

Abstract
Apolipoprotein E (apoE) is found in amyloid plaques and neurofibrillary tangles (NFTs) in Alzheimer's disease (AD) brains, but its role in their pathogenesis is unclear. Previously, we found C-terminal-truncated fragments of apoE in AD brains and showed that such fragments can cause neurodegeneration and can induce NFT-like inclusions in cultured neuronal cells and in transgenic mice. Here, we analyzed apoE fragmentation in brain tissue homogenates from transgenic mice expressing apoE3 or apoE4 in neurons [neuron-specific enolase (NSE)-apoE] or astrocytes [glial fibrillary acidic protein (GFAP)-apoE] by Western blotting. The C-terminal-truncated fragments of apoE accumulated, in an age-dependent manner, in the brains of NSE-apoE4 and, to a significantly lesser extent, NSE-apoE3 mice; however, no fragments were detected in GFAP-apoE3 or GFAP-apoE4 mice. In NSE-apoE mice, the pattern of apoE fragmentation resembled that seen in AD brains, and the fragmentation was specific for certain brain regions, occurring in the neocortex and hippocampus, which are vulnerable to AD-related neurodegeneration, but not in the less vulnerable cerebellum. Excitotoxic challenge with kainic acid significantly increased apoE fragmentation in NSE-apoE4 but not NSE-apoE3 mice. Phosphorylated tau (p-tau) also accumulated in an age-dependent manner in NSE-apoE4 mice and, to a much lesser extent, in NSE-apoE3 mice but not in GFAP-apoE3 or GFAP-apoE4 mice. Intraneuronal p-tau inclusions in the hippocampus were prominent in 21-month-old NSE-apoE4 mice but barely detectable in NSE-apoE3 mice. Thus, the accumulation of potentially pathogenic C-terminal-truncated fragments of apoE depends on both the isoform and the cellular source of apoE. Neuron-specific proteolytic cleavage of apoE4 is associated with increased phosphorylation of tau and may play a key role in the development of AD-related neuronal deficits.
Keywords: apolipoprotein E, Alzheimer's disease, excitotoxin, proteolysis, tau phosphorylation, transgenic mice
Introduction
Alzheimer's disease (AD) is one of the fastest growing neurological conditions in developed countries and is the leading cause of dementia (Hebert et al., 2003). The APOE ϵ4 allele has been genetically linked to late-onset familial and sporadic AD and has a gene dose effect on the risk and age of onset of the disease (Corder et al., 1993; Saunders et al., 1993; Roses, 1996; Tang et al., 1998; Romas et al., 2002). Individuals with two copies of the ϵ4 allele have a 50-90% chance of developing AD by the age of 85 years, and those with one copy have an ∼45% chance (Corder et al., 1993; Farrer et al., 1997). Only 20% of the general population will develop AD by the age of 85 years (Corder et al., 1993).
Several hypotheses have been proposed to explain the association of the APOE ϵ4 allele with AD. They include the modulation of the deposition and clearance of amyloid β (Aβ) peptides and the formation of plaques (Strittmatter et al., 1993b; LaDu et al., 1994; Ma et al., 1994; Wisniewski et al., 1994; Bales et al., 1999; Holtzman et al., 2000; Irizarry et al., 2000), impairment of the antioxidative defense system (Miyata and Smith, 1996), dysregulation of neuronal signaling pathways (Herz and Beffert, 2000), disruption of cytoskeletal structure and function (Nathan et al., 1994; Nathan et al., 1995), altered phosphorylation of tau and the formation of neurofibrillary tangles (NFTs; Strittmatter et al., 1994; Tesseur et al., 2000a; Huang et al., 2001; Ljungberg et al., 2002; Harris et al., 2003), depletion of cytosolic androgen receptor levels in the brain (Raber et al., 1998; Raber et al., 2002), potentiation of Aβ-induced lysosomal leakage and apoptosis in neuronal cells (Ji et al., 2002), and promotion of endosomal abnormalities linked to Aβ overproduction (Cataldo et al., 1997, 2000; Grbovic et al., 2003). However, the molecular mechanisms of these detrimental effects mediated by apolipoprotein E4 (apoE4) are still mostly unknown, and it is unclear whether these diverse effects reflect parallel processes or different components of a causal chain.
Although it was initially thought that apoE was synthesized in the brain by astrocytes but not by neurons (Boyles et al., 1985), numerous subsequent studies have demonstrated that CNS neurons can also express apoE, albeit at lower levels than astrocytes, under diverse physiological and pathological conditions (Diedrich et al., 1991; Poirier et al., 1991; Han et al., 1994; Bao et al., 1996; Beffert and Poirier, 1996; Metzger et al., 1996; Xu et al., 1996; Dupont-Wallois et al., 1997; Boschert et al., 1999; Xu et al., 1999a,b; Ferreira et al., 2000; Dekroon and Armati, 2001; Hartman et al., 2001; Aoki et al., 2003; Harris et al., 2004).
ApoE is also detected in amyloid plaques and NFTs, two neuropathological hallmarks of AD (Namba et al., 1991; Selkoe, 1991; Wisniewski and Frangione, 1992; Crowther, 1993; Strittmatter et al., 1993a; Roses, 1994; Tanzi and Bertram, 2001). However, the role of apoE in the pathogenesis of these two lesions is unclear. Histopathological and behavioral analyses of transgenic mice expressing different human apoE isoforms in the brain have provided clear evidence for a dominant adverse effect of apoE4 (Raber et al., 1998, 2000; Buttini et al., 1999), but the underlying mechanisms remain unknown. Recently, we demonstrated a biological event that could play a major causal role in apoE4-related neuropathology. Specifically, we found that apoE, especially apoE4, undergoes intracellular proteolytic cleavage, resulting in bioactive C-terminal-truncated fragments (Huang et al., 2001). These apoE fragments are generated inside cultured neurons and in AD brains; they can cause neurodegeneration and the formation of NFT-like intracellular inclusions in cultured neuronal cells as well as in CNS neurons of transgenic mice (Huang et al., 2001; Harris et al., 2003).
Here we demonstrate in apoE transgenic mice that production of apoE by neurons is associated with apoE fragmentation, whereas production of apoE by astrocytes is not. The intraneuronal proteolysis of apoE occurs preferentially in regions of the brain that are susceptible to AD-related neurodegeneration, affects apoE4 much more than apoE3, and is associated with enhanced phosphorylation of tau. Similar processes may contribute to neuronal deficits in AD patients and related animal models.
Materials and Methods
Reagents. Minimum essential medium, N2 medium supplements, and fetal bovine serum were purchased from Invitrogen (Rockville, MD). ECL was from Amersham Biosciences (Arlington Heights, IL). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was from Sigma (St. Louis, MO). Polyclonal goat anti-human apoE and kainic acid were purchased from Calbiochem (San Diego, CA). Anti-C-terminal apoE (amino acids 272-299) was prepared as described (Huang et al., 2001). Phosphorylation-dependent monoclonal tau antibodies AT8 (p-Ser202), AT100 (p-Ser212), AT180 (p-Thr231), and AT270 (p-Th181) were purchased from Endogen (Woburn, MA). Phosphorylation-dependent monoclonal neurofilament of high molecular weight (p-NF-H) antibody SM-31 was from Sternberger Monoclonals (Lutherville, MD). Fluorescein isothiocyanate (FITC)-, rodamine-, and Cy5-coupled anti-rabbit, anti-mouse, and anti-goat IgG were from Vector Laboratories (Burlingame, CA). Horseradish peroxidase-coupled anti-rabbit, anti-mouse, and anti-goat IgG were from Dako (Carpinteria, CA).
Transgenic mice expressing apoE3 or apoE4 in neurons or in astrocytes. We studied transgenic mice expressing human apoE3 or apoE4 in CNS neurons under the control of a neuron-specific enolase promoter (NSE-apoE3 or NSE-apoE4) on a mouse apoE-deficient background (Raber et al., 1998; Buttini et al., 1999). All NSE-apoE transgenic mice had been backcrossed >10 times with apoE-deficient mice (C57BL/6J-ApoetmlUnc; The Jackson Laboratory, Bar Harbor, ME). ApoE-deficient littermates of NSE-apoE3 or NSE-apoE4 mice served as controls.
Transgenic mice expressing human apoE3 or apoE4 in astrocytes driven by a glial fibrillary acidic protein promoter (GFAP-apoE3 or GFAP-apoE4) on a mouse apoE-deficient background (>10 times of backcross) were generated as described (Raber et al., 1998, 2002). Two sets of expression-matched GFAP-apoE3 and GFAP-apoE4 lines were analyzed. Cerebral expression levels of apoE were similar to those of our NSE-apoE3 and NSE-apoE4 mice (Raber et al., 1998; Buttini et al., 1999) in one set of GFAP-apoE mice and sixfold higher in the other set.
Because apoE4-induced behavioral deficits in NSE-apoE mice were gender-dependent (Raber et al., 1998, 2002), only female transgenic mice (1-21 months old) were used. Mice were kept under a 12 hr light/dark cycle with ad libitum access to sterile water and food (PicoLab Rodent Diet 20, 5053; PMI Nutrition International, St. Louis, MO).
Genotyping of transgenic mice. The five different genotypes analyzed in this study (apoE-deficient, NSE-apoE3, NSE-apoE4, GFAP-apoE3, and GFAP-apoE4) were identified by PCR using DNA purified from tail clips (Raber et al., 1998; Buttini et al., 1999). Because the human APOE intron 3 was included in the apoE4 but not in the apoE3 construct, the amplicon generated with intron 3-spanning primers (forward primer, nucleotides 3158-3175; reverse primer, nucleotides 3815-3834, GenBank accession number M10065) was 670 bp in apoE4 mice and 100 bp in apoE3 mice. The PCR reactions were run on a PTC-100 programmable thermal controller (MJ Research), and the products were analyzed on 1.5% agarose gels. To assess the Apoe knockout status of the mice, total plasma cholesterol levels were measured with a cholesterol measurement kit (Abbott Laboratories, Abbott Park, IL) as described (Huang et al., 1998).
Preparation of mouse brain tissues. Brains from apoE-deficient mice, hemizygous NSE-apoE3, NSE-apoE4, GFAP-apoE3, or GFAP-apoE4 transgenic mice, or doubly transgenic NSE-apoE4/NSE-apoE3 mice on a mouse apoE-deficient background were collected after a 2 min transcardial perfusion with PBS. One hemibrain from each mouse was homogenized and analyzed for apoE and phosphorylated tau (p-tau; Huang et al., 2001). In brief, brain tissues were homogenized in ice-cold lysis buffer I (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 0.1% SDS, 0.5% Nonidet P-40, 0.5% sodium deoxycholate, and a mixture of protease and phosphatase inhibitors). After centrifugation at 35,000 rpm for 30 min at 4°C using a TLA 100.3 rotor of an Optima TL Ultracentrifuge (Beckman) to separate the solubilized proteins (supernatant), the pellet was further homogenized in ice-cold lysis buffer II (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 4% SDS, 1% Nonidet P-40, 1% sodium deoxycholate, and a mixture of protease and phosphatase inhibitors) and centrifuged at 35,000 rpm for 30 min at 4°C using the TLA 100.3 rotor to obtain the second supernatant (solubilized pellet).
Western blotting and quantitative analysis of apoE and p-tau. The supernatant and the solubilized pellet (150-200 μg of protein) were subjected to SDS-PAGE and analyzed by Western blotting with antibodies against human full-length apoE, C-terminal apoE, or p-tau (Huang et al., 2001). The bands of full-length apoE, apoE fragments, or p-tau were scanned, and their intensities were calculated (Huang et al., 2001). ApoE fragmentation was expressed as the ratio of truncated apoE to full-length apoE, and p-tau was reported as arbitrary units.
Immunohistochemistry. The other hemibrain from each mouse was fixed in 3% paraformaldehyde, sectioned, and stained with anti-apoE and anti-p-tau (Buttini et al., 1999; Huang et al., 2001). Vibratome sections were incubated in 0.3% H2O2 in PBS for 20 min to quench endogenous peroxidase activity. To facilitate penetration of antibodies, sections used for immunoperoxidase staining were preincubated for 4 min in 1 μg/ml proteinase K in a buffer containing (in mm): 250 NaCl, 25 EDTA, and 50 Tris-HCl, pH 8. To block nonspecific reactions, all sections were incubated for 1 hr in 15% normal serum from the same species that produced the secondary antibodies (Jackson ImmunoResearch, West Grove, PA) in PBS or for 7 min in Superblock (Scytec, Logan, UT), followed by a 1 hr incubation in PBS with primary antibody (anti-apoE, anti-p-tau, or anti-p-NF-H). Sections were then washed three times in PBS and incubated for 1 hr with the corresponding secondary antibodies coupled to FITC (Jackson ImmunoResearch) or biotin (Vector Laboratories). After three washes in PBS, the sections were mounted in VectaShield (Vector Laboratories) and viewed with a Radiance 2000 laser-scanning confocal system (Bio-Rad, Hercules, CA) mounted on an Optiphot-2 microscope (Nikon, Tokyo, Japan). For immunoperoxidase staining, secondary antibody binding was detected with the ABC-Elite kit (Vector Laboratories). Six sections were stained for each antibody and each mouse brain.
Double labeling for human apoE and p-tau or p-NF-H was performed essentially as described above, except that brain sections were incubated with anti-apoE together with anti-p-tau. To detect primary antibody binding, sections were incubated for 1 hr in a mixture of secondary antibodies (FITC- and Cy5-conjugated antibodies, respectively). After three 10 min washes in PBS, sections were mounted under glass coverslips with VectaShield and viewed by confocal microscopy as described above. The Cy5 and FITC channels were viewed individually, and the resulting images were pseudocolored in red (Cy5) or green (FITC) with Photoshop version 6.0 (Adobe Systems, San Jose, CA). Six sections were stained for each antibody and each mouse brain.
Kainic acid injections. Kainic acid crosses the blood-brain barrier and induces excitotoxic CNS injury, particularly in the hippocampus and neocortex (Spinler and Cziraky, 1994; Masliah et al., 1997). At 7-8 months of age, NSE-apoE3 or NSE-apoE4 mice were injected intraperitoneally with kainic acid (Sigma) dissolved in saline (0.9%) at 18 mg/kg body weight in one dose, as described (Buttini et al., 1999). Within ∼15 min, all mice developed seizures. Seizure activity was assessed as described (Schauwecker and Steward, 1997). There were no differences in kainic acid-induced seizures across groups of mice with respect to overall incidence, period between injection and seizure onset, intensity, or duration of seizures (data not shown), suggesting that brain penetration of kainic acid was similar in all the mice. Mice in the control groups were injected intraperitoneally with saline and did not develop seizures. All mice were killed 6 d after the injection of kainic acid or saline.
Electron microscopy. Mouse brain tissues were sectioned with a vibratome, fixed in 2.5% glutaraldehyde for 1 hr and in 2% OsO4 for 1 hr, dehydrated, embedded, sectioned, and stained with uranyl acetate and lead citrate. Hippocampal neurons were examined using a JEOL JEM 100CX transmission electron microscope.
Statistical analysis. Results are reported as mean ± SD. Differences were evaluated by t test or ANOVA.
Results
Similar patterns of apoE fragmentation in NSE-apoE mice and humans with AD
We previously demonstrated that C-terminal-truncated 29 kDa and 14-20 kDa fragments accumulate in human AD brains, and APOE ϵ4 carriers have substantially higher levels of these fragments than people carrying other APOE alleles (Huang et al., 2001; Harris et al., 2003). Figure 1A illustrates typical levels of apoE fragmentation in an AD case with one APOE ϵ4 and one APOE ϵ3 allele. To determine whether apoE fragmentation also occurs in apoE transgenic mice, brain homogenates from singly (apoE3 or apoE4) or doubly (apoE4/3) transgenic NSE-apoE mice were analyzed by anti-apoE western blotting. Polyclonal anti-apoE revealed full-length 34 kDa apoE and apoE fragments in both NSE-apoE3 and NSE-apoE4 mouse brains (Fig. 1B), suggesting that apoE proteolysis also occurred in mouse brains. Importantly, the pattern of apoE fragmentation in NSE-apoE mice was similar to that in humans (Fig. 1, compare A, B). In doubly transgenic NSE-apoE4/3 mice and in apoE4/3 AD patients, both the 29 and 14-20 kDa apoE fragments were observed. In contrast, NSE-apoE3 mice and apoE3/3 nondemented humans primarily had the 29 kDa fragment and relatively few 14-20 kDa fragments.
Figure 1.

ApoE fragmentation occurs in brains of humans and NSE-apoE mice but not in brains of GFAP-apoE mice. Brain homogenates from a nondemented human control (age 73 years) and a case with AD (age 71 years; A), NSE-apoE mice (age 7-9 months; B), and GFAP-apoE mice (age 8-10 months; C) were analyzed by Western blotting with antibodies against full-length apoE or C-terminal apoE. In C, apoE in brains of GFAP-apoE mice expressing apoE at levels similar to (2 right lanes) or sixfold higher than (3 left lanes) those in NSE-apoE mice was analyzed.
The NSE-apoE4 and NSE-apoE4/3 mice had similar levels of the 14-20 kDa fragments, but NSE-apoE4 mice had lower levels of the 29 kDa fragment, suggesting that the 29 kDa fragment in the NSE-apoE4/3 mice might be primarily generated from apoE3. Our previous study demonstrated that the 29 kDa fragment generated from apoE4 is rapidly cleaved to generate the 14-20 kDa fragments, whereas the 29 kDa fragment generated from apoE3 appears to be resistant to further cleavage (Harris et al., 2003). All the apoE fragments in human and mouse brains were the C-terminal-truncated forms: anti-C-terminal apoE recognizing the last 27 amino acids failed to detect them (Fig. 1A,B). These results suggest that the NSE-apoE mouse represents a good animal model of apoE proteolysis in humans.
ApoE fragmentation does not occur in brains of GFAP-apoE mice
Next, we assessed apoE fragmentation in transgenic mice expressing apoE3 or apoE4 in astrocytes (GFAP-apoE). Surprisingly, no apoE fragmentation was detected in GFAP-apoE3 or GFAP-apoE4 mouse brains no matter whether apoE was expressed at a level similar to or sixfold higher than that in NSE-apoE mice (Fig. 1C). These results suggest that apoE undergoes neuron-specific proteolytic processing to generate the C-terminal-truncated fragments. Furthermore, because neurons take up apoE secreted from astrocytes in GFAP-apoE mice, these results also suggest that the apoE proteolysis occurs in the secretory pathway in neurons but not in the internalization pathway.
Region specificity of apoE fragmentation in NSE-apoE4 mouse brains
To determine whether apoE4 fragmentation occurs predominantly in specific brain regions, homogenates from the neocortex, hippocampus, or cerebellum of NSE-apoE4 mice were subjected to SDS-PAGE and analyzed by anti-apoE Western blotting. As shown in Figure 2, apoE4 fragmentation occurred in the neocortex and hippocampus but not in the cerebellum. These results indicate that the apoE4 fragmentation is brain region-dependent, occurring predominantly in areas vulnerable to AD-related neurodegeneration (Morrison and Hof, 2002).
Figure 2.

Region specificity of apoE fragmentation in NSE-apoE4 mouse brains. Brain tissues from NSE-apoE4 mice (n = 4; age 4-5 months) were dissected into neocortex, hippocampus, and cerebellum and homogenized. ApoE in the homogenates was detected by Western blotting with a polyclonal antibody against full-length apoE. A, Representative Western blot. B, The apoE fragmentation was quantified by measuring the ratio of apoE fragments to full-length apoE. Note that the apoE fragmentation is much more prominent in the neocortex and hippocampus than in the cerebellum (p < 0.001, neocortex or hippocampus vs cerebellum).
Excitotoxic challenge increases apoE fragmentation in NSE-apoE4 but not NSE-apoE3 mice
We have shown previously that kainic acid treatment at a relatively low dose (18 mg/kg of body weight) induces neurodegeneration in the neocortex and hippocampus of NSE-apoE4 but not NSE-apoE3 mice (Buttini et al., 1999, 2000). After such injections of kainic acid, apoE fragmentation increased significantly in brains of NSE-apoE4 mice but not NSE-apoE3 mice (Fig. 3A), whereas the total apoE levels were not altered significantly (Fig. 3B). These results suggest that the kainic acid treatment enhances the neuronal proteolysis of apoE4 but not apoE3 and raise the possibility that C-terminal-truncated apoE4 fragments contribute to the dominant adverse effects of apoE4 identified previously (Buttini et al., 2000).
Figure 3.

ApoE fragmentation in NSE-apoE mice challenged with kainic acid. At 7-8 months of age, NSE-apoE3 and NSE-apoE4 mice were injected intraperitoneally with saline or kainic acid (KA; n = 4 per genotype and treatment). They were analyzed 6 d later. ApoE in brain homogenates (150 μg of total proteins) was subjected to SDS-PAGE and detected by Western blotting with a polyclonal antibody against full-length apoE. A, The apoE fragmentation was quantified by measuring the ratios of all apoE fragments (14-30 kDa) to the full-length apoE (p < 0.01, KA vs saline for NSE-apoE4 mice). B, The total apoE levels were determined by comparing the bands of samples (full-length apoE plus fragments of apoE) with standards of various amounts of recombinant human apoE3 or apoE4 by densitometric analysis.
Age-dependent accumulation of the truncated apoE in brains of NSE-apoE4 mice
Anti-apoE immunoblotting also revealed an age-dependent accumulation of the C-terminal-truncated apoE in the brains of 1- to 9-month-old NSE-apoE4 mice (Fig. 4A). ApoE4 fragmentation increased progressively between the ages of 3 and 9 months, reaching a plateau at 7-9 months, an age at which neuronal and behavioral deficits have been observed in NSE-apoE4 mice (Raber et al., 1998; Buttini et al., 1999). Fewer apoE fragments were found in age-matched NSE-apoE3 mice (Fig. 4A). The ratios of apoE fragments to full-length apoE were significantly higher in NSE-apoE4 mice than in NSE-apoE3 mice at all ages (Fig. 4B). These results suggest that the generation or accumulation of apoE4 fragments in brains of transgenic mice is age-dependent.
Figure 4.

Age-dependent accumulation of C-terminal-truncated fragments of apoE in brains of NSE-apoE4 mice. A, ApoE in brain homogenates of NSE-apoE3 or NSE-apoE4 mice was detected by Western blotting with antibodies against full-length apoE. B, Quantitative analysis of the ratios of the C-terminal-truncated fragments of apoE to the full-length apoE in brain homogenates of NSE-apoE3 and NSE-apoE4 mice (n = 4-6 per genotype and age; p < 0.01, apoE3 vs apoE4 mice at all ages).
Age-dependent accumulation of p-tau in NSE-apoE4 but not GFAP-apoE4 mice
Because the C-terminal-truncated apoE induces the formation of NFT-like inclusions in cultured neuronal cells (Huang et al., 2001), we next examined whether there was an age-dependent accumulation of p-tau in the brains of NSE-apoE4 mice. Brain homogenates of 3- to 9-month-old NSE-apoE4 or NSE-apoE3 mice were analyzed by Western blotting with monoclonal antibody AT8, which recognizes p-tau. P-tau with a molecular mass of 55-60 kDa increased gradually with age in both groups but to a much greater extent in NSE-apoE4 mice (Fig. 5A). In addition, a small amount of SDS-resistant p-tau aggregate, which remained at the top of the gel, was found in 5- to 9-month-old NSE-apoE4 mice but not in age-matched NSE-apoE3 mice (Fig. 5A). At 18 months of age, NSE-apoE4 mice had much higher levels of the SDS-resistant p-tau aggregates than NSE-apoE3 or mice lacking both murine and human apoE (Fig. 5B). Densitometric analysis of Western blots revealed an age-dependent accumulation of SDS-resistant p-tau aggregates in both NSE-apoE3 and NSE-apoE4 mice that was much more prominent in NSE-apoE4 mice, especially in the oldest groups (Fig. 5D). The pattern of tau phosphorylation in brain homogenates of NSE-apoE4 mice, analyzed with monoclonal antibodies that recognize different phosphorylation sites in p-tau in NFTs of AD brains, showed that the increased p-tau was positive for AT8, AT100, and AT270 but negative for AT180, suggesting that tau was hyperphosphorylated at multiple sites in NSE-apoE4 mice. No significant increase in p-tau was found in brains of GFAP-apoE3 or GFAP-apoE4 mice at 8-21 months (Fig. 5C). These data suggest that neuronal accumulation of apoE fragments is associated with increased phosphorylation of tau in vivo, consistent with results obtained in cultured neuronal cells (Huang et al., 2001).
Figure 5.
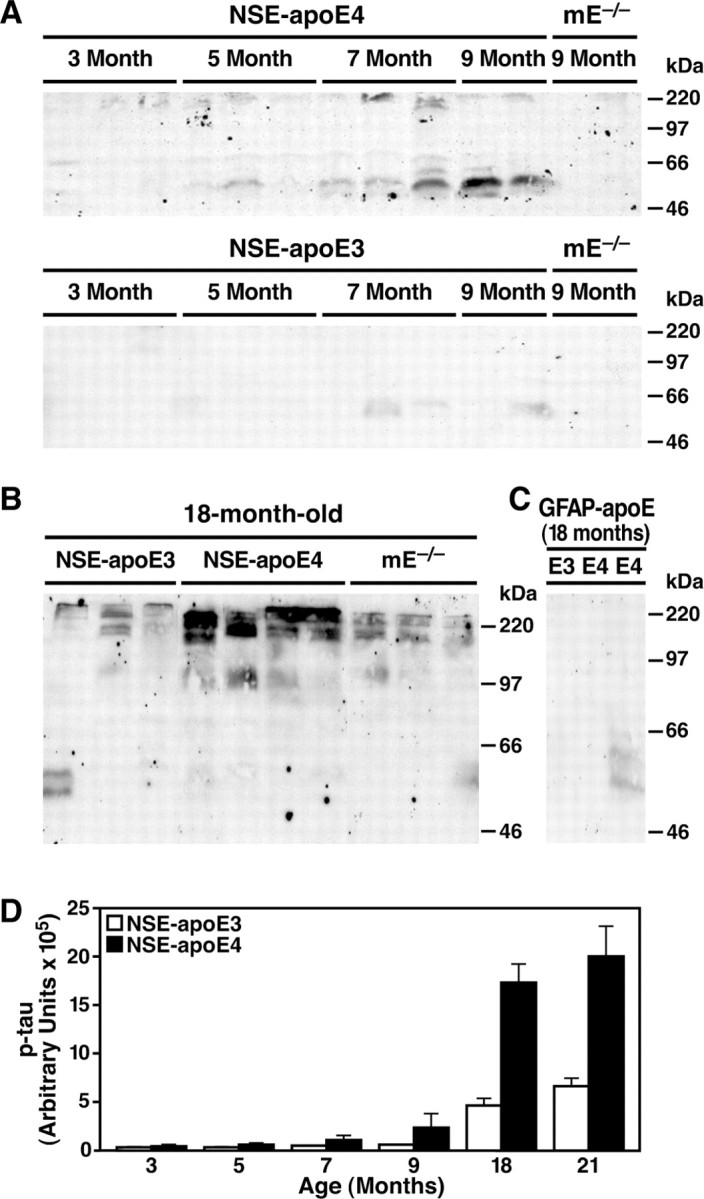
Age-dependent accumulation of p-tau in brains of NSE-apoE4 mice. p-Tau in brain homogenates of NSE-apoE mice (A, B) and GFAP-apoE mice (C) at different ages was analyzed by Western blotting with monoclonal antibody AT8. Note the higher levels of SDS-resistant p-tau aggregates at the top of the gel in aged NSE-apoE4 mice compared with aged NSE-apoE3 mice. D, Quantitative analysis of the SDS-resistant p-tau aggregates in brain homogenates of NSE-apoE3 and NSE-apoE4 mice (n = 4-6 per genotype and age; p < 0.01, apoE3 vs apoE4 mice at 7-21 months). mE, mouse apoE.
Formation of intraneuronal p-tau-containing inclusions in the hippocampus of aged NSE-apoE4 mice
Immunostaining of brain sections with monoclonal antibody AT8, which recognizes p-tau in NFTs, revealed p-tau-positive intraneuronal inclusions in the hippocampal CA3 region (Fig. 6A) and in the hilus of the dentate gyrus (data not shown) in 21-month-old NSE-apoE4 mice. Such p-tau-positive intraneuronal inclusions were detected much less frequently in age-matched NSE-apoE3 mice (Fig. 6B). Aged NSE-apoE4 mice had four times more neurons containing p-tau-positive inclusions than aged NSE-apoE3 mice (Fig. 6C). Double immunostaining revealed that p-tau and apoE were colocalized in the inclusions (Fig. 6D-F). The inclusions also contained phosphorylated neurofilament of high molecular weight as detected by monoclonal antibody SM31 (Fig. 6G-I). Electron microscopy revealed cytosolic straight filaments with diameters of 17 ± 3 nm in CA3 neurons of the hippocampus (Fig. 6J), resembling pre-NFTs in AD brains (Crowther, 1993). The filaments were densely packed and usually closely related to mitochondria (Fig. 6J). No p-tau-positive intraneuronal inclusions were found in the hippocampus of GFAP-apoE3 or GFAP-apoE4 mice (data not shown). These results suggest a neuron-specific association of apoE4 fragments with increased phosphorylation and aggregation of tau.
Figure 6.
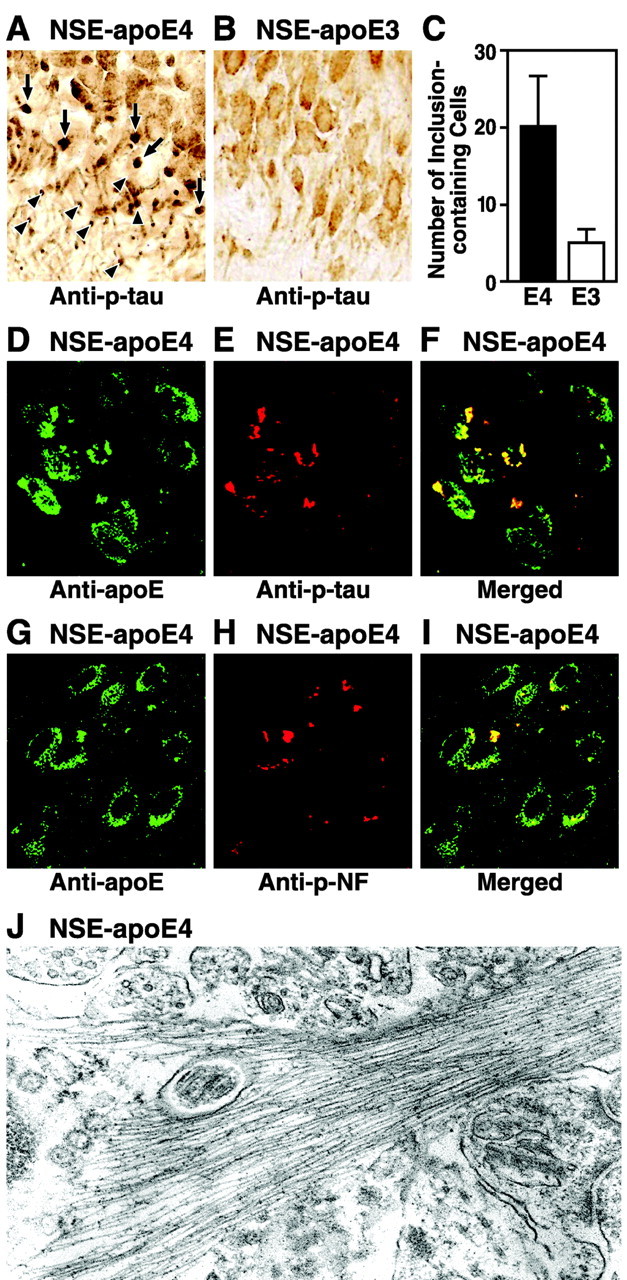
Formation of intraneuronal p-tau-containing inclusions in the hippocampus of aged NSE-apoE4 mice. Brain sections from 21-month-old NSE-apoE4 (A) and NSE-apoE3 (B) mice were immunostained with monoclonal antibody AT8. p-tau-positive intraneuronal inclusions were abundant in the CA3 region of NSE-apoE4 mice (A) but barely detected in the CA3 region of NSE-apoE3 mice (B). C, Quantitative analysis of neurons containing p-tau-positive inclusions in 21-month-old NSE-apoE4 and NSE-apoE3 mice (n = 5 per genotype; p < 0.01, apoE3 vs apoE4 mice). D-F, Colocalization of apoE4 and p-tau in intraneuronal inclusions in the hippocampus of a 21-month-old NSE-apoE4 mouse as determined by anti-apoE and anti-p-tau double immunofluorescent staining. G-I, Colocalization of apoE4 and phosphorylated neurofilament (p-NF) in intraneuronal inclusions in the hippocampus of a 21-month-old NSE-apoE4 mouse as determined by anti-apoE and anti-p-NF double immunofluorescence staining. J, Intraneuronal straight filaments with diameters of 17 ± 3 nm visualized by electron microscopy in the hippocampus of the NSE-apoE4 transgenic mice (magnification, 30,000×).
Discussion
This study demonstrates in vivo that the accumulation of pathogenic C-terminal-truncated apoE fragments in the brain depends on the isoform and cellular source of apoE, as well as on brain region, aging, and neural injury. ApoE proteolysis occurs in neurons but not in astrocytes, with more fragments being generated from apoE4 than from apoE3. Accumulation of apoE fragments increased with age and was exacerbated by kainic acid challenge in NSE-apoE4 but not NSE-apoE3 mice. Accumulation of apoE4 fragments was greater in the cortex and hippocampus, which are vulnerable to AD-related neurodegeneration, than in the cerebellum, which is less vulnerable. Our results also suggest that the neuron-specific production of C-terminal-truncated fragments of apoE4 stimulates tau phosphorylation and contributes to the formation of intraneuronal p-tau-containing filamentous inclusions in the hippocampus, consistent with previous observations in cultured neuronal cells and related transgenic models (Tesseur et al., 2000a,b; Huang et al., 2001; Harris et al., 2003). Interestingly, the intraneuronal p-tau-containing inclusions were found primarily in the hippocampal CA3 region and in the hilus of the dentate gyrus of aged NSE-apoE4 mice, although the apoE fragmentation occurred in both the cortex and the hippocampus. Our preliminary studies suggest that this brain region-specific accumulation of p-tau-containing inclusions might be related to a synergistic effect of apoE fragments and zinc (F. M. Harris and Y. Huang, unpublished observation), which accumulates significantly in the CA3 and the hilus regions of the hippocampus.
ApoE fragments similar to those observed in our study were generated in primary cultured cortical neurons but not in primary cultured astrocytes obtained from transgenic mice expressing apoE4 in both cell types (Lesuisse et al., 2001). Others have shown that the N-terminal 22 kDa thrombin cleavage fragment (amino acids 1-191) of apoE4 is neurotoxic in vitro (Tolar et al., 1997, 1999); however, this toxicity appears to require relatively high concentrations of the fragment and has not yet been confirmed in vivo. In addition, the lipid-binding domain of apoE (amino acids 244-272; Huang and Mahley, 1999; Mahley and Huang, 1999, 2000) appears to be essential for apoE fragments to have neurotoxic effects in vivo, as demonstrated in our previous study (Harris et al., 2003).
It is tempting to speculate that the isoform-dependent and excitotoxin-enhanced accumulation of pathogenic apoE fragments could contribute to the greater susceptibility of APOE ϵ4 carriers to AD (Corder et al., 1993; Saunders et al., 1993; Roses, 1996; Farrer et al., 1997; Tang et al., 1998; Romas et al., 2002). It is possible that the neuronal and behavioral deficits elicited by the expression of full-length apoE4 in neurons of transgenic mice (Raber et al., 1998, 2002; Buttini et al., 1999) are also related to the intraneuronal generation of apoE4 fragments. These hypotheses are supported by our recent observation that overexpression of the C-terminal-truncated apoE4 in neurons is sufficient to cause cortical and hippocampal neurodegeneration in transgenic mice (Harris et al., 2003). The greater susceptibility of apoE4 to proteolysis helps explain the dominant negative effects of apoE4 revealed in NSE-apoE4/3 doubly transgenic mice (Buttini et al., 2000). The dependence of apoE4 fragmentation on the production of apoE in neurons might also explain why NSE-apoE4 mice are susceptible to age-related neurodegeneration (Buttini et al., 1999, 2002), whereas GFAP-apoE4 mice are not (Hartman et al., 2001).
Neuronal expression of apoE, which can be induced in vivo under a variety of conditions (Diedrich et al., 1991; Poirier et al., 1991; Han et al., 1994; Bao et al., 1996; Beffert and Poirier, 1996; Metzger et al., 1996; Xu et al., 1996, 1999a,b; Dupont-Wallois et al., 1997; Boschert et al., 1999; Ferreira et al., 2000; Dekroon and Armati, 2001; Hartman et al., 2001; Aoki et al., 2003; Harris et al., 2004), may participate in neuronal repair and remodeling and may protect neurons from additional injury (Boschert et al., 1999; Huang and Mahley, 1999; Mahley and Huang, 1999, 2000; Harris et al., 2004). However, in neurons expressing apoE4, the proteolytic processing of apoE and fragment generation we have identified (Huang et al., 2001; Harris et al., 2003; this study) may turn this neuroprotective process into a pathogenic process.
Interestingly, apoE3 was also proteolytically cleaved, albeit less effectively than apoE4, especially in brains of NSE-apoE4/3 mice. This finding raises the possibility that apoE fragments could contribute to the development of neuronal deficits also in APOE ϵ3 carriers, although it would likely take longer for adverse effects to become apparent than in APOE ϵ4 carriers, consistent with the differential effects of apoE3 and apoE4 on AD risk and onset (Corder et al., 1993; Farrer et al., 1997). Thus, the putative protease that cleaves apoE at its C terminus may serve as a therapeutic target for prevention and treatment of neurodegenerative diseases in people with diverse apoE genotypes.
Footnotes
This work was supported in part by National Institutes of Health Grants P01 AG022074, R01 AG11385, and R21 NS046465 and by a MetLife Foundation award. We thank Dr. Thomas Innerarity for critical reading of this manuscript, Dr. Eliezer Masliah for analyzing the electron microscopic data, Jennifer Polizzotto and Sylvia Richmond for manuscript preparation, Stephen Ordway and Gary Howard for editorial assistance, John C. W. Carroll and John Hull for graphics, and Stephen Gonzales and Chris Goodfellow for photography.
Correspondence should be addressed to Dr. Yadong Huang, Gladstone Institute of Neurological Disease, P.O. Box 419100, San Francisco, CA 94141-9100. E-mail: yhuang@gladstone.ucsf.edu.
I. Tesseur's and T. Wyss-Coray's present address: Department of Neurology and Neurological Sciences, Stanford University Medical Center, Room A343, Stanford, CA 94305-5235.
M. Buttini's present address: Elan Pharmaceuticals, 800 Gateway Boulevard, South San Francisco, CA 94080.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242527-08$15.00/0
References
- Aoki K, Uchihara T, Sanjo N, Nakamura A, Ikeda K, Tsuchiya K, Wakayama Y (2003) Increased expression of neuronal apolipoprotein E in human brain with cerebral infarction. Stroke 34: 875-880. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM (1999) Apolipoprotein E is essential for amyloid deposition in the APPV717F transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 96: 15233-15238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Arai H, Matsushita S, Higuchi S, Sasaki H (1996) Expression of apolipoprotein E in normal and diverse neurodegenerative disease brain. NeuroReport 7: 1733-1739. [DOI] [PubMed] [Google Scholar]
- Beffert U, Poirier J (1996) Apolipoprotein E, plaques, tangles and cholinergic dysfunction in Alzheimer's disease. Ann NY Acad Sci 777: 166-174. [DOI] [PubMed] [Google Scholar]
- Boschert U, Merlo-Pich E, Higgins G, Roses AD, Catsicas S (1999) Apolipoprotein E expression by neurons surviving excitotoxic stress. Neurobiol Dis 6: 508-514. [DOI] [PubMed] [Google Scholar]
- Boyles JK, Pitas RE, Wilson E, Mahley RW, Taylor JM (1985) Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J Clin Invest 76: 1501-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Orth M, Bellosta S, Akeefe H, Pitas RE, Wyss-Coray T, Mucke L, Mahley RW (1999) Expression of human apolipoprotein E3 or E4 in the brains of Apoe-/- mice: isoform-specific effects on neurodegeneration. J Neurosci 19: 4867-4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini M, Akeefe H, Lin C, Mahley RW, Pitas RE, Wyss-Coray T, Mucke L (2000) Dominant negative effects of apolipoprotein E4 revealed in transgenic models of neurodegenerative disease. Neuroscience 97: 207-210. [DOI] [PubMed] [Google Scholar]
- Buttini M, Yu G-Q, Shockley K, Huang Y, Jones B, Masliah E, Mallory M, Yeo T, Longo FM, Mucke L (2002) Modulation of Alzheimer-like synaptic and cholinergic deficits in transgenic mice by human apolipoprotein E depends on isoform, aging, and overexpression of amyloid β peptides but not on plaque formation. J Neurosci 22: 10539-10548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Pieroni C, Nixon RA (1997) Increased neuronal endocytosis and protease delivery to early endosomes in sporadic Alzheimer's disease: neuropathologic evidence for a mechanism of increased β-amyloidogenesis. J Neurosci 17: 6142-6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA (2000) Endocytic pathway abnormalities precede amyloid β deposition in sporadic Alzheimer's disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. Am J Pathol 157: 277-286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 261: 921-923. [DOI] [PubMed] [Google Scholar]
- Crowther RA (1993) Tau protein and paired helical filaments of Alzheimer's disease. Curr Opin Struct Biol 3: 202-206. [Google Scholar]
- Dekroon RM, Armati PJ (2001) Synthesis and processing of apolipoprotein E in human brain cultures. Glia 33: 298-305. [DOI] [PubMed] [Google Scholar]
- Diedrich JF, Minnigan H, Carp RI, Whitaker JN, Race R, Frey II W, Haase AT (1991) Neuropathological changes in scrapie and Alzheimer's disease are associated with increased expression of apolipoprotein E and cathepsin D in astrocytes. J Virol 65: 4759-4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont-Wallois L, Soulié C, Sergeant N, Wavrant-de Wrieze N, Chartier-Harlin M-C, Delacourte A, Caillet-Boudin M-L (1997) ApoE synthesis in human neuroblastoma cells. Neurobiol Dis 4: 356-364. [DOI] [PubMed] [Google Scholar]
- Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, Van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease: a meta-analysis. JAMA 278: 1349-1356. [PubMed] [Google Scholar]
- Ferreira S, Dupire M-J, Delacourte A, Najib J, Caillet-Boudin M-L (2000) Synthesis and regulation of apolipoprotein E during the differentiation of human neuronal precursor NT2/D1 cells into postmitotic neurons. Exp Neurol 166: 415-421. [DOI] [PubMed] [Google Scholar]
- Grbovic OM, Mathews PM, Jiang Y, Schmidt SD, Dinakar R, Summers-Terio NB, Ceresa BP, Nixon RA, Cataldo AM (2003) Rab5-stimulated up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J Biol Chem 278: 31261-31268. [DOI] [PubMed] [Google Scholar]
- Han S-H, Einstein G, Weisgraber KH, Strittmatter WJ, Saunders AM, Pericak-Vance M, Roses AD, Schmechel DE (1994) Apolipoprotein E is localized to the cytoplasm of human cortical neurons: a light and electron microscopic study. J Neuropathol Exp Neurol 53: 535-544. [DOI] [PubMed] [Google Scholar]
- Harris FM, Brecht WJ, Xu Q, Tesseur I, Kekonius L, Wyss-Coray T, Fish JD, Masliah E, Hopkins PC, Scearce-Levie K, Weisgraber KH, Mucke L, Mahley RW, Huang Y (2003) Carboxyl-terminal-truncated apolipoprotein E4 causes Alzheimer's disease-like neurodegeneration and behavioral deficits in transgenic mice. Proc Natl Acad Sci USA 100: 10966-10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris FM, Tesseur I, Brecht WJ, Xu Q, Mullendorff K, Chang S, Wyss-Coray T, Mahley RW, Huang Y (2004) Astroglial regulation of apolipoprotein E expression in neuronal cells: implications for Alzheimer's disease. J Biol Chem 279: 3862-3868. [DOI] [PubMed] [Google Scholar]
- Hartman RE, Wozniak DF, Nardi A, Olney JW, Sartorius L, Holtzman DM (2001) Behavioral phenotyping of GFAP-apoE3 and -apoE4 transgenic mice: apoE4 mice show profound working memory impairments in the absence of Alzheimer's-like neuropathology. Exp Neurol 170: 326-344. [DOI] [PubMed] [Google Scholar]
- Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60: 1119-1122. [DOI] [PubMed] [Google Scholar]
- Herz J, Beffert U (2000) Apolipoprotein E receptors: linking brain development and Alzheimer's disease. Nat Rev Neurosci 1: 51-58. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D, Paul SM (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 97: 2892-2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mahley RW (1999). Apolipoprotein E and human disease. In: Plasma lipids and their role in disease (Barter PJ, Rye K-A, eds), pp 257-284. Amsterdam: Harwood.
- Huang Y, Liu XQ, Rall Jr SC, Taylor JM, von Eckardstein A, Assmann G, Mahley RW (1998) Overexpression and accumulation of apolipoprotein E as a cause of hypertriglyceridemia. J Biol Chem 273: 26388-26393. [DOI] [PubMed] [Google Scholar]
- Huang Y, Liu XQ, Wyss-Coray T, Brecht WJ, Sanan DA, Mahley RW (2001) Apolipoprotein E fragments present in Alzheimer's disease brains induce neurofibrillary tangle-like intracellular inclusions in neurons. Proc Natl Acad Sci USA 98: 8838-8843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, Cheung BS, Rebeck GW, Paul SM, Bales KR, Hyman BT (2000) Apolipoprotein E affects the amount, form, and anatomical distribution of amyloid β-peptide deposition in homozygous APPV717F transgenic mice. Acta Neuropathol (Berl) 100: 451-458. [DOI] [PubMed] [Google Scholar]
- Ji Z-S, Miranda RD, Newhouse YM, Weisgraber KH, Huang Y, Mahley RW (2002) Apolipoprotein E4 potentiates amyloid β peptide-induced lysosomal leakage and apoptosis in neuronal cells. J Biol Chem 277: 21821-21828. [DOI] [PubMed] [Google Scholar]
- LaDu MJ, Falduto MT, Manelli AM, Reardon CA, Getz GS, Frail DE (1994) Isoform-specific binding of apolipoprotein E to β-amyloid. J Biol Chem 269: 23403-23406. [PubMed] [Google Scholar]
- Lesuisse C, Xu G, Anderson J, Wong M, Jankowsky J, Holtz G, Gonzalez V, Wong PCY, Price DL, Tang F, Wagner S, Borchelt DR (2001) Hyper-expression of human apolipoprotein E4 in astroglia and neurons does not enhance amyloid deposition in transgenic mice. Hum Mol Genet 10: 2525-2537. [DOI] [PubMed] [Google Scholar]
- Ljungberg MC, Dayanandan R, Asuni A, Rupniak TH, Anderton BH, Lovestone S (2002) Truncated apoE forms tangle-like structures in a neuronal cell line. NeuroReport 13: 867-870. [DOI] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer Jr HB, Das S, Potter H (1994) Amyloid-associated proteins α1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer β-protein into filaments. Nature 372: 92-94. [DOI] [PubMed] [Google Scholar]
- Mahley R, Huang Y (1999) Apolipoprotein E: from atherosclerosis to Alzheimer's disease and beyond. Curr Opin Lipidol 10: 207-217. [DOI] [PubMed] [Google Scholar]
- Mahley R, Huang Y (2000). Apolipoprotein E: structure and function in lipid metabolism and neurobiology. In: The molecular and genetic basis of neurologic and psychiatric disease, Ed 3 (Rosenberg RN, Prusiner SB, DiMauro S, Barchi RL, Nestler EJ, eds), pp 565-573. Philadelphia: Butterworth Heinemann.
- Masliah E, Westland CE, Rockenstein EM, Abraham CR, Mallory M, Veinberg I, Sheldon E, Mucke L (1997) Amyloid precursor proteins protect neurons of transgenic mice against acute and chronic excitotoxic injuries in vivo. Neuroscience 78: 135-146. [DOI] [PubMed] [Google Scholar]
- Metzger RE, LaDu MJ, Pan JB, Getz GS, Frail DE, Falduto MT (1996) Neurons of the human frontal cortex display apolipoprotein E immunoreactivity: implications for Alzheimer's disease. J Neuropathol Exp Neurol 55: 372-380. [DOI] [PubMed] [Google Scholar]
- Miyata M, Smith JD (1996) Apolipoprotein E allele-specific antioxidant activity and effects on cytotoxicity by oxidative insults and β-amyloid peptides. Nat Genet 14: 55-61. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Hof PR (2002) Selective vulnerability of corticocortical and hippocampal circuits in aging and Alzheimer's disease. Prog Brain Res 136: 467-486. [DOI] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K (1991) Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res 541: 163-166. [DOI] [PubMed] [Google Scholar]
- Nathan BP, Bellosta S, Sanan DA, Weisgraber KH, Mahley RW, Pitas RE (1994) Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 264: 850-852. [DOI] [PubMed] [Google Scholar]
- Nathan BP, Chang K-C, Bellosta S, Brisch E, Ge N, Mahley RW, Pitas RE (1995) The inhibitory effect of apolipoprotein E4 on neurite outgrowth is associated with microtubule depolymerization. J Biol Chem 270: 19791-19799. [DOI] [PubMed] [Google Scholar]
- Poirier J, Hess M, May PC, Finch CE (1991) Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Mol Brain Res 11: 97-106. [DOI] [PubMed] [Google Scholar]
- Raber J, Wong D, Buttini M, Orth M, Bellosta S, Pitas RE, Mahley RW, Mucke L (1998) Isoform-specific effects of human apolipoprotein E on brain function revealed in ApoE knockout mice: increased susceptibility of females. Proc Natl Acad Sci USA 95: 10914-10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raber J, Wong D, Yu G-Q, Buttini M, Mahley RW, Pitas RE, Mucke L (2000) Apolipoprotein E and cognitive performance. Nature 404: 352-354. [DOI] [PubMed] [Google Scholar]
- Raber J, Bongers G, LeFevour A, Buttini M, Mucke L (2002) Androgens protect against apolipoprotein E4-induced cognitive deficits. J Neurosci 22: 5204-5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romas SN, Santana V, Williamson J, Ciappa A, Lee JH, Rondon HZ, Estevez P, Lantigua R, Medrano M, Torres M, Stern Y, Tycko B, Mayeux R (2002) Familial Alzheimer disease among Caribbean Hispanics: a reexamination of its association with APOE. Arch Neurol 59: 87-91. [DOI] [PubMed] [Google Scholar]
- Roses AD (1994) The Alzheimer diseases. Curr Neurol 14: 111-141. [Google Scholar]
- Roses AD (1996) Apolipoprotein E alleles as risk factors in Alzheimer's disease. Annu Rev Med 47: 387-400. [DOI] [PubMed] [Google Scholar]
- Saunders AM, Strittmatter WJ, Schmechel D, St George-Hyslop PH, Pericak-Vance MA, Joo SH, Rosi BL, Gusella JF, Crapper-MacLachlan DR, Alberts MJ, Hulette C, Crain B, Goldgaber D, Roses AD (1993) Association of apolipoprotein E allele e4 with late-onset familial and sporadic Alzheimer's disease. Neurology 43: 1467-1472. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O (1997) Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA 94: 4103-4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1991) The molecular pathology of Alzheimer's disease. Neuron 6: 487-498. [DOI] [PubMed] [Google Scholar]
- Spinler SA, Cziraky MJ (1994) Lipoprotein(a): physiologic function, association with atherosclerosis, and effects of lipid-lowering drug therapy. Ann Pharmacother 28: 343-351. [DOI] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD (1993a) Apolipoprotein E: high-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA 90: 1977-1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong L-M, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993b) Binding of human apolipoprotein E to synthetic amyloid β peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA 90: 8098-8102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Goedert M, Weisgraber KH, Dong L-M, Jakes R, Huang DY, Pericak-Vance M, Schmechel D, Roses AD (1994) Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: implications for Alzheimer disease. Proc Natl Acad Sci USA 91: 11183-11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M-X, Stern Y, Marder K, Bell K, Gurland B, Lantigua R, Andrews H, Feng L, Tycko B, Mayeux R (1998) The APOE-e4 allele and the risk of Alzheimer disease among African Americans, whites, and Hispanics. JAMA 279: 751-755. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L (2001) New frontiers in Alzheimer's disease genetics. Neuron 32: 181-184. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Spittaels K, Van den Haute C, Moechars D, Van Leuven F (2000a) Expression of human apolipoprotein E4 in neurons causes hyperphosphorylation of protein tau in the brains of transgenic mice. Am J Pathol 156: 951-964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Van Dorpe J, Bruynseels K, Bronfman F, Sciot R, Van Lommel A, Van Leuven F (2000b) Prominent axonopathy and disruption of axonal transport in transgenic mice expressing human apolipoprotein E4 in neurons of brain and spinal cord. Am J Pathol 157: 1495-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar M, Marques MA, Harmony JAK, Crutcher KA (1997) Neurotoxicity of the 22 kDa thrombin-cleavage fragment of apolipoprotein E and related synthetic peptides is receptor-mediated. J Neurosci 17: 5678-5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolar M, Keller JN, Chan S, Mattson MP, Marques MA, Crutcher KA (1999) Truncated apolipoprotein E (apoE) causes increased intracellular calcium and may mediate apoE neurotoxicity. J Neurosci 19: 7100-7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Frangione B (1992) Apolipoprotein E: a pathological chaperone protein in patients with cerebral and systemic amyloid. Neurosci Lett 135: 235-238. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Castaño EM, Golabek A, Vogel T, Frangione B (1994) Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol 145: 1030-1035. [PMC free article] [PubMed] [Google Scholar]
- Xu P-T, Schmechel D, Rothrock-Christian T, Burkhart DS, Qiu H-L, Popko B, Sullivan P, Maeda N, Saunders AM, Roses AD, Gilbert JR (1996) Human apolipoprotein E2, E3, and E4 isoform-specific transgenic mice: human-like pattern of glial and neuronal immunoreactivity in central nervous system not observed in wild-type mice. Neurobiol Dis 3: 229-245. [DOI] [PubMed] [Google Scholar]
- Xu P-T, Gilbert JR, Qiu H-L, Ervin J, Rothrock-Christian TR, Hulette C, Schmechel DE (1999a) Specific regional transcription of apolipoprotein E in human brain neurons. Am J Pathol 154: 601-611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P-T, Schmechel D, Qiu H-L, Herbstreith M, Rothrock-Christian T, Eyster M, Roses AD, Gilbert JR (1999b) Sialylated human apolipoprotein E (apoEs) is preferentially associated with neuron-enriched cultures from APOE transgenic mice. Neurobiol Dis 6: 63-75. [DOI] [PubMed] [Google Scholar]