

Abstract
Transgenic models of neurodegenerative disease have proved uniquely powerful for delineating pathways of neuronal dysfunction and cell death. We have developed a transgenic model of the polyglutamine disease spinal and bulbar muscular atrophy (SBMA), an adult-onset, slowly progressive motor neuron disease caused by polyglutamine expansion in the androgen receptor (AR). Mice bearing a human AR with 112 glutamines reproduce many aspects of SBMA, including slowly progressive, gender-specific motor deficits, and neuronal intranuclear inclusions. Despite substantial motor deficits in male AR112Q mice, no motor neuron loss was observed, indicating that neuronal dysfunction, rather than neuronal death, is central to disease. Moreover, reduced levels of unphosphorylated neurofilament heavy chain (NF-H) were observed in motor neurons, suggesting a role for NF-H in SBMA neuronal dysfunction. The elimination of androgens by surgical castration of severely affected, aged 112Q male mice partially restored motor function as well as NF-H levels. These data suggest that hormone-based therapies designed to treat SBMA patients, even with advanced disease, are likely to be effective.
Keywords: androgen, testosterone, motor neuron, neurofilament, polyglutamine, animal model, neurodegeneration, transgenic
Introduction
Polyglutamine expansion is responsible for a number of dominantly inherited neurodegenerative diseases, including Huntington's disease (HD), several spinocerebellar ataxias (types 1, 2, 3, 6, 7, and 17), dentatorubropallidoluysian atrophy (DRPLA), and spinal and bulbar muscular atrophy (SBMA) (Nakamura et al., 2001) (for review, see Cummings and Zoghbi, 2000; Zoghbi and Orr, 2000). SBMA, currently the only X-linked polyglutamine expansion disease, results from an expansion of >40 CAGs in the first exon of the androgen receptor (AR) gene (La Spada et al., 1991). Men affected with SBMA exhibit weakness and atrophy in the proximal limb muscles of the hip and shoulder girdles and the bulbar muscles associated with the jaw (Kennedy et al., 1968). Although patients exhibit mild androgen insensitivity, loss of androgen function alone is not responsible for the neuropathy seen in SBMA, as evidenced by the finding that individuals with complete androgen insensitivity syndrome (CAIS) do not manifest neurological symptoms (Pinsky et al., 1992; Quigley et al., 1992). Rather, polyglutamine expansion leads to the acquisition of a novel, toxic property of the mutant AR (Merry et al., 1998; Stenoien et al., 1999; Diamond et al., 2000). Despite this dominant toxic property, women rarely show symptoms of the disease, although some mild cases have been reported (Ferlini et al., 1995).
Pathological hallmarks of SBMA include loss of motor neurons in the brain stem and anterior horn of the spinal cord (Sobue et al., 1989) and the presence of both non-neuronal and neuronal intranuclear inclusions (NIIs), nuclear aggregates of mutant AR protein (Li et al., 1998a,b). In patients, NIIs contain only N-terminal portions of the androgen receptor, suggesting aberrant cleavage or degradation of the AR, resulting in an insoluble N-terminal protein fragment (Li et al., 1998a,b). In several polyglutamine diseases, NIIs have been found to colocalize with numerous cellular proteins, including molecular chaperones [such as heat shock protein (Hsp) 70, heat shock cognate protein (Hsc) 70, Hsp40, and Hsp90], proteasome pathway components (such as ubiquitin, 20S proteasome core, and the 19S component, Tat-binding protein 7), cAMP response element binding protein (CREB)-binding protein (CBP), and steroid receptor coactivator 1 (SRC-1) (Cummings et al., 1998; Chai et al., 1999a,b; Stenoien et al., 1999; Abel et al., 2001). Whether the NIIs are involved in pathogenesis or play a protective role by sequestering the mutant protein has not yet been determined.
A critical role for androgens in the development of SBMA has been demonstrated recently in both animal (Katsuno et al., 2002; Takeyama et al., 2002) and cell models (Stenoien et al., 1999; Becker et al., 2000; Darrington et al., 2002; Walcott and Merry, 2002). Although it is clear that female carriers of the SBMA mutation can be protected from the toxic effects of the mutant polyglutamine protein because of lyonization [although some mildly affected females show skewed X-inactivation (Ishihara et al., 2001)], it is now also clear that the lack of circulating androgens is responsible for sparing motor neurons in females. These findings have important implications for patient therapy and suggest that the reduction of androgens in symptomatic SBMA men may be an effective therapy, as shown recently in another mouse model (Katsuno et al., 2003).
Despite the widespread expression of many of the polyglutamine-containing proteins (for review, see Zoghbi and Orr, 2000), this family of diseases shows specific neuronal vulner-ability. In SBMA, lower motor neurons are exclusively affected despite the fact that AR is expressed at high levels, not only in motor neurons but also in neurons of the hypothalamus, midbrain, pons, and cerebellum (Sar and Stumpf, 1977). Previously, we created transgenic mice expressing a truncated AR with an expanded (112Q) polyglutamine tract (Abel et al., 2001). These mice developed severe neurologic disease with various symptoms not seen in SBMA, suggesting that lower motor neuron specificity may derive from biochemical and cellular events that depend on the context of the full-length AR.
An unexpected finding from previous mouse models of SBMA (Abel et al., 2001; Katsuno et al., 2002), as well as several mouse models of polyglutamine disease (Clark et al., 1997; Guidetti et al., 2001), was that very little neuronal degeneration was observed. In contrast, several transgenic models do reproduce the cell loss seen in human patients (McManamny et al., 2002; Li et al., 2003; Yu et al., 2003). Not withstanding these findings, the lack of substantial neuronal loss in the face of debilitating neurologic disease suggests that therapeutic approaches that promote the removal of the toxic protein, prevent its expression, or prevent downstream sequelae affecting neuronal function may be quite effective, even after the onset of symptoms. Indeed, the finding that symptoms in an inducible HD mouse model were reversed on cessation of mutant protein synthesis supports this concept (Yamamoto et al., 2000).
We have created a transgenic mouse model of SBMA in which a full-length, expanded androgen receptor containing 112 glutamines (112Q) is expressed via the prion protein (PrP) promoter. Mice develop neurologic symptoms that more closely reproduce those of human SBMA patients, suggesting that protein context contributes to the neuronal subtype specificity of this disease. The gender specificity of SBMA is also reproduced in this model, indicating a role for circulating androgens. Consistent with findings from previous SBMA models, little or no neuronal loss was observed in these transgenic mice, supporting the notion that the neurologic symptoms observed are a result of neuronal dysfunction rather than neuronal loss. This finding suggested that hormone manipulation, even after the onset of symptoms, might ameliorate disease symptoms. Indeed, we found that the removal of androgens via orchiectomy effected a partial, yet substantial, motor recovery, even in aged mice. These data suggest that hormone-based therapies may be efficacious in symptomatic SBMA patients. Furthermore, we found substantial decreases in the levels of nonphosphorylated neurofilament heavy chain in motor neuron soma; this change is reversed after castration, concomitant with motor recovery. This finding suggests that NF-H alterations may represent an integral pathological feature of SBMA.
Materials and Methods
Construct. The human androgen receptor gene, bearing either 24 CAG repeats (normal) or 112 CAG repeats (expanded), was cloned into the XhoI site of a PrP construct deleted of coding sequences (obtained from D. Borchelt, The Johns Hopkins University, Baltimore, MD). DNAs were injected into fertilized oocytes, derived from a C57BL/6 × SJL mating, by the Transgenic and Chimeric Mouse Facility at the University of Pennsylvania. Mice were maintained by breeding to C57BL/6–SJL F1 animals (Jackson Laboratory, Bar Harbor, ME).
Genotyping. DNA from mice was prepared from tail biopsies using a standard protocol (Miller et al., 1988). Transgenic animals were identified by PCR of the human AR: forward primer from the PrP promoter region (5′-ACTGAACCATTTCAACCGAGC-3′), coupled with a reverse primer from the AR sequence 5′ to the CAG repeat (5′-AGGTGCTGCGCTCGCGGCCTCT-3′).
RT-PCR. Freshly dissected mouse tissue was homogenized in Trizol reagent (Invitrogen, Gaithersburg, MD) and RNA extracted according to the manufacturer's specifications. One microgram of RNA was treated with RQ1 DNase (Promega, Madison, WI) according to the manufacturer's specifications to eliminate residual DNA from the samples. RT-PCR was performed using the one-step RT-PCR kit (Qiagen, Hilden, Germany), using the primers described above. GAPDH was amplified as a control using primers with the following sequences: 5′-CATGTGGGCCATGAGGTCCACCAC-3′ and 5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′ (Invitrogen). Samples were electrophoresed on a 2% agarose gel, and the bands were visualized with ethidium bromide. A reaction lacking reverse transcription was performed for each sample–primer pair combination as an additional control.
Western blot analysis. Freshly dissected tissue was flash-frozen in liquid nitrogen. Frozen sections were pulverized in a mortar and pestle on dry ice and homogenized in 10 vol of sample buffer (20 mm DTT, 4% SDS, 160 mm Tris-HCl, pH 6.9, 20% glycerol, 0.004% bromophenol blue). Lysates were then sonicated three times for 10 sec using a Branson cup sonifier. Aliquots (50 μl) of protein lysates were electrophoresed by SDS-PAGE and transferred to nitrocellulose (Immobilon-P) using a semidry transfer apparatus. Western hybridization was performed using antibodies to the AR (ARN-20, Santa Cruz Biotechnology, Santa Cruz, CA). Detection was performed with ECL (Amersham Biosciences, Arlington Heights, IL).
Behavioral analyses. Latency to fall off a steadily accelerating (4–40 rpm over 10 min) Rotamex rotarod (Columbus Instruments, Columbus, OH) was measured monthly in an age-matched cohort of mice. Mice were tested four times per day for 3 consecutive days during the light phase of a 12 hr light/dark cycle. Mice were allowed a rest period of at least 15 min between testing. Scores were analyzed by two-way ANOVA using SigmaStat software.
Clasping was quantified for castrated or sham-castrated animals. Clasping was defined as flexion of one of the limbs. Mice were suspended by the tail for 30 sec to assess duration and frequency of the clasping behavior. Mice were given a rank (0 = no clasp to 5 = full, sustained clasp) based on the severity of the clasp. Scores were analyzed using the Mann–Whitney rank sum test and SigmaStat software.
Vertical and horizontal motion in a novel environment were measured using a Versamax activity monitor (AccuScan Instruments, Columbus, OH). Mice were monitored for 5 min. Data were analyzed using Versamax software. Significance was assessed using Student's t test.
Forepaws were dipped in red, water-soluble tempera paint; hindpaws were dipped in blue paint. Footprints were left on a strip of paper while mice walked to the end of a narrow tunnel.
A grip strength meter (Columbus Instruments) measured the force exerted by a mouse as it was pulled from a grid by the tail. Grip strength was measured for forepaws only and hindpaws and forepaws together (all paws). Grip strength was measured six times for both forepaws and all paws; the lowest and highest scores for each animal were dropped. Significance was determined with Student's t test.
Immunofluorescence. Tissue was frozen in OCT before fixation. Frozen sections (7 μm) were fixed with 4% paraformaldehyde for 10 min, washed in PBS, blocked in 1.5% goat serum (Jackson ImmunoResearch, West Grove, PA) for 20 min, washed again in PBS, and incubated 60 min in primary antibody (Ab) diluted in 1.5% goat serum. Tissue was washed in PBS, incubated for 30 min in secondary (FITC or rhodamine-conjugated secondary Abs (Jackson ImmunoResearch, West Grove, PA), washed in PBS, incubated for 10 min with Hoechst (0.25 nm), washed in PBS, and mounted in Immumount (Vector Laboratories, Burlingame, CA). Fluorescence was visualized with a Leica microscope; images were captured with a Leica camera and compiled with IP Labs software. Antibodies used include AR N-20, AR C-19, AR 441, SRC-1, and CBP (Santa Cruz Biotechnology), anti-ubiquitin and anti-glial fibrillary acidic protein (GFAP) (Dako, High Wycombe, UK), F4/80 (Serotec), SMI32 (Sternberger Monoclonal), AR 318 (Novo Castra), the DNAJ homolog HDJ-2 (NeoMarkers), anti-Hsp90, anti-Hsp40, anti-Hsc70, and anti-Hsp70 (Stressgen).
Immunohistochemistry. Neuronal populations were assessed using NeuN, a nuclear neuronal stain. Slides were dehydrated briefly in an ethanol series and xylene and mounted in Vectamount (Vector Laboratories). Mice were perfused with 2.5% paraformaldehyde–0.5% glutaraldehyde, and brain and spinal cord tissue were embedded in paraffin. Seven micrometer sections were defatted in xylene and rehydrated through a series of ethanol concentrations, and finally in water. Antigen retrieval was performed by autoclaving for 10 min in water. Endogenous peroxides were quenched with 3% peroxide in PBS. Tissue was washed in PBS, incubated in NeuN, and developed using Vector's ABC kit according to manufacturer's specifications.
Histology. Cresyl violet stain was applied to unfixed frozen sections (7 μm) for 10 min. Slides were dehydrated briefly in an ethanol series and xylene and mounted in Vectamount (Vector Laboratories). Quadriceps, masseter, and bulbocavernosis were dissected and embedded in OCT in isopentane cooled in liquid nitrogen. Frozen muscle sections (7 μm) were fixed in 4% paraformaldehyde, and Harris's hematoxylin stain (Fisher Scientific, Houston, TX) was applied for 5–10 min. Muscle sections were dehydrated briefly in an ethanol series and then xylene and mounted in Vectamount (Vector Laboratories).
Quantification of motor neurons. Motor neuron number, area, and, in the case of immunofluorescence, intensity of signal, were quantified from five 7 μm sections of lumbar spinal cord, beginning at the rostral edge of the lumbar intumescence (70 μm between sections) using IP Labs software on images captured with a Leica microscope. Statistical significance was determined using Student's t test.
Surgery. Mice were anesthetized with inhaled isofluorane. The scrotum was shaved, and the surgical area was sterilized with Betadine. Using aseptic technique and sterile instruments, a small (1–2 cm) ventral midline incision was made in the scrotum, and the skin was retracted to expose the tunica. The tunica was pierced, and the opening was stretched with a blunt probe. The testes were pushed out by gentle pressure on the pelvic region. The spermatic artery was sutured, and the testis removed. The epididymus, deferential vessels, and ductus were replaced in the tunica. Surgical glue was used to close the incision. During recovery, animals were warmed with a 37°C heating pad. Buprenex was administered (0.1 ml/20 gm mouse, i.p.) for analgesia after surgery. All animal surgeries were conducted under the guidance of Thomas Jefferson University veterinary staff and performed according to the guidelines of the Office of Laboratory Animal Welfare and the Thomas Jefferson University Institutional Animal Care and Use Committee.
Results
Transgenic lines and transgene expression
We identified three founder animals bearing the PrP–AR112 construct and two carrying the PrP–AR24 construct. From these founders, four transgenic lines were established, two carrying the transgene with a highly expanded (112 CAG) polyglutamine tract and two carrying a normal length (24 CAG) tract. Neither of the 112Q lines showed a decrease in life span; mice from both lines frequently live past 24 months of age. Decreased fertility is apparent in males from one of the 112Q lines. These males cease to be fertile after 6 months of age. Mice from lines with a normal length polyglutamine expansion (24Q) show no deficits in fertility and have normal life spans.
Transgene expression was detected in all four lines by RT-PCR, Western blot, and immunostaining. A survey of several different tissues in one 112Q line revealed widespread expression of transgene RNA. Analysis of RNA levels revealed that transgene expression in lines with 24 CAGs and 112 CAGs were similar in brain and spinal cord (data not shown). Western blot analysis of brain and spinal cord tissue from transgenic males revealed similar levels of AR protein between 24Q and 112Q transgenic mice (Fig. 1). Low levels of endogenous mouse AR could also be observed in nontransgenic and 112Q lanes. In addition to the monomeric AR in AR112Q spinal cord and brain, a detergent-insoluble high molecular weight AR complex was also observed.
Figure 1.

AR protein expression in transgenic 112Q and 24Q transgenic male mice reveals detergent-soluble monomeric AR, as well as detergent-insoluble macromolecular complex in 112Q mice. Western blot of lysates from brain and spinal cord of 112Q, 24Q, and nontransgenic (ntg) male mice was hybridized with AR Ab N-20 (A). Lysate from AR112Q-expressing PC12 cells was used as a positive control. Western blot of lysates from brain of 112Q male and female, castrated (Cast.) 112Q male, 24Q male, and ntg male mice was hybridized with antibody N-20. Lysate from castrated 112Q male indicates a reduction in AR monomer as well as a decrease in insoluble macromolecular complex (B). Actin was used as a loading control.
Behavior
Male mice carrying the 112Q transgene displayed slowly progressive motor impairment in various behavioral assays. As early as 6 weeks of age, transgenic 112Q males displayed intermittent clasping of the forelimbs. Frequency and severity of clasping increased, and by 5 months of age, male 112Q transgenics had developed a full clasp, clutching all four limbs to the abdomen and curving the spine. Female 112Q transgenics developed intermittent forelimb clasping by 4 months of age, but this clasp did not progress to a full clasp until 16 months of age. Transgenic mice bearing a 24Q transgene rarely exhibited clasping behavior, as did nontransgenic animals.
Rotarod function was examined as a measure of motor ability. Latency to fall from a steadily accelerating rod was measured monthly in an age-matched cohort of mice. Statistically significant (p < 0.05) rotarod deficits became apparent in transgenic 112Q males (n = 7) at 3 months of age and from 4 months of age onward were consistently observed (Fig. 2A). Rotarod scores progressively declined in these mice, until by 8 months of age transgenic 112Q males were unable to remain on the rod. Rotarod function in female 112Q transgenics (n = 8) was somewhat decreased at 4 months of age; however, deficits were not consistently observed until 8 months of age (p < 0.05) (Fig. 2A). By 12 months of age, female 112Q transgenics were still able to remain on the rotarod, although for a decreased period of time. At 14 months of age, rotarod function in 24Q transgenic mice was comparable with that of nontransgenic animals (n = 8 per genotype) (data not shown).
Figure 2.
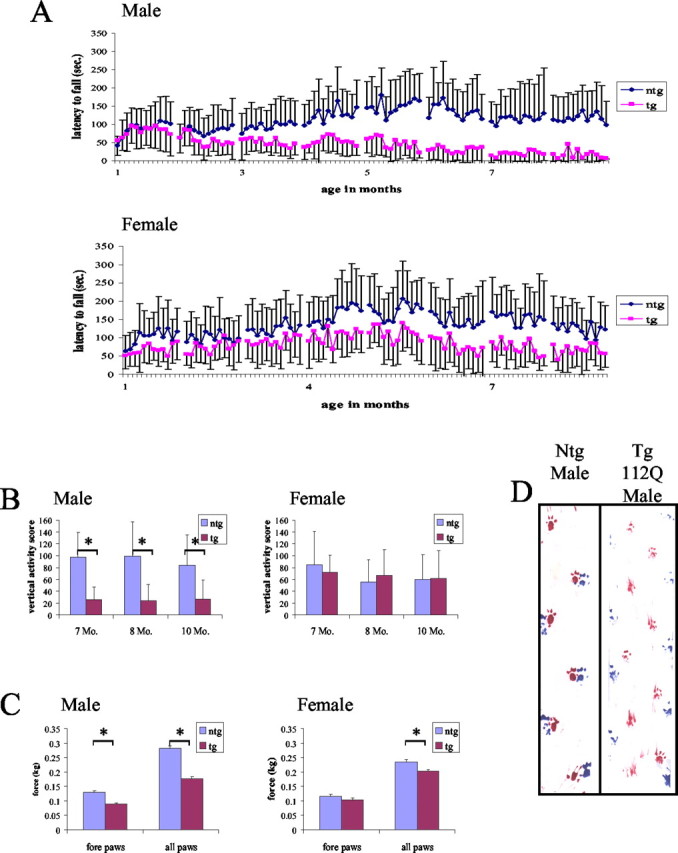
Male 112Q mice have more severe loss of motor function and earlier onset of deficiencies than their 112Q female littermates. 112Q males (n = 7) begin to show deficiencies in rotarod function beginning at 3 months of age; rotarod function continues to decline until ∼8 months of age, when 112Q mice are unable to stay on the rotarod (A). In contrast, 112Q females (n = 8) do not begin to show significant deficits in rotarod function until 8 months of age; although their rotarod function is impaired, 112Q females still retain the ability to stay on the rotarod, even at 12 months of age (A). 112Q Males also have decreased vertical activity, when compared with nontransgenic littermates (n = 8), whereas vertical activity in 112Q females is not significantly different from that of nontransgenic females (n = 12) (B); *p < 0.05. 112Q males have decreased grip strength with forepaws alone and all paws together, whereas female 112Q animals only show deficits in all paw grip strength (C), implying weakness of the hindlimbs. Error bars represent 1 SD. Male 112Q mice have an abnormal gait characterized by a widened, lower hindlimb posture and shorter hindlimb steps (D). The hindpaws were painted with blue paint, and the front paws were painted with red paint.
Mice were tested for changes in open field activity. Vertical and horizontal motion in a novel environment were measured for 5 min. Although female 112Q transgenics (n = 8) showed no significant changes in exploratory activity when compared with nontransgenic littermates (n = 12), transgenic 112Q males (n = 7) demonstrated significant decreases (p < 0.05) in both vertical and horizontal activity (Fig. 2B). By 7 months of age, male 112Q transgenics were rarely observed rearing without leaning against the cage wall, consistent with their hindlimb weakness.
Abnormalities in gait were also assessed. By 7 months of age, male 112Q mice (n = 7) displayed a distinct gait abnormality characterized by low posture and widened stance in the hindlimbs and shorter, “waddling” steps when compared with nontransgenic male littermates (Fig. 2D). Female transgenic 112Q mice (n = 8) did not display gait abnormalities until ∼14 months of age, whereas 24Q (n = 8) transgenics never developed gait abnormalities.
To ascertain whether the observed motor deficits were caused by muscle weakness or decreased coordination, grip strength was measured at 10 months of age. Grip strength was measured for forepaws only and hindpaws and forepaws together (all paws). Significant decreases in grip strength (p < 0.05) were observed when male 112Q transgenic mice (n = 7) gripped with all paws or forepaws alone (Fig. 2C). Transgenic 112Q female mice (n = 8) showed significantly decreased grip strength (p < 0.05) only when tested for all paws (Fig. 2C), suggesting that hindlimb strength is affected earlier in disease progression than that of the forelimbs.
Although none of the transgenic 112Q mice had lost significant weight by 14 months of age, male 112Q transgenic mice (n = 7) stopped gaining weight by 6 months of age. Consequently, the transgenic 112Q males weighed significantly less than their nontransgenic counterparts (n = 8) by 8 months of age (average weight for transgenic = 30.22 ± 2.48 gm vs average weight for nontransgenic = 32.23 ± 1.15 gm; p < 0.05). No significant weight differences were observed between transgenic and nontransgenic 112Q females (n = 8) or in 24Q transgenics (n = 8).
In summary, our data indicate that male transgenic mice develop a lower motor neuron disease primarily characterized by slowly progressive hindlimb muscle weakness. Male 112Q transgenic mice demonstrated greater motor deficits and earlier disease onset than female 112Q transgenic littermates, indicating that these mice recapitulate much of the gender specificity of the human disease. These data, coupled with the autosomal pattern of inheritance of the transgene, indicate that the disease process is dependent on the binding of circulating androgens.
Pathology
Male 112Q transgenic mice exhibited limited signs of neuronal pathology. NIIs were observed in several areas of the brain, particularly in the cortex and brain stem, and also in the spinal cord. Of note, NIIs were detected only with the N-terminal antibodies AR(N20) and AR(318) and were not seen with more C-terminal antibodies AR(C19) and AR(441) (Fig. 3). The presence of N-terminal portions of the androgen receptor in the NIIs in this model is consistent with studies of NIIs in patient tissue (Li, 1998). A progressive increase in the size of NIIs was also observed. As early as 6 weeks of age, multiple punctate NIIs could be detected in male 112Q transgenic spinal cord (Fig. 3C). By 4 months of age, single large NIIs were apparent (Fig. 3A,B). Unlike male 112Q transgenics, brain and spinal cord tissue from transgenic 112Q females rarely contained NIIs, and only at ≥10 months of age (Fig. 3E). No aggregates were seen in brain or spinal cord tissue from male nontransgenic animals (data not shown) or in neuronal tissue from 24Q transgenic male mice (Fig. 3D).
Figure 3.

NIIs are detected in 112Q male spinal cord. Shown are tissue sections from lumbar spinal cord. NIIs formed progressively in 112Q males from multiple, punctate inclusions at 6 weeks of age (C) to single, large inclusions by 4 months of age (A, B). Antibodies to N-terminal portions of the AR, such as AR-N20 (A) and AR-318 (B), detected NIIs, whereas C-terminal AR antibodies, such as AR-C19 (F) (12 months) and AR-441 (data not shown), did not. NIIs were not detected in spinal cord tissue from a nontransgenic male at 12 months of age (data not shown), in a 112Q female at 12 months of age (N20) (E), or in a 24Q male at 12 months of age (N20) (D).
On the basis of findings from previous mouse and cell culture models of polyglutamine diseases, NIIs were assessed for sequestration of various proteins. Using immunofluorescence, the chaperones HDJ-2 and HSC70, as well as ubiquitin, were found to colocalize with NIIs (data not shown). Other proteins such as HSP70, HSP90, SRC-1, and CBP did not colocalize with NIIs in this model at ages up to 10 months.
Brain and spinal cord tissue from 112Q transgenic males were assessed for signs of neuron degeneration using various assays. Cresyl violet staining did not reveal differences between the number of large motor neurons in transgenic and nontransgenic tissue (Fig. 4A,B) nor did staining with hematoxylin or the neuronal nuclear marker, NeuN (data not shown). Quantitative analysis of the number and size of motor neurons in the anterior horn of lumbar spinal cord in 14-month-old transgenic and nontransgenic males indicated no neuron loss or decrease in neuron size (Fig. 4G). In lumbar spinal cord of the same animals, however, quantitative image analysis revealed a decrease in the number of neurons immunostained with SMI32, which detects nonphosphorylated NF-H (p < 0.05). Image analysis also revealed a statistically significant decrease in the intensity of the SMI32 signal (p < 0.05) (Fig. 4I). No significant difference was observed between transgenic and nontransgenic males, however, in the size distribution of the neurons detected by the Ab SMI32 (Fig. 4H). Transgenic males bearing the 24Q AR transgene displayed wild-type levels of SMI32 staining (see Fig. 6A), indicating that expression of the AR transgene alone does not alter the presence of unphosphorylated NF-H.
Figure 4.

Unphosphorylated NF-H immunoreactivity is reduced in 112Q symptomatic males. Nissl staining reveals a similar number of motor neurons in the anterior horn of nontransgenic (A) and 112Q (B) males at 14 months of age, well after the onset of symptoms. Motor neuron size is also unaffected (G); however, immunostaining with SMI32, an Ab to unphosphorylated NF-H, reveals a reduction in both the number of immunoreactive neurons (p = 0.044) (H) and the intensity of staining (p = 0.038) (I) in 112Q males (D) when compared with age-matched nontransgenic littermates (C). The population of neurons detected with SMI32 did not differ significantly in size distribution (H). Error bars represent 1 SD. Quadriceps muscle from 14-month-old 112Q males (F) does not show signs of atrophy when compared with age-matched nontransgenic muscle (E).
Figure 6.
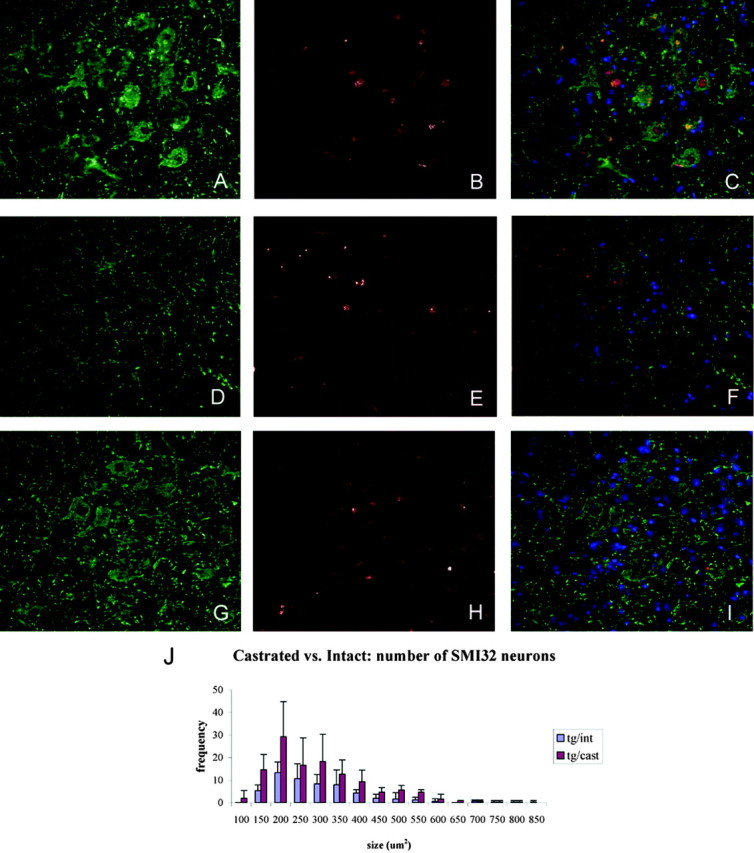
Castrated 112Q males show increased unphosphorylated NF-H immunoreactivity and decreased NII load. Aged (21 months) 112Q males demonstrated high NII load (E) in the spinal cord and a reduction of SMI32 immunoreactivity (D). Age-matched 24Q males had no NIIs (B) and normal amounts of SMI32 immunoreactivity (A), indicating that overexpression of AR is not responsible for this phenotype. Five months after castration, decreased NII load (H) and an increased number of SMI32-positive neurons (G) were observed in 23-month-old 112Q males (p < 0.05) (J). Error bars represent 1 SD. C, F, I, Merged images with the inclusion of DAPI stain.
Signs of muscle degeneration were absent in transgenic male animals up to 23 months of age. Tissue from quadriceps and masseter muscles appeared normal in both female and male 112Q transgenics (Fig. 4F). Bulbocavernosis muscle from 112Q transgenic males also appeared normal. Moreover, nicotinamide adenine dinucleotide staining did not reveal any fiber-type grouping (data not shown). These data suggest that the motor dysfunction in these animals is caused by deficits in neuronal function rather than neurogenic atrophy or myopathy.
Castration of symptomatic transgenic mice
Our finding that male mice transgenic for an expanded form of the AR developed more severe motor dysfunction than females and at a much earlier age indicates an important role for androgens in the pathogenesis of disease. Moreover, our finding of minimal neuronal loss in the face of substantial loss of motor function suggested that manipulation of androgens in males with progressed disease might be effective. To this end, symptomatic 9-month-old 112Q males were castrated, and their rotarod function was assessed. Beginning at 3 months after castration, castrated transgenic 112Q males (n = 9) showed significantly higher rotarod function than control 112Q transgenic littermates (n = 8) (p < 0.001) (Fig. 5A), although their rotarod function was still depressed when compared with nontransgenic castrated littermates (n = 9) (during the fifth month after castration: average nontransgenic castrated score = 146 ± 70.0 vs transgenic castrated score = 25.2 ± 32.1 vs transgenic sham score = 7.3 ± 4.7). Clasping behavior was milder in castrated 112Q transgenic animals (p < 0.05); forelimb-only clasping was observed in 112Q castrated males (average score = 3.33), whereas sham-castrated 112Q males exhibited a full clasp (average score = 4.67). The weight of transgenic castrated animals did not differ significantly from those of transgenic sham-castrated animals.
Figure 5.
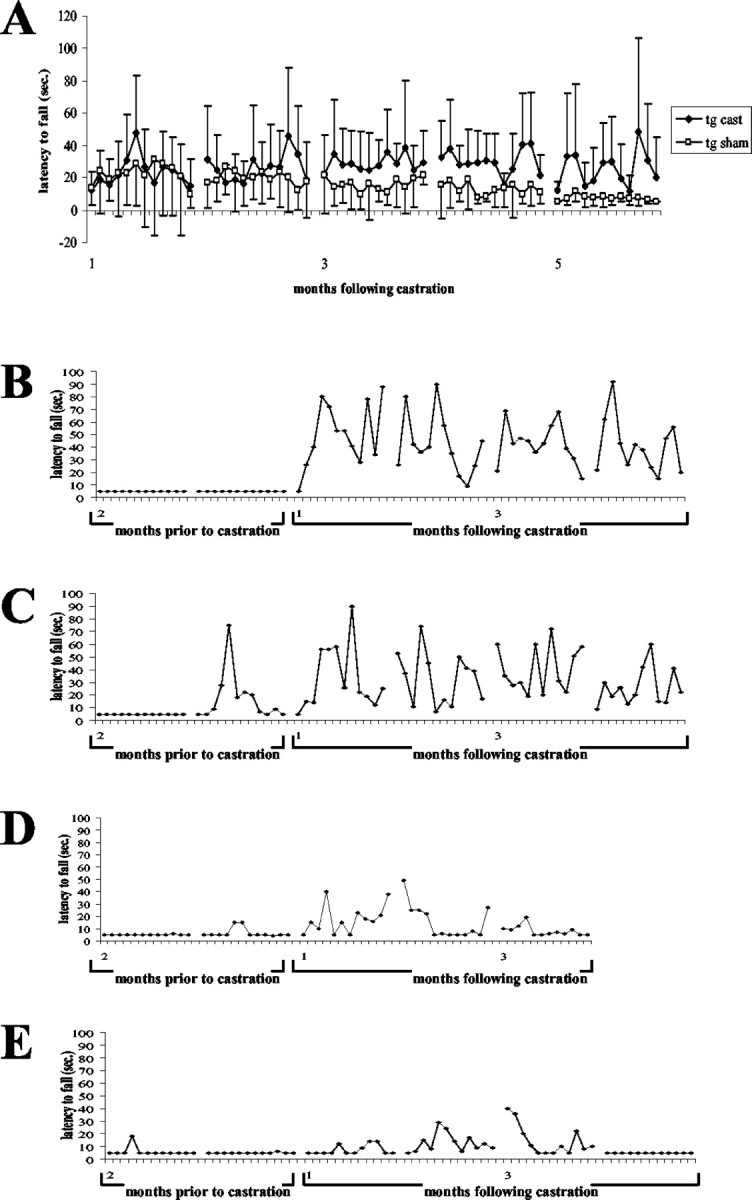
Castrated 112Q males show signs of recovery after surgery. Castration surgery was performed on a cohort of 9-month-old 112Q males. Three months after castration, rotarod function was significantly improved in castrated 112Q males (n = 9) when compared with intact 112Q males (n = 8), although still depressed in comparison with castrated nontransgenics (n = 8) (A). Error bars represent 1 SD. In a group of late-symptomatic 18-month-old males, rotarod function was severely depressed before castration. After surgery, two animals showed substantial improvement of rotarod function (B, C), whereas recovery was limited in the other two 112Q males (D, E).
Additionally, four 18-month-old 112Q mice that exhibited severely limited rotarod function for 10 months (shown for months 1–8 in Fig. 2A) were castrated. Of these animals, two showed substantial recovery of function (Fig. 5B,C), and two showed limited improvement (Fig. 5D,E).
NII load in brain and spinal cord tissue from castrated 112Q transgenics was reduced when compared with neuronal tissues from age-matched, intact 112Q transgenic males (Fig. 6E,H). In addition, brain tissue lysates from castrated 112Q males demonstrated a reduction in levels of insoluble macromolecular complex when compared with age-matched intact males (Fig. 1B). Also notable is the substantial reduction of monomeric AR in castrated males to levels even lower than those of an age-matched female mouse. This is not surprising given the fact that ligand binding is known to stabilize the AR (Kemppainen et al., 1992). Taken together, these data indicate that even after a prolonged disease course neurons are capable of clearing the mutant protein in the absence of hormone. Analysis of unphosphorylated NF-H levels revealed an increase in the number of neurons containing this neurofilament protein after castration (p < 0.05) (Fig. 6G). Although castration caused the restoration of unphosphorylated NF-H in spinal motor neurons, the intensity of the restored signal was still somewhat depressed when compared with nontransgenic or 24Q transgenic mice. The concomitant restoration of unphosphorylated NF-H levels with motor function after surgical castration suggests an important role for this axonal protein in SBMA pathogenesis and recovery. Further analysis of neurofilament levels, and of axonal trafficking, should illuminate this issue.
Discussion
We have demonstrated that transgenic mice expressing an expanded full-length AR driven by a strong neuronal promoter replicate several important features of SBMA. These include a slowly progressing motor dysfunction characterized by muscle weakness and gait abnormalities. Like SBMA patients, mice do not become paralyzed, nor is death a necessary endpoint of disease progression. Also like SBMA patients, the disease is restricted primarily to males, although females develop a substantially milder, less progressive disease. Histological analysis reveals that our transgenic mice also exhibit an important pathological feature of SBMA, the formation of NIIs.
The male specificity of these mice resembles that described for another mouse model of SBMA (Katsuno et al., 2002). In an elegant study, these mice were shown to be absolutely dependent on the presence of male hormones for disease onset and progression; however, these mice, which develop both myopathic and neurogenic muscle atrophy, have severely diminished life spans, making them refractory to therapeutic approaches beyond disease onset (Katsuno et al., 2003). Our mouse model of SBMA replicates both the gender specificity and slow disease progression seen in human SBMA. Although transgenic males have greater severity and earlier onset of symptoms than do transgenic females, their life span appears relatively unaffected and their motor function becomes severely impaired later in life. In addition, few, if any, signs of muscle degeneration were seen in our transgenic males, even well after the onset of symptoms, indicating that neither myopathy nor neurodegeneration is central to the neurologic phenotype. Data from our model support the findings from other models of SBMA (Abel et al., 2001; Katsuno et al., 2002), as well as models of other polyglutamine diseases (Clark et al., 1997; Guidetti et al., 2001), that neuron dysfunction rather than neuron loss may be responsible for all but the latest disease symptoms. Reduction of unphosphorylated NF-H may serve as a marker for such dysfunction or suggest a pathway that may be impaired in disease.
In addition to similarities in disease progression, pathologic features of human SBMA are present in our mouse model. NIIs are present and detected by antibodies to the N-terminal but not the C-terminal regions of the AR. The presence of only N-terminal epitopes in these NIIs is similar to NIIs in patient tissue (Li et al., 1998a) and indicates that the same process producing this phenomenon is at work in our mice as well. NIIs were observed in several areas of the brain, but particularly in the cortex, brain stem, and spinal cord. This is in contrast to the transgenic mice created with the truncated AR containing 112 CAGs, in which NIIs were observed in nearly every neuron of the brain and spinal cord (Abel et al., 2001). This finding suggests that the context of the full-length AR protein contributes to the specificity of the neuronal populations that is affected. The context of the full-length protein may also contribute to the population of proteins sequestered in the NIIs. Although NIIs in this model were found to contain ubiquitin, Hdj-2, and Hsc70, other proteins that were detected in NIIs in previous truncated AR mouse models, such as CBP, were not detected. Whether the protein determinants that contribute to cell type specificity do so through cell-specific interactions or through motor neuron-specific hormone metabolism remains to be determined.
Data from several recent models have indicated that the presence of ligand has a substantial effect on the gender specificity of SBMA (Darrington et al., 2002; Katsuno et al., 2002; Takeyama et al., 2002; Walcott and Merry, 2002). This explains why previous models, which used AR transgenes that lacked the C-terminal ligand-binding domain, failed to show phenotypic gender differences. The neurologic phenotype in our AR112Q mice is gender specific, despite an autosomal pattern of inheritance, supporting the idea that the levels of circulating AR ligand contribute to disease progression.
Understanding the role of ligand in SBMA may suggest mechanisms that can be exploited for treatment of the disease. Surgical and chemical castration, both preceding and shortly after the onset of symptoms, have been shown to ameliorate disease in transgenic mice (Katsuno et al., 2002, 2003); however, these treatments were ineffective in late-symptomatic animals. The fact that, in several models, motor neurons are still present after symptoms become severe suggests that restoring function to the motor neuron will allow the rescue of at least some motor function. This idea is supported by data from our transgenic AR112Q model, in which castrated late-symptomatic transgenic males retain rotarod function, whereas the performance of shamcastrated transgenic males continues to decline. Furthermore, castration resulted in recovery of function for a few 18-month-old 112Q animals with severe loss of rotarod function. The amount of recovery varied substantially among animals, with two animals showing a marked improvement of function after castration and two showing little recovery. It is likely that the hybrid genetic background of these animals contributes to the variability in their recovery.
The mechanism by which hormone ablation ameliorates disease is undetermined. Androgens have been shown to extend the half-life of AR protein (Kemppainen et al., 1992); therefore, removing testosterone may increase turnover of mutant AR, preventing its accumulation to toxic levels or shortening the exposure of the cell to its toxic effects. (Indeed, the manifestation of mild disease in very aged transgenic females may result from long-term accumulation of 112Q AR in the nucleus caused by small amounts of circulating testosterone.) Alternatively, eliminating hormone prevents the ligand–receptor binding necessary for the translocation of AR into the nucleus. It has been demonstrated that nuclear translocation is an important step in disease pathogenesis in a fly model of SBMA (Takeyama et al., 2002); thus blocking translocation may slow disease progression. In either case, the data from both our 9- and 18-month-old AR112Q animals indicate that removal of androgens ameliorates disease phenotype well after the onset of symptoms. This is particularly relevant when considering treatment of human disease; patients with progressed SBMA may still gain benefits from hormone ablation therapy.
Although the mechanisms that contribute to neuronal dysfunction are unclear, several possibilities exist, including disruption of transcriptional regulation, abnormal proteolysis, inability to launch a heat shock response, or a combination of these factors. It is likely that changes in unphosphorylated NF-H serve as a marker for neuronal dysfunction and may indicate a pathway that is affected in this process. The decrease in unphosphorylated NF-H observed in intact, symptomatic 112Q males may be the result of upstream abnormalities resulting in hyperphosphorylation of NF-H, a decrease in NF-H expression, or a change in unphosphorylated NF-H localization. No increase was detected in the level of phosphorylated NF-H in spinal motor neurons (data not shown), suggesting that reduced SMI32 staining represents a decrease in NF-H expression or a change in NF-H localization. Indeed, preliminary data suggest an increase in SMI32 stain in axonal tracts within the brain of 112Q transgenic males (data not shown). Western blot analyses of NF-H expression from spinal cord lysates in 112Q males have not revealed substantial changes. This is not entirely unexpected, because the affected neurons contribute only a small percentage to the total cellular mass of the spinal cord. It is possible that changes in NF-H regulation could have substantial downstream repercussions. Changes in NF-H expression have been shown to affect expression of other neurofilament subunits (Tu et al., 1995), whereas changes in NF-H phosphorylation have been shown to modulate proteolysis (Huh et al., 2002), association with microtubules (Saito et al., 1995), and axonal transport (Jung et al., 2000). Such disruption could affect axonal transport or cytoskeletal integrity. Abnormalities in anterograde transport have been reported in a cellular model of SBMA (Piccioni et al., 2002), and there is increasing evidence that deficits in transport contribute to other motor neuron diseases, such as amyotrophic lateral sclerosis (Williamson and Cleveland, 1999; LaMonte et al., 2002; Puls et al., 2003). It is notable that defects in retrograde axonal transport produce a slowly progressing form of human lower motor neuron disease with bulbar manifestations (Puls et al., 2003), suggesting that SBMA, another lower motor neuron disease, may involve similar defects in transport. In addition, recent studies have demonstrated that abnormal expression of huntingtin causes transport deficits in Drosophila (Gunawardena et al., 2003), whereas overexpression of expanded huntingtin or expanded AR disrupts fast axonal transport in squid giant axons (Szebenyi et al., 2003). Our finding of NF-H changes supports the involvement of axonal transport in the progression of SBMA. An increase in unphosphorylated NF-H immunoreactivity is observed in castrated 112Q males, concomitant with partial functional motor recovery and decreased NII load. This finding further supports the notion that NF-H levels may contribute to motor neuron dysfunction in this disease. Additional studies of the nature of NF-H alterations in SBMA will increase our understanding of the role of this change in disease.
Footnotes
This work was supported by the Muscular Dystrophy Association and the National Institutes of Health (NIH) (Grant R01 NS32214) (D.E.M.), NIH Signal Transduction Training Grant T05210 (E.S.C.-L.), and a Michigan Alzheimer's Disease Research Center grant (NIH Grant P50 AG08671) (A.P.L.). E.S.C.-L. was supported in part by a Farber Institute for Neurosciences Fellowship from Thomas Jefferson University. We thank Dr. David Borchelt for the prion protein promoter construct, Rodney Bermudez and Marianne Hamel for technical assistance, and Dr. Jean Richa (University of Pennsylvania Transgenic and Chimeric Mouse Facility) for transgene injections.
Correspondence should be addressed to Dr. Diane E. Merry, Thomas Jefferson University, 208 Bluemle Life Sciences Building, 233 South 10th Street, Philadelphia, PA 19107. E-mail: diane.merry@jefferson.edu.
DOI:10.1523/JNEUROSCI.0808-04.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244778-09$15.00/0
References
- Abel A, Walcott J, Woods J, Duda J, Merry DE (2001) Expression of expanded repeat androgen receptor produces neurologic disease in transgenic mice. Hum Mol Genet 10: 107–116. [DOI] [PubMed] [Google Scholar]
- Becker M, Elke M, Schneikert J, Krug HF, Cato ACB (2000) Cytoplasmic localization and the choice of ligand determine aggregate formation by androgen receptor with amplified polyglutamine stretch. J Cell Biol 149: 255–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Bonini NM, Paulson HL (1999a) Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci 19: 10338–10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL (1999b) Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet 8: 673–682. [DOI] [PubMed] [Google Scholar]
- Clark HB, Burright EN, Yunis WS, Larson S, Wilcox C, Hartman B, Matilla A, Zoghbi HY, Orr HT (1997) Purkinje cell expression of a mutant allele of SCA1 in transgenic mice leads to disparate effects on motor behaviors, followed by a progressive cerebellar dysfunction and histological alterations. J Neurosci 17: 7385–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings C, Zoghbi H (2000) Fourteen and counting: unraveling trinucleotide repeat diseases. Hum Mol Genet 9: 909–916. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY (1998) Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet 19: 148–154. [DOI] [PubMed] [Google Scholar]
- Darrington RS, Butler R, Leigh PN, McPhaul MJ, Gallo JM (2002) Ligand-dependent aggregation of polyglutamine-expanded androgen receptor in neuronal cells. NeuroReport 13: 2117–2120. [DOI] [PubMed] [Google Scholar]
- Diamond MI, Robinson MR, Yamamoto KR (2000) Regulation of expanded polyglutamine protein aggregation and nuclear localization by the glucocorticoid receptor. Proc Natl Acad Sci USA 97: 657–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlini A, Patrosso MC, Guidetti D, Merlini L, Uncini A, Ragno M, Plasmati R, Fini S, Repetto M, Vezzoni P, Forabosco A (1995) Androgen receptor gene (CAG)n repeat analysis in the differential diagnosis between Kennedy disease and other motoneuron disorders. Am J Med Genet 55: 105–111. [DOI] [PubMed] [Google Scholar]
- Guidetti P, Charles V, Chen EY, Reddy PH, Kordower JH, Whetsell Jr WO, Schwarcz R, Tagle DA (2001) Early degenerative changes in transgenic mice expressing mutant huntingtin involve dendritic abnormalities but no impairment of mitochondrial energy production. Exp Neurol 169: 340–350. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her L-S, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LSB (2003) Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila Neuron 40: 25–40. [DOI] [PubMed] [Google Scholar]
- Huh JW, Laurer HL, Raghupathi R, Helfaer MA, Saatman KE (2002) Rapid loss and partial recovery of neurofilament immunostaining following focal brain injury in mice. Exp Neurol 175: 198–208. [DOI] [PubMed] [Google Scholar]
- Ishihara H, Kanda F, Nishio H, Sumino K, Chihara K (2001) Clinical features and skewed X-chromosome inactivation in female carriers of X-linked recessive spinal and bulbar muscular atrophy. J Neurol 248: 856–860. [DOI] [PubMed] [Google Scholar]
- Jung C, Yabe JT, Shea TB (2000) C-terminal phosphorylation of the high molecular weight neurofilament subunit correlates with decreased neurofilament axonal transport velocity. Brain Res 856: 12–19. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G (2002) Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron 35: 843–854. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G (2003) Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med 9: 768–773. [DOI] [PubMed] [Google Scholar]
- Kemppainen JA, Lane MV, Sar M, Wilson EM (1992) Androgen receptor phosphorylation, turnover, nuclear transport and transcriptional activation: specificity for steroids and antihormones. J Biol Chem 267: 968–974. [PubMed] [Google Scholar]
- Kennedy WR, Alter M, Sung JH (1968) Progressive proximal spinal and bulbar muscular atrophy of late onset: a sex-linked recessive trait. Neurology 18: 671–680. [DOI] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbaur EL (2002) Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron 34: 715–727. [DOI] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991) Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 353: 77–79. [DOI] [PubMed] [Google Scholar]
- Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G (1998a) Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol 44: 249–254. [DOI] [PubMed] [Google Scholar]
- Li M, Nakagomi Y, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Mitsuma T, Hashizume Y, Fischbeck KH, Sobue G (1998b) Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol 153: 695–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Yu ZX, Li CL, Nguyen HP, Zhou YX, Deng C, Li XJ (2003) Lack of huntingtin-associated protein-1 causes neuronal death resembling hypothalamic degeneration in Huntington's disease. J Neurosci 23: 6956–6964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManamny P, Chy HS, Finkelstein DI, Craythorn RG, Crack PJ, Kola I, Cheema SS, Horne MK, Wreford NG, O'Bryan MK, De Kretser DM, Morrison JR (2002) A mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet 11: 2103–2111. [DOI] [PubMed] [Google Scholar]
- Merry DE, Kobayashi Y, Bailey CK, Taye AA, Fischbeck KH (1998) Cleavage, aggregation and toxicity of the expanded androgen receptor in spinal and bulbar muscular atrophy. Hum Mol Genet 7: 693–701. [DOI] [PubMed] [Google Scholar]
- Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16: 1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I (2001) SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet 10: 1441–1448. [DOI] [PubMed] [Google Scholar]
- Piccioni F, Pinton P, Simeoni S, Pozzi P, Fascio U, Vismara G, Martini L, Rizzuto R, Poletti A (2002) Androgen receptor with elongated polyglutamine tract forms aggregates that alter axonal trafficking and mitochondrial distribution in motor neuronal processes. FASEB J 16: 1418–1420. [DOI] [PubMed] [Google Scholar]
- Pinsky L, Trifiro M, Kaufman M, Beitel LK, Mhatre A, Kazemi-Esfarjani P, Sabbaghian N, Lumbroso R, Alvarado C, Vasiliou M, Gottlieb B (1992) Androgen resistance due to mutation of the androgen receptor. Clin Invest Med 15: 456–472. [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown Jr RH, Ludlow CL, Fischbeck KH (2003) Mutant dynactin in motor neuron disease. Nat Genet 33: 455–456. [DOI] [PubMed] [Google Scholar]
- Quigley CA, Friedman KJ, Johnson A, Lafreniere RG, Silverman LM, Lubahn DB, Brown TR, Wilson EM, Willard HF, French FS (1992) Complete deletion of the androgen receptor gene: definition of the null phenotype of the androgen insensitivity syndrome and determination of carrier status. J Clin Endocrinol Metab 74: 927–933. [DOI] [PubMed] [Google Scholar]
- Saito T, Shima H, Osawa Y, Nagao M, Hemmings BA, Kishimoto T, Hisanaga S (1995) Neurofilament-associated protein phosphatase 2A: its possible role in preserving neurofilaments in filamentous states. Biochemistry 34: 7376–7384. [DOI] [PubMed] [Google Scholar]
- Sar M, Stumpf WE (1977) Androgen concentration in motor neurons of cranial nerves and spinal cord. Science 19: 77–79. [DOI] [PubMed] [Google Scholar]
- Sobue G, Hashizume Y, Mukai E, Hirayama M, Mitsuma T, Takahashi A (1989) X-linked recessive bulbospinal neuronopathy: a clinicopathological study. Brain 112: 209–232. [DOI] [PubMed] [Google Scholar]
- Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA (1999) Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet 8: 731–741. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST (2003) Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron 40: 41–52. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S (2002) Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila Neuron 35: 855–864. [DOI] [PubMed] [Google Scholar]
- Tu PH, Elder G, Lazzarini RA, Nelson D, Trojanowski JQ, Lee VM (1995) Overexpression of the human NFM subunit in transgenic mice modifies the level of endogenous NFL and the phosphorylation state of NFH subunits. J Cell Biol 129: 1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcott JL, Merry DE (2002) Ligand promotes intranuclear inclusions in a novel cell model of spinal and bulbar muscular atrophy. J Biol Chem 277: 50855–50859. [DOI] [PubMed] [Google Scholar]
- Williamson TL, Cleveland DW (1999) Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat Neurosci 2: 50–56. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JL, Hen R (2000) Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101: 57–66. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ (2003) Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci 23: 2193–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoghbi HY, Orr HT (2000) Glutamine repeats and neurodegeneration. Annu Rev Neurosci 23: 217–247. [DOI] [PubMed] [Google Scholar]