of Nav1.3 MM had no effect on withdrawal latency, whereas administration of Nav1.3 AS resulted in withdrawal latencies that were significantly (*p < 0.05) increased in the ipsilateral paw. Cessation of AS administration resulted in restoration of thermal hyperalgesia.
Administration of Nav1.3 AS starting on day 11 decreased the mechanical allodynia and resulted in an increase in the withdrawal threshold and a reduction of the difference score between ipsilateral and contralateral limbs (Fig. 8A). By day 4 of Nav1.3 AS administration, the ipsilateral (withdrawal threshold, 17.4 ± 4.9) and contralateral (withdrawal threshold, 21.0 ± 1.6) difference score was reduced to 3.6 ± 3.3 gm. Three days after cessation of AS administration (day 17), allodynia was restored to pre-AS levels in these animals (difference score, 18.2 ± 5.6). Administration of MM had no effect on CCI animals (difference score remained at 20.1 ± 1.7), and AS had no effect on intact animals (data not shown). Nav1.3 AS administration decreased thermal hyperalgesia and reduced the difference score to 0.15 ± 1.52 sec (withdrawal latencies: ipsilateral, 10.23 ± 1.49 sec; contralateral, 10.38 ± 1.56 sec) by day 4 of administration (Fig. 8B). Three days after AS withdrawal, thermal hyperalgesia reappeared and the difference score for ipsilateral (withdrawal latency, 6.61 ± 0.95 sec) and contralateral (withdrawal latency, 10.61 ± 0.88 sec) sides was restored to 3.99 ± 0.91 sec. No effects were observed in CCI animals receiving MM (difference score, 3.75 ± 1.01 sec) or in uninjured animals receiving AS (data not shown).
Discussion
In the present study, we show for the first time that after peripheral nerve injury, the expression of the voltage-gated sodium channel Nav1.3 is upregulated in higher-order sensory (dorsal horn) neurons. Because these results suggest an association between Nav1.3 upregulation, hyperresponsiveness of dorsal horn sensory neurons, and neuropathic pain, we used a selective antisense strategy to show that targeted knockdown of Nav1.3 results in downregulation of Nav1.3 mRNA and protein, reduction in hyperresponsiveness of dorsal horn neurons, and reversal of pain-related behaviors after injury.
There may be many possible explanations for the increases in spontaneous and evoked firing of MR neurons after injury, including presynaptic and local network alterations after CCI. Our results strongly suggest that the increased neuronal responsiveness is attributable, at least in part, to sodium channel upregulation. After injury to their axons, first-order spinal sensory neurons (DRG neurons) demonstrate increased sodium conductance, which has been associated with increased expression of voltage-gated sodium channels (Devor et al., 1989; Matzner and Devor, 1994; Rizzo et al., 1995; Cummins and Waxman, 1997; Zhang et al., 1997; Novakovic et al., 1998; Black et al., 1999), implicating a link between sodium channel activity within DRG neurons and hyperresponsiveness of these neurons associated with pain. Consistent with this, experimental and clinical observations have demonstrated partial efficacy of sodium channel blocking agents in neuropathic pain (Chabal et al., 1989; Devor et al., 1992; Omana-Zapata et al., 1997; Rizzo, 1997),
Spinal expression of Nav1.3 is detectable at embryonic day 17 and postnatal day 2 (P2) but is downregulated with further development, exhibiting very low levels of expression by P30 (Beckh et al., 1989; Felts et al., 1997). Upregulation of Nav1.3 is functionally important because, Nav1.3 recovers (reprimes) rapidly from inactivation, a property that poises neurons to produce high-frequency activity (Cummins and Waxman, 1997; Black et al., 1999; Cummins et al., 2001). Increased expression of Nav1.3 occurs in DRG neurons after injury to the sciatic nerve (Waxman et al., 1994; Dib-Hajj et al., 1996; Black et al., 1999; Kim et al., 2001) and in facial motor neurons after transection of the facial nerve (Iwahashi et al., 1994), but expression of Nav1.3 is not increased in spirally axotomized cortical pyramidal neurons (Hains et al., 2002). Upregulation of Nav1.3 expression in DRG neurons is associated with allodynia and hyperalgesia (Dib-Hajj et al., 1999). Recently, we showed that Nav1.3 expression is upregulated in dorsal horn neurons after spinal cord contusion injury, and that Nav1.3 AS reduces neuronal hyperresponsiveness and pain-related behaviors (Hains et al., 2003c).
Increased firing activity of dorsal horn neurons after peripheral nerve injury could be driven at several sites. First, as early as 3 d after injury, ectopic discharges and abnormal responses to stimuli originate in the injured axons and their cells of origin within the DRG (Wall and Gutnick, 1974; Govrin-Lippmann and Devor, 1978; Scadding, 1981; Wall and Devor, 1983; Kajander et al., 1992; Kral et al., 1999) and are expected to drive dorsal horn neurons to relay higher-frequency afferent information supraspinally. Second, dorsal horn neurons undergo reactive changes that make them hyperresponsive, and abnormal firing has been shown to originate from within the dorsal horn after peripheral injury (Basbaum and Wall, 1976; Woolf, 1983; Woolf and Wall, 1986; Palecek et al., 1992; Sotgiu et al., 1992; Laird and Bennett, 1993). Activity-dependent central sensitization (heterosynaptic facilitation) is evident within seconds of a nociceptive conditioning stimulus and can outlast the stimulus for hours (Woolf and Wall, 1986). Within the dorsal horn, increases in excitatory amino acid concentrations (Somers et al., 2002) and reduction of GABA concentrations (Ibuki et al., 1997; Stiller et al., 1996) may also contribute to allodynia and hyperalgesia after nerve injury. The present results also suggest that upregulated expression of Nav1.3 contributes to hyperresponsiveness of these central neurons after nerve injury. We cannot rule out a contribution of other species of voltage-gated sodium channel isoforms that could effect firing thresholds, however.
We have shown previously (Hains et al., 2003c) that intrathecal administration of Nav1.3 knocks down Nav1.3 expression within dorsal horn neurons, but not within DRG neurons, after SCI, and we confirm this observation in the present study. Lack of penetration of Cy3-labeled AS into the DRG (in contrast to its clear penetration into the dorsal horn) may be attributable to differences in ensheathment that limit access to DRG neurons. Although it might be argued that upregulated Nav1.3 expression within dorsal horn neurons could be a response to high-frequency firing in nociceptive pathways after nerve injury, our results demonstrate the opposite (i.e., that high-frequency firing is attenuated when Nav1.3 is knocked down in these neurons). Our observation, in the present study, of reduced pain-related behaviors and reduced hyperresponsiveness during Nav1.3 AS administration as well as return of pain-related behaviors when AS is withdrawn implicates upregulation of Nav1.3 as a factor contributing to pain after nerve injury. Our observations lead us to propose that upregulated expression of Nav1.3 within dorsal horn sensory neurons contributes to hyperresponsiveness of these neurons and to resultant allodynia and hyperalgesia.
Although the upregulated expression of Nav1.3 within DRG neurons after axotomy appears to be attributable, at least in part, to deprivation from peripheral pools of neurotrophic factors (Black et al., 1997; Boucher et al., 2000; Leffler et al., 2002), the signals that trigger Nav1.3 upregulation within the dorsal horn have not been identified. Moreover, the presence of upregulated levels of Nav1.3 within the dorsal horn raises the question of whether peripheral nerve injury is associated with changes in ion channel expression within still higher-order sensory neurons at supraspinal levels. Future studies will be needed to examine this possibility.
Regardless of this, our results show for the first time that expression of sodium channel Nav1.3 is upregulated in secondary spinal sensory neurons after peripheral nerve injury and suggest a link between upregulated Nav1.3 expression within dorsal horn neurons, hyperresponsiveness of these higher-order sensory neurons, and pain-related behaviors.
Footnotes
This work was supported in part by grants from the Medical Research Service and Rehabilitation Research Service of the Department of Veterans Affairs of the United States. We also thank the Paralyzed Veterans of America and the United Spinal Association for support. B.C.H. was funded by The Christopher Reeve Paralysis Foundation (HB1-0304-2) and the National Institutes of Health–National Institute of Neurological Disorders and Stroke (1 F32 NS046919-01). We thank Dr. Joel Black for valuable experimental advice and Bart Toftness for technical assistance.
Correspondence should be addressed to Dr. Stephen G. Waxman, Department of Neurology, Yale University School of Medicine, 333 Cedar Street, LCI-707, New Haven, CT 06510. E-mail: stephen.waxman@yale.edu.
DOI:10.1523/JNEUROSCI.0300-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244832-08$15.00/0
References
- Basbaum AI, Wall PD (1976) Chronic changes in the response of cells in adult cat dorsal horn following partial deafferentation: the appearance of responding cells in a previously non-responsive region. Brain Res 116: 181–204. [DOI] [PubMed] [Google Scholar]
- Beckh S, Noda M, Lubbert H, Numa S (1989) Differential regulation of three sodium channel messenger RNAs in the rat central nervous system during development. EMBO J 8: 3611–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33: 87–107. [DOI] [PubMed] [Google Scholar]
- Black JA, Dib-Hajj S, McNabola K, Jeste S, Rizzo MA, Kocsis JD, Waxman SG (1996) Spinal sensory neurons express multiple sodium channel alpha-subunit mRNAs. Res Mol Brain Res 43: 117–131. [DOI] [PubMed] [Google Scholar]
- Black JA, Langworthy K, Hinson AW, Dib-Hajj SD, Waxman SG (1997) NGF has opposing effects on Na+ channel III and SNS gene expression in spinal sensory neurons. NeuroReport 8: 2331–2335. [DOI] [PubMed] [Google Scholar]
- Black JA, Cummins TR, Plumpton C, Chen YH, Hormuzdiar W, Clare JJ, Waxman SG (1999) Upregulation of a silent sodium channel after peripheral, but not central, nerve injury in DRG neurons. J Neurophysiol 82: 2776–2785. [DOI] [PubMed] [Google Scholar]
- Boucher TJ, Okuse K, Bennett DL, Munson JB, Wood JN, McMahon SB (2000) Potent analgesic effects of GDNF in neuropathic pain states. Science 290: 124–127. [DOI] [PubMed] [Google Scholar]
- Chabal C, Jacobson L, Little J (1989) Intrathecal fentanyl depresses nociceptive flexion reflexes in patients with chronic pain. Anesthesiology 70: 226–229. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Waxman SG (1997) Down-regulation of tetrodotoxin-resistant sodium currents and up-regulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons following nerve injury. J Neurosci 17: 3503–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib-Hajj SD, Waxman SG (2001) Nav1.3 sodium channels: rapid repriming and slow closed-state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21: 5952–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M, Keller CH, Deerinck TJ, Levinson SR, Ellisman MH (1989) Na+ channel accumulation on axolemma of afferent endings in nerve end neuromas in Apteronotus Neurosci Lett 102: 149–154. [DOI] [PubMed] [Google Scholar]
- Devor M, Wall PD, Catalan N (1992) Systemic lidocaine silences ectopic neuroma and DRG discharge without blocking nerve conduction. Pain 48: 261–268. [DOI] [PubMed] [Google Scholar]
- Dib-Hajj S, Black JA, Felts P, Waxman SG (1996) Down-regulation of transcripts for Na channel alpha-SNS in spinal sensory neurons following axotomy. Proc Natl Acad Sci USA 93: 14950–14954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib-Hajj SD, Fjell J, Cummins TR, Zheng Z, Fried K, LaMotte R, Black JA, Waxman SG (1999) Plasticity of sodium channel expression in DRG neurons in the chronic constriction model of neuropathic pain. Pain 83: 591–600. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL (1997) Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods 76: 183–191. [DOI] [PubMed] [Google Scholar]
- Dixon WJ (1980) Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol 20: 441–462. [DOI] [PubMed] [Google Scholar]
- Felts PA, Yokoyama S, Dib-Hajj S, Black JA, Waxman SG (1997) Sodium channel alpha-subunit mRNAs I, II, III, NaG, Na6 and hNE (PN1): different expression patterns in developing rat nervous system. Brain Res Mol Brain Res 45: 71–82. [DOI] [PubMed] [Google Scholar]
- Govrin-Lippmann R, Devor M (1978) Ongoing activity in severed nerves: source and variation with time. Brain Res 159: 406–410. [DOI] [PubMed] [Google Scholar]
- Hains BC, Black JA, Waxman SG (2002) Primary motor neurons fail to up-regulate voltage-gated sodium channel Na(v)1.3/brain type III following axotomy resulting from spinal cord injury. J Neurosci Res 70: 546–552. [DOI] [PubMed] [Google Scholar]
- Hains BC, Eaton MJ, Willis WD, Hulsebosch CE (2003a) Engraftment of serotonergic precursors amends hyperexcitability of dorsal horn neurons after spinal hemisection-induced central sensitization. Neuroscience 116: 1097–1110. [DOI] [PubMed] [Google Scholar]
- Hains BC, Willis WD, Hulsebosch CE (2003b) Temporal plasticity of dorsal horn somatosensory neurons after acute and chronic spinal cord hemisection in rat. Brain Res 970: 238–241. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG (2003c) Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci 23: 8881–8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibuki T, Hama AT, Wang XT, Pappas GD, Sagen J (1997) Loss of GABA-immunoreactivity in the spinal dorsal horn of rats with peripheral nerve injury and promotion of recovery by adrenal medullary grafts. Neuroscience 76: 845–858. [DOI] [PubMed] [Google Scholar]
- Ishigooka M, Zermann DH, Doggweiler R, Schmidt RA, Hashimoto T, Nakada T (2001) Spinal NK1 receptor is upregulated after chronic bladder irritation. Pain 93: 43–50. [DOI] [PubMed] [Google Scholar]
- Iwahashi Y, Furuyama T, Inagaki S, Morita Y, Takagi H (1994) Distinct regulation of sodium channel types I, II and III following nerve transection. Brain Res Mol Brain Res 22: 341–345. [DOI] [PubMed] [Google Scholar]
- Kajander KC, Wakisaka S, Bennett GJ (1992) Spontaneous discharge originates in the dorsal root ganglion at the onset of a painful peripheral neuropathy in the rat. Neurosci Lett 138: 225–228. [DOI] [PubMed] [Google Scholar]
- Kim CH, Oh Y, Chung JM, Chung K (2001) The changes in expression of three subtypes of TTX sensitive sodium channels in sensory neurons after spinal nerve ligation. Brain Res Mol Brain Res 95: 153–161. [DOI] [PubMed] [Google Scholar]
- Kral MG, Xiong Z, Study RE (1999) Alteration of Na+ currents in dorsal root ganglion neurons from rats with a painful neuropathy. Pain 81: 15–24. [DOI] [PubMed] [Google Scholar]
- Laird JM, Bennett GJ (1993) An electrophysiological study of dorsal horn neurons in the spinal cord of rats with an experimental peripheral neuropathy. J Neurophysiol 69: 2072–2085. [DOI] [PubMed] [Google Scholar]
- Leffler A, Cummins TR, Dib-Hajj SD, Hormuzdiar WN, Black JA, Waxman SG (2002) GDNF and NGF reverse changes in repriming of TTX-sensitive Na+ currents following axotomy of dorsal root ganglion neurons. J Neurophysiol 88: 650–658. [DOI] [PubMed] [Google Scholar]
- Matzner O, Devor M (1994) Hyperexcitability at sites of nerve injury depends on voltage-sensitive Na+ channels. J Neurophysiol 72: 349–359. [DOI] [PubMed] [Google Scholar]
- Novakovic SD, Tzoumaka E, McGivern JG, Haraguchi M, Sangameswaran L, Gogas KR, Eglen RM, Hunter JC (1998) Distribution of the tetrodotoxin-resistant sodium channel PN3 in rat sensory neurons in normal and neuropathic conditions. J Neurosci 18: 2174–2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omana-Zapata I, Khabbaz MA, Hunter JC, Bley KR (1997) QX-314 inhibits ectopic nerve activity associated with neuropathic pain. Brain Res 771: 228–237. [DOI] [PubMed] [Google Scholar]
- Palecek J, Paleckova V, Dougherty PM, Carlton SM, Willis WD (1992) Responses of spinothalamic tract cells to mechanical and thermal stimulation of skin in rats with experimental peripheral neuropathy. J Neurophysiol 67: 1562–1573. [DOI] [PubMed] [Google Scholar]
- Rizzo MA (1997) Successful treatment of painful traumatic mononeuropathy with carbamazepine: insights into a possible molecular pain mechanism. J Neurol Sci 152: 103–106. [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Kocsis JD, Waxman SG (1995) Selective loss of slow and enhancement of fast Na+ currents in cutaneous afferent dorsal root ganglion neurones following axotomy. Neurobiol Dis 2: 87–96. [DOI] [PubMed] [Google Scholar]
- Scadding JW (1981) Development of ongoing activity, mechanosensitivity, and adrenaline sensitivity in severed peripheral nerve axons. Exp Neurol 73: 345–364. [DOI] [PubMed] [Google Scholar]
- Somers DL, Clemente FR (2002) Dorsal horn synaptosomal content of aspartate, glutamate, glycine and GABA are differentially altered following chronic constriction injury to the rat sciatic nerve. Neurosci Lett 323: 171–174. [DOI] [PubMed] [Google Scholar]
- Sotgiu ML, Biella G (2000) Contribution of central sensitization to the pain-related abnormal activity in neuropathic rats. Somatosens Mot Res 17: 32–38. [DOI] [PubMed] [Google Scholar]
- Stiller CO, Cui JG, O'Connor WT, Brodin E, Meyerson BA, Linderoth B (1996) Release of gamma-aminobutyric acid in the dorsal horn and suppression of tactile allodynia by spinal cord stimulation in mononeuropathic rats. Neurosurgery 39: 367–375. [DOI] [PubMed] [Google Scholar]
- Study RE, Kral MG (1996) Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain 65: 235–242. [DOI] [PubMed] [Google Scholar]
- Wall PD, Devor M (1983) Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 17: 321–339. [DOI] [PubMed] [Google Scholar]
- Wall PD, Gutnick M (1974) Properties of afferent nerve impulses originating from a neuroma. Nature 248: 740–743. [DOI] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD, Black JA (1994) Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is re-expressed following axotomy. J Neurophysiol 72: 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ (1983) Evidence for a central component of post-injury pain hypersensitivity. Nature 306: 686–688. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Wall PD (1986) Relative effectiveness of C primary afferent fibers of different origins in evoking a prolonged facilitation of the flexor reflex in the rat. J Neurosci 6: 1433–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Donnelly DF, Song XJ, Lamotte RH (1997) Axotomy increases the excitability of dorsal root ganglion cells with unmyelinated axons. J Neurophysiol 78: 2790–2794. [DOI] [PubMed] [Google Scholar]