

Abstract
Memory retrieval is not a passive phenomenon. Instead, it triggers a number of processes that either reinforce or alter stored information. Retrieval is thought to activate a second memory consolidation cascade (reconsolidation) that requires protein synthesis. Here, we show that the temporal dynamics of memory reconsolidation are dependent on the strength and age of the memory, such that younger and weaker memories are more easily reconsolidated than older and stronger memories. We also report that reconsolidation and extinction, two opposing processes triggered by memory retrieval, have distinct biochemical signatures: pharmacological antagonism of either cannabinoid receptor 1 or L-type voltage-gated calcium channels blocks extinction but not reconsolidation. These studies demonstrate the dynamic nature of memory processing after retrieval and represent a first step toward a molecular dissection of underlying mechanisms.
Keywords: consolidation, reconsolidation, extinction, fear conditioning, spatial memory, protein synthesis
Introduction
Memory consolidation is often thought of as a process of fixation (i.e., memories become increasingly immune to disruption as they mature) (McGaugh, 1966). This process of consolidation requires gene expression and de novo protein synthesis (Flexner et al., 1965; Davis and Squire, 1984; Abel et al., 1997; McGaugh, 2000). Although it was previously thought that this consolidation occurs just once, there is growing evidence that memory retrieval is a dynamic process that either reinforces or alters memory (Misanin et al., 1968; Schneider and Sherman, 1968; Lewis, 1979; Mactutus et al., 1979; Judge and Quartermain, 1982; Przybyslawski and Sara, 1997; Przybyslawski et al., 1999; Nader et al., 2000a; Sara, 2000; Anokhin et al., 2002; Pedreira et al., 2002). For example, inhibition of protein synthesis before or immediately after retrieval of a fear memory disrupts the subsequent expression of that memory, suggesting that retrieval triggers a wave of protein synthesis required for the reconsolidation of memory (Nader et al., 2000a; Taubenfeld et al., 2001; Debiec et al., 2002; Kida et al., 2002). Memory reconsolidation after retrieval may be used to update or integrate new information into long-term memories (LTMs) (Nader et al., 2000b; Sara, 2000; Dudai, 2002). The finding that LTM may be more dynamic and plastic than previously thought may have important clinical implications for the treatment of emotional disorders.
Cued recall of the original memory is the key event that initiates reconsolidation. In pavlovian fear conditioning, a conditioned stimulus (CS; such as a context) is paired with an unconditioned stimulus (US; such as footshock). When placed back in the training context, the animal shows conditioned fear responses such as freezing. Blocking protein synthesis around the time of training blocks long-term consolidation of the contextual fear memory (Abel et al., 1997; Schafe et al., 1999; Kida et al., 2002). Experimentally, cued recall typically involves reexposing subjects to the CS without the US. This reminder is akin to an extinction trial in which the CS comes to predict no US and loses its ability to evoke a conditioned response (Pavlov, 1927; Baum, 1988; Bouton, 1993; Myers and Davis, 2002). Experimental extinction does not reflect forgetting of the original memory trace but rather reflects new learning. That is, in addition to the previously acquired CS–US association, a new CS–no US association is formed that is thought to inhibit the conditioned response (Konorksi, 1967; Rescorla and Heth, 1975; Robbins, 1990; Rescorla, 2001). Therefore, memory retrieval may initiate two potentially dissociable but opposite processes: reconsolidation and extinction. Reconsolidation acts to stabilize, whereas extinction tends to weaken, the expression of the original memory. Recent studies using crab and medaka fish have shown that the duration of a reminder event may be an important determinant of subsequent memory processing: brief reminders lead to reconsolidation, whereas longer reminders result in memory extinction (Debiec et al., 2002; Eisenberg et al., 2003; Pedreira and Maldonado, 2003).
Here, we systematically explore both the temporal dynamics of memory processing after retrieval and the biochemical signatures of these processes in mice.
Materials and Methods
Mice. All experiments were conducted according to the Guide for the Care and Use of Laboratory Animals, Japan Neuroscience Society. Male C57BL/6 mice (Charles River, Yokohama, Japan) were housed in cages of five or six, maintained on a 12 hr light/dark cycle, and allowed ad libitum access to food and water. Mice were at least 8 weeks of age when tested. Testing was performed during the light phase of the cycle. All experiments were conducted blind to the treatment condition of the mouse.
Drugs. Because the neural sites critically responsible for acquisition, extinction, and reconsolidation of fear memories may be different, we used systemic rather than intracranial injections of a protein synthesis inhibitor [anisomysin (ANI)], a cannabinoid receptor 1 (CB1) antagonist (SR141716A), an L-type voltage-gated calcium channel (LVGCC) antagonist (nimodipine), and an NMDA receptor (NMDAR) antagonist [d(–)-3-(2-carboxypiperazine-4-yl)-propyl-1-phosphonic acid (CPP)]. ANI (150 mg/kg, i.p.; Sigma, St. Louis, MO) was dissolved in PBS (pH adjusted to 7.0–7.4) and administered to mice 30 min before the behavioral manipulation. At this dose, ANI inhibits >90% of protein synthesis in the brain during the first 2 hr (Flood et al., 1973). SR141716A was synthesized (Rinaldi-Carmona et al., 1994). SR141716A (1, 3, or 10 mg/kg, i.p.) was dissolved in solution (1 drop of Tween 80 in 3 ml of 2.5% dimethylsulphoxide and 10% Cremophor in saline) and administered to mice 20 min before the behavioral manipulation. Nimodipine (8, 16, or 32 mg/kg, i.p.; Sigma) was sonicated into 100% Cremophor EL (Sigma) and then diluted to make the final vehicle 10% Cremophor in solution (1 drop of Tween 80 in 3 ml of 2.5% dimethylsulphoxide). Nimodipine was administered to mice 20 min before the behavioral manipulation. CPP (10 or 20 mg/kg, i.p.; Sigma) was dissolved in saline solution (1 drop of Tween 80 in 3 ml of 2.5% dimethylsulphoxide and 10% Cremophor in saline) and administered to mice 60 min before the behavioral manipulation. Drug doses were determined according to previous reports showing effective doses for the blockade of long-term extinction (Santini et al., 2001; Cain et al., 2002; Marsicano et al., 2002).
Contextual fear conditioning. Mice were trained and tested in conditioning chambers (17.5 × 17.5 × 15 cm) that had a stainless steel rod floor through which footshocks could be delivered. Training consisted of placing the mice in the chamber and delivering an unsignaled footshock (2 sec duration; 0.75 mA) 148 sec later. Under more intense training conditions, mice received a series of three footshocks at 30 sec intervals (the first footshock starting 148 sec after placement in the chamber). Mice were returned to the home cage 30 sec after the final footshock.
Twenty-four hours after training, mice were reexposed to the training context (reexposure) for varying lengths of time (0, 1, 3, or 30 min). Memory was assessed 24 hr later (test) as the percentage of time mice spent freezing when replaced in the training context. Freezing behavior (defined as complete lack of movement, except for respiration) was assessed at 5 sec intervals (Blanchard and Blanchard, 1969; Fanselow, 1980).
For the first experiment (effects of CS reexposure duration on subsequent memory expression), mice were treated with saline or ANI 30 min before training with a single footshock. Memory was assessed 2 or 24 hr later [short-term memory (STM) and LTM, respectively). For experiments involving reexposure, mice were trained drug free with a single footshock and 24 hr later replaced back in the training context for 0, 1, 3, or 30 min, during which freezing behavior was assessed (reexposure). Mice were treated with saline or ANI 30 min before reexposure. Twenty-four hours later, mice were once again placed in the training context (5 min) and freezing behavior was assessed (test). In the case of 0 min reexposure, mice remained in the home cage (not reexposed to training chamber) but were treated with the ANI or saline. For studies examining the time course of reconsolidation, mice were trained with a single footshock and 24 hr later replaced in the training context for 3 min (reexposure). Before reexposure, mice received saline or ANI. Two hours (Fig. 1G) or 1 week (Fig. 1 H) after reexposure, mice were placed back in the training context and memory was assessed for 5 min.
Figure 1.

Effects of CS reexposure duration on stability of reactivated memory and extinction. A–E, G, H, Experimental design used with data presented below. A, Effects of ANI injection on STM and LTM (control group, n = 10; ANI-injected group, n = 10). B, Effects of 0 min reexposure (no reexposure; saline, n=10; ANI, n=10). C, Effects of 1 min reexposure (saline, n=10; ANI, n= 10). D, Effects of 3 min reexposure (saline, n = 10; ANI, n = 10). E, Effects of 30 min reexposure (saline, n = 10; ANI, n = 10). During reexposure, freezing score in 3 min blocks are presented. F, Summary of the relationship between the duration of reexposure at reexposure and freezing scores at test (B–E). G, Disruption of reactivated memory is not observed 2 hr after reexposure (saline, n=10; ANI, n=10). H, Disruption of reactivated memory by anisomycin is observed 1 week after reexposure (saline, n= 10; ANI, n = 10).
For the second experiment (effect of strength of the memory on memory reconsolidation), mice were trained using a stronger training protocol. Mice were trained with three footshocks and 24 hr later replaced in the training context for 3, 5, or 10 min (reexposure). Mice received saline or ANI 30 min before reexposure. Twenty-four hours after this, mice were replaced in the training context and memory was assessed for 5 min (test).
For the third experiment (effect of age of memory on subsequent memory expression), mice were trained with a single footshock and replaced in the training context (for 3 min; reexposure) either 1, 3, or 8 weeks later. Mice received saline or ANI 30 min before reexposure. Twenty-four hours after this, mice were replaced in the training context and memory was assessed for 5 min (test).
For the fourth experiment (molecular differences between reconsolidation and extinction), the effects of the CB1 antagonist SR141716A (1-10 mg/kg), nimodipine (8-32 mg/kg), CPP (10 or 20 mg/kg), and ANI (150 mg/kg) on LTM were assessed (as above). To assess the effects of these drugs on reconsolidation, mice were trained as in the reconsolidation experiments above and treated with these drugs before a 3 min reexposure.
Morris water maze. The water maze apparatus and procedures have been described (Bourtchaleze et al., 1994). Spatial learning and memory were assessed in probe trials with the platform removed from the pool. In probe trials, mice were allowed to swim for 60 sec and the time spent in each quadrant of the pool was scored. Mice were trained with 6 trials per day for 2 d (at 1 min intervals). Twenty-four hours later, mice were given either a single probe trial or a series of 10 probe trials (at 3 min intervals; reexposure). Mice were treated with saline or ANI 30 min before reexposure. A final probe trial was given 24 hr later (test).
Data analysis. Data were analyzed with ANOVA. A single-factor ANOVA and post hoc comparisons were used to analyze the difference between probe trials in the Morris water maze and the effects of drugs in contextual fear conditioning. Planned comparisons, using a nonpaired t test, were used to analyze differences of the time spent in the target quadrant (TQ) in the Morris water maze between two groups. Planned comparisons using a paired t test were used to analyze differences of freezing scores from the contextual conditioning of each group between the first 3 min reexposure during 30 min reexposure and test. All values in the text and figure legends are means ± SEM.
Results
Effects of CS reexposure duration on subsequent memory expression
To test whether under our conditions protein synthesis is critical for the stabilization of LTM but not STM, we first administered the protein synthesis inhibitor ANI or saline 30 min before contextual fear training and assessed memory in separate groups of mice either 2 or 24 hr later. In agreement with other studies (Kida et al., 2002), we found that ANI blocked memory assessed 24 hr (F(1,18) = 33.89; p < 0.05), but not 2 hr (F(1,18) = 0.42; p > 0.05), after contextual conditioning (Fig. 1A).
Previous studies suggest that protein synthesis may also be required for restabilizing memories after retrieval, a process referred to as reconsolidation. We examined the importance of three variables on reconsolidation: (1) duration of memory reactivation (CS reexposure); (2) strength of memory; and (3) age of memory.
In the first experiment, we examined the effects of CS duration on reconsolidation. Mice were trained with a single footshock and 24 hr later reexposed to the training context (30 min after ANI or saline treatment) for different amounts of time (0, 1, 3, or 30 min; reexposure). Memory was assessed 24 hr later as drug-free mice were once again placed in the training context (test). We found that memory (freezing) tested 24 hr after reexposure was not affected by ANI treatment after very short (0 or 1 min) reexposures (0 min: F(1,18) = 0.72, p > 0.05; 1 min: F(1,18) = 0.73, p > 0.05) (Fig. 1B,C), suggesting that reconsolidation is not affected under these conditions. In contrast, as shown previously (Kida et al., 2002), reconsolidation after a longer reexposure (3 min) was disrupted by ANI (F(1,18) = 27.83; p < 0.05) (Fig. 1D). When longer reexposures were used (30 min), conditioned freezing levels decreased over time within the session in both the saline- and ANI-treated groups (saline: F(1,9) = 24.04, p < 0.05; ANI: F(1,9) = 13.05, p < 0.05). Measurements 24 hr after this 30 min reexposure confirmed that the extinction of conditioned freezing was persistent in saline-treated mice (first block in reexposure session versus test; t(9) = 7.325; p < 0.05) (Fig. 1E). However, extinction tested 24 hr after the 30 min reexposure was blocked by ANI (F(1,18) = 19.37; p < 0.05), indicating that the long-term stability of extinction requires protein synthesis. Because freezing levels during the reexposure were not affected by ANI treatment, our results also show that protein synthesis is not required for recall of a conditioned fear memory. The effects of reexposure duration on protein synthesis-dependent reconsolidation of contextual fear memory are summarized in Figure 1F.
Together, these results indicate that under certain circumstances, ANI produces amnesia for previously consolidated conditioned fear memories. To further investigate the temporal dynamics of this impairment, we varied the length of time between reexposure and test (2 hr, 24 hr, or 1 week). ANI did not impair memory tested 2 hr after reexposure (F(1,18) = 1.03; p > 0.05) (Fig. 1G) but did block memory tested 24 hr or 1 week after reexposure [24 hr (Fig. 1D); 1 week: F(1,18) = 18.06, p < 0.05 (Fig. 1H)]. Therefore, the amnestic effects of ANI on reconsolidation are observed 24 hr after training and are stable for at least 1 week.
These results revealed the presence of three distinct time-dependent phases of memory processing after memory retrieval: in the first phase, the memory trace is immune to the effects of protein synthesis inhibition. With our experimental conditions, this phase lasts approximately 1 min. Further extending reexposure triggers a second wave of memory consolidation processes (reconsolidation) required for the stability of the memory trace. Hence, blocking protein synthesis during this second phase compromises the long-term stability of the trace. Finally, prolonged reexposures to the CS in the absence of the US trigger the formation of a new “extinction” trace (CS–no US). Inhibition of protein synthesis at this stage blocks the formation of the new extinction memory, leaving expression of the old memory unchanged. It is important to note that our results indicate that there is an interaction between the extinction and reconsolidation phases, because blocking both reconsolidation and extinction with protein synthesis inhibitors appears to leave the original memory unaffected, whereas blocking protein synthesis during the reconsolidation phase alone disrupts the original memory.
Effects of strength of the memory on memory reconsolidation
It is possible that stronger memories are more resistant to reconsolidation than weaker memories. To test this possibility, we trained mice with three, rather than one, footshocks. Twenty-four hours after training, mice were reexposed to the training context for 3 min (30 min after ANI or saline treatment; reexposure). Memory was assessed 24 hr after reexposure (test). In contrast to the results of our previous experiments with one footshock (Fig. 1D), after training with three footshocks, ANI did not block reconsolidation (F(1,19) = 0.14; p > 0.05) (Fig. 2A), suggesting that stronger memories are more resistant to disruption, perhaps because they are more widely represented in neural networks. However, it is possible that longer reexposures can trigger the reconsolidation of stronger memories. To test this, we trained mice as described above but increased the duration of reexposure to the training context from 3 min to either 5 or 10 min. ANI did not block reconsolidation after a 5 min reexposure (F(1,18) = 0.01; p > 0.05) (Fig. 2B) but blocked it after a 10 min reexposure (F(1,18) = 16.63; p < 0.05) (Fig. 2C), indicating that longer reexposures are required to reconsolidate stronger memories.
Figure 2.

Effects of memory strength on the stability of reactivated memories. A–C, Experimental design used with data presented below. Mice were trained with three footshocks. A, Effects of 3 min reexposure on freezing during test (saline, n = 10; ANI, n = 11). B, Effects of 5 min reexposure on freezing during test (saline, n = 10; ANI, n = 10). C, Effects of 10 min reexposure on freezing during test (saline, n = 10; ANI, n = 11).
Effects of age of the memory on subsequent memory expression
Another important variable that may determine the stability of a retrieved memory is its age (Milekic and Alberini, 2002). To test whether older memories are less likely to undergo reconsolidation, we trained separate groups of mice with a single footshock and reexposed them to the training context (for 3 min) 1, 3, or 8 weeks later. As in previous experiments, memory was assessed 24 hr after reexposure (test). Young (1 or 3 weeks) but not old (8 weeks) memories were disrupted by ANI treatment given before reexposure (1 week: F(1,18) = 20.31, p < 0.05; 3 weeks: F(1,18) = 153.45, p < 0.05; 8 weeks: F(1,18) = 0.72, p > 0.05) (Fig. 3A–C). Consistent with previous findings (Milekic and Alberini, 2002), these results indicate that older memories are immune to the disruptive effects of protein synthesis inhibition during retrieval. However, it is possible that, like stronger memories, older memories undergo reconsolidation with longer reexposure. To test this, we trained another group of mice and reexposed them to the training context 8 weeks after training, but this time we used a 10 min, rather than 3 min, reexposure. ANI disrupted reconsolidation after this longer duration reexposure (F(1,18) = 13.95, p < 0.05) (Fig. 3D). Whereas previous studies suggested that older memories do not undergo protein synthesis-dependent reconsolidation (Milekic and Alberini, 2002), our results show that older memories do require protein synthesis for re-storage after longer duration reexposure.
Figure 3.

Effect of memory age on the stability of reactivated memories. A–D, Experimental design used with data presented below. A, Effect of 1 week retention on freezing during test between reexposure (3 min) and test (control, n = 10; ANI, n = 10). B, Effect of 3 week retention on freezing during test between reexposure (3 min) and test (control, n = 10; ANI, n = 10). C, Effect of 8 week retention on freezing during test between reexposure (3 min) and test (control, n = 10; ANI, n = 10). D, Effect of 8 week retention on freezing during test between reexposure (10 min) and test (control, n = 10; ANI, n = 10).
Molecular differences between reconsolidation and extinction
Recent studies using pharmacology or mouse genetics identified two genes (CB1 and LVGCCs) that may be required for extinction but not initial memory consolidation (Cain et al., 2002; Marsicano et al., 2002; but see Bauer et al., 2002). To compare the molecular mechanisms underlying consolidation, reconsolidation, and extinction, we examined the effects of antagonists for CB1, LVGCCs, and NMDAR on these three processes. First, we examined the effects of these drugs on initial memory consolidation (LTM). Mice received the CB1 antagonist SR141716A (1–10 mg/kg), LVGCC inhibitor (nimodipine; 8–32 mg/kg), NMDAR antagonist (CPP; 10–20 mg/kg), or ANI (150 mg/kg; positive control) before contextual fear training (as described above). ANI and CPP disrupted initial memory consolidation, whereas nimodipine (8–32 mg/kg) or SR141716A (1–10 mg/kg) did not (F(9, 90) = 22.01, p < 0.05) (Fig. 4A). The post hoc Newman–Keuls test showed that the ANI and CPP groups froze significantly less than the other groups 24 hr after training. These results suggest that both protein synthesis and NMDAR function, but not LVGCCs or CB1 receptor function, are required for initial memory consolidation.
Figure 4.

Effects of CB1, LVGCC, and NMDAR antagonists on reconsolidation and extinction. A–C, Experimental design used with data presented below. A, Effects of ANI, SR141716A, nimodipine, and CPP on memory consolidation (n = 10 for all groups). B, Effects of ANI, SR141716A, nimodipine, and CPP on memory reconsolidation (n = 10 for all groups). C, Effects of ANI, SR141716A, nimodipine, and CPP on memory extinction. Freezing responses during reexposure are shown only for the highest dose of nimodipine (32 mg/kg), SR141716A (10 mg/kg), and CPP (20 mg/kg). Subsequent test data are shown in the right panel (n = 12 for all groups). C, Control; A, anisomycin; Nimo, nimodipine; SR, SR141716A.
In the second experiment, we examined the effects of these drugs on reconsolidation after a 3 min reexposure. ANI and CPP disrupted reconsolidation, whereas nimodipine or SR141716A did not (F(9,90) = 7.23, p < 0.05) (Fig. 4B). The post hoc Newman–Keuls test showed that the ANI and CPP groups froze significantly less than the other groups.
To examine the effects of these drug treatments on extinction, we reexposed separate groups of mice to the context for 30 min. As above, saline-treated mice showed decreasing levels of freezing over the 30 min reexposure. This within-session reduction in freezing (or short-term extinction) was blocked by nimodipine (0 vs 32 mg/kg; drug × time: F(9,198) = 2.85, p < 0.05; drug: F(1,22) = 9.49, p < 0.05; time: F(9,198) = 18.68, p < 0.05) (Fig. 4C). However, all drug treatments impaired the stability of extinction measured 24 hr after reexposure (F(9,110) = 6.72, p < 0.05) (Fig. 4C). The post hoc Newman–Keuls test showed that all drug groups froze significantly more than control groups [control vs ANI, CB1 blocker (3 or 10 mg/kg), nimodipine (16 or 32 mg/kg), or CPP (10 or 20 mg/kg): p < 0.001; control vs CB1 blocker (1 mg/kg): p < 0.05; control vs nimodipine (8 mg/kg): p < 0.01]. Together, these results indicate that extinction depends on protein synthesis and intact function of NMDAR, CB1, and LVGCCs. Therefore, although LVGCCs and CB1 receptor function are not required for consolidation and reconsolidation, they are necessary for extinction. The molecular underpinnings of consolidation and reconsolidation, consequently, differ from that of extinction.
Relationship between reconsolidation and extinction in the Morris water maze
Recent studies of reconsolidation and extinction have focused on contextual fear conditioning. To test whether our results generalize to another hippocampal-dependent task with different sensory, motivational, and performance demands, we trained mice in the Morris water maze. Training took place over 2 d, during which mice were given six trials to find a hidden platform (Fig. 5B). In our contextual fear conditioning experiments, memory was reactivated by reexposure to the training context. Similarly, 24 hr after training in the water maze, mice were given a probe trial in which the platform was removed from the pool (reexposure). Thirty minutes before this probe trial, mice were treated with ANI or saline. As before, memory was subsequently assessed 24 hr later in a probe trial (test). As expected, pretreatment with ANI did not affect recall of the spatial memory; both saline- and ANI-treated mice searched selectively in the quadrant of the pool where the platform was previously located (the TQ) (saline: F(3,32) = 11.27, p < 0.05; ANI: F(3,32) = 6.80, p < 0.05; comparable searching scores in the TQ between saline and ANI: t(16) = 0.34, p > 0.05) (Fig. 5C). However, when these mice were tested 24 hr later (test), the saline-treated, but not ANI-treated, mice searched selectively in the TQ (saline: F(3,32) = 5.67, p < 0.05; ANI: F(3,32) = 2.29, p > 0.05) (Fig. 5D). Therefore, these data suggest that reexposure to the training environment triggers memory reconsolidation of spatial memory and that this process is protein synthesis dependent.
Figure 5.

Effects of protein synthesis inhibition during a probe trial on reactivated memory for platform position in the Morris water maze. A, Experimental design. B, Time to find the hidden platform (saline, n = 9; ANI, n = 9). Data are indicated in blocks of two trials. C, Probe trial after 24 hr last training (reexposure). Mice were given injections of ANI or saline 30 min before reexposure. D, Probe trial conducted 24 hr after reexposure (test). C, D, Time spent (seconds) in target (T), adjacent left (L), adjacent right (R), and opposite (O) quadrants during the probe trial (60 sec) is shown.
The amnestic effects of ANI in our fear conditioning experiments were contingent on reexposure to the training context (see above). To test whether this holds true for spatial memories, mice were trained in the water maze as above but were treated with ANI or saline in the home cage 24 hr later (but not reexposed to the pool) and then given a probe test 24 hr later. ANI failed to disrupt subsequent expression of the spatial memory (saline: F(3,36) = 12.76, p < 0.05; ANI: F(3,36) = 16.66, p < 0.05; comparable searching scores in the TQ between saline and ANI: t(18) = –1.94, p > 0.05) (Fig. 6B), suggesting that the amnestic effects of ANI are contingent on reexposure to the training environment in spatial memory as well as fear conditioning.
Figure 6.

Memory reactivation during a probe trial is necessary for the disruption of the reactivated memory by anisomycin. A, Experimental design. B, Probe trial 48 hr after the last training (saline, n = 10; ANI, n = 10). Mice were given injections of anisomycin or saline 24.5 hr before the probe trial. Time (seconds) spent in target (T), adjacent left (L), adjacent right (R), and opposite (O) quadrants during the probe trial (60 sec) is shown.
In our fear conditioning experiments, prolonged reexposure initiated extinction; consolidation of this extinction memory was blocked by ANI treatment. To test whether this holds true for spatial memory, mice were trained as described above, but reexposure was extended (a greater number of probe trials). Thirty minutes after pretreatment with ANI or saline, mice were given a series of 10 probe trials (spaced 3 min apart). Both ANI- and saline-treated mice searched selectively in the TQ in the first probe trial (reexposure) (saline: F(3,32) = 46.15, p < 0.05; ANI: F(3,36) = 68.67, p < 0.05) (Fig. 7B) but showed a progressive loss of this selectivity over successive probe trials (saline: F(1,8) = 12.74, p < 0.05; ANI: F(1,9) = 26.60, p < 0.05) (Fig. 7C). In the final probe trial, neither the ANI- nor saline-treated mice showed a preference for the TQ, indicating extinction of the spatial memory (saline: F(3,32) = 1.70, p > 0.05; ANI: F(3,36) = 0.59, p > 0.05) (Fig. 7D). Therefore, ANI did not disrupt short-term extinction of spatial memories (for drug: F(1,17) = 2.07, p > 0.05; for time: F(9,153) = 35.24, p < 0.05; for drug × time: F(9,153) = 1.20, p > 0.05) (Fig. 7C). However, when these mice were tested 24 hr later, the ANI-treated, but not the saline-treated, mice searched selectively in the quadrant in which the platform was previously located (saline: F(3,32) = 1.32, p > 0.05; ANI: F(3,36) = 17.22, p < 0.05) (Fig. 7E). These results show that long-term extinction of a spatial memory is blocked by protein synthesis inhibition.
Figure 7.
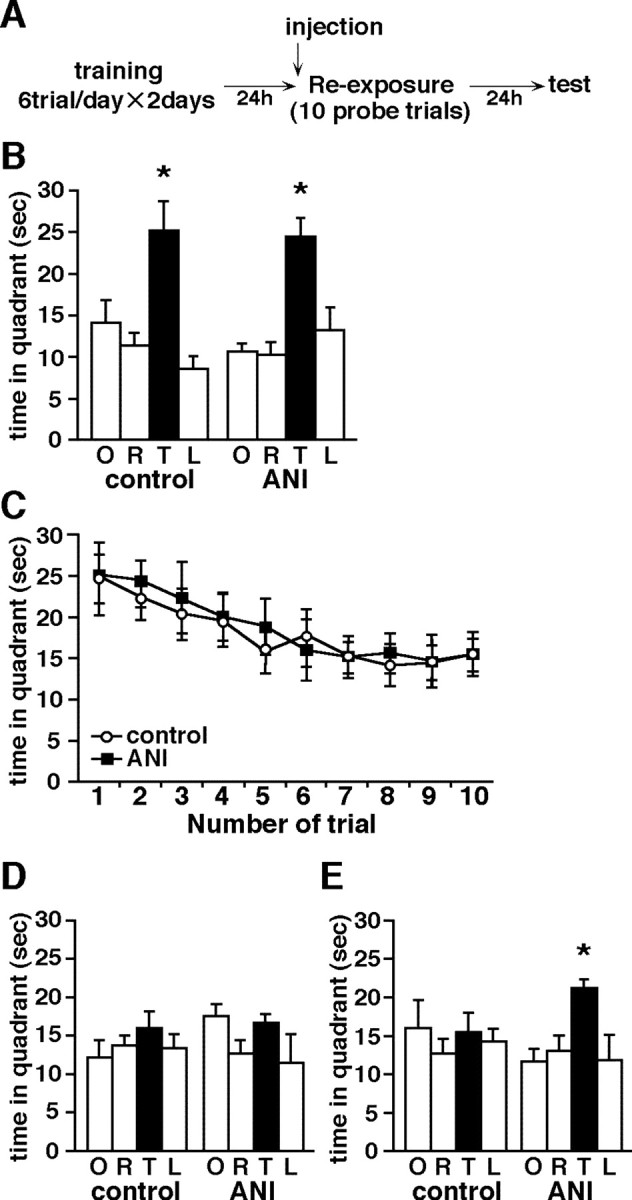
Effects of protein synthesis inhibition during 10 probe trials on memory extinction for platform position in the Morris water maze. A, Experimental design. B, First probe trial during 10 trials after 24 hr last training (reexposure; saline, n = 9; ANI, n = 10). Mice were given injections of ANI or saline 30 min before reexposure. C, Extinction curve of time spent swimming in the TQ during 10 probe trials. D, Last probe trial during 10 trials at reexposure. E, Probe trial 24 hr after reexposure (test). B, D, E, Time (seconds) spent in target (T), adjacent left (L), adjacent right (R), and opposite (O) quadrants during the probe trial (60 sec) is shown.
Discussion
There has been renewed interest in memory processing after retrieval: brief exposure to the CS seems to trigger a second wave of memory consolidation (reconsolidation), whereas prolonged exposure to the CS leads to the formation of a new memory that competes with the original memory (extinction). However, many of the findings supporting current views on reconsolidation, and extinctions remain controversial. For example, although several investigators found that inhibition of protein synthesis before or immediately after memory retrieval disrupts the subsequent expression of memory, suggesting that reconsolidation is protein synthesis dependent (Nader et al., 2000a; Taubenfeld et al., 2001; Debiec et al., 2002; Kida et al., 2002; Milekic and Alberini, 2002), others did not (Lattal and Abel, 2001). Similarly, there is disagreement about the role of protein synthesis in memory extinction: some groups found that protein synthesis inhibition at the time of memory retrieval impairs memory extinction (Berman and Dudai, 2001; Vianna et al., 2001; Eisenberg et al., 2003; Pedreira and Maldonado, 2003), whereas others did not (Lattal and Abel, 2001). These discrepancies suggest that there may be fundamental methodological variables that influence the memory processes activated during retrieval.
In this series of experiments, we identified three key variables that control subsequent behavior after memory retrieval. We provided a systematic demonstration of how reexposure duration, the age of the memory, and the strength of the memory interact to influence behavior in tasks that model declarative memory in mice. As proposed previously (Debeic et al., 2002; Nader, 2003), our results show that the duration of reexposure is a key variable that controls subsequent behavior. Memory retrieval initiates two competing processes [reconsolidation (CS–US) and extinction (CS–no US)], and the duration of reexposure helps determine which of these processes is dominant in the control of subsequent behavior. The present results are consistent with previous results using crab, medaka fish, and snail (Eisenberg et al., 2003; Pedreira and Maldonado, 2003; Sangha et al., 2003a,b). Furthermore, we showed that these competing processes have distinct temporal and biochemical signatures.
The results presented here reveal three distinct time-dependent phases of memory processing after memory retrieval. During the first phase, the retrieved memory is in a state that precedes both the reconsolidation and extinction processes. Further extending reexposure, however, initiates the protein synthesis-dependent reconsolidation processes required for the stability of the memory trace. Hence, blocking protein synthesis during this second phase compromises the long-term stability of the trace. Finally, prolonged reexposures to the CS in the absence of the US trigger the formation of a new memory trace that encodes the dissociation between the CS and the US (CS–no US; extinction memory), therefore competing with the original memory (CS–US). Inhibition of protein synthesis at this stage blocks the formation of this new extinction memory, leaving expression of the original memory unchanged. It is important to note that our results also indicate that there is an interaction between the extinction and reconsolidation processes: although blocking protein synthesis during short reexposures (reconsolidation) disrupts the original memory, blocking protein synthesis during prolonged reexposure, conditions in which both reconsolidation and extinction would be expected to be initiated, leaves the original memory unaffected.
Lattel and Abel (2001) found that inhibiting protein synthesis at the time of CS reexposure in contextual fear conditioning and the Morris water maze produced no effect on subsequent memory expression. This lack of effect may be attributed to the duration of reexposure used in those experiments. Accordingly, we found that ANI treatments (Fig. 1) failed to either disrupt the reactivated memory or block long-term extinction (data not shown) with certain reexposure durations.
Our results indicate that after retrieval behavior is controlled by ensuing competing processes: reconsolidation and extinction. The process that prevails and dominates subsequent behavior is determined in part by the duration of the reexposure. Whereas reconsolidation acts on the original memory (Nader et al., 2000a; Taubenfeld et al., 2001; Anokhin et al., 2002; Debiec et al., 2002; Kida et al., 2002; Kraus et al., 2002; Milekic and Alberini, 2002; Pedreira et al., 2002), extinction involves the formation of distinct alternative (CS–no US) memories (Quirk et al. 2000). Our data suggest that these two processes have different temporal profiles. After retrieval, there is a brief time window for reconsolidation, whereas extinction only occurs after prolonged reexposure. These two processes may be dissociable at the anatomical level. In fear conditioning, for example, extinction learning is thought to depend on the medial prefrontal cortex (Teich et al., 1989; Morgan et al., 1993; Morgan and LeDoux, 1995, 1999; Morrow et al., 1999; Quirk et al., 2000). In contrast, reconsolidation of fear memories is thought to be mediated by the amygdala (Nader et al., 2000a) or hippocampus (Debiec et al., 2002).
Recent reports using pharmacology or mouse genetics indicate that the downstream signaling transduction pathways of LVGCCs and CB1 are essential for long-term extinction but not initial learning (Cain et al., 2002; Marsicano et al., 2002). Because reconsolidation and extinction are dissociable both temporally and anatomically, this raises the possibility that the two processes are also dissociable biochemically. Consistent with this, we found that pharmacological blockade of either CB1 or LVGCCs blocked extinction but not reconsolidation. In contrast, blocking NMDARs or protein synthesis disrupts both reconsolidation and extinction, as well as initial consolidation. These findings are the first to demonstrate that the competing processes of reconsolidation and extinction require different molecular components. Furthermore, the observation that the inhibition of LVGCCs impairs both short- and long-term extinction processes indicates that LVGCCs function upstream of protein synthesis mechanisms required for the stability of the extinction memory. Additionally, we identified a role for CB1 receptors in long-term extinction of contextual, as well as cued (Marsicano et al., 2002), fear memory, suggesting a general role of CB1 in extinction of fear memories.
We also examined how reexposure duration interacts with two other characteristics of a memory: its age and strength. Our results show that memories become increasingly resistant to disruption with age. After contextual fear conditioning, ANI treatment before CS reexposure (3 min) disrupted the subsequent expression of 1- and 3-week-old memories but not 8-week-old memories. However, increasing the duration of the reexposure made even 8-week-old memories susceptible to disruption by ANI. These results suggest that there is a time-dependent change in the activation of reconsolidation processes, as if it were easier to activate the reconsolidation of younger than older memories. Whereas initial encoding and storage of fear memories depend on the hippocampus, permanent storage is proposed to depend on the neocortex (Kim and Fanselow, 1992; Eichenbaum et al., 1994; McClelland et al., 1995; Squire and Alvarez, 1995; Anagnostaras et al., 1999, 2001; Quevedo et al., 1999; LeDoux, 2000; McGaugh, 2000; Frankland et al., 2001). Therefore, the reactivation of an old memory should activate a cortical, rather than hippocampal, trace. This cortical trace is more resistant to disruption by protein synthesis inhibitors, perhaps because it is broadly distributed across different cortical regions (McClelland et al., 1995; Bontempi et al., 1999). Of interest are the findings using inhibitory avoidance that show that an old memory is immune to protein synthesis inhibition after retrieval using short-duration reexposure (Taubenfeld et al., 2001). Our data indicate that longer duration reexposures are necessary to destabilize older memories.
Surprisingly, we also found that memory strength affected the ease of reconsolidation. Increasing the strength of memory (by increasing the number of shocks during training), increased the resistance of these fear memories to disruption by ANI. Similar to the studies with older memories, longer reexposures were required to induce reconsolidation of stronger memories. Therefore, the activation of reconsolidation processes seems to be dependent both on the age and the strength of the memory.
The majority of previous studies have used conditioned fear to examine reconsolidation (Nader et al., 2000a; Taubenfeld et al., 2001; Anokhin et al., 2002; Debiec et al., 2002; Kida et al., 2002; Kraus et al., 2002; Milekic and Alberini, 2002; Pedreira et al., 2002). To test whether these results generalize to another hippocampal-dependent task with different sensory, motivational, and performance demands, we trained mice in the Morris water maze. Again, we found that memory retrieval initiates two competing processes (reconsolidation and extinction) and that the length of reexposure (in this case, the number of reinforced probe trials) determines in part which process is dominant: one probe trial (similar to a brief reexposure to the context in fear conditioning) initiates reconsolidation of a spatial memory that is blocked by ANI treatment. Ten probe trials (similar to a more prolonged reexposure to the context in fear conditioning) initiate extinction. As with fear conditioning, ANI treatment blocks the long-term consolidation of this extinction memory. These findings indicate contextual fear conditioning and spatial learning have similar reconsolidation and extinction processes, extending previous studies using the radial arm maze (Przybyslawski and Sara, 1997; Przybyslawski et al., 1999).
The studies presented here demonstrate the dynamic nature of memory processing after retrieval. They also identify three fundamental variables (duration of CS exposure, age of memory, strength of memory) affecting the stability of reactivated memories and show how these variables interact to determine the fate of the memory trace.
Footnotes
This work was supported by Grants-in Aid for Scientific Research and High Technology Research from the Ministry of Education, Science and Culture, Japan. We thank R. Costa for helpful discussions, invaluable comments, and critical reading of this manuscript.
Correspondence should be addressed to Dr. Satoshi Kida, Department of Bioscience, Faculty of Applied Bioscience, Tokyo University of Agriculture, 1-1-1 Sakuragaoka, Setagaya-ku, Tokyo 156-8502, Japan. E-mail: kida@nodai.ac.jp.
DOI:10.1523/JNEUROSCI.5491-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244787-09$15.00/0
References
- Abel T, Nguye PV, Barad M, Deuel TA, Kandel ER, Bourtchouladze R (1997) Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88: 615–626. [DOI] [PubMed] [Google Scholar]
- Anagnostaras SG, Maren S, Fanselow MS (1999) Temporally graded retrograde amnesia of contextual fear after hippocampus damage in rat: within-subjects examination. J Neurosci 19: 1106–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anagnostaras SG, Gale GD, Fanselow MS (2001) hippocampus and contextual fear conditioning: recent controversies and advances. Hippocampus 11: 8–17. [DOI] [PubMed] [Google Scholar]
- Anokhin KV, Tiunova AA, Rose SPR (2002) Reminder effects–reconsolidation or retrieval deficit? Pharmachological dissection with protein synthesis inhibitors following reminder for a passive-avoidance task in young chicks. Eur J Neurosci 15: 1759–1765. [DOI] [PubMed] [Google Scholar]
- Bauer EP, Schafe GE, LeDoux JE (2002) NMDA receptors and L-type voltage-gated calcium channels contribute to long-term potentiation and different components of fear memory formation in the lateral amygdala. J Neurosci 22: 5239–5249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum M (1988) Spontaneous recovery from the effects of flooding (exposure) in animals. Behav Res Ther 26: 185–186. [DOI] [PubMed] [Google Scholar]
- Berman DE, Dudai Y (2001) Memory extinction, learning anew, and learning the new: dissociation in the molecular machinery of learning in cortex. Science 291: 2417–2419. [DOI] [PubMed] [Google Scholar]
- Blanchard RJ, Blanchard DC (1969) Passive and active reactions to fear-eliciting stimuli. J Comp Physiol Psychol 68: 129–135. [DOI] [PubMed] [Google Scholar]
- Bontempi B, Laurent-Demir C, Destrade C, Jaffard R (1999) Time-dependent reorganization of brain circuitry underlying long-term memory strage. Nature 400: 671–675. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79: 59–68. [DOI] [PubMed] [Google Scholar]
- Bouton ME (1993) Context, time, and memory retrieval in the interference paradigms of Pavlovian learning. Psychol Bull 114: 80–99. [DOI] [PubMed] [Google Scholar]
- Cain C, Blouin A, Barad MG (2002) L-type voltage-gated calcium channels are required for extinction, but not for acquisition or expression, of conditioned fear in mice. J Neurosci 22: 9113–9121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HP, Squire LR (1984) Protein synthesis and memory. Psychol Bull 96: 518–559. [PubMed] [Google Scholar]
- Debiec J, LeDoux JE, Nader K (2002) Cellular and systems reconsolidation in the hippocampus. Neuron 36: 527–538. [DOI] [PubMed] [Google Scholar]
- Dudai Y (2002) Molecular bases of long-term memories: a question of persistence. Curr Opin Neurobiol 12: 211–216. [DOI] [PubMed] [Google Scholar]
- Eichenbaum H, Otto T, Cohen NJ (1994) Two functional components of the hippocampal memory system. Behav Brain Sci 17: 449–518. [Google Scholar]
- Eisenberg M, Kobilo T, Berman DE, Dudai Y (2003) Stability of retrieved memory: inverse correlation with trace dominance. Science 301: 1102–1104. [DOI] [PubMed] [Google Scholar]
- Fanselow MS (1980) Conditioned and unconditional components of post-shock freezing. Pavlov J Biol Sci 15: 177–182. [DOI] [PubMed] [Google Scholar]
- Flexner LB, Flexner JB, Stellar E (1965) Memory and cerebral protein synthesis in mice as affected by graded amounts of puromycin. Exp Neurol 13: 264–272. [DOI] [PubMed] [Google Scholar]
- Flood JF, Rosenzweig MR, Bennett EL, Orme AE (1973) The influence of duration of protein synthesis inhibition on memory. Physiol Behav 10: 555–562. [DOI] [PubMed] [Google Scholar]
- Frankland PW, O'Brien C, Ohno M, Kirkwood A, Silva AJ (2001) Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature 411: 309–313. [DOI] [PubMed] [Google Scholar]
- Judge ME, Quartermain D (1982) Alleviation of anisomycin-induced amnesia by pre-test treatment with lysine-vasopressin. Pharmacol Biochem Behav 16: 463–466. [DOI] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, deOrtiz SP, Kogan JH, Chevere I, Masushige S, Silva AJ (2002) CREB required for the stability of new and reactivated fear memories. Nat Neurosci 5: 348–355. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Fanselow MS (1992) Modality-specific retrograde amnesia of fear. Science 256: 675–677. [DOI] [PubMed] [Google Scholar]
- Konorski J (1967) Some new ideas concerning the physiological mechanisms of perception. Acta Biol Exp (Warsz) 27: 147–161. [PubMed] [Google Scholar]
- Kraus M, Schicknick H, Wetzel W, Ohl F, Staak S, Tischmeyer W (2002) Memory consolidation for the discrimination of frequency-modulated tones in mongolian gerbils is sensitive to protein-synthesis inhibitors applied to the auditory cortex. Learn Mem 9: 293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Abel T (2001) Different requirements for protein synthesis in acquisition and extinction of spatial preferences and context-evoked fear. J Neurosci 21: 5773–5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux JE (2000) Emotion circuits in the brain. Annu Rev Neurosci 23: 155–184. [DOI] [PubMed] [Google Scholar]
- Lewis DJ (1979) Psychobiology of active and inactive memory. Psychol Bull 86: 1054–1083. [PubMed] [Google Scholar]
- Mactutus CF, Riccio DC, Ferek JM (1979) Retrograde amnesia for old (reactivated) memory: some anomalous characteristics. Science 204: 1319–1320. [DOI] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgansberger W, Di Marzo V, Lutz B (2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418: 530–534. [DOI] [PubMed] [Google Scholar]
- McClelland JL, McNaughton BL, O'Reilly RC (1995) Why there are complementary learning systems in the hippocampus and neocortex: insights from the successes and failures of connectionist models of learning and memory. Psychol Rev 102: 419–457. [DOI] [PubMed] [Google Scholar]
- McGaugh JL (1966) Time-dependent processes in memory storage. Science 153: 1351–1358. [DOI] [PubMed] [Google Scholar]
- McGaugh JL (2000) Memory—a century of consolidation. Science 287: 248–251. [DOI] [PubMed] [Google Scholar]
- Milekic MH, Alberini CM (2002) Temporally graded requirement for protein synthesis following memory reactivation. Neuron 36: 521–525. [DOI] [PubMed] [Google Scholar]
- Misanin JR, Miller RR, Lewis DJ (1968) Retrograde amnesia produced by electroconvulsive shock after reactivation of a consolidated memory trace. Science 160: 554–555. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE (1995) Differential contribution of dorsal and ventral medial prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Behav Neurosci 109: 681–688. [DOI] [PubMed] [Google Scholar]
- Morgan MA, LeDoux JE (1999) Contribution of ventrolateral prefrontal cortex to the acquisition and extinction of conditioned fear in rats. Neurobiol Learn Mem 72: 244–251. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Romanski LM, LeDoux JE (1993) Extinction of emotional learning: contribution of medial prefrontal cortex. Neurosci Lett 163: 109–113. [DOI] [PubMed] [Google Scholar]
- Morrow BA, Elsworth JD, Rasmusson AM, Roth RH (1999) The role of mesoprefrontal dopamine neurons in the acquisition and expression of conditioned fear in the rat. Neuroscience 92: 553–564. [DOI] [PubMed] [Google Scholar]
- Myers KM, Davis M (2002) Behavioral and neural analysis of extinction. Neuron 36: 567–584. [DOI] [PubMed] [Google Scholar]
- Nader K (2003) Memory traces unbound. Trends Neurosci 26: 65–72. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE (2000a) Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature 406: 722–726. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, Le Doux JE (2000b) The labile nature of consolidation theory. Nat Rev Neurosci 1: 216–219. [DOI] [PubMed] [Google Scholar]
- Pavlov IP (1927) Conditioned reflexes. London: Oxford UP.
- Pedreira ME, Maldonado H (2003) Protein synthesis subserves reconsolidation or extinction depending on reminder duration. Neuron 38: 863–869. [DOI] [PubMed] [Google Scholar]
- Pedreira ME, Perez-Cuesta LM, Maldonado H (2002) Reactivation and reconsolidation of long-term memory in the crab Chasmagnathus: protein synthesis requirement and mediation by NMDA-type glutamatergic receptors. J Neurosci 22: 8305–8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyslawski J, Sara SJ (1997) Reconsolidation of memory after its reactivation. Behav Brain Res 84: 241–246. [DOI] [PubMed] [Google Scholar]
- Przybyslawski J, Roullet P, Sara SJ (1999) Attenuation of emotional and nonemotional memories after their reactivation: role of β adrenergic receptors. J Neurosci 19: 6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quevedo J, Vianna MR, Roesler R, de-Paris F, Izquierdo I, Rose SP (1999) Two time windows of anisomycin-induced amnesia for inhibitory avoidance training in rats: protection from amnesia by pretraining but not pre-exposure to the task apparatus. Learn Mem 6: 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk GJ, Russo GK, Barron JL, Lebron K (2000) The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J Neurosci 20: 6225–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rescorla RA (2001) Experimental extinction. In: Handbook of contemporary learning theories (Mowrer RR, Klein S, eds), pp 119–154. Mahwah, NJ: Erlbaum.
- Rescorla RA, Heth CD (1975) Reinstatement of fear to an extinguished conditioned stimulus. J Exp Psychol Anim Behav Process 1: 88–96. [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Heaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Neliat G, Caput D, Ferrara P, Soubrie P, Breliere JC, Le Fur G (1994) SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 350: 240–244. [DOI] [PubMed] [Google Scholar]
- Robbins SJ (1990) Mechanisms underlying spontaneous recovery in autoshaping. J Exp Psychol Anim Behav Process 16: 235–249. [Google Scholar]
- Sangha S, Scheibenstock A, Lukowiak K (2003a) Reconsolidation of a longterm memory in Lymnaea requires new protein and RNA synthesis and the soma of right pedal dorsal 1. J Neurosci 23: 8034–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangha S, Scheibenstock A, Morrow R, Lukowiak K (2003b) Extinction requires new RNA and protein synthesis and the soma of the cell right pedal dorsal 1 in Lymnaea stagnalis J Neurosci 23: 9842–9851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Muller RU, Quirk GJ (2001) Consolidation of extinction learning involves transfer from NMDA-independent to NMDA-dependent memory. J Neurosci 21: 9009–9017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sara SJ (2000) Retrieval and reconsolidation: toward a neurobiology of remembering. Learn Mem 7: 73–84. [DOI] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, LeDoux JE (1999) Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem 6: 97–110. [PMC free article] [PubMed] [Google Scholar]
- Schneider AM, Sherman W (1968) Amnesia: a function of the temporal relation of footshock to electroconvulsive shock. Science 159: 219–221. [DOI] [PubMed] [Google Scholar]
- Squire LR, Alvarez P (1995) Retrograde amnesia and memory consolidation: a neurobiological perspective. Curr Opin Neurobiol 5: 169–177. [DOI] [PubMed] [Google Scholar]
- Taubenfeld SM, Milekic MH, Monti B, Alberini CM (2001) The consolidation of new but not reactivated memory requires hippocampal C/EBP-beta. Nat Neurosci 4: 813–818. [DOI] [PubMed] [Google Scholar]
- Teich AH, McCabe PM, Gentile CC, Schneiderman LS, Winters RW, Liskowsky DR, Schneiderman N (1989) Auditory cortex lesions prevent the extinction of Pavlovian differential heart rate conditioning to tonal stimuli in rabbits. Brain Res 480: 210–218. [DOI] [PubMed] [Google Scholar]
- Vianna MR, Szapiro G, McGaugh JL, Medina JH, Izquierdo I (2001) Retrieval of memory for fear-motivated training initiates extinction requiring protein synthesis in the rat hippocampus. Proc Natl Acad Sci USA 98: 12251–12254. [DOI] [PMC free article] [PubMed] [Google Scholar]