

Abstract
Progressive deposition of senile plaques (SPs) is one of the major neuropathological features of Alzheimer's disease (AD) that precedes cognitive decline. Noninvasive detection of SPs could, therefore, be a potential diagnostic test for early detection of AD patients. For imaging SPs in the living brain, we have developed a series of styrylbenzoxazole derivatives that achieve high binding affinity for amyloid-β (Aβ) fibrils. One of these compounds, 6-(2-Fluoroethoxy)-2-[2-(4-methylaminophenil) ethenyl]benzoxazole (BF-168), selectively binds SPs in AD brain sections and recognizes Aβ1-42-positive diffuse plaques as well as neuritic plaques in AD brain sections. Intravenous injection of BF-168 in PS1/APP and APP23 transgenic mice resulted in specific in vivo labeling to both compact and diffuse amyloid deposits in the brain. In addition, 18F-radiolabeled BF-168 demonstrated abundant initial brain uptake (3.9% injected dose/gm at 2 min after injection) and fast clearance (t1/2 = 24.7 min) after intravenous administration in normal mice. Furthermore, autoradiograms of brain sections from APP23 transgenic mice at 180 min after intravenous injection of [18F]BF-168 showed selective labeling of brain amyloid deposits with little nonspecific binding. These findings strongly suggest that styrylbenzoxazole derivatives are promising candidate probes for positron emission tomography and single-photon emission computed tomography imaging for early detection of amyloid plaque formation.
Keywords: amyloid-β protein, Alzheimer's disease, senile plaques, neurofibrillary tangles, imaging, positron emission tomography
Introduction
Alzheimer's disease (AD) is the commonest cause of dementia in the elderly. Currently, clinical diagnosis of AD is based on clinical criteria that require the presence of memory impairment and at least one of several other cognitive dysfunctions (McKhann et al., 1984; American Psychiatric Association, 1994). Although these criteria have been well accepted by many clinicians, detection of AD patients in early, or at least mild, stage of the disease is still problematic. To overcome this problem, many kinds of diagnostic tests have been proposed for early detection of AD (Okamura et al., 2002; Riemenschneider et al., 2002). Nevertheless, it is still impossible to detect individuals with no clinical symptom who are destined to progress AD.
The pathological hallmark of AD is a substantial neuronal loss accompanied by deposition of senile plaques (SPs) and neurofibrillary tangles (NFTs) (Esiri, 2001). SPs are composed of the amyloid-β (Aβ) peptide, which is proteolytically cleaved from amyloid precursor proteins (Shoji et al., 1992). It has been shown that in early dementia brain levels of Aβ are elevated and correlate with cognitive decline (Naslund et al., 2000). In addition, extensive deposition of extracellular amyloid plaques has been regarded as a critical event for the pathogenesis of AD (Hardy and Selkoe, 2002). NFTs, in contrast, are composed of hyperphosphorylated tau (Lee et al., 1991). Early formation of NFTs in the entorhinal cortex has been reported to be closely related to neuronal loss in patients with very mild AD (Gomez-Isla et al., 1996). From the evidence above, it is suggested that noninvasive detection of SPs and NFTs has a potential merit in early and accurate diagnosis of AD.
For in vivo detection of brain amyloid deposits, compounds that have high binding affinity for amyloid fibrils and adequate permeability of blood brain barrier (BBB) are required for the probe of positron emission tomography (PET) and single-photon emission computed tomography (SPECT) (Bacskai et al., 2002). Accordingly, several promising agents have recently been developed for the imaging probe of SPs and NFTs (Skovronsky et al., 2000; Agdeppa et al., 2001; Klunk et al., 2002, 2003; Kung et al., 2002; Shimadzu et al., 2003). Previous neuropathological studies have suggested that substantial deposition of diffuse plaques is considered to be the initial pathological change in AD that precedes cognitive deterioration (Morris et al., 1996; Price and Morris, 1999). Thus, in vivo imaging probes that can detect diffuse plaques may be ideal for presymptomatic detection of AD patients.
To develop such unique probes for PET and SPECT imaging, we have synthesized a series of promising styrylbenzoxazole derivatives that target SPs, including diffuse plaques. One of these compounds, 6-(2-Fluoroethoxy)-2-[2-(4-methylaminophenil) ethenyl]benzoxazole (BF-168), was found to specifically recognize both neuritic and diffuse plaques in vivo and, thus, was selected as a candidate probe for PET imaging. Here, we report the in vitro and in vivo properties of this compound.
Materials and Methods
Radiolabeling. [125I]BF-180 was synthesized by reacting a tributylstannyl derivative with sodium [125I]iodide and 3% H2O2, followed by HPLC purification (Fig. 1). [18F]BF-168 was also synthesized by reacting 2-(4-methylaminophenyl)-6-(2-tosyloxyethoxy)benzoxazole (Tanabe R&D Service, Osaka, Japan) with [18F]KF and Kryptofix 222 (Merck, Whitehouse Station, NJ) in acetonitrile at 80°C for 20 min, followed by HPLC purification (Fig. 1). The fraction containing the desired product was collected, concentrated, and transferred into an injection vial. The radiochemical yield was 30-40%, and the radiochemical purity was >95%. The maximum specific activity of [18F]BF-168 was 106 TBq/mmol at the end of synthesis.
Figure 1.

Chemical structure and radiolabeling of BF-168 and BF-180.
In vitro binding assays. Styrylbenzoxazole derivatives (Table 1) were purchased from Tanabe R&D Service. A solid form of Aβ1-42 and Aβ1-40 (Peptide Institute, Osaka, Japan) was dissolved in 10 mm potassium phosphate buffer (pH 7.4; 60 μm) containing 1 mm EDTA and incubated at 37°C for 40 hr with gentle shaking. For saturation studies, a solution of [125I]BF-180 (final concentration, 0.15-25.6 nm) was prepared by mixing 0.05 nm [125I]BF-180 and nonradioactive BF-180. The binding assay was performed by mixing 100 μl of aggregated Aβ1-42 or Aβ1-40 fibrils (100 nm in the final assay mixture), the appropriate concentration of 100 μl of [125I]BF-180, and 800 μl of 8% ethanol. After incubation for 4 hr at room temperature, the binding mixture was filtered through GF/B filters (Whatman, Kent, UK) using a M-24 cell harvester (Brandel, Gaithersburg, MD). Filters containing the bound 125I ligand were counted in a gamma counter. The dissociation constant (Kd) and maximum specific binding (Bmax) of BF-180 were determined by Scatchard analysis using GraphPad Prism (GraphPad Software, San Diego, CA). Data are the average ± SD of the results from three independent experiments. For inhibition studies, binding studies were performed using synthetic Aβ1-42 aggregates, because our probe was targeted at the detection of diffuse plaques that were mainly composed of Aβ1-42. A mixture containing 50 μl of test compounds (0.1-25.6 nm in 40% ethanol), 50 μl of 0.05 nm [125I]BF-180, 100 μl of 100 nm Aβ1-42 and 800 μl of 8% ethanol was incubated at room temperature for 4 hr. The mixture was then filtered through Whatman GF/B filters using a Brandel M-24 cell harvester, and the filters containing the bound 125I ligand were counted in a gamma counter. Values for half-maximal inhibitory concentration (IC50) were determined from displacement curves of three independent experiments using GraphPad Prism, and those for the inhibition constant (Ki) were determined using the Cheng-Prusoff equation (Cheng and Prusoff, 1973): Ki = IC50/(1 + [L]/Kd), where [L] is the concentration of [125I]BF-180 used in the assay, and Kd is the dissociation constant of BF-180.

Neuropathological staining. Postmortem brain tissues from four autopsy-confirmed AD cases (67-, 78-, and 79-year-old females and a 69-year-old male) and one physiological aging case (81-year-old male) were obtained from Fukushimura Hospital (Aichi, Japan). Experiments were performed under regulations of the ethics committee of BF Research Institute. Six-micrometer-thick serial sections of frozen or paraffin-embedded blocks of hippocampus and temporal cortex were used for staining. Paraffin sections were first taken through two 10 min washes in xylene, two 5 min washes in 100% ethanol, two 5 min washes in 95% ethanol/H2O, and a 10 min wash under running tap water. Before the staining of BF-168, quenching of autofluorescence was performed by the following method: after immersing in 10% formalin neutral buffer solution for 60 min and washing in PBS, the sections were blanched in 0.25% potassium permanganate solution for 20 min. The sections were then washed in PBS and treated with 0.1% potassium metabisulfite and 0.1% oxalic acid, followed by washing in PBS. Quenched tissue sections were immersed in 100 μm BF-168 solution containing 50% ethanol or PBS for 10 min. Finally, the sections were dipped briefly into water, rinsed in PBS for 60 min, coverslipped with FluorSave reagent (Calbiochem, Darmstadt, Germany), and examined using an Eclipse E800 microscope (Nikon, Tokyo, Japan) equipped with a V-2A filter set (excitation, 380-420 nm; dichroic mirror, 430 nm; longpass filter, 450 nm). BF-168 staining was also performed in AD brain sections pretreated with 90% formic acid for 5 min. Immunostaining was performed using monoclonal antibodies (mAbs) against Aβ (6F/3D; Dako, Glostrup, Denmark) at a dilution of 1:50: Aβ1-40 (BA27; Wako, Osaka, Japan), Aβ1-42 (BC05; Wako), and Tau (pSer422; Wako). Sections were placed in blocking buffer for 30 min. After incubation at 37°C in the primary antibodies for 60 min, sections were processed by the avidin-biotin method using a Pathostain ABC-POD(M) kit (Wako) and DAB as a chromogen.
Fluorescent labeling of Aβ deposits in living transgenic mice. In vivo plaque labeling with BF-168 was evaluated using PS1/APPsw double transgenic mice (n = 2) and one wild-type mouse (male; 32 weeks of age) (Holcomb et al., 1998). The experiments were also performed using young APP23 single transgenic mice (n = 2) and one wild-type mouse (female; 13 months of age) (Sturchler-Pierrat et al., 1997). A BF-168 solution containing 10% polyethylenglycol 400 and 0.1 m HCl was administered into the tail vein in a dose of 4 mg/kg. Two hours after injection of BF-168, the mice were anesthetized with sodium pentobarbital, perfused transcardially with ice-cold saline, followed by 4% paraformaldehyde in 0.1 m PBS, and their brains were removed. After cryoprotection in 30% sucrose/0.1 m PBS, 6 μm frozen sections were cut by an OTF cryostat (Bright Instruments, Huntingdon, UK) and imaged with no additional staining for fluorescent microscopy using a V-2A filter set. The same sections were immunostained with a mAb against Aβ (6F/3D), as described above.
Biodistribution of [18F]BF-168. [18F]BF-168 (0.24-0.38 MBq) was administered into the tail vein of ICR mice (n = 23; males; average weight, 28-32 gm). The mice were then killed by decapitation at 2, 10, 30, 60, 120, and 180 min after injection. The brain and blood were removed and weighed, and the radioactivity was counted with an automatic gamma counter (Wizard 1480; Wallac, Turku, Finland). The percentage of injected dose per gram (%ID/g) was calculated by comparison of tissue count to tissue weight. Each %ID/g value is an average ± SD of three or four separate experiments. The half-clearance time in the brain was defined as the time required for the %ID/g value to decline by 50%.
Autoradiography of Aβ deposits in living transgenic mice. APP23 transgenic mice (n = 3; females; 22 months of age) (Sturchler-Pierrat et al., 1997) and one age-matched control mouse (female; 22 months of age) were used for the autoradiographic study. [18F]BF-168 (2 MBq) in 200 μl of saline was administered into the tail vein. The mice were killed by decapitation at 180 min after injection, and their brains were removed and frozen. Frozen sections were cut (30 μm) using an OTF cryostat (Bright Instruments), dried, and exposed to a BAS-III imaging plate (Fuji Film, Tokyo, Japan) for 18 hr. Autoradiographic images were obtained using a BAS2000 scanner system (Fuji Film). After autoradiographic examination, the same sections were stained with thioflavin-S to confirm the presence of amyloid plaques. For the staining with thioflavin-S, sections were immersed in 0.125% thioflavin-S solution containing 50% ethanol for 3 min and dipped briefly five times in tap water, followed by differentiation in 50% ethanol for 2 min. The sections were then placed in PBS for 60 min, coverslipped with FluorSave reagent (Calbiochem), and examined using an Eclipse E800 microscope (Nikon) equipped with a BV-2A filter set (excitation, 400-440 nm; dichroic mirror, 455 nm; longpass filter, 470 nm). To confirm whether the amount of BF-168 uptake correlates with the amount of Aβ deposition, the areas of [18F]BF-168 accumulation and positive staining with thioflavin-S were measured in nine coronal brain sections of an APP23 mouse. First, the images of autoradiography and thioflavin-S staining were digitized. The percentage of areas of positive accumulation and staining were then automatically measured using Scion Image software (Scion, Frederick, MD). Correlation of the areas of [18F]BF-168 uptake with the areas of positive thioflavin-S staining wasassessed by Pearson's simple correlation methods.
Results
In vitro binding affinity for Aβ fibrils
[125I]BF-180 showed excellent binding affinity for both Aβ1-40 (Kd = 6.8 ± 1.4 nm; Bmax = 3.2 ± 0.55 pmol/mg) and Aβ1-42 (Kd = 10.6 ± 1.5 nm; Bmax = 4.1 ± 1.3 pmol/mg) aggregates (Fig. 2). As shown in Table 1, most styrylbenzoxazole derivatives also showed high binding affinity for Aβ1-42 aggregates, except for BF-208 and BF-191, which displayed low binding affinity, indicating that an amino group in the R2 site is necessary for binding to Aβ aggregates. Furthermore, the styrylbenzothiazole derivative (BF-124) displayed equipotent binding affinity for Aβ1-42 aggregates (Table 1).
Figure 2.
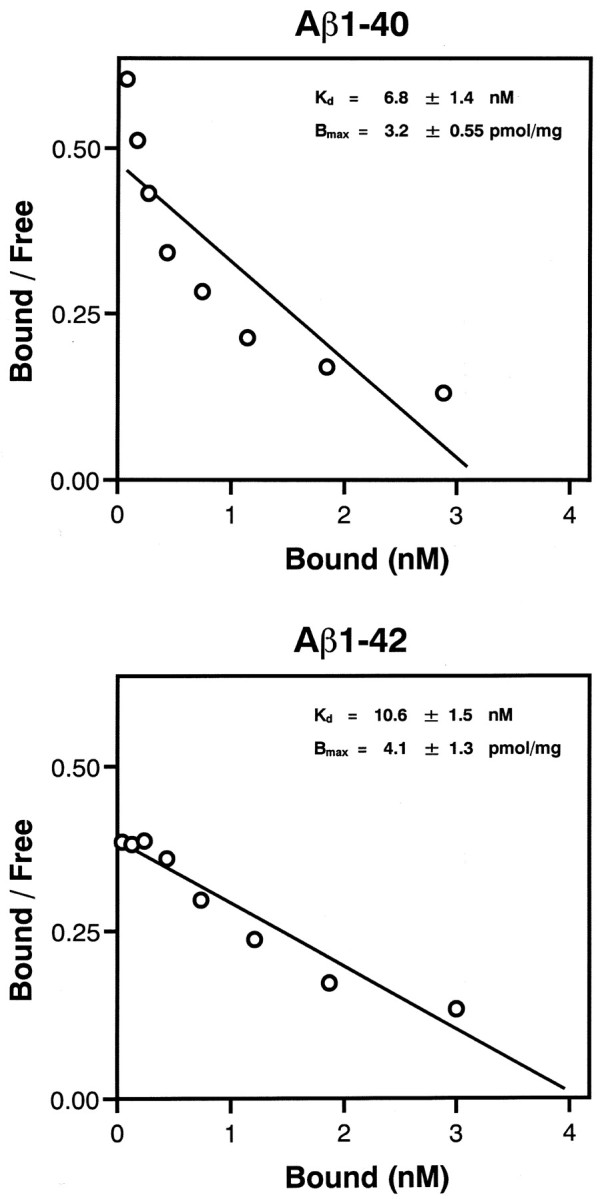
Scatchard plots of [125I]BF-180 binding to aggregates of Aβ1-40 and Aβ1-42. A Kd value of 6.8 nm (Aβ1-40) and 10.6 nm (Aβ1-42) indicate high binding affinity for Aβ aggregates.
Neuropathological staining of SPs and NFTs in AD brain sections
Because BF-168 showed high binding affinity for Aβ aggregates and could easily be 18F-radiolabelled (Fig. 1), we selected this compound for additional studies and assessed its neuropathological staining of SPs and NFTs in AD brain sections. BF-168 clearly stained both neuritic (Fig. 3A) and diffuse (Fig. 3C) amyloid plaques in AD temporal brain sections. Notably, high-intensity fluorescence was observed in the core region of the neuritic plaques. These staining patterns corresponded well to those of Aβ immunostaining in the serial sections (Fig. 3B,D). A comparison of serial sections with Aβ1-40 and Aβ1-42 immunostaining (Fig. 3E-G) demonstrated that BF-168 bound both Aβ1-40-positive and Aβ1-42-positive plaques. Moreover, BF-168 stained NFTs (Fig. 3H) and cerebrovascular amyloids (Fig. 3J). The distribution of BF-168 staining corresponded well to those of tau immunostaining in the serial sections (Fig. 3I). Other styrylbenzoxazole and styrylbenzothiazole derivatives, except for BF-191 and BF-208 (Table 1), showed nearly the same staining pattern of SPs and NFTs as BF-168 (data not shown). To assess whether the solvent of BF-168 solution affects BF-168 staining of SPs in AD brain sections, staining with BF-168 in ethanol was compared with that in PBS. As shown in Fig. 3, K and L, no apparent difference was observed in the BF-168 staining ability of AD brain amyloid plaques. This result indicates that BF-168 high-contrast staining of SPs is not attributable to the reduction of nonspecific binding by ethanol. Furthermore, BF-168 staining of SPs and NFTs was eliminated by pretreatment with 90% formic acid for 5 min (Fig. 3M,N). Because pretreatment with formic acid is considered to disrupt the β-pleated sheet structure of Aβ (Kitamoto et al., 1987), our result from pretreatment with formic acid suggests that BF-168 recognizes β-pleated sheet structure of SPs and NFTs but not nonfibrillar forms of Aβ and tau. Finally, no apparent staining of BF-168 was observed in aged normal brain sections (Fig. 3O), indicating little nonspecific binding of BF-168 in the brain.
Figure 3.

Neuropathological staining of 6 μm AD brain sections from the temporal cortex (A-G, M, N) and hippocampus (H-L) and an aged normal temporal brain section (O). A-D are frozen sections, and E-O are paraffin-embedded sections. Many neuritic plaques are clearly stained with BF-168 (A). Intense fluorescence can be seen in the core of neuritic plaques. Aβ immunostaining with antibodies 6F/3D (B) in the adjacent section show an identical staining pattern of plaques. The brain section stained with BF-168 (C) and the adjacent section immunostained with a mAb against Aβ (D) demonstrate specific binding of BF-168 to diffuse amyloid plaques. Aβ1-40 (E) and Aβ1-42 (G) immunostaining and BF-168 staining (F) in serial sections reveal specific binding of BF-168 to not only Aβ1-40-positive plaques (arrows) but also Aβ1-42-positive diffuse plaques (arrowheads). H, BF-168-stained NFTs, and this staining correlated well with tau immunostaining (I) in the adjacent section. Clear staining of cerebrovascular amyloid (J) can also be observed. There is no obvious difference in the BF-168 staining ability in ethanol solution (K) and in the PBS solution (L) in the serial sections. BF-168 could not stain any plaques and tangles after the treatment with formic acid (N), in contrast to the staining without pretreatment with formic acid (M). Finally, no apparent staining was observed in aged normal brain sections (O). Scale bars: A-D, 50 μm; E-O, 100 μm.
In vivo binding to Aβ deposits in transgenic mice brain
To test the in vivo binding of BF-168 to amyloid plaques, brain sections were examined after intravenous injection of 4 mg/kg BF-168 solution to PS1/APPsw double transgenic mice. In brain sections of the transgenic mice, numerous fluorescent spots with very low background fluorescence were observed in the neocortex and hippocampus of the brain (Fig. 4A). In contrast, no fluorescence was observed in the brains of the wild-type mouse (Fig. 4B). To confirm whether these fluorescent spots reflect specific binding of BF-168 to amyloid deposits, the same brain sections were immunostained with anti-Aβ antibodies. As shown in Fig. 4, C and D, the distribution of fluorescent spots corresponded well to that in Aβ immunostaining. More noteworthy is the fact that Aβ deposits labeled with intravenously administered BF-168 comprised not only compact Aβ deposits but also diffuse Aβ deposits. The same experiments were performed using young APP23 single transgenic mice (13 months of age) that develop only mild depositions of amyloid plaques in the neocortex. Even in these young transgenic mice, BF-168 successfully detected early plaque formation in the entorhinal cortex and hippocampus (Fig. 5A). No fluorescence was detected in the brain slices of the wild-type mouse (data not shown). The fluorescent spots of BF-168 in the entorhinal cortex (Fig. 5B) corresponded well to Aβ immunostaining (Fig. 5C) in the same brain section. These findings are in agreement with the neuropathological data of BF-168 staining in the AD brain sections described above.
Figure 4.

Brain sections from a PS1/APPsw transgenic mouse (A, C, D) and a wild-type mouse (B) after intravenous administration of 4 mg/kg BF-168, showing specific binding of BF-168 to amyloid deposits (A, C) and no nonspecific binding in the brain of the wild-type mouse (B). The same brain section from the transgenic mouse was subsequently immunostained with Aβ-specific mAb 6F/3D (D), revealing specific binding of BF-168 to amyloid deposits in vivo. Both compact amyloid deposits (arrows) and diffuse deposits (arrowhead) were labeled with intravenously administered BF-168. Scale bar, 50 μm.
Figure 5.
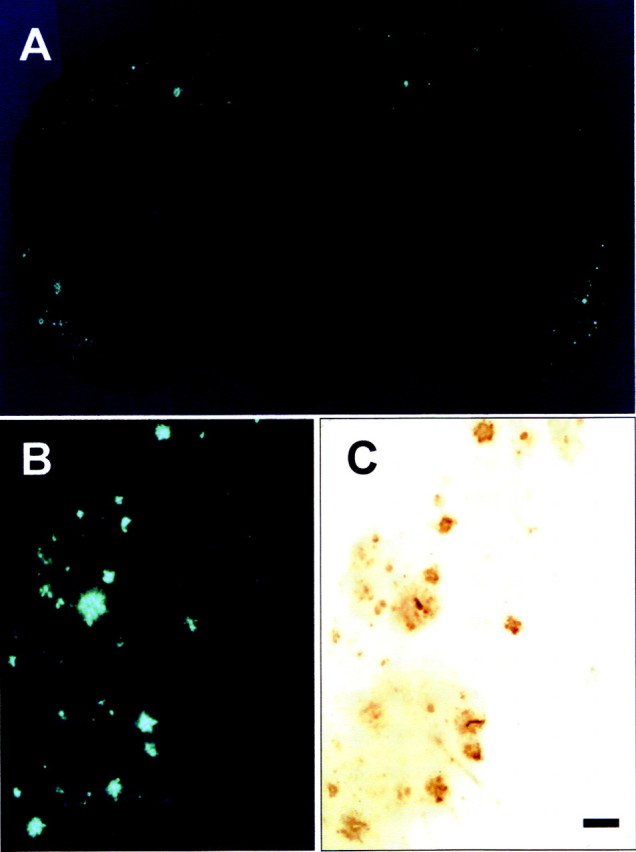
Brain sections from an APP23 transgenic mouse (13 months of age) after intravenous administration of 4 mg/kg BF-168, showing specific binding of BF-168 to early amyloid deposits in the entorhinal cortex and hippocampus (A). The fluorescence of BF-168 in the entorhinal cortex (B) showed identical distribution to immunostaining with Aβ-specific mAb 6F/3D (C) in the same section. Scale bar, 100 μm.
Brain uptake of [18F]BF-168 in normal mice
Next, we examined in vivo brain uptake of 18F-radiolabeled BF-168 using normal (ICR) mice. Brain uptake at 2 min after intravenous injection of [18F]BF-168 (0.24-0.38 MBq) was 3.9 ± 0.22%ID/g, indicating a level sufficient for imaging probe of the brain. In addition, [18F]BF-168 displayed good clearance from normal brain with 3.2 ± 0.35, 1.6 ± 0.0071, 1.3 ± 0.040, 0.93 ± 0.12, and 0.76 ± 0.13%ID/g remaining at 10, 30, 60, 120, and 180 min after injection, respectively (Fig. 6). [18F]BF-168 half-clearance time in the brain was sufficient for the PET imaging probe (24.7 min), and its brain/blood ratio dropped rapidly from 2.0 at 10 min to 0.72 at 60 min after injection in normal mice. These results indicate that [18F]BF-168 has ideal kinetic properties, including substantial brain uptake and fast brain washout.
Figure 6.

Brain uptake and brain/blood ratio of [18F]BF-168 in normal mice. Each data point was obtained from three or four separate experiments and plotted as value of mean ± SD.
In vivo plaque labeling with [18F]BF-168 in transgenic mice
[18F]BF-168 in vivo labeling ability of amyloid plaques was further evaluated using 22-month-old APP23 transgenic mice. Autoradiographic images of APP23 mice brains 180 min after intravenous injection of [18F]BF-168 displayed high uptakes of this labeling compound in the cerebral cortex, hippocampus, and entorhinal cortex (Fig. 7A). In addition, nonspecific accumulation of [18F]BF-168 in the white matter areas of the brain was unremarkable. In contrast, wild-type mouse brain showed no remarkable accumulation of [18F]BF-168 in the brain (Fig. 7B). Furthermore, the hot spots of [18F]BF-168 in APP23 mice brains (Fig. 7A) closely corresponded with those of in vitro thioflavin-S staining in the same brain section (Fig. 7C), indicating that intravenously administered [18F]BF-168 specifically labels amyloid deposits in transgenic mice brains. Finally, the areas of [18F]BF-168 uptake in autoradiographic images were significantly correlated with the areas of in vitro thioflavin-S staining in the same sections (r = 0.923; p < 0.001) (Fig. 7D), suggesting little nonspecific binding of [18F]BF-168.
Figure 7.

In vivo labeling of amyloid deposits in brain sections from an APP23 transgenic mouse (A) and a wild-type mouse (B) with [18F]BF-168. An autoradiographic study was performed using brain sections at 180 min after intravenous injection of [18F]BF-168. Numerous hot spots of [18F]BF-168 can be detected in the cerebral cortex, hippocampus, and entorhinal cortex of transgenic mouse brain (A), in contrast to no apparent uptake in wild-type mouse brain (B). Fluorescences of amyloid deposits appeared after in vitro staining with thioflavin-S in the same section (C). Arrows show amyloid deposits labeled with both [18F]BF-168 and thioflavin-S, indicating specific labeling of amyloid plaques with intravenously administered [18F]BF-168. The percentage of areas of [18F]BF-168 uptake in nine autoradiographic images were significantly correlated with the percentage of areas of in vitro thioflavin-S staining in the same sections (r = 0.923; p < 0.001) (D), suggesting high binding specificity of [18F]BF-168 to amyloid plaques.
Discussion
The results of the present study indicate that BF-168, a styrylbenzoxazole derivative, is a potent agent that selectively recognizes SPs and NFTs in AD brain. Indeed, BF-168 and other benzoxazole derivatives showed high binding affinity for synthetic Aβ aggregates. In addition, [18F]BF-168 exhibited high initial brain uptake in normal mice (3.9%ID/g at 2 min). This value, which is equivalent to a 117% injected dose index in a 30 gm mouse, clearly meets the prerequisites for useful PET imaging agents (Klunk et al., 2002). Moreover, both autoradiography and fluorescent microscopy of brain sections from two different transgenic mouse models demonstrated that BF-168 specifically labels brain amyloid deposits in vivo. These findings strongly suggest that styrylbenzoxazole derivatives are potentially useful for in vivo imaging of amyloid plaques.
It is especially noteworthy that BF-168 binds not only neuritic plaques but also diffuse plaques that were predominantly positive for Aβ1-42 immunostaining. Because diffuse plaques are exclusively positive for Aβ1-42 (Iwatsubo et al., 1994) and considered to be the initial pathological lesion of AD (Morris et al., 1996), probes that can detect Aβ1-42-positive diffuse plaques are better suited for presymptomatic detection of AD-related pathology. Several compounds, such as [18F]FDDNP and [125I]TZDM, have been reported to have affinity for diffuse plaques or Aβ1-42-positive plaques (Agdeppa et al., 2001; Kung et al., 2003). However, these compounds have some limitations in their practical use as probes for in vivo imaging, because of their delayed washout and nonspecific accumulation in the brain white matter (Bacskai et al., 2002). Nonspecific binding of imaging probes leads to high background activity and low-contrast images of target structures, resulting in difficult early detection of plaque deposits. In contrast to these compounds, [18F]BF-168 showed short clearance half-life (24.7 min) in normal mouse brain and mild nonspecific accumulation in the white matter of transgenic mice brains. In addition, BF-168 could be labeled with18F, which shows longer half-life (109.0 min) than 11C (20.3 min). It is advantageous when it takes longer time to reach the binding equilibrium. Its longer half-life also permits the shipment of this tracer to multiple PET centers. These characteristics are the advantage of 18F-labeled compounds. Therefore, it is believed that [18F]BF-168 is a practical imaging probe for amyloid plaques and may be useful for early detection of AD-related brain changes.
It must be noted that the deposition of diffuse plaques is frequently observed in nondemented normal brain. This fact calls in the question whether BF-168 can distinguish normal aging from the AD pathological process. As Morris et al. (1996) pointed out, the neuropathological characteristics that distinguish cases with AD from nondemented aging are the deposition of widespread neocortical diffuse plaques and the presence of neuritic plaques (Morris et al., 1996). Our neuropathological staining data and in vivo experiment using PS1/APP and APP23 transgenic mice indicate that BF-168 can visualize early amyloid formation in the brain. In addition, the fluorescence intensity of BF-168 was higher in mature plaques, especially in the fibril-rich core region, than in diffuse plaques, suggesting that the amount of BF-168 binding reflects the density of Aβ fibrils. Thus, an increase in the deposition of fibril-rich neuritic plaques in the course of AD progression would be reflected by a drastic increase in the total binding ability of BF-168 in the neocortex and limbic regions.
In addition, BF-168 labeled not only SPs but also NFTs in AD brain sections. This finding indicates that BF-168 recognizes the β-pleated sheet structure of SPs and NFTs. This speculation is supported by the finding that neuropathological staining of SPs and NFTs with BF-168 was eliminated by pretreatment with formic acid. A marked increase in the amount of NFT accumulation has also been observed in preclinical AD stage, although deposition of NFTs in the hippocampus and entorhinal cortex is known to gradually increase with age (Price and Morris, 1999). Thus, because BF-168 can detect NFT accumulation, an increased uptake of [18F]BF-168 in the hippocampus and entorhinal cortex would discriminate between normal aging and AD. Considering all these various factors together, quantitative evaluation of BF-168 binding in the whole brain may provide useful information on Aβ and tau pathology and discriminate progression to AD from normal aging, although additional studies are required to determine the watershed between normal aging and AD. Moreover, BF-168 specificity for SPs and NFTs should be investigated further to determine whether [18F]BF-168 binding truly reflects the amount of SPs and NFTs.
In our autoradiographic study, [18F]BF-168 succeeded in labeling amyloid deposits in transgenic mouse brain, suggesting that BBB permeability of [18F]BF-168 is sufficient for in vivo imaging of amyloid deposits. However, it must be noted that the injected dose of [18F]BF-168 in our autoradiographic study was ∼20-fold higher than that usually used in clinical PET studies. Thus, whether the pharmacokinetics of BF-168 is sufficient for PET imaging remains to be determined using either in vivo micro-PET in a transgenic mouse model or in vivo PET in aged dogs or humans. [18F]BF-168 clearance from normal brain was 0.93%ID/g remaining at 120 min after injection. This value was equal to a 24% peak brain uptake at 2 min. This is speculated to be attributable to the persistence of [18F]BF-168 or its metabolites in the plasma, because the brain/blood ratio is stable over 1 hr after injection. To overcome this issue, our compound should be optimized for future clinical application.
In conclusion, we introduced here novel styrylbenzoxazole derivatives that have high in vitro binding affinity for Aβ fibrils as candidate probes for imaging amyloid deposits. BF-168, one of these compounds, specifically binds both neuritic and diffuse plaques in AD brain sections in vitro and in two different transgenic mouse brains in vivo. In addition, 18F-labeled BF-168 displayed excellent BBB permeability and specific in vivo labeling of amyloid deposits. Although recently, amyloid-lowering agents such as γ-secretase inhibitors and Aβ vaccine have been developed for the treatment of AD, these agents are subjected to preventive trials in presymptomatic subjects who have a high risk of progression to AD. Thus, sensitive and accurate methods to detect and monitor the progression of Aβ deposition are desperately needed. The styrylbenzoxazole derivatives described here are promising candidate probes for PET and SPECT imaging that can detect high-risk AD patients in their presymptomatic stage and monitor the formation of amyloid fibrils in vivo.
Footnotes
This work was supported by the Organization for Pharmaceutical Safety and Research of Japan and the Japan Foundation for Aging and Health. We thank N. Ejima for technical assistance in radioisotope experiments.
Correspondence should be addressed to Dr. Nobuyuki Okamura, Department of Pharmacology, Tohoku University School of Medicine, 2-1, Seiryo-machi, Aoba-ku, Sendai 980-8575, Japan. E-mail: oka@mail.tains.tohoku.ac.jp.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242535-07$15.00/0
References
- Agdeppa ED, Kepe V, Liu J, Flores-Torres S, Satyamurthy N, Petric A, Cole GM, Small GW, Huang SC, Barrio JR (2001) Binding characteristics of radiofluorinated 6-dialkylamino-2-naphthylethylidene derivatives as positron emission tomography imaging probes for beta-amyloid plaques in Alzheimer's disease. J Neurosci 21: RC189(1-5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association (1994) Diagnostic and statistical manuals of mental disorders, Ed 4. Washington, DC: American Psychiatric Association.
- Bacskai BJ, Klunk WE, Mathis CA, Hyman BT (2002) Imaging amyloid-beta deposits in vivo. J Cereb Blood Flow Metab 22: 1035-1041. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (Ki) and the concentration of an inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22: 3099-3108. [DOI] [PubMed] [Google Scholar]
- Esiri MM (2001) The neuropathology of Alzheimer's disease. In: Neurobiology of Alzheimer's disease, Ed 2 (Dawbarn D, Allen SJ, eds), pp 33-53. New York: Oxford UP.
- Gomez-Isla T, Price JL, McKeel Jr DW, Morris JC, Growdon JH, Hyman BT (1996) Profound loss of layer II entorhinal cortex neurons occurs in very mild Alzheimer's disease. J Neurosci 16: 4491-4500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med 4: 97-100. [DOI] [PubMed] [Google Scholar]
- Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y (1994) Visualization of A beta 42(43) and A beta 40 in senile plaques with end-specific A beta monoclonals: evidence that an initially deposited species is A beta 42(43). Neuron 13: 45-53. [DOI] [PubMed] [Google Scholar]
- Kitamoto T, Ogomori K, Tateishi J, Prusiner SB (1987) Formic acid pretreatment enhances immunostaining of cerebral and systemic amyloids. Lab Invest 57: 230-236. [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, Mathis CA, Kajdasz ST, McLellan ME, Frosch MP, Debnath ML, Holt DP, Wang Y, Hyman BT (2002) Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J Neuropathol Exp Neurol 61: 797-805. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA (2003) The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci 23: 2086-2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung MP, Hou C, Zhuang ZP, Zhang B, Skovronsky D, Trojanowski JQ, Lee VM, Kung HF (2002) IMPY: an improved thioflavin-T derivative for in vivo labeling of beta-amyloid plaques. Brain Res 956: 202-210. [DOI] [PubMed] [Google Scholar]
- Kung MP, Skovronsky DM, Hou C, Zhuang ZP, Gur TL, Zhang B, Trojanowski JQ, Lee VM, Kung HF (2003) Detection of amyloid plaques by radioligands for Abeta40 and Abeta42: potential imaging agents in Alzheimer's patients. J Mol Neurosci 20: 15-24. [DOI] [PubMed] [Google Scholar]
- Lee VM, Balin BJ, Otvos Jr L, Trojanowski JQ (1991) A68: a major subunit of paired helical filaments and derivatized forms of normal Tau. Science 251: 675-678. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 34: 939-944. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, McKeel Jr DW, Rubin EH, Price JL, Grant EA, Berg L (1996) Cerebral amyloid deposition and diffuse plaques in “normal” aging: evidence for presymptomatic and very mild Alzheimer's disease. Neurology 46: 707-719. [DOI] [PubMed] [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD (2000) Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 283: 1571-1577. [DOI] [PubMed] [Google Scholar]
- Okamura N, Arai H, Maruyama M, Higuchi M, Matsui T, Tanji H, Seki T, Hirai H, Chiba H, Itoh M, Sasaki H (2002) Combined analysis of CSF tau levels and [(123)I]Iodoamphetamine SPECT in mild cognitive impairment: implications for a novel predictor of Alzheimer's disease. Am J Psychiatry 159: 474-476. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 45: 358-368. [DOI] [PubMed] [Google Scholar]
- Riemenschneider M, Lautenschlager N, Wagenpfeil S, Diehl J, Drzezga A, Kurz A (2002) Cerebrospinal fluid tau and beta-amyloid 42 proteins identify Alzheimer disease in subjects with mild cognitive impairment. Arch Neurol 59: 1729-1734. [DOI] [PubMed] [Google Scholar]
- Shimadzu H, Suemoto T, Suzuki M, Shiomitsu T, Okamura N, Kudo Y, Sawada T (2003) A novel probe for imaging amyloid-β: Synthesis of F-18 labelled BF-108, an Acridine Orange analog. J Label Comp Radiopharm 46: 765-772. [Google Scholar]
- Shoji M, Golde TE, Ghiso J, Cheung TT, Estus S, Shaffer LM, Cai XD, McKay DM, Tintner R, Frangione B, Younkin SG (1992) Production of the Alzheimer amyloid beta protein by normal proteolytic processing. Science 258: 126-129. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Zhang B, Kung MP, Kung HF, Trojanowski JQ, Lee VM (2000) In vivo detection of amyloid plaques in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97: 7609-7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B (1997) Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 94: 13287-13292. [DOI] [PMC free article] [PubMed] [Google Scholar]