

Abstract
A fundamental question about the early pathogenesis of Alzheimer's disease (AD) concerns how toxic aggregates of amyloid β protein (Aβ) are formed from its nontoxic soluble form. We hypothesized previously that GM1 ganglioside-bound Aβ (GAβ) is involved in the process. We now examined this possibility using a novel monoclonal antibody raised against GAβ purified from an AD brain. Here, we report that GAβ has a conformation distinct from that of soluble Aβ and initiates Aβ aggregation by acting as a seed. Furthermore, GAβ generation in the brain was validated by both immunohistochemical and immunoprecipitation studies. These results imply a mechanism underlying the onset of AD and suggest that an endogenous seed can be a target of therapeutic strategy.
Keywords: Alzheimer's disease, amyloid, amyloid β protein, seed, ganglioside, raft
Introduction
The lifelong expression of genetic mutations responsible for familial Alzheimer's disease (AD) appears to induce the formation of neurotoxic aggregates of amyloid β protein (Aβ) by accelerating cellular Aβ generation (Selkoe, 1997). However, there is currently no evidence that Aβ generation is enhanced in sporadic, late-onset AD, the principal form of the disease. Thus, it is reasonable to assume that Aβ aggregation in conventional AD may be induced by unknown posttranslational modification(s) of Aβ and/or by altered clearance mechanisms.
We previously identified a novel Aβ species, characterized by its tight binding to GM1 ganglioside (GM1), in human brains that exhibited early pathological changes associated with AD (Yanagisawa et al., 1995, 1997; Yanagisawa and Ihara, 1998). On the basis of the molecular characteristics of the GM1-bound Aβ (GAβ), including its altered immunoreactivity and a strong tendency to form aggregates of Aβ, we hypothesized that Aβ adopts an altered conformation by binding to GM1 and initiates the aggregation of soluble Aβ by acting as a seed (Yanagisawa et al., 1995, 1997; Yanagisawa and Ihara, 1998). Evidence that supports our hypothesis is growing from in vitro studies by our group and other groups (McLaurin and Chakrabartty, 1996; Choo-Smith and Surewicz, 1997; Choo-Smith et al., 1997; McLaurin et al., 1998; Matsuzaki and Horikiri, 1999; Ariga et al., 2001; Kakio et al., 2001, 2002).
In the present study, we directly characterize GAβ at the molecular level using a novel monoclonal antibody raised against GAβ purified from an AD brain with the aim of validating our hypothesis. Here we show that GAβ has a conformation distinct from that of soluble Aβ and initiates Aβ aggregation by acting as a seed. Importantly, we successfully verified GAβ generation in the brain by both immunohistochemical and immunoprecipitation methods.
Materials and Methods
Preparation of seed-free Aβ solutions. Aβ solutions were prepared essentially according to a previous report (Naiki and Gejyo, 1999). Briefly, synthetic Aβ (Aβ40 and Aβ42) (Peptide Institute, Osaka, Japan) was dissolved in 0.02% ammonia solution at 500 μm for Aβ40. Aβ42 was dissolved at 250 μm because it has a higher potential to form aggregates rapidly than Aβ40. To obtain seed-free Aβ solutions, the prepared solutions were centrifuged at 540,000 × g for 3 hr using the Optima TL Ultracentrifuge (Beckman Instruments, Fullerton, CA) to remove undissolved peptide aggregates, which can act as preexisting seeds. The supernatant was collected and stored in aliquots at –80°C until use. Immediately before use, the aliquots were thawed and diluted with Tris-buffered saline (TBS) (150 mm NaCl and 10 mm Tris-HCl, pH 7.4). In the present study, we used the seed-free Aβ solutions, except in the preparation of Aβ fibrils used as the seeds (see below).
Preparation of Aβ40 seeds. Aβ40 fibrils used as exogenous seeds were prepared essentially according to the method reported previously (Naiki and Nakakuki, 1996). Briefly, Aβ40 solution was incubated at 500 μm and 37°C for 72 hr without previous removal of the preexisting seeds. After incubation, newly formed Aβ40 fibrils were precipitated by ultracentrifugation. The resultant pellets were subjected to sonication. Protein concentrations of the solutions containing Aβ40 fibrils were determined using a protein assay kit (Bio-Rad, Hercules, CA) as described previously (Bradford, 1976). The Aβ40 solution quantified by amino acid analysis was used as the standard. Aliquots of the solution were stored at –80°C until use.
Preparation of liposomes. Cholesterol and sphingomyelin (Sigma, St. Louis, MO) and GM1 (Wako, Osaka, Japan) were dissolved in chloroform/methanol (1:1) at a molar lipid ratio of 2:2:1 to generate GM1-containing liposomes. GM1-lacking liposomes were prepared by mixing cholesterol and sphingomyelin at a molar lipid ratio of 1:1. The mixtures were stored at –80°C until use. Immediately before use, the lipids were resuspended in TBS at a GM1 concentration of 2.5 mm and subjected to freezing and thawing. The lipid suspension was centrifuged once at 15,000 × g for 15 min, and the resultant pellet was resuspended in TBS. Finally, the suspension was subjected to sonication on ice.
Thioflavin T assay. The assay was performed according to a method described previously (Naiki and Gejyo, 1999) on a spectrofluorophotometer (RF-5300PC; Shimadzu, Tokyo, Japan). Optimum fluorescence measurements of amyloid fibrils were obtained at the excitation and emission wavelength of 446 and 490 nm, respectively, with the reaction mixture (1.0 ml) containing 5 μm thioflavin T (ThT) (Sigma) and 50 mm glycine-NaOH buffer, pH 8.5. Fluorescence was measured immediately after making the mixture. The Aβ (Aβ40 and Aβ42) solution described above was incubated at 37°C with liposomes at an Aβ concentration of 50 μm at a GM1/Aβ molar ratio of 10:1.
Detection of Aβ oligomers by SDS-PAGE. The Aβ40 solution described above was incubated with liposomes at an Aβ concentration of 50 μm at a GM1/Aβ molar ratio of 10:1 for 24 hr at 37°C and then centrifuged at 100,000 × g for 15 min. The supernatant was subjected to SDS-PAGE after glutaraldehyde treatment (LeVine, 1995) to stabilize oligomeric Aβ during SDS-PAGE. The Aβ in the gel was detected by Western blotting using BAN052, a monoclonal antibody specific to the N terminus of Aβ (Suzuki et al., 1994), as reported previously (Yanagisawa et al., 1995).
Experiments using animal models. All experiments using animals were performed in compliance with existing laws and institutional guidelines. For experiments using nonhuman primates, animals were anesthetized with pentobarbital (25 mg/kg, i.p.) and killed by draining blood from the heart.
Cell culture. Cerebral cortical neuronal cultures were prepared from Sprague Dawley rats at embryonic day 17 as described previously (Michikawa and Yanagisawa, 1998). The dissociated single cells were suspended in a feeding medium and plated onto poly-d-lysine-coated 12-well plates at a cell density of 2.5 × 105/well. The feeding medium, N2, consisted of DMEM–F-12 containing 0.1% bovine serum albumin fraction V solution (Invitrogen, Gaithersburg, MD) and N2 supplements. The reagents examined, including TBS, Aβ40, GM1-lacking liposome, and GM1-containing liposome in the absence or presence of Aβ40, were prepared as described above. For treatment, at 24 hr after plating, the culture medium was changed with fresh N2 medium diluted with the same volume of each reagent solution. At 48 hr after the commencement of the treatment, phase-contrast photomicrographs of each culture were taken, and the cells were stained with propidium iodide, which selectively permeates the broken membranes of dying cells and stains their nuclei, and with a viable cell-specific marker, calcein AM, as described previously (Michikawa and Yanagisawa, 1998). Photomicrographs were taken by laser confocal microscopy (Zeiss, Oberkochen, Germany) and the number of viable neurons on each micrograph was determined in each microscope field (40× objective). Two hundred to 500 cells were counted for each determination of cell viability. Statistical analysis was performed using ANOVA.
Production of 4396C. The IgG monoclonal antibody 4396C was produced by the genetical class-switch technique (Binding and Jones, 1996) from IgM hybridomas that were raised against GAβ purified from an AD brain. The procedures for the generation and characterization of the original IgM monoclonal antibody 4396 were reported previously (Yanagisawa et al., 1997).
Immunoelectron microscopy. GM1-containing and GM1-lacking liposomes were mixed with soluble Aβ40 on ice for 5 sec at a weight ratio of lipid/Aβ40 at 100:3. The mixtures were immediately ultracentrifuged to remove unbound Aβ40, and liposome pellets were fixed for immunoelectron microscopy of 4396C or isotype-matched control IgG staining. They were then incubated with gold-tagged goat anti-mouse IgG. Aβ40 fibrils formed by the extension reaction of Aβ40 seeds (10 μg/ml) with seed-free Aβ40 (50 μm) as described above, were also subjected to immunoelectron microscopy of 4396C, isotype-matched control IgG, or 4G8 (Kim et al., 1988) staining. The first antibodies and control IgG were diluted at 1:100 using PBS containing 1% bovine serum albumin (PBS-BSA).
Quantitative binding assay. GM1-containing liposomes and GM1-lacking liposomes were mixed with soluble Aβ40 at various concentrations (2.5–25 μm) by vortexing for 60 sec, and the mixtures were ultracentrifuged at 540,000 × g for 10 min to remove unbound Aβ40. Then, 4396C and isotype-matched control IgG, at a concentration of 10 μg/ml in PBS-BSA, were incubated with the liposomes at room temperature for 60 min. After incubation, the mixtures were ultracentrifuged at 540,000 × g for 20 min, and the resultant pellets were washed with PBS-BSA to remove the unbound antibody. The levels of IgG bound to the liposomes were determined after the incubation of liposomes with peroxidase-conjugated goat anti-mouse IgG using cyanogen bromide Sepharose CL4B-bound 4396C as the standard.
Dot blot analysis. Liposomes carrying GAβ were prepared by mixing GM1-containing liposomes with soluble Aβ (Aβ40 and Aβ42) on ice for 5 sec at a weight ratio of lipid/Aβ at 100:3. The liposomes, and Aβ and GM1, in amounts equal to those contained in blotted liposomes (300 and 600 ng of Aβ; 2 and 4 μg of GM1), were blotted. The blots were incubated with 4396C (1:1000), BAN052 (1:5000), HRP-conjugated cholera toxin subunit B (CTX) (1:20,000), or the isotype-matched control IgG (1: 1000). The blots incubated with 4396C, BAN052, or control IgG were then incubated with horseradish peroxidase-conjugated anti-mouse IgG (Invitrogen). The bound-enzyme activities were visualized with an ECL system (Amersham Biosciences, Buckinghamshire, UK).
Inhibition assay of Aβ aggregation. Soluble Aβ40 (50 μm) was incubated at 37°C with GM1-containing liposomes (GM1/Aβ molar ratio of 10:1) or preformed Aβ40 seeds (10 μg/ml), which were prepared as described above, and an antibody (4396C or 4G8) at various concentrations. Aβ40 aggregation levels in the mixtures were determined by ThT assay.
Immunohistochemistry. Sections of cerebral cortices of human brains, which were fixed in 4% formaldehyde or Kryofix (Merck, Darmstadt, Germany) and embedded in paraffin, were immunolabeled with 4396C (10 μg/ml) or 4G8 (1 μg/ml) after pretreatment with formic acid (99%) or SDS (4%). Sections of cerebral cortices of nonhuman primates at various ages (4, 5, 17, 19, 20, 30, and 36 years old) were fixed with 1% paraformaldehyde and subjected to immunohistochemistry with 4396C (10 μg/ml). For double staining with 4396C and BAN052, we first labeled 4396C with FITC to distinguish its reaction from that of BAN052. The binding of BAN052 was detected using Alexa 568-conjugated anti-mouse IgG (Molecular Probes, Eugene, OR). For double staining with 4396C and CTX, we used a zenon one-mouse IgG2a labeling kit (Molecular Probes) for the previous conjugation of 4396C with Alexa 568. For GM1 detection, we used FITC-conjugated cholera toxin subunit B (Sigma). Autofluorescence was blocked by pretreatment with Sudan Black B (Tokyo Kasei Kogyo Company, Tokyo, Japan).
Immunoprecipitation. Immunoprecipitation of GAβ from nonhuman primate brains was performed as described previously (Yanagisawa and Ihara, 1998). Briefly, cerebral cortices of primates were Dounce homogenized with 9 vol of TBS, pH 7.6. Homogenates were subjected to sucrose density gradient fractionation to obtain the membrane fraction. The membrane fraction was dried and then directly labeled with 4396C (5 μg/ml) after sonication on ice. After 1 hr incubation, the mixtures were centrifuged at 15,000 × g for 10 min to remove the unbound antibody. The pellets were washed with TBS and solubilized in radioimmunoprecipitation assay buffer (0.1% SDS, 0.5% deoxycholic acid, and 1% NP-40) for 5 min and then centrifuged at 15,000 × g for 10 min. Supernatants were collected and diluted with TBS. Protein G Sepharose (PGS) (Amersham Biosciences, Piscataway, NJ), which had been precoated with goat anti-mouse IgG, was added to the supernatants. Finally, PGS pellets were thoroughly washed with TBS containing 0.05% Tween 20.
Results
Kinetics of Aβ fibrillogenesis in the presence of GM1 ganglioside
As reported previously (Naiki et al., 1998; Ding and Harper, 1999), the removal of preexisting seeds is critical in the kinetic study of Aβ fibril formation in vitro. The ThT fluorescence intensity of Aβ42 solutions without removing undissolved peptide aggregates, which can act as preexisting seeds, started to increase as early as 1 hr of incubation at 50 μm and 37°C (Fig. 1a). In contrast, the ThT fluorescence intensity of seed-free Aβ42 solutions did not increase as long as 6 hr of incubation under the same conditions (Fig. 1a). The ThT fluorescence intensity of Aβ40 solutions without removing undissolved peptide aggregates started to increase after 4 d of incubation at 50 μm and 37°C (Fig. 1a); however, the seed-free Aβ40 solutions incubated under the same conditions did not show any increase in the ThT fluorescence intensity at this point (Fig. 1a). Thus, to investigate the molecular process of GAβ-initiated Aβ aggregation, we incubated the seed-free Aβ (Aβ40 and Aβ42) solutions at 50 μm and 37°C under various conditions, as long as 48 and 6 hr for Aβ40 and Aβ42, respectively, in the following experiments.
Figure 1.
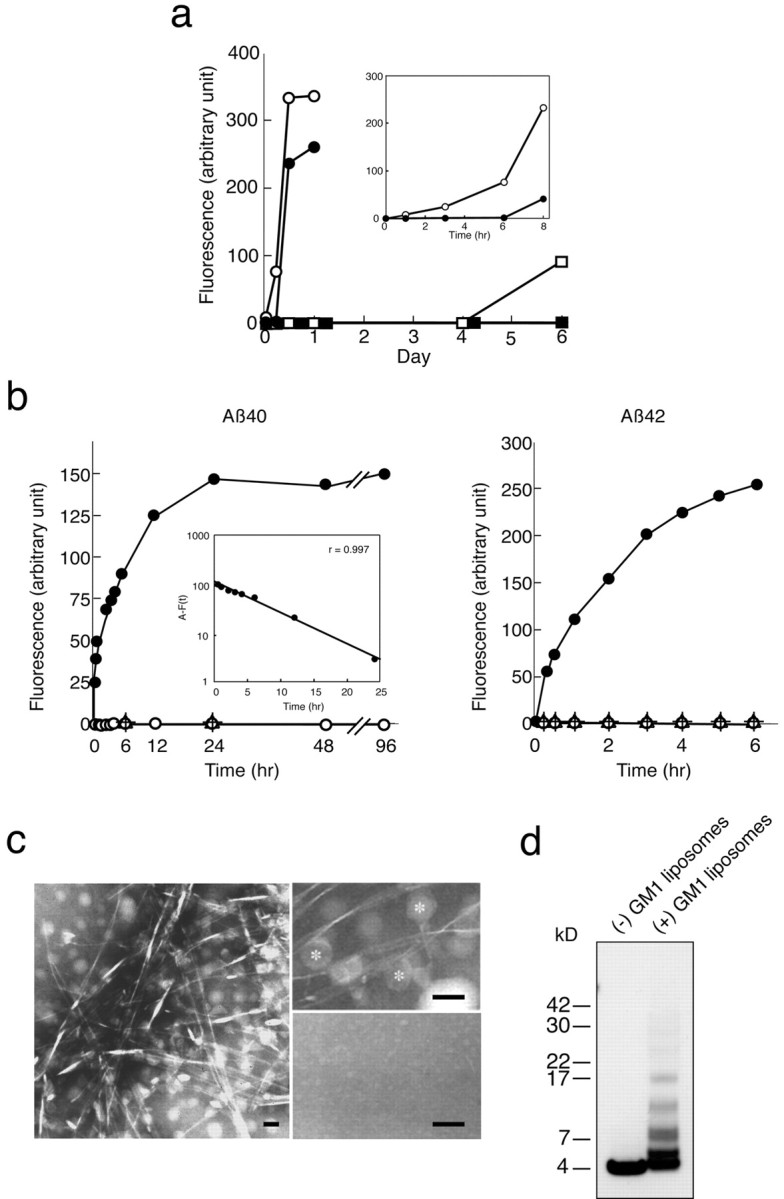
Amyloid fibril formation from soluble Aβ in the absence or presence of GM1-containing liposomes. a, Kinetics of Aβ fibrillogenesis using Aβ (Aβ40 and Aβ42) solutions, with or without removing undissolved peptide aggregates, which can act as preexisting seeds. Aβ solutions were incubated at 50 μm and 37°C. Open and filled circles indicate ThT fluorescence intensities of Aβ42 solutions without and with removing undissolved peptide aggregates, respectively. Open and filled squares indicate ThT fluorescence intensities of Aβ40 solutions without and with removing undissolved peptide aggregates, respectively. b, Kinetics of Aβ fibrillogenesis. Aβ (Aβ40 and Aβ42) solutions, after removal of undissolved peptide aggregates, were incubated at 50 μm and 37°C in the presence of GM1-containing liposomes (filled circles) or GM1-lacking liposomes (plus signs) or were incubated in the absence of liposomes (open circles). The GM1-containing liposomes alone were also incubated in the absence of Aβ (triangles). The fluorescence intensity of thioflavin T was obtained by excluding background activity at 0 hr. Inset, Semilogarithmic plot of the difference, A – F(t), versus incubation time (0–24 hr). F(t) represents the increase in fluorescence intensity as a function of time in the case of Aβ incubated with GM1-containing liposomes, and A is tentatively determined as F (infinity). Linear regression and correlation coefficient values were calculated (r = 0.997). F(t) is described by a differential equation: F′ (t) = B – CF(t). c, Electron micrographs of the Aβ40 solutions incubated at 50 μm and 37°C for 24 hr with GM1-containing liposomes (left and right top panels) or without liposomes (right bottom panel). The liposomes are indicated by asterisks. Scale bars, 50 nm. d, Westernblot of Aβ40 solutions incubated at 50 μm and 37°C for 24 hr in the presence or absence of GM1-containing liposomes. The incubated Aβ solutions were centrifuged at 100,000 × g for 15 min. Ten nanograms of Aβ in the supernatant were subjected to SDS-PAGE (4–20%) after glutaraldehyde treatment. The Aβ in the gel was detected by Westernblotting using BAN052. Aβ oligomers were detected in the Aβ solution incubated in the presence of GM1-containing liposomes but not detected in the incubation mixture containing Aβ alone.
We incubated Aβ40 solutions in the presence or absence of GM1-containing liposomes. The ThT fluorescence intensity increased without a lag phase after the addition of GM1-containing liposomes to the Aβ40 solutions (Fig. 1b). In contrast, there was no increase in the ThT fluorescence intensity of the solutions containing Aβ40 alone or Aβ40 plus GM1-lacking liposomes during an incubation period of as long as 96 hr (Fig. 1b). By a semilogarithmic calculation, a perfect linear plot (r = 0.997) was obtained for the experiment using GM1-containing liposomes (Fig. 1b, inset). The fluorescence intensity of ThT also increased in seed-free Aβ42 solution after the addition of GM1-containing liposomes (Fig. 1b). Under an electron microscope, typical amyloid fibrils were observed in the Aβ40 solution after 24 hr incubation at 50 μm and 37°C in the presence of GM1-containing liposomes (Fig. 1c). These results suggest that Aβ binds to GM1, leading to the generation of GAβ, and then induces Aβ fibrillogenesis in the manner of a first-order kinetic model (Naiki and Nakakuki, 1996) by acting as a seed; that is, the extension of fibrils is likely to proceed via consecutive binding of soluble Aβ first onto GAβ and then onto the ends of growing fibrils.
We then investigated whether the formation of Aβ oligomers is also acclerated in the presence of GM1 ganglioside. We perfomed SDS-PAGE of the Aβ40 solutions incubated at 50 μm and 37°C for 24 hr. Notably, Aβ oligomers were detected by Western blotting of the SDS-PAGE in the incubation mixture with GM1-containing liposomes but was not detected in the incubation mixture containing Aβ40 alone (Fig. 1d).
Neurotoxicity of Aβ incubated in the presence of GM1 ganglioside
We then investigated whether Aβ aggregates formed in the presence of GM1-containing liposomes are neurotoxic. We incubated Aβ40 solutions in the presence or absence of GM1-containing liposomes for 24 hr and then applied them to a primary neuronal culture after dilution with N2 media. In this experiment conducted 48 hr after the commencement of the treatment, significant neuronal death was observed only in the culture treated with the Aβ40 solution incubated in the presence of GM1-containing liposomes (Fig. 2).
Figure 2.
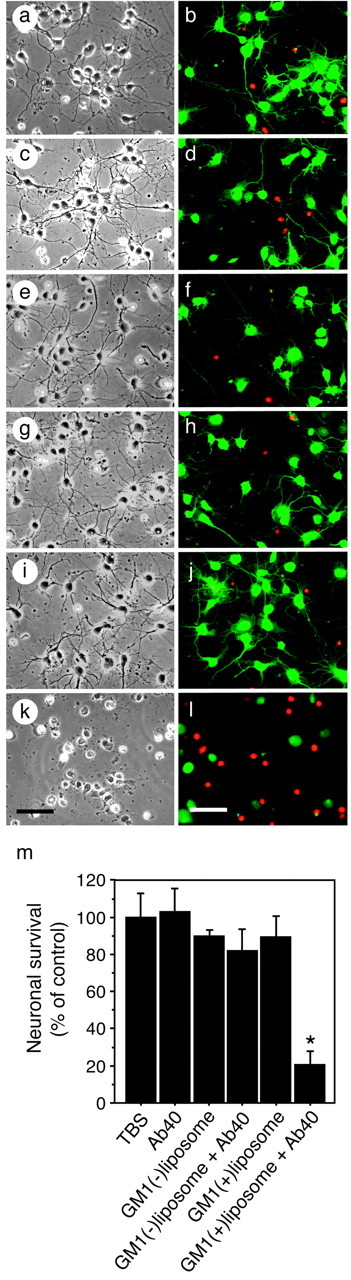
Viability of neurons treated with Aβ40 incubated in the presence of GM1 ganglioside. Phase-contrast and calsein AM–ethidium homodimer-stained photographs of cultured neurons were taken after treatment with TBS (a, b), Aβ40 (c, d), GM1-lacking liposomes (e, f), GM1-lacking liposomes plus Aβ40 (g, h), GM1-containing liposomes (i, j), and GM1-containing liposomes plus Aβ40 (k, l). m, Viable cells stained with calcein AM were counted. The data represent means ± SE for triplicate samples. *p < 0.003 versus other treatments. Scale bars, 20 μm.
Molecular characterization of GAβ
To characterize GAβ at the molecular level and to clarify the process of Aβ aggregation in the presence of GM1, we raised a monoclonal antibody against natural GAβ purified from an AD brain. In an experiment using immunoelectron microscopy, the antibody (4396C), but not the isotype-matched control IgG, recognized artificially generated GAβ on liposomes (Fig. 3a). The specificity of 4396C to GAβ was confirmed by quantitative binding assay (Fig. 3b) and dot blot analysis (Fig. 3c, left). Notably, 4396C did not recognize the unbound forms of Aβ40 and GM1, whereas BAN052, a monoclonal antibody specific to the N terminus of Aβ (Suzuki et al., 1994), recognized both GAβ40 and Aβ (Fig. 3c, left). The 4396C antibody reacted with GM1-bound forms of two Aβ isoforms (Aβ40 and Aβ42) (Fig. 3c, right). In the inhibition assay of Aβ fibrillogenesis, the increase in the fluorescence intensity of ThT of the Aβ40 plus GM1-containing liposomes was suppressed by 4396C in a dose-dependent manner (Fig. 3d, left). In contrast, the increase in the fluorescence intensity of ThT was not affected by 4G8, a monoclonal antibody specific to amino acid residues 17–24 of Aβ (Kim et al., 1988) (Fig. 3d, left). We then examined the possibility that Aβ fibrillogenesis proceeds with consecutive conformational alteration of Aβ at the ends of growing fibrils; that is, GAβ and Aβ at the ends of growing fibrils share a specific conformation that is required for Aβ fibrillogenesis in the manner of a first-order kinetic model. We incubated Aβ40 solutions with 4396C in the presence of preformed Aβ40 seeds instead of GM1-containing liposomes. Notably, 4396C inhibited the increase in the fluorescence intensity of ThT under this condition in a dose-dependent manner (Fig. 3d, right). To further examine this possibility, we performed immunoelectron microscopy using preformed Aβ40 fibrils. In this experiment, 4396C bound only to the ends but not to the lateral sides of the fibrils, whereas 4G8 bound only to the lateral sides (Fig. 3e).
Figure 3.
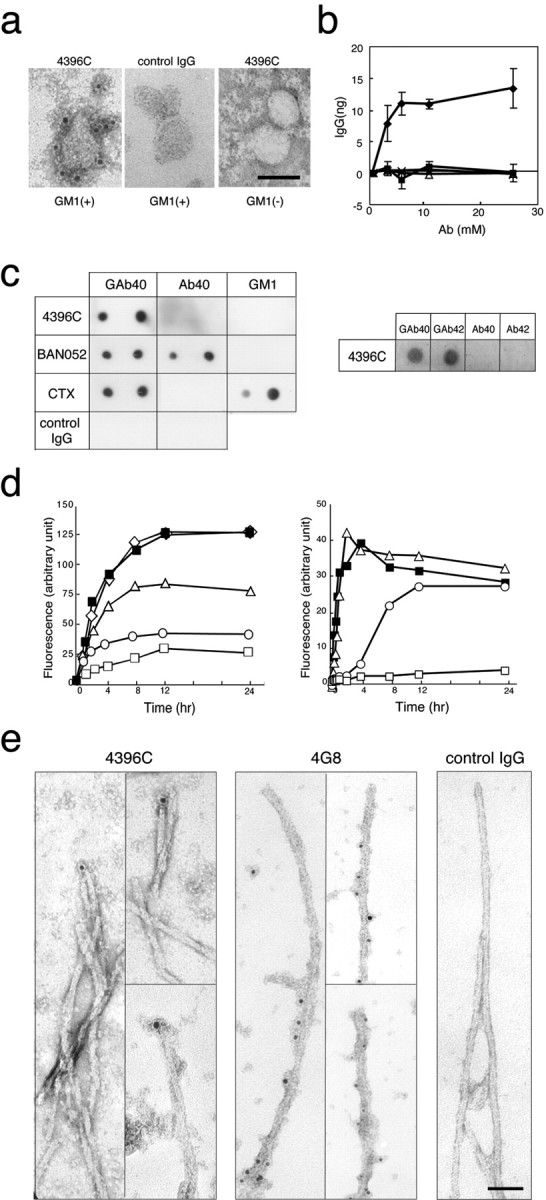
Characterization of the binding specificity of 4396C. a, Immunoelectron micrographs of liposomes. GM1-containing and GM1-lacking liposomes were subjected to immunoelectron microscopy of 4396C or isotype-matched control IgG staining after incubation with soluble Aβ40. GM1(+), GM1-containing liposomes; GM1(–), GM1-lacking liposomes. Scale bar, 50 nm. b, Quantitative assay of binding of 4396C to liposomes. GM1-containing liposomes were incubated with 4396C (diamonds) or isotype-matched control IgG (filled squares) after their mixing with soluble Aβ40 at indicated concentrations. GM1-lacking liposomes were also incubated with 4396C (triangles) or isotype-matched control IgG (× symbols). Ab, Antibody. c, Dot blot analysis. Left, Liposomes carrying GAβ (GAβ40), Aβ40, and GM1 in amounts equal to those contained in blotted liposomes (300 and 600 ng of Aβ40; 2 and 4 μg of GM1) were blotted. The blots were incubated with 4396C, BAN052, HRP-conjugated CTX, or isotype-matched control IgG. Right, Liposomes carrying GAβ (GAβ40 and GAβ42), prepared using Aβ40 or Aβ42, and Aβ40 and Aβ42 in amounts equal to those contained in GAβ40 and GAβ42 (600 ng of each peptide) were blotted. The blots were incubated with 4396C. d, Inhibition of amyloid fibril formation from soluble Aβ40 by 4396C. Left, Soluble Aβ40 was incubated with GM1-containing liposomes in the absence (filled squares) or presence of an antibody (4396C or 4G8). The molar ratios of 4396C to soluble Aβ40 were 0.3:50 (triangles), 1.3:50 (circles), and 4:50 (open squares) and that of 4G8 to Aβ40 was 4:50 (diamonds). Right, Soluble Aβ40 was incubated with preformed Aβ40 fibrils in the absence of an antibody (filled squares) or in the presence of 4396C. The molar ratios of 4396C to soluble Aβ40 were 0.3:50 (triangles), 1.3:50 (circles), and 4:50 (open squares). e, Immunoelectron micrographs of preformed Aβ40 fibrils. Aβ40 fibrils were formed by the extension reaction of Aβ40 seeds (10 μg/ml) with seed-free Aβ40 (50 μm), as described in Materials and Methods, and subjected to immunoelectron microscopy of 4396C, 4G8, or isotype-matched control IgG staining. Scale bar, 50 nm.
GAβ generation in the brain
Having established the specificity of 4396C, we then aimed to detect GAβ in the brain by immunohistochemistry. We first performed routine immunohistochemistry: that is, fixation in formaldehyde and enhancement of immunoreactivity by formic acid or microwave treatment of brain sections. Under these conditions, no immunostaining by 4396C was observed in AD brains (data not shown), suggesting that the conformation of GAβ is sensitive to the procedures of conventional immunohistochemistry. Thus, we used an alternative fixation procedure using Kryofix, because it eliminates the possibility of obtaining false-negative results that usually occur when using formaldehyde-fixed sections (Boon and Kok, 1991), and we also pretreated sections with SDS, which is known to improve immunostaining (Barrett et al., 1999). With these procedures, neurons in the cerebral cortices of AD brains were immunostained by 4396C (Fig. 4a). Although neuropil staining was rather strong, plaques were not recognized by 4396C (Fig. 4a). In contrast to staining by 4396C, 4G8 immunostained plaques but did not react with neurons (Fig. 4a). We performed immunohistochemistry of cerebral cortices of 22 nondemented control individuals (36–91 years old), which included one amyotrophic lateral sclerosis patient (44 years old) and two Down's syndrome (DS) patients (47 and 52 years old), in addition to five AD patients (65–96 years old). The most intense neuronal staining by 4396C was observed in the brains of both DS patients (Fig. 4b). In the sections of control brains, neuronal staining was absent (Fig. 4b) or at comparable levels with those of AD brains (Table 1). These results suggest that GAβ can be detected by immunohistochemistry, but neuronal staining by 4396C under these conditions can also be nonspecifically induced, probably because of changes that occur during the agonal and/or postmortem state. Thus, to confirm the immunohistochemical detection of GAβ, we then examined fresh nonhuman primate (Macaca fascicularis) brains, which naturally and consistently develop Aβ deposition at ages >25 years (Nakamura et al., 1998). Cerebral cortices of seven animals at different ages (4, 4, 5, 17, 19, 30, and 36 years old) were fixed with paraformaldehyde, because it also preserves tissue and cell surface antigens (Smit et al., 1974), and were subjected to 4396C immunostaining without pretreatment with SDS. In the sections obtained from the two older animals, that is, 30 years old (data not shown) and 36 years old (Fig. 5a), a number of neurons were strongly immunostained by 4396C with a granular pattern (Fig. 5a, inset). In these sections, plaques were immunostained by 4G8 but not by 4396C (data not shown). In the sections of cerebral cortices of the five animals at ages <20 years old, which showed no plaques, the neuronal staining by 4396C was generally at negligible levels, and the strong staining was only occasionally observed in the sections from the 17-year-old (Fig. 5a) and 19-year-old (data not shown) animals. In double immunostaining of the sections obtained from the 36-year-old animal, intraneuronal staining by 4396C was distinctly colocalized with that by BAN052 or cholera toxin, a natural ligand specific to GM1 (Fig. 5b). To verify GAβ generation in the brain, we performed an immunoprecipitation study using 4396C. GAβ was immunoprecipitated by 4396C only from the cerebral cortex of the older animal (Fig. 5c). These results indicate that GAβ is generated in the brain.
Figure 4.
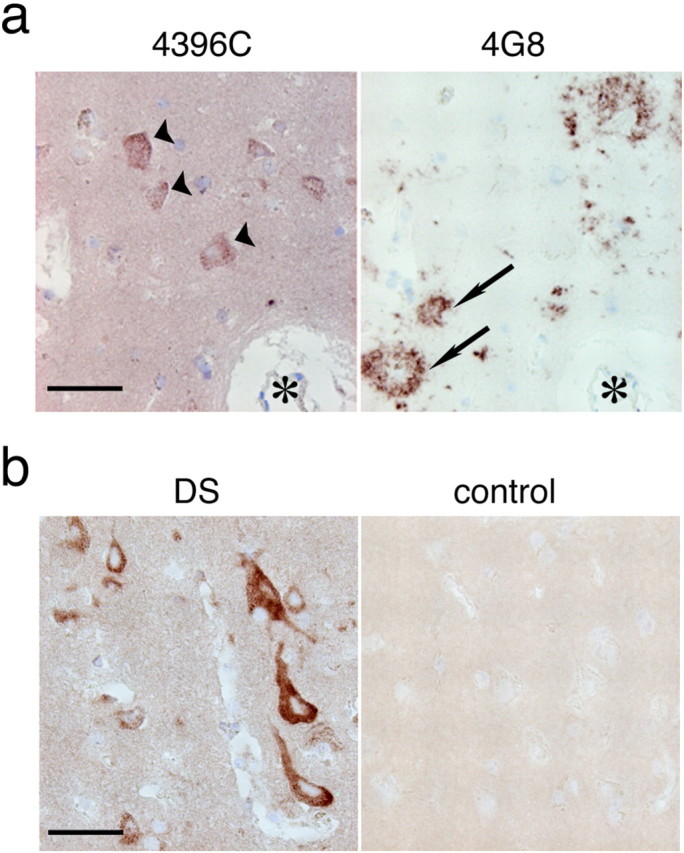
Immunohistochemistry of GAβ in sections of human brains. a, Immunostaining of serial sections of the cerebral cortex of an AD brain fixed in Kryofix and pretreated with SDS. Neurons (arrowheads) were immunostained by 4396C but not by 4G8, whereas plaques (arrows) were immunostained by 4G8 but not by 4396C. The asterisks indicate the same blood vessel in the serial sections. Scale bar, 50 μm. b, Immunostaining by 4396C of sections of cerebral cortices of DS (left) and control (right) brains fixed in Kryofix and pretreated with SDS (DS, 47 years old; control, 65 years old). Scale bar, 50 μm.
Table 1.
Neuronal immunoreactivity to 4396C in human cerebral cortices
|
|
Score |
|
|
|
|
|---|---|---|---|---|---|
|
|
0 |
1 |
2 |
3 |
4 |
| Control (22) | 4 | 5 | 12 | 1 | 0 |
| AD (5) | 0 | 2 | 2 | 1 | 0 |
| DS (2) |
0 |
0 |
0 |
0 |
2 |
Intensities of neuronal immunoreactivity to 4396C were semiquantitatively assessed in cerebral cortices obtained from nondemented individuals (Control), patients with AD, and those with DS. The numbers in parentheses indicate the number of cases. 0, Absent; 1, weak; 2, moderate; 3, intense; 4, most intense. Difference in the neuronal immunoreactivities between control and AD plus DS was significant (p < 0.05, Mentel-Haenszel χ2 test; p < 0.05, Cochran-Armitage trend test).
Figure 5.
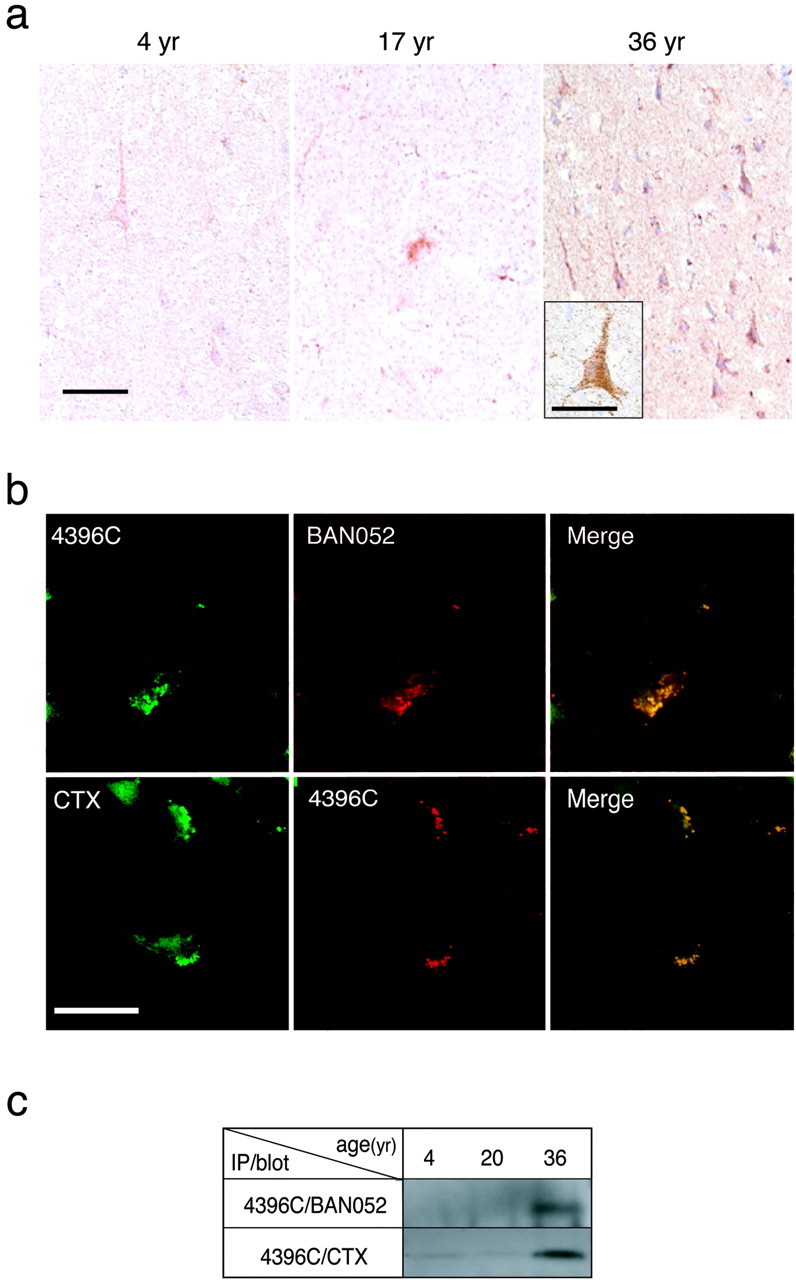
Immunohistochemistry and immunoprecipitation of GAβ in sections of nonhuman primate brains. a, Immunostaining by 4396C of sections of the cerebral cortices of primate brains, which were fixed in paraformaldehyde, from animals of different ages. Scale bar, 50 μm. Inset, Higher magnification. Scale bar, 20 μm. b, Double immunostaining of sections of the cerebral cortex of a 36-year-old primate brain, which was fixed in paraformaldehyde, after the blocking of autofluorescence by pretreatment with Sudan Black B. Colocalization of immunostaining by 4396C and that by BAN052 or CTX is shown in the merged image. Scale bar, 25 μm. c, Immunoprecipitation of GAβ by 4396C from cerebral cortices of primates at different ages. Immunoprecipitates were blotted and reacted with BAN052 or HRP-conjugated CTX.
Discussion
In regard to the formation of pathogenic aggregates of constituent proteins, including Aβ and prion proteins, the seeded polymerization theory was proposed previously (Harper and Lansbury, 1997). For the transition of the nontoxic monomeric form of Aβ to its toxic aggregated form, the template-dependent dock-lock mechanism was reported previously (Esler et al., 2000). The present study supports these possibilities and also indicates that a seed can be endogenously generated by the binding of an aggregating protein to another molecule as was suggested in the mechanism underlying the aggregation of prion proteins (Telling et al., 1995; Deleault et al., 2003).
To understand the early events in the development of AD and also to develop tharapeutic strategies, clarification of the time course of Aβ fibrillogenesis is fundamentally important. Previously, Harper et al. analyzed the process of in vitro Aβ assembly using an atomic force microscope at a fine resolution. They reported that protofibrils, transient species of Aβ assembly, are formed during the first week of incubation of Aβ40 before mature fibrils are generated (Harper et al., 1997a,b, 1999; Ding and Harper, 1999). This model of Aβ assembly was supported by a recent study of Nichols et al. (2002). In the present study, we examined the acceleration of Aβ aggregation in the presence of GM1 ganglioside. In the EM examination of the present study, we observed mature fibrils in Aβ40 solutions incubated at 50 μm and 37°C for 24 hr, but protofibrils were hardly recognized (Fig. 1c). This discrepancy is likely to mainly stem from the presence or absence of GM1-containing liposomes and the differences in incubation period and peptide concentrations. Additional careful examination at a much greater resolution is required; however, the molecular process of Aβ fibrillogenesis in the presence of GM1 ganglioside may be different from that initiated by spontaneous nucleation from seed-free Aβ solution.
From the results of molecular characterization of GAβ in the present study, it seems likely that Aβ adopts an altered conformation at its midportion through binding to GM1 because GAβ was readily recognized by BAN052, an antibody specific to the N terminus of Aβ, and 4396C comparably recognized two Aβ isoforms with different lengths at their C termini. This possibility is supported by the following: first, 4G8, an antibody specific to the midportion of Aβ, failed to react with GAβ in the inhibition assay of Aβ aggregation; and second, BAN052, but not 4G8, immunoprecipitated GAβ from the cerebral cortex of an AD brain in our previous study (Yanagisawa and Ihara, 1998). The conformational epitope for 4396C on the Aβ molecule remains to be determined; however, the results of the present study suggest that the 4396C-reactive conformation, which is shared by GAβ and Aβ at the ends of fibrils, is necessary for Aβ fibrillogenesis. Because amyloid fibrils composed of various proteins share a common structure that is readily recognized by Congo red, it may be interesting to study in the future whether the 4396C-reactive conformation is shared by seed or oligomer of other amyloidogenic proteins. Regarding this, we must draw attention to a recent report by Glabe and his colleagues (Kayed et al., 2003): they have successfully generated an antibody that potentially recognizes the common structure of soluble amyloid oligomers. Their results suggest that the oligomers have a conformation that is distinct from those of soluble monomers and amyloid fibrils.
In this study, only careful immunohistochemistry allowed us to visualize seed molecules, suggesting that we may likely fail to detect some population of seed molecules unless their conformations are preserved during immunohistochemistry. This may also be the case with other neurodegenerative diseases in which seed molecules are likely to play a critical role in the initiation of aggregation of soluble proteins. In this study, it remains to be clarified why 4396C did not immunostain plaques in which the ends of amyloid fibrils were supposed to exist. Possible explanations for this failure are as follows; first, the number of epitopes that can be recognized by the antibody may be limited, as was clearly indicated by immunoelectron microscopy using fibrils; second, we may have lost their immunoreactivities because of their higher susceptibility to treatment in immunohistochemistry than GAβ; and third, the ends of amyloid fibrils may have been masked or modified in the brain (Shapira et al., 1988; Roher et al., 1993).
A challenging subject of studies in the future is determining how and where GAβ is generated in the brain. Regarding this issue, we favor the possibility that GAβ is generated in GM1- and cholesterol-rich microdomains such as lipid rafts (Parton, 1994; Simons and Ikonen, 1997), because of the following: (1) lipid rafts contain soluble and insoluble Aβs under physiological (Lee et al., 1998; Morishima-Kawashima and Ihara, 1998) and pathological (Sawamura et al., 2000) conditions, respectively; (2) amyloidogenic processing of the amyloid precursor protein is associated with lipid rafts (Ehehalt et al., 2003); and (3) the aggregation of soluble Aβ is readily induced by its interaction with lipid raft-like model membranes (Kakio et al., 2003). We found recently that Aβ binding to GM1 is markedly accelerated in a cholesterol-rich environment and that, in such an environment, GM1 forms a cluster that can be recognized by soluble Aβ as a receptor (Kakio et al., 2001). The alteration of cholesterol content in the AD brain is a controversial issue; however, it is noteworthy that cholesterol content in the outer leaflet of the synaptic plasma membrane can be increased in association with risk factors for the development of AD, including aging (Igbavboa et al., 1996) and the expression of apolipoprotein E allele ϵ4 (Hayashi et al., 2002). Thus, altogether, one possible scenario may be as follows: GAβ is generated in the lipid rafts or lipid raft-like microdomains in the neuron because of an increase in the local concentration of cholesterol, and then, GAβ initiates its seed activity to accelerate Aβ aggregation after its transport to the neuronal surface and/or shedding into the extracellular space. Alternatively, GAβ itself can be noxious per se because it has been reported previously that the disruption of membranes (McLaurin and Chakrabartty, 1996) and alteration of bilayer organization (Matsuzaki and Horikiri, 1999) can be induced by the generation of GAβ on the membranes. Thus, it may also be worthy to examine in future studies whether GAβ causes impairment of neuronal, particularly lipid raft-related, functions before extraneuronal Aβ deposition.
Several studies have suggested that therapeutically useful antibodies can be generated (Solomon et al., 1997; Bard et al., 2000; Hock et al., 2002; McLaurin et al., 2002; Lombardo et al., 2003). Together with the finding that GAβ has a conformation distinct from that of soluble Aβ, it may be possible to develop a novel therapeutic strategy to specifically inhibit the initiation of oligomerization–polymerization of Aβ in the brain.
Footnotes
This work was supported by research grants for Brain Research Science from the Ministry of Health and Welfare and the Core Research for Evolutional Science and Technology and by a Grant-in-Aid for Scientific Research on Priority Area (C) from the Ministry of Education, Culture, Sports, Science, and Technology (Japan). We thank Y. Ihara for critically reading this manuscript, A. Kakio for assisting in the supplementary experiment, H. Shimokata for performing the statistical analysis, Y. Hanai for assisting in the preparation of this manuscript, and Takeda Chemical Industries Ltd. for providing the antibodies (BAN052).
Correspondence should be addressed to Dr. Katsuhiko Yanagisawa, Department of Dementia Research, National Institute for Longevity Sciences, 36-3 Gengo, Morioka, Obu, 474-8522, Japan. E-mail: katuhiko@nils.go.jp.
H. Hayashi's present address: Department of Medicine, University of Alberta, Edmonton, T6G 2S2, Canada.
DOI:10.1523/JNEUROSCI.0861-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244894-09$15.00/0
References
- Ariga T, Kobayashi K, Hasegawa A, Kiso M, Ishida H, Miyatake T (2001) Characterization of high-affinity binding between gangliosides and amyloid beta-protein. Arch Biochem Biophys 388: 225–230. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. (2000) Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916–919. [DOI] [PubMed] [Google Scholar]
- Barrett JE, Wells DC, Conrad GW (1999) Pretreatment methods to improve nerve immunostaining in corneas from long-term fixed embryonic quail eyes. J Neurosci Methods 92: 161–168. [DOI] [PubMed] [Google Scholar]
- Binding MM, Jones ST (1996) Rodent to human antibodies by CDR grafting. In: Antibody engineering (Mcmafferty J, Hoogenboon HR, Chiswell DJ, eds), pp 147–168. Oxford: Oxford UP.
- Boon ME, Kok LP (1991) Formalin is deleterious to cytoskeleton proteins: do we need to replace it by formalin-free Kryofix? Eur J Morphol 29: 173–180. [PubMed] [Google Scholar]
- Bradford MM (1976) A rapid and sensitive method for the quantitaion of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- Choo-Smith LP, Surewicz WK (1997) The interaction between Alzheimer amyloid β(1–40) peptide and ganglioside GM1-containing membranes. FEBS Lett 402: 95–98. [DOI] [PubMed] [Google Scholar]
- Choo-Smith LP, Garzon-Rodriguez W, Glabe CG, Surewicz WK (1997) Acceleration of amyloid fibril formation by specific binding of Aβ-(1–40) peptide to ganglioside-containing membrane vesicles. J Biol Chem 272: 22987–22990. [DOI] [PubMed] [Google Scholar]
- Deleault NR, Lucassen RW, Supattapone S (2003) RNA molecules stimulate prion protein conversion. Nature 425: 717–720. [DOI] [PubMed] [Google Scholar]
- Ding TT, Harper JD (1999) Analysis of amyloid-β assemblies using tapping mode atomic force microscopy under ambient conditions. In: Amyloid, prions, and other proteinaggregates (Wetzel R, ed), pp 510–525. New York: Academic. [DOI] [PubMed]
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K (2003) Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esler WP, Stimson ER, Jennings JM, Vinters HV, Ghilardi JR, Lee JP, Mantyh PW, Maggio JE (2000) Alzheimer's disease amyloid propagation by a template-dependent dock-lock mechanism. Biochemistry 39: 6288–6295. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lansbury Jr PT (1997) Models of amyloid seeding in Alzheimer's disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu Rev Biochem 66: 385–407. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury Jr PT (1997a) Observation of metastable Aβ amyloid protofibrils by atomic force microscopy. Chem Biol 4: 119–125. [DOI] [PubMed] [Google Scholar]
- Harper JD, Lieber CM, Lansbury Jr PT (1997b) Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-β protein. Chem Biol 4: 951–959. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury Jr PT (1999) Assembly of Aβ amyloid protofibrils: an in vitro model for a possible early event in Alzheimer's disease. Biochemistry 38: 8972–8980. [DOI] [PubMed] [Google Scholar]
- Hayashi H, Igbavboa U, Hamanaka H, Kobayashi M, Fujita SC, Wood WG, Yanagisawa K (2002) Cholesterol is increased in the exofacial leaflet of synaptic plasma membranes of human apolipoprotein E4 knock-in mice. NeuroReport 13: 383–386. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM (2002) Generation of antibodies specific for β-amyloid by vaccination of patients with Alzheimer disease. Nat Med 8: 1270–1275. [DOI] [PubMed] [Google Scholar]
- Igbavboa U, Avdulov NA, Schroeder F, Wood WG (1996) Increasing age alters transbilayer fluidity and cholesterol asymmetry in synaptic plasma membranes of mice. J Neurochem 66: 1717–1725. [DOI] [PubMed] [Google Scholar]
- Kakio A, Nishimoto SI, Yanagisawa K, Kozutsumi Y, Matsuzaki K (2001) Cholesterol-dependent formation of GM1 ganglioside-bound amyloid β-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem 276: 24985–24990. [DOI] [PubMed] [Google Scholar]
- Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, Matsuzaki K (2002) Interactions of amyloid β-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 41: 7385–7390. [DOI] [PubMed] [Google Scholar]
- Kakio A, Nishimoto S, Kozutsumi Y, Matsuzaki K (2003) Formation of a membrane-active form of amyloid β-protein in raft-like model membranes. Biochem Biophys Res Commun 303: 514–518. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300: 486–489. [DOI] [PubMed] [Google Scholar]
- Kim KS, Miler DL, Sapienza VJ, Chen C-MJ, Bai C, Grundke-Iqbal I, Currie JR, Wisniewski HM (1988) Production and characterization of monoclonal antibodies reactive to synthetic cerebrovascular amyoid peptide. Neurosci Res Commun 2: 121–130. [Google Scholar]
- Lee SJ, Liyanage U, Bickel PE, Xia W, Lansbury Jr PT, Kosik KS (1998) A detergent-insoluble membrane compartment contains A beta in vivo. Nat Med 4: 730–734. [DOI] [PubMed] [Google Scholar]
- LeVine III H (1995) Soluble multimeric Alzheimer β (1–40) preamyloid complexes in dilute solution. Neurobiol Aging 16: 755–764. [DOI] [PubMed] [Google Scholar]
- Lombardo JA, Stern EA, McLellan ME, Kajdasz ST, Hickey GA, Bacskai BJ, Hyman BT (2003) Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J Neurosci 23: 10879–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki K, Horikiri C (1999) Interactions of amyloid β-peptide (1–40) with ganglioside-containing membranes. Biochemistry 38: 4137–4142. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Chakrabartty A (1996) Membrane disruption by Alzheimer β-amyloid peptides mediated through specific binding to either phospholipids or gangliosides. Implications for neurotoxicity. J Biol Chem 271: 26482–26489. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Franklin T, Fraser PE, Chakrabartty A (1998) Structural transitions associated with the interaction of Alzheimer beta-amyloid peptides with gangliosides. J Biol Chem 273: 4506–4515. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Ambermon MH, Darabie AA, Brown ME, Janus C, Chishti MA, Horne P, Westaway D, Fraser PE, Mount HT, Przybylski M, St. George-Hyslop P (2002) Therapeutically effective antibodies against amyloid-beta peptide target amyloid-β residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med 8: 1263–1269. [DOI] [PubMed] [Google Scholar]
- Michikawa M, Yanagisawa K (1998) Apolipoprotein E4 induces neuronal cell death under conditions of suppressed de novo cholesterol synthesis. J Neurosci Res 54: 58–67. [DOI] [PubMed] [Google Scholar]
- Morishima-Kawashima M, Ihara Y (1998) The presence of amyloid β-protein in the detergent-insoluble membrane compartment of human neuroblastoma cells. Biochemistry 37: 15247–15253. [DOI] [PubMed] [Google Scholar]
- Naiki H, Gejyo F (1999) Kinetic analysis of amyloid fibril formation. Methods Enzymol 309: 305–318. [DOI] [PubMed] [Google Scholar]
- Naiki H, Nakakuki K (1996) First-order kinetic model of Alzheimer's β-amyloid fibril extension in vitro. Lab Invest 74: 374–383. [PubMed] [Google Scholar]
- Naiki H, Hasegawa K, Yamaguchi I, Nakamura H, Gejyo F, Nakakuki K (1998) Apolipoprotein E and antioxidants have different mechanisms of inhibiting Alzheimer's β-amyloid fibril formation in vitro. Biochemistry 37: 17882–17889. [DOI] [PubMed] [Google Scholar]
- Nakamura S, Nakayama H, Goto N, Ono F, Sakakibara I, Yoshikawa Y (1998) Histopathological studies of senile plaques and cerebral amyloidosis in cynomolgus monkeys. J Med Primatol 27: 244–252. [DOI] [PubMed] [Google Scholar]
- Nichols MR, Moss MA, Reed DK, Lin W-L, Mukhopadhyay R, Hoh JH, Rosenberry TL (2002) Growth of β-amyloid (1–40) protofibrils by monomer elongation and lateral association. Characterization of distinct products by light scattering and atomic force microscopy. Biochemistry 41: 6115–6127. [DOI] [PubMed] [Google Scholar]
- Parton RG (1994) Ultrastructural localization of gangliosides; GM1 is concentrated in caveolae. J Histochem Cytochem 42: 155–166. [DOI] [PubMed] [Google Scholar]
- Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zurcher-Neely HA, Heinrikson RL, Ball MJ, Greenberg BD (1993) Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer's disease. J Biol Chem 268: 3072–3083. [PubMed] [Google Scholar]
- Sawamura N, Morishima-Kawashima M, Waki H, Kobayashi K, Kuramochi T, Frosch MP, Ding K, Ito M, Kim TW, Tanzi RE, Oyama F, Tabira T, Ando S, Ihara Y (2000) Mutant presenilin 2 transgenic mice. A large increase in the levels of Aβ42 is presumably associated with the low density membrane domain that contains decreased levels of blycerophospho-lipids and sphingomyelin. J Biol Chem 275: 27901–27908. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ (1997) Alzheimer's disease: genotypes, phenotypes, and treatments. Science 275: 630–631. [DOI] [PubMed] [Google Scholar]
- Shapira R, Austin GE, Mirra SS (1988) Neuritic plaque amyloid in Alzheimer's disease is highly racemized. J Neurochem 50: 69–74. [DOI] [PubMed] [Google Scholar]
- Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387: 569–572. [DOI] [PubMed] [Google Scholar]
- Smit JW, Meijer CJ, Decary F, Feltkamp-Vroom TM (1974) Paraformaldehyde fixation in immunofluorescence and immunoelectron microscopy. Preservation of tissue and cell surface membrane antigens. J Immunol Methods 6: 93–98. [DOI] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frankel D, Hanan-Aharon E (1997) Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc Natl Acad Sci USA 94: 4109–4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Iwatsubo T, Odaka A, Ishibashi Y, Kitada C, Ihara Y (1994) High tissue content of soluble β 1–40 is linked to cerebral amyloid angiopathy. Am J Pathol 145: 452–460. [PMC free article] [PubMed] [Google Scholar]
- Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB (1995) Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83: 79–90. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Ihara Y (1998) GM1 ganglioside-bound amyloid β-protein in Alzheimer's disease brain. Neurobiol Aging 19: S65–S67. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Odaka A, Suzuki N, Ihara Y (1995) GM1 ganglioside-bound amyloid β-protein (Aβ): a possible form of preamyloid in Alzheimer's disease. Nat Med 1: 1062–1066. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, McLaurin J, Michikawa M, Chakrabartty A, Ihara Y (1997) Amyloid β-protein (Aβ) associated with lipid molecules: immunoreactivity distinct from that of soluble Aβ. FEBS Lett 420: 43–46. [DOI] [PubMed] [Google Scholar]