

Abstract
Human herpesvirus 6 (HHV-6), a common resident virus of the human CNS, has been implicated in both acute and chronic inflammatory–demyelinating diseases. Although HHV-6 persists within the human CNS and has been described to infect mature oligodendrocytes, nothing is known about the susceptibility of glial precursors, the ancestors of myelin-producing oligodendrocytes, to viral infection.
We show that HHV-6 infects human glial precursor cells in vitro. Active infection was demonstrated by both electron microscopy and expression of viral gene transcripts and proteins, with subsequent formation of cell syncytia. Infection leads to alterations in cell morphology and impairment of cell replication but not increased cell death. Infected cells showed decreased proliferation as measured by bromodeoxyuridine uptake, which was confirmed by blunting of the cell growth rate of infected cells compared with uninfected controls over time. The detailed analysis using novel, fluorescent-labeled HHV-6A or HHV-6B reagents demonstrated strong G1/S phase inhibition in infected precursor cells. Cell cycle arrest in HHV-6-infected cells was associated with a profound decrease in the expression of the glial progenitor cell marker A2B5 and a corresponding increase in the oligodendrocyte differentiation marker GalC.
These data demonstrate for the first time that infection of primary human glial precursor cells with a neurologically relevant human herpesvirus causes profound alterations of critical precursor cell properties. In light of recent observations that repair of CNS demyelination is dependent on the generation of mature oligodendrocytes from the glial precursor cell pool, these findings may have broad implications for both the ineffective repair seen in demyelinating diseases and the disruption of normal glial maturation.
Keywords: glial precursors, HHV-6, cell cycle, proliferation, differentiation, human
Introduction
Infections of the human CNS caused by a wide variety of viruses result in substantial morbidity and mortality worldwide (Whitley and Gnann, 2002; Johnson, 2003; McCarthy, 2003). Among these, the eight members of the human herpesvirus family are the most frequent and significant causes of CNS infection; cytomegalovirus (CMV), for example, is the most commonly recognized cause of congenital anomalies of the human CNS (Stagno et al., 1986; Demmler, 1991; McCutchan, 1995; Mamidi et al., 2002). However, human herpesvirus (HHV) 6, the closest relative to CMV, is the most abundant resident HHV in the human brain, detectable in up to 70% of human brain tissues at autopsy (Sanders et al., 1996). CNS infection with HHV-6 may cause leukoencephalitis in both immunocompetent and immunocompromised patients, accompanied by demyelinative lesions that fail to repair (Carrigan et al., 1996; Novoa et al., 1997; Ito et al., 2000). Typically, CNS infections by HHV-6 lead to benign viral latency, a strategy for persistence used by all members of the herpesvirus family. HHV-6 has been recently demonstrated to infect primary human oligodendrocytes in vitro and in vivo (Albright et al., 1998; Mock et al., 1999; Blumberg et al., 2000; Goodman et al., 2003), as well as human astrocytes (He et al., 1996; Donati et al., 2003), but virtually nothing is known about its effects on glial precursor cells, from which both oligodendrocytes and astrocytes originate.
Oligodendrocyte precursors (OPCs) are of particular interest in the context of demyelinating diseases because of an accumulating consensus that most, if not all, remyelinating cells originate from this precursor cell pool (Blakemore and Keirstead, 1999; Franklin, 2002; Bruck et al., 2003). Although normal numbers of OPCs are frequently present within early demyelinating lesions, cells within the precursor pool appear to remain quiescent and unable to appropriately proliferate and/or differentiate (Wolswijk, 1998; Chang et al., 2000, 2002). The exact mechanisms that contribute to this repair failure remain mostly speculative.
The observation that aberrant precursor cell functions may contribute to ineffective repair strategies, together with the fact that HHV-6 is the most abundant herpesvirus in the human brain, led us to examine the possibility that HHV-6 can infect human glial precursors and alter critical precursor cell functions. However, the study of virus interactions with human glial precursor cells has been hampered by the lack of a renewable source of such cells. Recently, human A2B5+ glial precursor cells have been isolated from fetal brain (Dietrich et al., 2002). These primary cells are capable of continuous replication in culture in defined medium and can be induced to differentiate into oligodendrocytes and astrocytes. Here, we show for the first time that HHV-6 infection of human glial precursors induces profound alterations of the critical progenitor cell properties of self-renewal and differentiation.
Materials and Methods
Human glial precursor cell cultures. Human glial precursor cells were isolated and purified as described previously (Dietrich et al., 2002). Briefly, 18- to 20-week-old human fetal brain neural progenitor cells, obtained from Cambrex (Baltimore, MD), were plated on fibronectin/laminin (20 and 5 μg/ml, respectively)-coated plastic tissue culture flasks (Nunc, Naperville, IL). Cells were grown in DMEM/F-12 (Invitrogen, Gaithersburg, MD) supplemented with additives described by Bottenstein and Sato (1979), basic FGF (bFGF; 10 ng/ml) and chick embryo extract (10%), and prepared as described by Stemple and Anderson (1992). A2B5+/neural cell adhesion molecule-negative glial precursor cells were subsequently purified by immunopanning and fluorescence-activated cell sorting (FACS) as described previously (Dietrich et al., 2002). All experiments were performed on cells exposed to oligodendrocyte-promoting conditions in the presence of bFGF (10 ng/ml) and platelet-derived growth factor-AA (PDGF-AA; 10 ng/ml), or bFGF alone. Human astrocytes, obtained from ScienCell (San Diego, CA), were grown in the presence of bFGF (10 ng/ml) and bone morphogenetic protein-4 (10 ng/ml). Astrocytic phenotypes were confirmed by GFAP expression using a human specific anti-GFAP monoclonal antibody (mAb) (see below also).
Virus propagation. HHV-6 virus stocks were produced by serial propagation in permissive human lymphoblastoid cell lines as described previously (Rotola et al., 1998; Soldan et al., 2000). The U1102 strain of HHV-6A was passaged in the J-JAHN cell line, whereas the Z29 strain of HHV-6B was grown in SupT1 cells. Both cell lines were maintained in RPMI 1640 media supplemented with 10% FCS at a density of ∼5 × 105 cells/ml. Cytopathological effects were noted typically within 5–7 d and were manifested by vacuolization and “ballooning” of cells. At this point, fresh cells were added in a ratio of 2:1 together with fresh media. Infection was confirmed for all experiments by an immunofluorescent assay (IFA) using monoclonal antibodies (ABI, Columbia, MD) to either the viral proteins gp82/105 (A strain specific) or p101 (B strain specific). Both antibodies were diluted 1:50 and incubated overnight at 4°C before labeling for 1 hr at room temperature with FITC-conjugated horse anti-mouse secondary antibody diluted at 1:500. Cells were counted on a fluorescent microscope, aliquoted at 1 × 107 cells/ml in 1 ml cryotubes, and frozen in liquid nitrogen. For all experiments, ∼40–60% of cells were IFA positive.
Preparation of cell-free virus. Cell-free viral inocula were prepared by pelleting 60–70 × 106-infected J-JAHN (U1102 strain) or SupT1 (Z29 strain) cells at 400 × g for 15 min to remove cells. The supernatant was removed and centrifuged again at 3000 × g for 15 min. The resulting supernatant, entirely cell free as noted by microscopic observation, was then centrifuged in a J-21 rotor for 3 hr at 16,000 × g to pellet virus. Supernatants were removed, and viral pellets were resuspended in 1 ml of DMEM/F-12 with Sato additives (same medium as used for glial progenitor cells). Uninfected inocula (controls) were prepared in the same manner from uninfected cells, and aliquots were frozen at –80°C until use.
HHV-6 infection of human glial precursor cells. Infections were performed by adding 100 μl of the cell-free virus preparations to one 80 cm2 tissue culture flask of human glial precursor cells containing ∼2 × 106 cells. Because HHV-6 is exceedingly cell associated with a strong tendency for self-aggregation and clumping (Padilla et al., 2003), a true multiplicity of infection (MOI) could not be obtained; rather, virus was first titered in permissive SupT1 or J-JAHN cells, and the TCID50 (tissue culture infectious dose required for infection of 50% of the susceptible population of the permissive indicator cell line) was determined for each batch by the method of Reed and Muench (1938). Infection was confirmed by examination of the appropriate lymphoblastoid cell line for cytopathic effect and by IFA as described above.
To study the effects of HHV-6 infection on glial precursors, the protocol described above was followed, using the same ratio of virus to cells. Control or infected human glial precursor cells were plated on fibronectin/laminin-coated plastic flasks or coverslips in 24-well tissue culture plates, as described previously (Dietrich et al., 2002). Cells were incubated either 2 or5din humidified chambers at 37°C before fixation in 2% paraformaldehyde for 10 min.
Immunocytochemistry. Antigen expression of glial precursor cells was assessed using the following antibodies: anti-A2B5 mouse IgM mAb to label glia precursor cells (Rao et al., 1998; Scolding et al., 1999; Lee et al., 2000; Dietrich et al., 2002); and anti-galactocerebroside mouse IgG3 (GalC, 1:50; Developmental Hybridoma Bank, Iowa City, IA) (Lee et al., 2000), anti-myelin/oligodendrocyte-specific protein IgM (1:100; Chemicon, Temecula, CA) (Dyer et al., 1991), and anti-MBP mouse IgG1 mAb (1:100; Chemicon) to assess oligodendrocyte differentiation (Warrington et al., 1992; Duchala et al., 1995). GFAP polyclonal rabbit Ig (1:400; Dako, Carpinteria, CA) and GFAP IgG1 mAb (1:200; Sternberger Monoclonals, Lutherville, MA) were used to identify astrocytic differentiation. Anti-CD46 mouse IgG1 mAb (1:100; Chemicon) was used to assess receptor expression for HHV-6 (Santoro et al., 1999) and anti-HHV-6 gp116 mouse IgG1 mAb (1:50; Chemicon) to visualize HHV-6-infected cells (Cardinali et al., 1998). Anti-CD21 mouse IgG2a (1:100; Serotec, Oxford, UK) was used to confirm the presence of a main cellular receptor for Epstein–Barr virus. Cell proliferation was assessed by bromodeoxyuridine (BrdU) incorporation and by using the mouse anti-BrdU mAb IgG1 (1:100; Sigma, St. Louis, MO) to label dividing cells. In addition, Ki-67 staining (mouse anti-Ki67 IgM, 1:100; Sigma) was used to assess the proportion of proliferating cells (Endl and Gerdes, 2000). Stained cells on coverslips were rinsed two times in 1× PBS, counter-stained with 4′6-diamidino-2-phenylindole (DAPI; Molecular Probes, Eugene, OR) and mounted on glass slides with Fluoromount (Molecular Probes). Staining against surface proteins was performed on cultures of living cells or on cells fixed with 2% paraformaldehyde. Staining with intracellular antibodies was performed by permeabilizing cells with ice-cold methanol for 4 min or by using 0.5% Triton for 15 min on 2% paraformaldehyde-fixed cells. Antibody binding was detected with appropriate fluorescent dye-conjugated secondary antibodies at 10 μg/ml (Southern Biotech, Birmingham, AL) or Alexa Fluor-coupled antibodies at a concentration of 1 μg/ml (Molecular Probes), applied for 20 min.
Fluorophore labeling and titration of HHV-6A and HHV-6B. Infectious, fluorescently labeled HHV-6 was prepared in a three-step procedure. First, virus grown in permissive lymphoid cells and isolated as described above was resuspended in 1× PBS and passed through a Sephadex G-50 spin column equilibrated with 1× PBS to remove small fragments that might interfere with labeling. Second, the virus preparation was adjusted to pH 9 with sodium bicarbonate and reacted with lyophilized succinimidyl esters (1 mg/ml virus) of either FITC (Roche, Indianapolis, IN) or ALEXA 488 (Molecular Probes) for 2–4 hr at room temperature. Third, the labeled virus was passed through a second Sephadex G-50 spin column equilibrated with DMEM/F-12 medium.
The labeled virus was immediately titrated in a simplified Reed–Muench procedure by the addition of 1–20 μl aliquots to 2.5 × 105 permissive SupT1 or J-JAHN cells suspended in 100 μl of RPMI medium and incubation for 8 hr at 37°C in 5% CO2 humidified incubator, before examination on a fluorescent microscope (Eclipse E400 upright microscope; Nikon). Typically, a maximum of 40–60% of cells are labeled (TCID50) using 2–5 μl of virus per 250,000 cells. Because both HHV-6A and -6B produce clumps of virus in permissive cells (Padilla et al., 2003) and also give rise to syncytia and clumping of both SupT1 and glial precursor cells, it is not possible to calculate a precise MOI. In over 20 experiments, we found that using 50 μl of virus per 5 × 105 glial precursors in a 80 cm2 flask (an estimated five times the TCID50) is optimal.
Flow cytometry and cell cycle analysis. Human glial precursor cells were harvested and analyzed by direct immunofluorescence using FITC-labeled HHV-6 together with propidium iodide (PI) staining to analyze DNA content as described (Zhu et al., 2001). The cells were detached with trypsin/EDTA, washed three times with 10 ml of 1× PBS, fixed with 0.25% paraformaldehyde in 1× PBS for 1 hr, and stained with PI solution containing 20 μg/ml PI, 0.2% Triton X-100, and 11.25 Kunitz units of RNase A per milliliter in 1× PBS. Flow cytometric analysis was performed in an Epics Elite ESP analyzer (Coulter, Hialeah, FL). Gating of FITC-positive (i.e., HHV-6-FITC labeled) cells were established using mock-infected cells as background. Because electronic settings varied from experiment to experiment, gates were defined such that the percentage of false positive events was not higher than 0.3% in the mock-infected population. Cell cycle analysis was performed using Multicycle AV software (Phoenix Flow, San Diego, CA). Percentages of FITC-positive cells in various stages of the cell cycle were compared with FITC-negative cells from the same flask. All experiments were repeated at least five times, with representative experiments shown. A schematic outline of experiments using the fluorescently labeled virus is provided in supplemental Figure 1 (available at www.jneurosci.org).
Fluorescence-activated cell sorting. FITC-positive and -negative cells were isolated using the BD FACSVantage SE cell sorter (Becton Dickinson, Palo Alto, CA). FITC fluorescence was excited by an argon ion laser at an emission wavelength of 488 nm, and fluorescence emission was collected using a 530 +/– 30 nm bandpass filter. A two-parameter sorting window (forward light scattering and FITC fluorescent intensity) was used to define the FITC-positive and -negative cell populations. The cells were sorted through a flow chamber with an 80 μm nozzle tip under 12 psi sheath fluid pressure. The sorted cells were collected into 15 ml conical tubes filled with sterile media for biochemical analysis, and the brightest 10% of the FITC-positive population were collected for the study. On average, ∼2 × 105 cells were collected for each experiment.
Reverse transcription-PCR. Total DNA and RNA were extracted from HHV-6-infected and control glial precursor cells using TRIzol (Invitrogen) according to the manufacturer's instructions. Reverse transcription was accomplished with Superscript (Invitrogen) in a 20 μl volume using the manufacturer's recommended buffers. One microgram of total cellular RNA was denatured for 5 min at 65°C with 1 μl each of 50 μm random hexamers and 10 mm dNTPs in a 12 μl volume under mineral oil. After incubation for 25 min at 25°C, 4 μlof5× first-strand buffer, 2 μl of DTT, 1 μl of RNase inhibitor, and 1 μl of enzyme (200 U/μl) were added, and incubation was continued for 50 min at 37°C, followed by 60 min at 42°C. Reverse transcriptase was then inactivated by heating 15 min at 70°C, and 2 μl of this reaction was added to 50 μl PCR reactions, which were performed in standard PCR buffer (Invitrogen) with 2 mm MgCl2, 200 μm dNTPs, 1 μm primers, and1Uof Platinum Taq (Invitrogen). Thermocycling in an MJ model 100 (MJ Research, Waltham, MA) followed a touch-down protocol, with an initial denaturation of 30 sec at 95°C, 6 cycles of 95°C/30 sec, 69°C/1 min; 8 cycles of 94°C/1 min, 66°C/1 min, 72°C/1 min; 10 cycles of 94°C/1 min, 63°C/1 min, 72°C/1 min; 12 cycles of 94°C/1 min, 60°C/1 min, 72°C/1 min; 10 cycles of 94°C/1 min, 57°C/1 min, 72°C/1 min; and a final extension for 5 min at 72°C. The PCR product was analyzed by agarose gel electrophoresis. U47 primers were chosen to distinguish A and B strains (cf. Isegawa et al., 1999). The following forward/reverse primer pairs were used (expected band size): host glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene, 5′-accacagtccatgccatcac/5′-tccaccaccctgttgctgta (550 bp); HHV-6 IE gene U86, 5′-gcacatcctctgcttccaatctac/5′-gcggatctgtctgagcaatcg (370 bp); HHV-6 late gene U47, 5′-acaccgaacacaccactccgat/5′-tgtacttcagttttggcagttac (A strain, 350 bp; B strain, 500 bp).
Electron microscopy. The cells were fixed on glass slides in 2.5% phosphate-buffered glutaraldehyde, pH 7.4, and postfixed in 1.0% phosphate-buffered osmium tetroxide. The slides were passed through a graded series of ethanol to 100%, infiltrated with liquid Spurr epoxy resin, and embedded on the glass surface with inverted capsular molds containing fresh resin. After polymerization at 70°C, the hardened capsules containing the cells of interest were removed from the glass slide by dipping into liquid nitrogen. The capsules were examined with a light microscope to determine the area to be trimmed and sectioned with a diamond knife onto 200 mesh copper grids. The grids were contrasted with aqueous uranyl acetate for 10 min, lead citrate for 15 min, and examined and photographed with a 7100 transmission electron microscope (Hitachi).
Lactate dehydrogenase assay for cell death. Glial precursor cell death was measured by assaying lactate dehydrogenase (LDH) released from dead cells using the Cytotoxicity Detection kit (Roche) per the manufacturer's instructions. All assays were run in triplicate with results read on a Benchmark Elisa plate reader (Bio-Rad, Hercules, CA) using absorbance at 490 nm. Data are expressed as relative absorbance units.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling assay for cell death. To detect nuclear DNA fragmentation, one criterion of apoptosis, we used the terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick-end labeling (TUNEL) assay on cells fixed with 2% paraformaldehyde. The ApopTag In Situ Cell Death Detection kit (Intergene, Purchase, NY) was used according to the manufacturer's recommendations. The TUNEL assay was followed by DAPI counterstaining to visualize nuclear morphology. Experiments were performed in quadruplicate, and data were averaged to represent the mean from three independent experiments.
Image and data analysis. Digital images were captured on a Eclipse E400 upright microscope (Nikon) using the spot camera and the spot-advanced software for Macintosh (Diagnostic Instruments). Quantitative analysis was performed counting the total number of immunoreactive cells per total number of viable cells as determined by phase-contrast microscopy and/or immunostaining using DAPI (Molecular Probes) to visualize cell nuclei. Each experiment was performed in quadruplicate in at least three independent experiments. Photomicrographs were processed on a Macintosh G4 and assembled with Adobe Photoshop 6.0.
Statistical analysis. In all comparisons, unpaired, two-tailed Student's t tests were used. Statistical significance was considered to be at a level of p < 0.05.
Results
Human (A2B5+) glial precursor cells can be infected by HHV-6
HHV-6 (A and B strain) infection is thought to occur primarily through binding to the cell surface receptor CD46, a complement regulatory protein that is widely expressed throughout the human CNS (Santoro et al., 1999). To determine whether human glial precursor cells may represent a target for HHV-6 virus infection, the expression of CD46 was analyzed by immunocytochemistry. As a positive control, we used human astrocytes that have been described to be a target for HHV-6 infection. As shown in Figure 1A, ∼70–80% of A2B5+ precursors express CD46, suggesting that these cells should allow virus attachment (as expected, CD-46 was also localized to GFAP+astrocytes (supplemental Fig. 2). To evaluate whether CD46 expression on precursor cells leads to virus attachment and subsequent infection and expression of viral genes and proteins, we exposed glial precursors to cell-free HHV-6A or -6B strain viruses at an estimated five times the TCID50. Typically, 10–20% of precursor cells were infected by either virus strain as judged by immunocytochemical staining using specific antibodies against early and late structural proteins of HHV-6A and -6B (p41 and gp116, respectively) and the HHV-6B-specific mAb to the late protein p101. Figure 1, B and C, demonstrates expression of viral proteins p41 and p101 antigens in glial precursor cells after infection with either HHV-6A or HHV-6B, respectively. Glial precursors exposed to control medium did not express these viral proteins (Fig. 1D).
Figure 1.

Human glial precursor cells express the surface receptor CD46 and can be infected with HHV-6. Glial precursors were adhered overnight to fibronectin/laminin-coated coverslips before being fixed and immunostained with antibodies to CD46 (red) and counterstained with DAPI (blue) to reveal cell nuclei (A). Approximately 70–80% of cells demonstrated expression of CD46. Glial precursors were infected with HHV-6A (B) or HHV-6B (C), stained with antibodies to the HHV-6 p41 and p101 proteins (green), and counterstained with DAPI to highlight cell nuclei (blue). Approximately 10–20% of cells exposed to either virus strain expressed viral proteins. No viral protein expression could be detected in unexposed precursor cells (D). Scale bars, 15 μm.
The type of viral infection in human glial precursor cells was also confirmed at the mRNA level by reverse transcription (RT)-PCR (Fig. 2). Transcripts corresponding to regions of a viral late gene (U47) and an immediate early (IE) gene (U86) were compared with a similarly sized region of the host housekeeping gene GAPDH. A touchdown PCR protocol optimized for GAPDH and primers capable of distinguishing A and B strains of HHV-6 were used (see Materials and Methods). Strong bands representing both IE (U86) and late (U47) viral transcripts were readily detectable in cells exposed to virus (lanes 3 and 4 and lanes 6 and 7, respectively) but not in uninfected cells (lanes 5 and 8). Taken together, the above results show that human glial precursor cells support active infection with both strains of HHV-6.
Figure 2.

HHV-6-infected and uninfected control precursor cells express IE (U86) and late (U47) gene transcripts as determined by RT-PCR. Lanes 3 and 4 show a 370 bp HHV-6 amplicon from the U86 gene. Lanes 6 and 7 show amplicons from gene U47 of HHV-6 strains A and B, respectively. Note that the U47 A gene contains a 150 bp deletion with respect to the U47 B gene within the region defined by the primers. No amplicons are generated from uninfected precursor cells as shown in lanes 5 and 8.
Effects of HHV-6 infection on cell morphology
HHV-6 infection in human lymphoid cells has been shown to result in morphological changes characterized by syncytia formation and vacuolization [known as “ballooning degeneration” (Salahuddin et al., 1986)]. Therefore, we examined whether HHV-6-infected precursors might exhibit similar morphological changes. Cells were plated on fibronectin/laminin-coated dishes and exposed to cell-free HHV-6B for 5 d, fixed with 2% paraformaldehyde, immunostained with a mAb against the viral gp116 (ALEXA green), and counterstained with DAPI (blue) to label cell nuclei. Uninfected cells demonstrated characteristic polygonal cell morphology with relatively few processes, as described previously (Fig. 3A) (Dietrich et al., 2002). In contrast, HHV-6B-infected cells showed morphological changes characterized by the development of increased numbers of radial processes accompanied by formation of multinucleated syncytia (Fig. 3B). Similar results were also found after infection with HHV-6A (data not shown).
Figure 3.

HHV-6B-infected human glial precursor cells show morphological changes as determined by light and electron microscopy. Cells were grown on fibonectin/laminin-coated coverslips for 5 d before being fixed and stained with antibodies against HHV-6 gp 116 (green) and counterstained with DAPI (blue). Uninfected cells demonstrated characteristic polygonal cell morphology with relatively few processes characteristic of normal human glial precursor cells (A). In contrast, HHV-6B-infected cells examined at the same 5 d time point exhibited morphological changes characterized by development of increased numbers of radial processes accompanied by multinucleated syncytia (B). Morphological changes in HHV-6B-infected precursor cells examined by electron microscopy show extensive vacuolization and contain nests of dense core particles not seen in controls (magnification, 4000×) (C). Later in infection with HHV-6B (or HHV6-A; data not shown), glial precursors formed multinucleated syncytia (magnification, 1500×) (D). A high-power image of an HHV-6B-infected vacuole (magnification, 40,000×) shows virions consistent in size and morphology with HHV-6 (E); spherical intracellular viral particles possessing an electron-dense core, tegument, and capsid were identified (arrow). No vacuolization or cell syncytia were seen in uninfected glial precursors (magnification, 2500×) (F). Scale bar (in A): A, B, 15 μm.
These morphological alterations were more closely examined using electron microscopy on selected fields of precursors infected with FITC-labeled HHV-6A or HHV-6B 2 d after infection. Infected cells were identified by fluorescence microscopy, and the areas of interest were marked before detailed examination by electron microscopy was performed. Multiple intracytoplasmic vacuoles filled with dense core particles were seen in infected precursor cells (Fig. 3C). In contrast to uninfected cells, infected precursors formed multinucleated syncytia (Fig. 3D). In addition, spherical intracellular herpes-like viral particles possessing an electron-dense core, tegument, and capsid were identified (Fig. 3E). No syncytia or extensive vacuolization was seen in uninfected cells (Fig. 3F). The appearance of the infected cells was consistent with the well described cytopathological changes associated with herpes family virus infections of lymphocytes (Cermelli et al., 1997).
HHV-6 infection impairs cell division of human glial precursors
Subsequent experiments used FACS analyses and sorting of FITC-labeled HHV-6-infected glial precursors, as outlined in supplemental Figure 1. One of the key characteristics of glial precursor cells is their ability to self-renew. To determine whether virus infection affects the ability of glial precursors to divide, we compared BrdU incorporation in infected and uninfected glial precursor cells (Fig. 4A). FAC-sorted HHV-6A, HHV-6B, and uninfected precursor cells serving as controls were incubated for 48 hr and pulsed with BrdU for 8 hr before fixation with 2% paraformaldehyde and immunostaining. Quantification of BrdU+ cells as a percentage of the total number of cells demonstrated significant suppression of BrdU uptake in precursors infected with either HHV-6A or HHV-6B when compared with controls. Overall, HHV-6-infected cells demonstrated an >30% decrease in BrdU uptake (p < 0.0016 for HHV-6A and p < 0.0013 for HHV-6B) when compared with uninfected controls. Similar results were obtained with Ki-67 as a proliferation marker (data not shown). To confirm that a decrease of BrdU labeling coincides with a suppressed cell growth over time in infected cells compared with controls, FAC-sorted HHV-6A, HHV-6B, or uninfected control human glial precursors were plated at the same densities in 24-well plates in the presence of optimal growth conditions. All conditions were performed in triplicate, cells were stained with DAPI on the day indicated, and unbiased counting of cell numbers was performed. As shown in Figure 4B, HHV-6A- or HHV-6B-infected precursors showed markedly diminished cell numbers at day 5 compared with uninfected controls.
Figure 4.

HHV-6-infected glial precursor cells showed a decrease in BrdU incorporation that was accompanied by reduced cell growth in culture over time. A, Precursor cells show decreased BrdU incorporation after HHV-6 infection. Quantification of BrdU-positive glial precursor cells was expressed as a percentage of total cells. Precursor cells were either exposed or sham exposed to HHV-6 overnight, fluorescence-activated cell sorted, and incubated for 48 hr before being pulsed with BrdU for the final 8 hr. Proliferation of precursor cells was significantly reduced by infection with both the A and B strains of virus. p values are expressed relative to uninfected control precursor cells (*p < 0.0016; **p < 0.0013). B, Glial precursor cells infected with HHV-6A or HHV-6B showed markedly diminished cell numbers over time compared with uninfected controls. Cells were infected with HHV-6A or HHV-6B overnight before being fluorescence-activated cell sorted in parallel with uninfected controls and plated at the same density on coverslips in optimal growth conditions in triplicate. Medium was changed every other day, cells were stained with DAPI on the day indicated, and unbiased counting of all cells was performed. Cell numbers of infected populations were ∼55–60% of controls by day 5 after infection.
HHV-6-infected human glial precursor cells are inhibited during the G1/S phase of the cell cycle
The finding of decreased DNA synthesis and cell growth in HHV-6-infected precursor cells prompted us to examine whether infection is associated with an arrest of cell division during a specific phase in the cell cycle (Fig. 5). Compared with controls (Fig. 5A,A′), changes in precursor cell cycle profile attributable to infection with either HHV-6A (Fig. 5B) or HHV-6B (Fig. 5B′) could not be readily discerned because of the normal predominance of cells in G1/S. Therefore, because it is not possible to synchronize primary cells in G0/G1, we exposed precursor cells to the G2/M blocking agent nocodazole [as has been done to demonstrate G1/S arrest in a variety of tumor cell lines after infection with the closely related HHV and CMV (Wiebusch and Hagemeier, 1999)]. In normal, uninfected cells, this treatment blocks spindle formation and cell cycle progression leading to accumulation of cells in G2/M (Fig. 5C,C′). If cells were unable to progress through the G1/S phase as a result of preceding virus infection, we would expect infected cells to be unable to progress to G2/M in the presence of nocodazole, but rather to remain and accumulate in G1/S.
Figure 5.

HHV-6A and HHV-6B infection of human glial precursor cells result in cell cycle arrest. Human glial precursors were infected with FITC–HHV-6 for 72 hr, fixed, and analyzed for DNA content by FACS. Uninfected controls show the characteristic profile of proliferating glial precursors, with the majority of cells in G0 /G1 (A, A′). The effects of HHV6-A (B) or HHV-6B (B′) infection on precursor cell cycling cannot be visually discriminated from each other or the uninfected controls. The G2 /M blocking agent nocodazole caused uninfected precursors to accumulate in G2 /M (C, C′). In D and E, as well as (D′) and (E′), precursors were exposed to FITC–HHV-6A and FITC–HHV-6B, respectively, followed by a terminal 24 hr incubation in nocodazole. HHV-6-negative (D, D′) or HHV-6-positive (E, E′) cells from the same flask were then analyzed for DNA content. The nocodazole treatment permits the discrimination of the G1 /S block induced by prior infection with either HHV-6A or HHV-6B (E and E′, respectively) in comparison with uninfected controls (D and D′). F shows a summary of the results of HHV-6A and HHV-6B infection on glial precursor cell cycling from five independent experiments. The error bars indicate SEM.
We infected glial precursor cells (which are predominantly in G1/S) with FITC-labeled HHV-6A or HHV-6B and cultured them for 48 hr. Then, we exposed all the cells (infected and uninfected) to nocodazole for an additional 24 hr. This mixed population was then fixed and analyzed by FACScan for both the DNA content and the presence or absence of virus infection (FITC positive or FITC negative). Figure 5E demonstrates that FITC–HHV-6A-infected precursor cells strongly accumulate in G1/S, whereas the uninfected, non-FITC-labeled cells progress normally to G2/M, where they are then arrested, as expected, because of the presence of nocadozole (Fig. 5D). The effect of the FITC–HHV-6B strain is somewhat less pronounced (Fig. 5E′), but the number of cells arrested in G1/S (18.9 + 21.0% = 39.9%) is still more than double the number of uninfected FITC-negative cells (4.5 + 11.9% = 16.4% in Fig. 5D′). Exposure of precursor cells to heat-inactivated HHV-6A or HHV-6B (56°C for 30 min) produced no evidence of cell cycle arrest (data not shown). The graph in Fig. 5F represents the average of five cell cycle experiments, each with HHV-6A and HHV-6B. Glial precursor cell infection with either strain of HHV-6 resulted in significant accumulation of infected cells in G1/S and a corresponding decrease in cells in G2/M compared with uninfected controls.
HHV-6 infection is not associated with increased glial precursor cell death
The impairment of cell proliferation of infected precursors could result in cell death. The LDH release assay was, therefore, used as an overall measure of cell death in infected and uninfected precursors (Fig. 6A). The LDH content in the supernatant of control and infected cultures at the same density were compared from 48 to 96 hr after infection. Although overall cell death increased over time, no significant differences were apparent between infected and control cells up to 96 hr after infection (p values ranged from 0.09 to 0.88). In addition, TUNEL staining was used as corroborating evidence that glial precursor cells were not dying and did not meet at least one criterion of apoptotic cell death, DNA fragmentation (Fig. 6B). Consistent with the LDH analysis, no statistically significant differences were found in the percentages of apoptotic control cells compared with HHV-6A- or HHV-6B-infected cells (p > 0.11 for HHV-6A and p > 0.19 for HHV-6B, relative to uninfected controls).
Figure 6.

HHV-6-infection of human glial precursor cells was not associated with increased cell death. LDH assay of precursor cell supernatants demonstrates no significant differences in LDH leakage attributable to cell membrane damage between HHV-6-infected and uninfected control cells (A). TUNEL assay for apoptotic cell death also showed no significant difference between HHV-6-infected and uninfected control cells examined 5 d after infection (B).
Human glial precursor cell infection with HHV-6 increases expression of the oligodendrocyte differentiation marker GalC
To determine whether HHV-6-induced cell cycle arrest affects human precursor cell differentiation, infected cells were plated at a density of 1000 cells/coverslip and incubated an additional 48 hr in the presence of both bFGF and PDGF-AA, a medium that allows for oligodendrocytic differentiation. Cells were then fixed in 2% paraformaldehyde before immunostaining for either the glial progenitor cell marker A2B5 or the oligodendrocyte differentiation marker GalC. Figure 7A shows a representative fluorescence microscopic field from uninfected versus HHV-6B-infected precursor cells immunostained for GalC (red) and counter-stained with DAPI (blue) to identify cell nuclei. Consistently more GalC+ cells were seen in infected glial precursors when compared with uninfected controls. These results are quantified in Figure 7B, showing the percentages of GalC+, A2B5+, or double positive cells. Infected precursor cell cultures contained 65–70% GalC+ cells compared with 40–45% GalC+ cells in control cultures (p = 0.035 for HHV-6A; p = 0.028 for HHV-6B). Conversely, the appearance of A2B5+ precursor cells was markedly decreased, to ∼25% in infected cells compared with 55–60% in uninfected controls (p = 0.012 for HHV-6A; p = 0.005 for HHV-6B). Staining for GFAP to reveal astrocytic differentiation under these conditions did not show a significant change in the number of astrocytes (data not shown).
Figure 7.
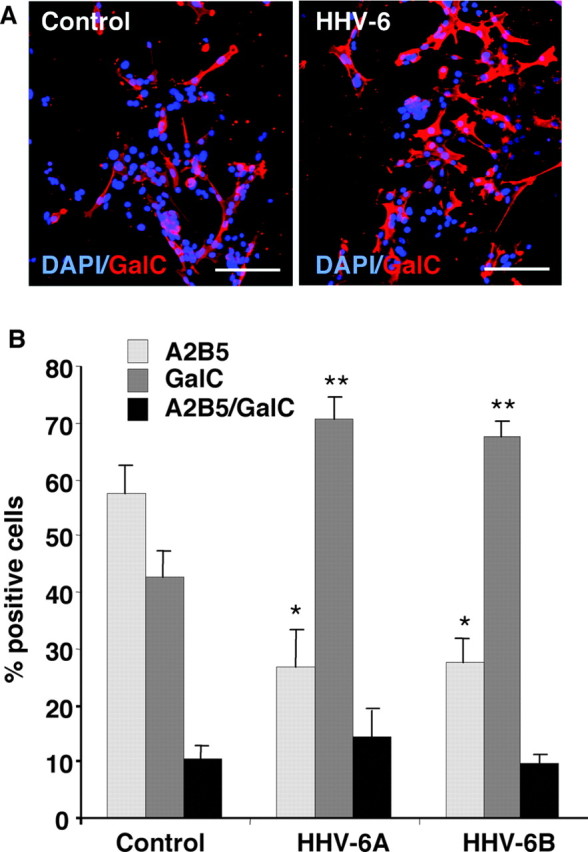
Infection with HHV-6A and -6B inhibits self-renewal and promotes differentiation of human glial progenitor cells. A representative fluorescence microscopic field from uninfected versus HHV-6B-infected precursor cells immunostained for GalC (red) and counterstained with DAPI (blue) for cell nuclei (A) is shown. Human glial progenitors were exposed overnight to either FITC-labeled HHV-6A or HHV-6B before being separated on a fluorescence-activated cell sorter and plated on coverslips. After a 48 hr incubation in a medium that allows for oligodendrocytic differentiation, cells were fixed and immunostained. The histogram (B) shows that a higher percentage of GalC+ cells was seen in the HHV-6-infected cell population. Uninfected precursor cell cultures (controls) contained ∼55–60% A2B5+ progenitor cells compared with only 25–30% in either HHV-6A- or HHV-6B-infected cells. Conversely, in the HHV-6A- or HHV-6B-infected populations, ∼70% of cells expressed the oligodendrocyte differentiation antigen GalC, compared with 40–45% in the uninfected controls. The percentage of cells coexpressing both A2B5 and GalC was small and did not change significantly with infection. *p < 0.01; **p < 0.035). Scale bars, 75 μm.
Discussion
The present study is the first to examine the interaction of a common CNS-resident HHV with primary human glial precursor cells and demonstrates profound virus-induced impairment in critical precursor cell properties. Human glial precursors were found to express surface CD46, the major cell surface receptor for HHV-6. HHV-6 infection of glial precursors resulted in early productive infection as evidenced by the demonstration of late viral transcripts by RT-PCR, the expression of viral structural proteins by immunocytochemistry, and the identification of characteristic herpesvirus virions within infected glial precursors by electron microscopy.
More prolonged infection with HHV-6 resulted in extensive morphological changes, including cytoplasmic vacuolization and the formation of multinucleated syncytia that showed multiple radial processes. Our findings are consistent with previously published morphological changes observed after HHV-6 infection of permissive lymphoid cells (Cermelli et al., 1997; Mayne et al., 2001), primary mature human astrocytes (He et al., 1996), and oligodendrocytes (Albright et al., 1998). These cells form enlarged cells and multinucleated syncytia after HHV-6 infection in vitro, a morphological change we also observed in infected glial precursor cells.
The observation that glial precursor cells can be productively infected with HHV-6 prompted us to analyze the biological consequences of such an infection. One of the most pronounced characteristics of glial precursor cells is their ability to self-renew. We demonstrate that HHV-6 viral infection leads to a marked suppression of precursor cell proliferation, as measured by decreased uptake of BrdU and Ki-67 immunostaining and a suppression of cell growth over time compared with uninfected controls. Subsequent flow cytometric studies identified a specific G1/S phase arrest with both A and B strains of HHV-6. FAC-sorted HHV-6A or -6B-infected precursors also displayed a highly significant loss of the self-renewing glial precursor cellular pool, as assayed by expression of the glial precursor cell marker A2B5. The loss of A2B5 was accompanied by a corresponding induction of GalC+ oligodendrocytes. Therefore, virus-mediated suppression of human glial precursor cell proliferation and cell cycle arrest is accompanied by loss of the self-renewing glial progenitor pool and an increase in differentiated oligodendroglial cell types.
The inhibition of proliferation and cell cycle arrest observed in our studies are consistent with the previously reported ability of HHVs to cause cell cycle arrest in a variety of cell types (Flemington, 2001). Studies of human CMV, a β-herpesvirus that is 67% homologous to HHV-6 at the protein level (Dominguez et al., 1999), as well as murine CMV have demonstrated an inhibition of the G1/S phase transition in several distinct cell systems (Wiebusch and Hagemeier, 1999; Kosugi et al., 2000; Flemington, 2001; Castillo and Kowalik, 2002). HHV-6 has been demonstrated to be able to infect and arrest human bone marrow progenitor cells in vitro (Luppi et al., 1999; Isomura et al., 2003) and in vivo (Wilborn et al., 1994; Rosenfeld et al., 1995; Secchiero et al., 1995) and to suppress proliferative responses to antigens and mitogens (Burd and Carrigan, 1993; Horvat et al., 1993; Carrigan and Knox, 1995). Our studies now extend the importance of viral-mediated cell cycle arrest to relevant neural precursor cells and show that this arrest is coupled with alterations in their differentiation potential.
The disruption of precursor cell proliferation attributable to HHV-6 infection might limit the availability of precursor cells for recruitment to the site of injury. In addition, remyelination might fail as a result of inappropriate differentiation of precursor cells that are already present at the lesion site. In fact, our results suggest that HHV-6 infection of precursor cells leads to increased differentiation of precursor cells toward GalC+ oligodendrocytes compared with noninfected controls. These prematurely differentiated oligodendrocytes may be more vulnerable to the inflammatory environment generated by viral infection than precursor cells. One of the major inflammatory cytokines, tumor necrosis factor α (TNF-α) has been described in demyelinated lesions (Raine et al., 1998; Bitsch et al., 2000) and is highly toxic to GalC+ human oligodendrocytes in vivo and in vitro (Selmaj and Raine, 1988; McLarnon et al., 1993; McLaurin et al., 1995). Therefore, the premature differentiation of glial progenitors in a TNF-α-rich environment could result in the relative loss of the more resilient precursor pool, replacing them with a more vulnerable population of precursor cells or GalC+ cells that are unable to effectively remyelinate.
Interestingly, our results show that direct infection of the precursor population with virus is not accompanied by increased cytotoxicity compared with uninfected control cells. This is in direct contrast to findings in primary lymphocytes, in which HHV-6 infection induced apoptotic cell death (Inoue et al., 1997). Both the LDH assay, which measures cell membrane leakage, and the TUNEL assay, which measures DNA fragmentation as a criterion of apoptotic cell death, showed no differences in cell death between HHV-6-infected and uninfected precursors. If anything, there was a weak trend toward enhanced survival in infected cells. This finding may be of interest in light of recent reports that the U45 gene product from both human and murine CMV has been reported to confer a weak antiapoptotic effect in cultured cell lines (Brune et al., 2001; Patrone et al., 2003).
The human CNS contains multiple resident viruses, in addition to herpes viruses, that have the potential to fundamentally interfere with normal cellular functions without destroying the infected cells (Haase et al., 1984; Johnson, 2003; Kennedy, 2003). It has been shown that benign or latent infection by neurotropic viruses may cause the loss of more differentiated cell functions such as myelin formation (Oldstone et al., 1977, 1982; Oldstone, 2002). The results of this study now show that infection of glial precursor cells by HHV-6 causes a disruption of basic and critical properties of these cells. In addition, we have preliminary evidence that human glial precursor cells might provide a target for other known resident CNS viruses. In our experiments, we observed that a significant number of glial precursor cells express CD21, a known receptor for the γ-HHV Epstein–Barr virus (Tsoukas and Lambris, 1993) (data not shown). The presence of the viral receptor CD21 on precursor cells raises the interesting possibility that effects on glial progenitors, similar to those observed in the present study, may not be limited to HHV-6.
Taken together, these findings raise the question of whether infection of glial progenitor cells by resident viruses of the human CNS might contribute to the yet unexplained incompetence of myelin repair seen in human demyelinating diseases (Wolswijk, 1998; Franklin, 2002). It is clear that CNS disease represents a dynamic process between mechanisms leading to cellular damage and those participating in repair. The present study presents a novel potential mechanism, suggesting that endogenous CNS-resident herpes family viruses may be able to alter the dynamics of repair in ways that may cause clinically apparent disease in the setting of demyelinative or other neuroinflammatory or neurodegenerative processes. Interestingly, both clinicopathological and magnetic resonance imaging studies clearly demonstrate that a substantial segment of the population harbors clinically silent lesions of white matter demyelination (Stewart et al., 1987; Engell, 1989).
A viral-induced dysregulation of critical neural precursor properties such as proliferation and differentiation could contribute to the extent or progression of a variety of neurological diseases. In light of the abundance of HHV-6 and other herpesviruses in the human brain, our data raise the concern that incompetence of repair might be a more common event than previously recognized.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (J.D.), the Wilmot Cancer Foundation (J.D.), the National Multiple Sclerosis Society (B.M.B., D.J.M.), the Multiple Sclerosis Society of Canada (M.M.-P.), and National Institutes of Health Grant RO1 NS-044220-01A1 (M.M.-P.). We thank Karen Jensen-Bentley for technical assistance with the electron microscopy, Peter Keng for assistance with the cell cycle analysis and cell sorting, and Andrew Goodman for continuous support of our work. We also thank Mark Noble and Hartmut Land for helpful comments on this manuscript.
Correspondence should be addressed to either of the following: Dr. David J. Mock, Department of Neurology, University of Rochester, 601 Elmwood Avenue, Box 605, Rochester, NY 14642, E-mail: david_mock@URMC.rochester.edu; or Dr. Margot Mayer-Pröschel, Department of Biomedical Genetics, University of Rochester, 601 Elmwood Avenue, Box 633, Rochester, NY 14642, E-mail: margot_mayer-proschel@URMC.rochester.edu.
DOI:10.1523/JNEUROSCI.5584-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/244875-09$15.00/0
References
- Albright AV, Lavi E, Black JB, Goldberg S, O'Connor MJ, Gonzalez-Scarano F (1998) The effect of human herpesvirus-6 (HHV-6) on cultured human neural cells: oligodendrocytes and microglia. J Neurovirol 4: 486–494. [DOI] [PubMed] [Google Scholar]
- Bitsch A, Kuhlman T, Da Costa C, Bunkowski S, Polak T, Bruck W (2000) Tumoure necrosis factor alpha mRNA expression in early multiple sclerosis lesions: correlation with demyelinating activity and oligodendrocyte pathology. Glia 29: 366–375. [DOI] [PubMed] [Google Scholar]
- Blakemore WF, Keirstead HS (1999) The origin of remyelinating cells in the central nervous system. J Neuroimmunol 98: 69–76. [DOI] [PubMed] [Google Scholar]
- Blumberg BM, Mock DJ, Powers JM, Ito M, Assouline JG, Baker JV, Chen B, Goodman AD (2000) The HHV6 paradox: ubiquitous commensal or insidious pathogen? A two-step in situ PCR approach. J Clin Virol 16: 159–178. [DOI] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH (1979) Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci USA 76: 514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruck W, Kuhlmann T, Stadelmann C (2003) Remyelination in multiple sclerosis. J Neurol Sci 206: 181–185. [DOI] [PubMed] [Google Scholar]
- Brune W, Menard C, Heesemann J, Koszinowski UH (2001) A ribonucleotide reductase homolog of cytomegalovirus and endothelial cell tropism. Science 291: 303–305. [DOI] [PubMed] [Google Scholar]
- Burd EM, Carrigan DR (1993) Human herpesvirus 6 (HHV-6)-associated dysfunction of blood monocytes. Virus Res 29: 79–90. [DOI] [PubMed] [Google Scholar]
- Cardinali G, Gentile M, Cirone M, Zompetta C, Frati L, Faggioni A, Torrisi MR (1998) Viral glycoproteins accumulate in newly formed annulate lamellae following infection of lymphoid cells by human herpesvirus 6. J Virol 72: 9738–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrigan DR, Knox KK (1995) Bone marrow suppression by human herpesvirus-6: comparison of the A and B variants of the virus. Blood 86: 835–836. [PubMed] [Google Scholar]
- Carrigan DR, Harrington D, Knox KK (1996) Subacute leukoencephalitis caused by CNS infection with human herpesvirus-6 manifesting as acute multiple sclerosis. Neurology 47: 145–148. [DOI] [PubMed] [Google Scholar]
- Castillo JP, Kowalik TF (2002) Human cytomegalovirus immediate early proteins and cell growth control. Gene 290: 19–34. [DOI] [PubMed] [Google Scholar]
- Cermelli C, Pietrosemoli P, Meacci M, Pecorari M, Sabbatini AM, Colombari B, Portolani M (1997) SupT-1: a cell system suitable for an efficient propagation of both HHV-7 and HHV-6 variants A and B. New Microbiol 20: 187–196. [PubMed] [Google Scholar]
- Chang A, Nishiyama A, Peterson J, Prineas J, Trapp BD (2000) NG2-positive oligodendrocyte progenitor cells in adult human brain and multiple sclerosis lesions. J Neurosci 20: 6404–6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD (2002) Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 346: 165–173. [DOI] [PubMed] [Google Scholar]
- Demmler GJ (1991) Infectious Diseases Society of America and Centers for Disease Control. Summary of a workshop on surveillance for congenital cytomegalovirus disease. Rev Infect Dis 13: 315–329. [DOI] [PubMed] [Google Scholar]
- Dietrich J, Noble M, Mayer-Proschel M (2002) Characterization of A2B5+ glial precursor cells from cryopreserved human fetal brain progenitor cells. Glia 40: 65–77. [DOI] [PubMed] [Google Scholar]
- Dominguez G, Dambaugh TR, Stamey FR, Dewhurst S, Inoue N, Pellett PE (1999) Human herpesvirus 6B genome sequence: coding content and comparison with human herpesvirus 6A. J Virol 73: 8040–8052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donati D, Akhyani N, Fogdell-Hahn A, Cermelli C, Cassiani-Ingoni R, Vortmeyer A, Heiss JD, Cogen P, Gaillard WD, Sato S, Theodore WH, Jacobson S (2003) Detection of human herpesvirus-6 in mesial temporal lobe epilepsy surgical brain resections. Neurology 61: 1405–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchala CS, Asotra K, Macklin WB (1995) Expression of cell surface markers and myelin proteins in cultured oligodendrocytes from neonatal brain of rat and mouse: a comparative study. Dev Neurosci 17: 70–80. [DOI] [PubMed] [Google Scholar]
- Dyer CA, Hickey WF, Geisert Jr EE (1991) Myelin/oligodendrocyte-specific protein: a novel surface membrane protein that associates with microtubules. J Neurosci Res 28: 607–613. [DOI] [PubMed] [Google Scholar]
- Endl E, Gerdes J (2000) The Ki-67 protein: fascinating forms and an unknown function. Exp Cell Res 257: 231–237. [DOI] [PubMed] [Google Scholar]
- Engell T (1989) A clinical patho-anatomical study of clinically silent multiple sclerosis. Acta Neurol Scand 79: 428–430. [DOI] [PubMed] [Google Scholar]
- Flemington EK (2001) Herpesvirus lytic replication and the cell cycle: arresting new developments. J Virol 75: 4475–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ (2002) Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci 3: 705–714. [DOI] [PubMed] [Google Scholar]
- Goodman AD, Mock DJ, Powers JM, Baker JV, Blumberg BM (2003) Human herpesvirus 6 genome and antigen in acute multiple sclerosis lesions. J Infect Dis 187: 1365–1376. [DOI] [PubMed] [Google Scholar]
- Haase AT, Stowring L, Ventura P, Burks J, Ebers G, Tourtellotte W, Warren K (1984) Detection by hybridization of viral infection of the human central nervous system. Ann NY Acad Sci 436: 103–108. [DOI] [PubMed] [Google Scholar]
- He J, McCarthy M, Zhou Y, Chandran B, Wood C (1996) Infection of primary human fetal astrocytes by human herpesvirus 6. J Virol 70: 1296–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvat RT, Parmely MJ, Chandran B (1993) Human herpesvirus 6 inhibits the proliferative responses of human peripheral blood mononuclear cells. J Infect Dis 167: 1274–1280. [DOI] [PubMed] [Google Scholar]
- Inoue Y, Yasukawa M, Fujita S (1997) Induction of T-cell apoptosis by human herpesvirus 6. J Virol 71: 3751–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isegawa Y, Mukai T, Nakano K, Kagawa M, Chen J, Mori Y, Sunagawa T, Kawanishi K, Sashihara J, Hata A, Zou P, Kosuge H, Yamanishi K (1999) Comparison of the complete DNA sequences of human herpesvirus 6 variants A and B. J Virol 73: 8053–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isomura H, Yoshida M, Namba H, Yamada M (2003) Interaction of human herpesvirus 6 with human CD34 positive cells. J Med Virol 70: 444–450. [DOI] [PubMed] [Google Scholar]
- Ito M, Baker JV, Mock DJ, Goodman AD, Blumberg BM, Shrier DA, Powers JM (2000) Human herpesvirus 6-meningoencephalitis in an HIV patient with progressive multifocal leukoencephalopathy. Acta Neuropathol (Berl) 100: 337–341. [DOI] [PubMed] [Google Scholar]
- Johnson RT (2003) Emerging viral infections of the nervous system. J Neurovirol 9: 140–147. [DOI] [PubMed] [Google Scholar]
- Kennedy P (2003) Reviews in neurovirology: an introduction. J Neurovirol 9: 139–283. [Google Scholar]
- Kosugi I, Shinmura Y, Kawasaki H, Arai Y, Li RY, Baba S, Tsutsui Y (2000) Cytomegalovirus infection of the central nervous system stem cells from mouse embryo: a model for developmental brain disorders induced by cytomegalovirus. Lab Invest 80: 1373–1383. [DOI] [PubMed] [Google Scholar]
- Lee JC, Mayer-Proschel M, Rao MS (2000) Gliogenesis in the central nervous system. Glia 30: 105–121. [DOI] [PubMed] [Google Scholar]
- Luppi M, Barozzi P, Morris C, Maiorana A, Garber R, Bonacorsi G, Donelli A, Marasca R, Tabilio A, Torelli G (1999) Human herpesvirus 6 latently infects early bone marrow progenitors in vivo. J Virol 73: 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamidi A, DeSimone JA, Pomerantz RJ (2002) Central nervous system infections in individuals with HIV-1 infection. J Neurovirol 8: 158–167. [DOI] [PubMed] [Google Scholar]
- Mayne M, Cheadle C, Soldan SS, Cermelli C, Yamano Y, Akhyani N, Nagel JE, Taub DD, Becker KG, Jacobson S (2001) Gene expression profile of herpesvirus-infected T cells obtained using immunomicroarrays: induction of proinflammatory mechanisms. J Virol 75: 11641–11650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy M (2003) Newer viral encephalitides. Neurology 9: 189–199. [DOI] [PubMed] [Google Scholar]
- McCutchan JA (1995) Clinical impact of cytomegalovirus infections of the nervous system in patients with AIDS. Clin Infect Dis 21 [Suppl 2]: S196–S201. [DOI] [PubMed] [Google Scholar]
- McLarnon JG, Michikawa M, Kim SU (1993) Effects of tumor necrosis factor on inward potassium current and cell morphology in cultured human oligodendrocytes. Glia 9: 120–126. [DOI] [PubMed] [Google Scholar]
- McLaurin J, D'Souza S, Stewart J, Blain M, Beaudet A, Nalbantoglu J, Antel JP (1995) Effect of tumor necrosis factor alpha and beta on human oligodendrocytes and neurons in culture. Int J Dev Neurosci 13: 369–381. [DOI] [PubMed] [Google Scholar]
- Mock DJ, Powers JM, Goodman AD, Blumenthal SR, Ergin N, Baker JV, Mattson DH, Assouline JG, Bergey EJ, Chen B, Epstein LG, Blumberg BM (1999) Association of human herpesvirus 6 with the demyelinative lesions of progressive multifocal leukoencephalopathy. J Neurovirol 5: 363–373. [DOI] [PubMed] [Google Scholar]
- Novoa LJ, Nagra RM, Nakawatase T, Edwards-Lee T, Tourtellotte WW, Cornford ME (1997) Fulminant demyelinating encephalomyelitis associated with productive HHV-6 infection in an immunocompetent adult. J Med Virol 52: 301–308. [DOI] [PubMed] [Google Scholar]
- Oldstone MB (2002) Travels along the viral-immunobiology highway. Immunol Rev 185: 54–68. [DOI] [PubMed] [Google Scholar]
- Oldstone MB, Holmstoen J, Welsh Jr RM (1977) Alterations of acetylcholine enzymes in neuroblastoma cells persistently infected with lymphocytic choriomeningitis virus. J Cell Physiol 91: 459–472. [DOI] [PubMed] [Google Scholar]
- Oldstone MB, Sinha YN, Blount P, Tishon A, Rodriguez M, von Wedel R, Lampert PW (1982) Virus-induced alterations in homeostasis: alteration in differentiated functions of infected cells in vivo. Science 218: 1125–1127. [DOI] [PubMed] [Google Scholar]
- Padilla JA, Nii S, Grose C (2003) Imaging of the varicella zoster virion in the viral highways: comparison with herpes simplex viruses 1 and 2, cytomegalovirus, pseudorabies virus, and human herpes viruses 6 and 7. J Med Virol 70 [Suppl 1]: S103–S110. [DOI] [PubMed] [Google Scholar]
- Patrone M, Percivalle E, Secchi M, Fiorina L, Pedrali-Noy G, Zoppe M, Baldanti F, Hahn G, Koszinowski UH, Milanesi G, Gallina A (2003) The human cytomegalovirus UL45 gene product is a late, virion-associated protein and influences virus growth at low multiplicities of infection. J Gen Virol 84: 3359–3370. [DOI] [PubMed] [Google Scholar]
- Raine CS, Bonetti B, Cannella B (1998) Multiple sclerosis: expression of molecules of the tumor necrosis factor ligand and receptor families in relationship to the demyelinated plaque. Rev Neurol (Paris) 154: 577–585. [PubMed] [Google Scholar]
- Rao M, Noble M, Mayer-Proschel M (1998) A tripotential glial precursor cell is present in the developing spinal cord. Proc Natl Acad Sci USA 95: 3996–4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Muench H (1938) A simple method of estimating fifty percent endpoints. Am J Hyg 27: 493–497. [Google Scholar]
- Rosenfeld CS, Rybka WB, Weinbaum D, Carrigan DR, Knox KK, Andrews DF, Shadduck RK (1995) Late graft failure due to dual bone marrow infection with variants A and B of human herpesvirus-6. Exp Hematol 23: 626–629. [PubMed] [Google Scholar]
- Rotola A, Ravaioli T, Gonelli A, Dewhurst S, Cassai E, Di Luca D (1998) U94 of human herpesvirus 6 is expressed in latently infected peripheral blood mononuclear cells and blocks viral gene expression in transformed lymphocytes in culture. Proc Natl Acad Sci USA 95: 13911–13916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahuddin SZ, Ablashi DV, Markham PD, Josephs SF, Sturzenegger S, Kaplan M, Halligan G, Biberfeld P, Wong-Staal F, Kramarsky B, Gallo RC (1986) Isolation of a new virus, HBLV, in patients with lymphoproliferative disorders. Science 234: 596–601. [DOI] [PubMed] [Google Scholar]
- Sanders VJ, Felisan S, Waddell A, Tourtellotte WW (1996) Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol 2: 249–258. [DOI] [PubMed] [Google Scholar]
- Santoro F, Kennedy PE, Locatelli G, Malnati MS, Berger EA, Lusso P (1999) CD46 is a cellular receptor for human herpesvirus 6. Cell 99: 817–827. [DOI] [PubMed] [Google Scholar]
- Scolding NJ, Rayner PJ, Compston DA (1999) Identification of A2B5-positive putative oligodendrocyte progenitor cells and A2B5-positive astrocytes in adult human white matter. Neuroscience 89: 1–4. [DOI] [PubMed] [Google Scholar]
- Secchiero P, Carrigan DR, Asano Y, Benedetti L, Crowley RW, Komaroff AL, Gallo RC, Lusso P (1995) Detection of human herpesvirus 6 in plasma of children with primary infection and immunosuppressed patients by polymerase chain reaction. J Infect Dis 171: 273–280. [DOI] [PubMed] [Google Scholar]
- Selmaj K, Raine CS (1988) Tumor necrosis factor mediates myelin damage in organotypic cultures of nervous tissue. Ann NY Acad Sci 540: 568–570. [DOI] [PubMed] [Google Scholar]
- Soldan SS, Leist TP, Juhng KN, McFarland HF, Jacobson S (2000) Increased lymphoproliferative response to human herpesvirus type 6A variant in multiple sclerosis patients. Ann Neurol 47: 306–313. [PubMed] [Google Scholar]
- Stagno S, Pass RF, Cloud G, Britt WJ, Henderson RE, Walton PD, Veren DA, Page F, Alford CA (1986) Primary cytomegalovirus infection in pregnancy. Incidence, transmission to fetus, and clinical outcome. JAMA 256: 1904–1908. [PubMed] [Google Scholar]
- Stemple DL, Anderson DJ (1992) Isolation of a stem cell for neurons and glia from the mammalian neural crest. Cell 71: 973–985. [DOI] [PubMed] [Google Scholar]
- Stewart JM, Houser OW, Baker Jr HL, O'Brien PC, Rodriguez M (1987) Magnetic resonance imaging and clinical relationships in multiple sclerosis. Mayo Clin Proc 62: 174–184. [DOI] [PubMed] [Google Scholar]
- Tsoukas CD, Lambris JD (1993) Expression of EBV/C3d receptors on T cells: biological significance. Immunol Today 14: 56–59. [DOI] [PubMed] [Google Scholar]
- Warrington AE, Barbarese E, Pfeiffer SE (1992) Stage specific, (04+GalC-) isolated oligodendrocyte progenitors produce MBP+ myelin in vitro. Dev Neurosci 14: 93–97. [DOI] [PubMed] [Google Scholar]
- Whitley RJ, Gnann JW (2002) Viral encephalitis: familiar infections and emerging pathogens. Lancet 359: 507–513. [DOI] [PubMed] [Google Scholar]
- Wiebusch L, Hagemeier C (1999) Human cytomegalovirus 86-kilodalton IE2 protein blocks cell cycle progression in G(1). J Virol 73: 9274–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilborn F, Brinkmann V, Schmidt CA, Neipel F, Gelderblom H, Siegert W (1994) Herpesvirus type 6 in patients undergoing bone marrow transplantation: serologic features and detection by polymerase chain reaction. Blood 83: 3052–3058. [PubMed] [Google Scholar]
- Wolswijk G (1998) Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci 18: 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Gelbard HA, Roshal M, Pursell S, Jamieson BD, Planelles V (2001) Comparison of cell cycle arrest, transactivation, and apoptosis induced by the simian immunodeficiency virus SIVagm and human immunodeficiency virus type 1 vpr genes. J Virol 75: 3791–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]