

Abstract
The hippocampus, being sensitive to stress and glucocorticoids, plays significant roles in certain types of learning and memory. Therefore, the hippocampus is probably involved in the increasing drug use, drug seeking, and relapse caused by stress. We have studied the effect of stress with morphine on synaptic plasticity in the CA1 region of the hippocampus in vivo and on a delayed-escape paradigm of the Morris water maze. Our results reveal that acute stress enables long-term depression (LTD) induction by low-frequency stimulation (LFS) but acute morphine causes synaptic potentiation. Remarkably, exposure to an acute stressor reverses the effect of morphine from synaptic potentiation (∼20%) to synaptic depression (∼40%), precluding further LTD induction by LFS. The synaptic depression caused by stress with morphine is blocked either by the glucocorticoid receptor antagonist RU38486 or by the NMDA-receptor antagonist d-APV. Chronic morphine attenuates the ability of acute morphine to cause synaptic potentiation, and stress to enable LTD induction, but not the ability of stress in tandem with morphine to cause synaptic depression. Furthermore, corticosterone with morphine during the initial phase of drug use promotes later delayed-escape behavior, as indicated by the morphine-reinforced longer latencies to escape, leading to persistent morphine-seeking after withdrawal. These results suggest that hippocampal synaptic plasticity may play a significant role in the effects of stress or glucocorticoids on opiate addiction.
Keywords: hippocampus, long-term depression, associative learning, stress, morphine-seeking, opiate addiction
Introduction
The molecular and cellular changes that occur with the transition from drug taking to compulsive use are understood only partially. Addictive drugs likely engage learning mechanisms in brain regions including the hippocampus (White, 1996; Koob et al., 1998; Robbins and Everitt, 1999; Berke and Hyman, 2000; Hyman and Malenka, 2001; Nestler, 2001a). Drug-associated memories may be encoded in the hippocampus (Holden, 2001; Nestler, 2001b), and thus low-frequency electric stimulation in the hippocampus may read out the memories and induce relapse (Vorel et al., 2001). On the other hand, evidence has revealed that stress or glucocorticoids play a significant role in determining the propensity of an individual to drug abuse, and in increasing drug seeking and relapse (Erb et al., 1996; Piazza and Le Moal, 1996; Shaham et al., 2000; Sinha, 2001; Deroche-Gamonet et al., 2003). Drug seeking and relapse also can be triggered by cues or drugs, but the underlying mechanisms may differ from stress (Sutton et al., 2003; Stewart, 2003). Stress may interact with drug addiction through a common mechanism of synaptic plasticity in the ventral tegmental area (VTA) (Saal et al., 2003; Kauer, 2003). However, the role of the hippocampus in the interaction between stress and drug addiction has been studied only rarely.
Activity-dependent hippocampal synaptic plasticity, e.g., long-term potentiation (LTP), is believed to underlie certain types of learning and memory (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Martin et al., 2000). Previous studies have shown that stress or glucocorticoids block or facilitate memory and affect LTP and long-term depression (LTD) in the hippocampus (Shors et al., 1989; McEwen, 1994; Diamond et al., 1996; Kim et al., 1996; Xu et al., 1997, 1998a; de Quervain et al., 1998; Conrad et al., 1999; Mizoguchi et al., 2000). Similarly, opiates affect cognitive function (Beatty, 1983; Classen and Mondadori, 1984; Guerra et al., 1987; Li et al., 2001) facilitate (Mansouri et al., 1999) or inhibit (Terman et al., 1994; Pu et al., 2002) LTP, and facilitate LTD (Wagner et al., 2001) in the hippocampus. Furthermore, stress modifies morphine responses in behavior (Olley et al., 1990; Deroche et al., 1992), and morphine influences the effect of stress on synaptic plasticity and behavior (Fratta et al., 1977; Shors et al., 1990; Scheggi et al., 2000).
The hypothalamo-pituitary-adrenocortical axis is the common pathway activated by addictive drugs and stress. Morphine induces stress levels of corticosterone (Buckingham and Cooper, 1984; Pirnik et al., 2001), but chronic morphine use leads to a complete tolerance of this effect (Buckingham and Cooper, 1984; Pechnick, 1999). Here, stress or glucocorticoids may have made the difference between drug taking and drug addiction because evidence has demonstrated that the glucocorticoid receptor is a major substrate for drug abuse (De Vries et al., 1996; Deroche-Gamonet et al., 2003). Thus, we have hypothesized that stress or glucocorticoids during the initial phase of opiate use may lead to some adaptations of the hippocampal synaptic plasticity related to the effects of stress or glucocorticoids on opiate addiction.
Materials and Methods
Animals
Experiments were performed on male Sprague Dawley rats (inbred strain, Animal House Center, Kunming General Hospital, Kunming, or BioResources Unit, Institute of Psychology, Beijing), weighing 200-250 gm. Animals were group-housed, with ad libitum access to water and food in the established animal houses, with a 12 hr light/dark cycle and a thermoregulated environment. The animal care and experimental protocol was approved by The Chinese Academy of Sciences.
Chronic morphine in electrophysiological studies
Animals were chronically treated with morphine (10 mg/kg, s.c.) twice per day at 12 hr intervals for 12 d as described previously (Trujillo and Akil, 1991; Pu et al., 2002). A lower dose of morphine (2 mg · kg-1 · d-1, s.c.) was chronically administered to two groups of animals using the same regimen.
Electrophysiological studies
Recordings of the field EPSPs were made from the CA1 stratum radiatum of the hippocampus in response to ipsilateral stimulation of the Schaffer collateral/commissural pathway using techniques similar to those described previously (Xu et al., 1998a; Wei et al., 2002; Xiong et al., 2003). Experiments were performed under pentobarbital sodium (50-60 mg/kg, i.p.) anesthesia, and the core temperature was maintained at 37 ± 0.5°C. Animals were ventilated with 95% O2 and 5% CO2 to avoid the possible depressive effect of morphine on respiration. Recording and stimulating electrodes were made by gluing together a pair of twisted Teflon-coated 90% platinum and 10% iridium wires (50 μm inner diameter, 75 μm outer diameter; World Precision Instruments, Sarasota, FL). Test EPSPs were evoked at a frequency of 0.033 Hz and at a stimulation intensity adjusted to give an EPSP amplitude of 50% maximum response. The low-frequency stimulation protocol for inducing LTD consists of 900 pulses at 3 Hz. The LTD was measured as the mean percentage ± SEM of the baseline EPSP amplitude recorded over at least a 40 min baseline period.
Stress and nonstress protocol in electrophysiological studies.
Behavioral stress was evoked by elevated platform stress as described previously (Xu et al., 1997, 1998a; Xiong et al., 2003; Rocher et al., 2004). The animals were anesthetized immediately after the stress procedure. Nonstressed animals were carefully taken out of their home cage and anesthetized immediately.
Behavior studies: apparatus
The Morris water maze consists of a circular pool (200 cm diameter, 75 cm depth) filled with milk-diluted water at 21.0 ± 0.5°C. An automatic tracking system was used to record latencies and swimming distances for offline analysis.
Training procedures
Stage 1: addictive training (days 1-5). Animals rapidly adapt to the elevated platform stress, used in the present electrophysiological studies, in a few days (Xiong et al., 2003). Corticosterone (5 mg · kg-1 · d-1 or 1.66 mg · kg-1 · trial-1, i.p.), the principal glucocorticoid hormone in the rat, was used in behavioral studies. Low doses of morphine (2 mg · kg-1 · d-1 or 0.66 mg · kg-1 · trial-1, i.p.) were used to avoid the impairment of motor activity and cognitive function. A low dose of morphine is also crucial in determining the sensitivity of the delayed-escape paradigm in the Morris water maze. Fifty-five rats were divided into four treatment groups (supplemental Table 1, available at) during this stage: saline (Sal), corticosterone (Cor), morphine (Mor), corticosterone with morphine (C-M; morphine immediately after corticosterone).
Addictive training was conducted in the absence of a hidden platform. Animals were allowed free swimming in the water maze for three trials with 2 hr intertrial intervals per day. Each trial lasted 120 sec on the first day and 60 sec on the next 4 d. Each animal was administered a treatment immediately after each trial. Addictive training allows an animal to adapt to the water maze rapidly. Animals showed mild stress (defecation) on the first trial of the whole training. To minimize behavioral stress during training, each animal was handled for 3 d before training, each training trial was reduced to 60 sec, and each animal was dried with a towel after each trial.
Stage 2: reinforced or nonreinforced training (days 6-10). The four groups were then divided into 10 subgroups (supplemental Table 1, available at) and trained to find the hidden platform (7.5 × 7.5 cm, 1.5 cm below the surface of the water) in 60 sec in three trials with 2 hr intertrial intervals per day for 5 d. An animal was given a reward in the reinforced training if it did not climb onto the hidden platform in 60 sec; an animal was treated with saline if it climbed onto the hidden platform in 60 sec. For comparison, an animal was randomly given a reward or saline in the nonreinforced training regardless of whether it climbed onto the hidden platform in 60 sec.
The reward was either morphine (Mor) or corticosterone with morphine (C-M, morphine immediately after corticosterone). Total morphine exposure was normalized between groups by supplemental injections in the evening (20:00) each day. Total corticosterone was also normalized between treated groups using the same procedure each day.
A low dose of morphine effectively rewarded the escape with a delay in the reinforced training. That is, an animal was able to escape but performed a delayed escape (delayed-escape behavior). The nonreinforced group received the same treatment as the reinforced group, providing comparable information about motor activity and cognitive function for its counterpart, the reinforced group.
Stage 3: extinction test procedures (days 15, 20, and 29). Extinction of the learned delayed-escape was tested by escape latencies to the hidden platform, three trials with 2 hr intertrial intervals. Each trial lasted 120 sec, and all animals escaped to the hidden platform during this period. Morphine-seeking is the delayed-escape behavior without reward, as indicated by longer latencies to escape. The first extinction test was conducted on day 15 after withdrawal for 5 d. The second and third extinction tests were conducted on days 20 and 29 after withdrawal for 10 and 19 d, respectively. Each animal was given a priming injection of morphine (0.01 mg/kg, i.p.) 10 min before the last extinction test on day 29.
Data analysis
Statistical comparisons in electrophysiological and behavioral studies were made by using the t test or the least significance difference test of one-way ANOVA (SPSS 10.0; SPSS, Chicago, IL). The significance level was set at p < 0.05.
Results
Effect of stress or acute morphine on hippocampal synaptic efficacy
The first set of experiments determined the effect of stress or acute morphine on the field EPSP in the CA1 region of the hippocampus of anesthetized rats. As expected, low-frequency stimulation (LFS) failed to induce LTD in nonstressed animals (Fig. 1a, white circles) (n = 4; 102.5 ± 0.7% of baseline 60 min after LFS; p > 0.05 compared with baseline). However, stressing rats by placing them on an elevated platform for 30 min immediately before anesthesia enabled LFS to induce LTD (Fig. 1a, black circles) (n = 5; 85.5 ± 1.0% of baseline 60 min after LFS; p < 0.05 compared with baseline), in agreement with previous reports in vivo (Xu et al., 1997, 1998a; Xiong et al., 2003) and in vitro (Kim et al., 1996). The stress-enabled LTD induction was blocked by the glucocorticoid receptor antagonist RU38486 (20 mg/kg, s.c., immediately after stress; n = 4; 100.3 ± 1.5% of baseline 40 min after LFS; p > 0.05 compared with baseline) (Fig. 1b), in agreement with a previous report (Xu et al., 1998a). On the other hand, a single exposure to morphine of nonstressed animals (Fig. 1c, arrow) (3 mg/kg, i.p.) induced a synaptic potentiation of the field EPSP amplitude (Fig. 1c, white circles) (n = 5; 119.3 ± 1.2% of baseline 60 min after morphine injection; p < 0.05 compared with baseline or saline), significantly different from saline control (Fig. 1c, white diamonds) (n = 4; 99.5 ± 1.4% of baseline 60 min after saline injection; p > 0.05 compared with baseline; p < 0.05 compared with the potentiation after morphine injection). Moreover, the morphine-induced synaptic potentiation was maintained for at least 400 min and then tended to decline (n = 5; 122.4 ± 1.0% of baseline 400 min after morphine injection and 105.8 ± 2.4% of baseline 560 min after morphine injection; both p < 0.05 compared with baseline) (Fig. 1d). The result is consistent with the synaptic plasticity changes described in the VTA by a single exposure of cocaine (Ungless et al., 2001; Saal et al., 2003). Low-frequency stimulation 30 min after morphine injection was able to induce LTD in nonstressed animals (n = 5; 88.9 ± 5.3% of baseline 60 min after LFS; p < 0.05 compared with baseline) (Fig. 1e), in agreement with the finding that morphine facilitated LTD induction by LFS (Wagner et al., 2001).
Figure 1.

Effect of stress or acute morphine on hippocampal synaptic efficacy. a, LFS (3 Hz; bar) failed to induce LTD in nonstressed animals (white circles; n = 4; 102.5 ± 0.7% of baseline 60 min after LFS; p > 0.05 compared with baseline). However, stress before anesthesia enabled LTD induction by LFS (3 Hz, bar; black circles; n = 5; 85.5 ± 1.0% of baseline 60 min after LFS; p < 0.05 compared with baseline). b, The stress-enabled LTD was blocked by the glucocorticoid antagonist RU38486 (20 mg/kg, s.c., immediately after behavioral stress; n = 4; 100.3 ± 1.5% of baseline 40 min after LFS; p > 0.05 compared with baseline). c, A single exposure to morphine (arrow; 3 mg/kg, i.p.) of nonstressed animals elicited synaptic potentiation (white circles; n = 5; 119.3 ± 1.2% of baseline 60 min after morphine injection; p < 0.05 compared with baseline). A saline control (arrow) did not affect the field EPSP amplitude (white diamonds; n = 4; 99.5 ± 1.4% of baseline 60 min after saline injection; p > 0.05 compared with baseline). d, The morphine-induced potentiation was maintained for at least 400 min and then tended to decline (n = 5; 122.4 ± 1.0% of baseline 400 min after morphine injection and 105.8 ± 2.4% of baseline 560 min after morphine injection; both p < 0.05 compared with baseline). e, LFS (3 Hz; bar), 30 min after morphine exposure (arrow; 3 mg/kg, i.p), enabled LTD induction (n = 5; 88.9 ± 5.3% of baseline 60 min after LFS; p < 0.05 compared with baseline).
Stress in tandem with acute morphine on hippocampal synaptic efficacy
Because the interaction between stress and drug addiction is evident, we have studied the effect of stress with morphine on hippocampal synaptic efficacy. A single exposure to morphine (Fig. 2a, arrow) (3 mg/kg, i.p.) of stressed animals induced a remarkable depression of the field EPSP amplitude (n = 5; 64.0 ± 2.5% of baseline 90 min after morphine exposure; p < 0.05 compare with baseline). The synaptic depression precluded further LTD induction by LFS (n = 5; 74.8 ± 1.0% of baseline 60 min after LFS and 90 min after morphine exposure; p < 0.05 compared with baseline; p > 0.05 compared with Fig. 2a,b). Although this synaptic depression is large (∼40%) compared with numerous reported LTDs in vivo, it was blocked by the glucocorticoid receptor antagonist RU38486 (20 mg/kg, s.c., immediately after stress; n = 7; 98.7 ± 0.6% of baseline 90 min after morphine exposure; p > 0.05 compared with baseline) (Fig. 2c), and by the NMDA-receptor antagonist d-APV (bar; 6 μl; 120 nm, i.c.v.) given 6-8 min before morphine injection (n = 5; 100.8 ± 0.6% of baseline 90 min after morphine exposure; p > 0.05 compared with baseline) (Fig. 2d). These results suggested that the stress-activated glucocorticoid receptor was interacting with the effect of acute morphine and then elicited a novel form of the NMDA-receptor-dependent synaptic plasticity.
Figure 2.
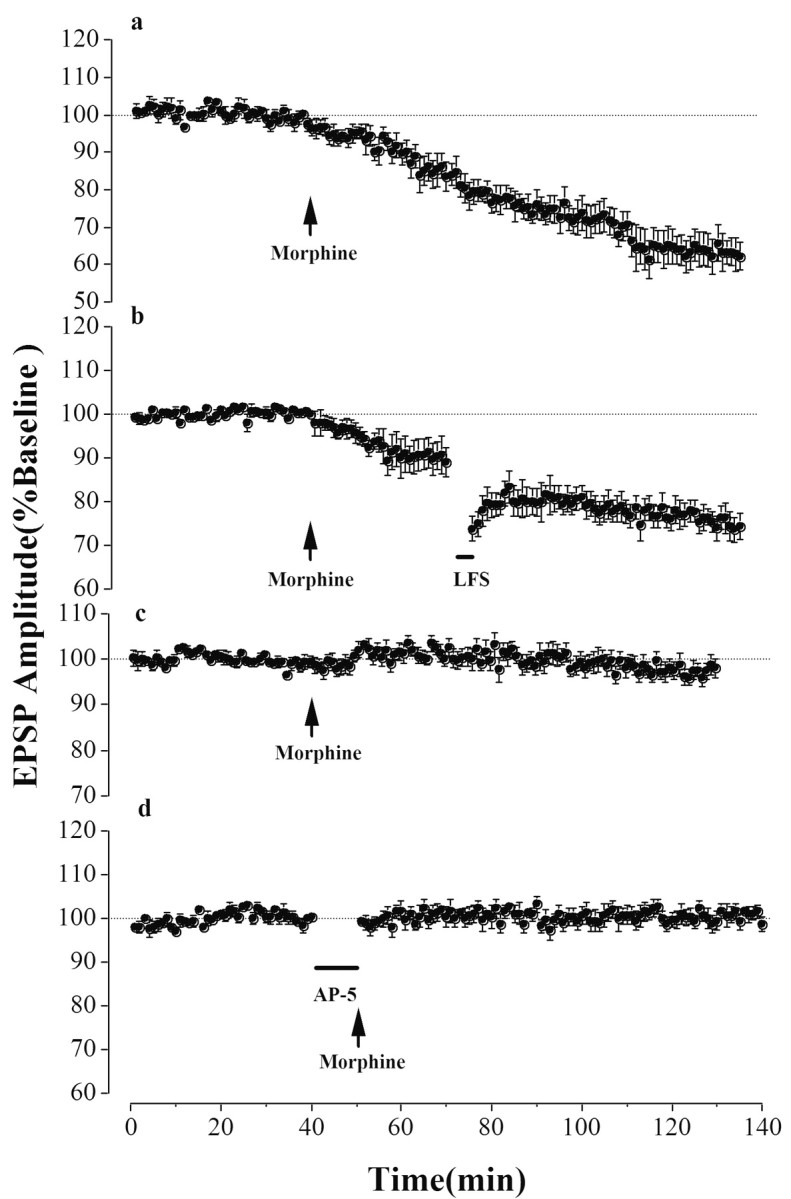
Stress in tandem with acute morphine on hippocampal synaptic efficacy. a, A single exposure of morphine (arrow; 3 mg/kg, i.p.) to stressed animals elicited a remarkable synaptic depression (n = 5; 64.0 ± 2.5% of baseline 90 min after morphine exposure; p < 0.05 compared with baseline). b, The synaptic depression precluded further LTD induction by LFS (3Hz; bar) 30 min after morphine exposure (arrow; 3 mg/kg, i.p.; n = 5; 74.8 ± 1.0% of baseline 60 min after LFS and 90 min after morphine exposure; p < 0.05 compared with baseline; p > 0.05 compared with a). c, The synaptic depression was blocked by the glucocorticoid receptor antagonist RU38486 (20 mg/kg, s.c., immediately after stress; n = 7; 98.7 ± 0.6% of baseline 90 min after morphine exposure; p > 0.05 compared with baseline). d, The synaptic depression was also blocked by the NMDA-receptor antagonist d-APV (6 μl; 120 nm, i.c.v.; bar) given 6-8 min before morphine exposure (arrow; 3 mg/kg, i.p.; n = 5; 100.8 ± 0.6% of baseline 90 min after morphine exposure; p > 0.05 compared with baseline).
Stress in tandem with morphine re-exposure on hippocampal synaptic efficacy after chronic morphine
The results that exposure to an acute stressor reversed the effect of acute morphine from synaptic potentiation to synaptic depression led us to examine the effect of stress in tandem with morphine re-exposure after chronic morphine treatment. Animals were chronically treated with morphine (10 mg/kg, s.c.) twice per day for 12 d, a procedure known to produce significant tolerance to and dependence on the drug (Trujillo and Akil, 1991; Pu et al., 2002). Two groups of animals were chronically treated with a lower dose of morphine (1 mg/kg, s.c.) using the same procedure.
First, we have studied the effect of stress or morphine re-exposure on synaptic efficacy after chronic morphine. A single re-exposure to morphine (Fig. 3a, arrow) (3 mg/kg, i.p.) of nonstressed animals failed to induce synaptic potentiation (2 mg · kg-1 · d-1 chronic morphine, s.c.; n = 4; 102.1 ± 0.4% of baseline 90 min after morphine re-exposure; p > 0.05 compared with baseline). Similarly, morphine re-exposure failed to induce synaptic potentiation after a higher dose of chronic morphine (Fig. 3b, arrow) (20 mg · kg-1 · d-1, s.c.; 3 mg/kg of morphine in re-exposure, i.p.), and subsequent LFS failed to induce LTD (n = 5; 100.8 ± 0.6% of baseline 30 min after morphine re-exposure; 101.3 ± 0.6% of baseline 60 min after LFS; p > 0.05 compared with baseline) (Fig. 3b). Moreover, stress no longer enabled LTD induction by LFS in the addicted animals (20 mg · kg-1 · d-1 chronic morphine, s.c.; n = 7; 97 ± 0.6% of baseline 60 min after LFS; p > 0.05 compared with baseline) (Fig. 3c).
Figure 3.

Effect of stress or morphine re-exposure on hippocampal synaptic efficacy after chronic morphine. a, Morphine re-exposure (arrow; 3 mg/kg, i.p.) to nonstressed animals failed to induce synaptic potentiation after chronic morphine (2 mg · kg-1 · d-1, s.c.; n = 4; 102.1 ± 0.4% of baseline 90 min after morphine re-exposure; p > 0.05 compared with baseline). b, Morphine re-exposure (arrow; 3 mg/kg, i.p.) also failed to induce synaptic potentiation after a higher dose of chronic morphine (20 mg · kg-1 · d-1, s.c.), and subsequent LFS (3 Hz; bar) failed to induce LTD (n = 5; 100.8 ± 0.6% of baseline 30 min after morphine re-exposure; 101.3 ± 0.6% of baseline 60 min after LFS; p > 0.05 compared with baseline). c, Stress also failed to induce LTD by LFS (3 Hz; bar) after chronic morphine (20 mg · kg-1 · d-1, s.c.; n = 7; 97 ± 0.6% of baseline 60 min after LFS; p > 0.05 compared with baseline).
Second, the combinatorial effect of stress with morphine was studied. After chronic morphine (20 mg · kg-1 · d-1, s.c.), a single re-exposure of morphine (Fig. 4a, arrow) (3 mg/kg, i.p.) to stressed animals still elicited a remarkable synaptic depression (n = 4; 70.2 ± 1.7% of baseline 90 min after morphine re-exposure; p < 0.05 compared with baseline). The synaptic depression also precluded further LTD induction by LFS (n = 5; 61.0 ± 3.8% of baseline 60 min after LFS and 90 min after morphine re-exposure; p < 0.05 compared with baseline; p > 0.05 compared with Fig. 4a,b). A similar synaptic depression was induced by morphine re-exposure (Fig. 4c, arrow; 3 mg/kg, i.p.) after a lower dose of chronic morphine (2 mg · kg-1 · d-1, s.c.), and maintained for at least 400 min (n = 6; 62.1 ± 1.9% of baseline 400 min after morphine re-exposure; p < 0.05 compared with baseline; p > 0.05 compared with Fig. 4a,b).
Figure 4.

Stress in tandem with morphine re-exposure on hippocampal synaptic efficacy after chronic morphine. a, A single re-exposure to morphine (arrow; 3 mg/kg, i.p.) of stressed animals after chronic morphine (20 mg · kg-1 · d-1, s.c.) still elicited a remarkable synaptic depression (n = 4; 70.2 ± 1.7% of baseline 90 min after morphine re-exposure; p < 0.05 compared with baseline). b, The synaptic depression also precluded further LTD induction by LFS (3 Hz; bar) 30 min after morphine re-exposure (arrow; 3 mg/kg, i.p.; n = 5; 61.0 ± 3.8% of baseline 60 min after LFS and 90 min after morphine re-exposure; p < 0.05 compared with baseline; p > 0.05 compared with a). c, A similar synaptic depression was also induced by morphine re-exposure (arrow; 3 mg/kg, i.p.) to stressed animals after a lower dose of chronic morphine (2 mg · kg-1 · d-1, s.c.), and maintained for at least 400 min (n = 6; 62.1 ± 1.9% of baseline 400 min after morphine re-exposure; p < 0.05 compared with baseline, p > 0.05 compared with a and b).
These results suggested that chronic morphine led to the tolerance of morphine to induce synaptic potentiation and morphine to enable LTD induction by LFS. Chronic morphine also led to the loss of stress to enable LTD induction by LFS. However, the ability of stress with morphine after chronic morphine treatment remained unchanged in eliciting synaptic depression.
Multiple comparisons
Additional comparisons of these results between naive and chronic morphine in the stress-enabled LTD induction by LFS, morphine-induced synaptic potentiation, and stress with morphine-induced synaptic depression are shown in Figure 5a. Stress enabled LTD induction by LFS in naive (Fig. 1a, black circles) but not after chronic morphine (Fig. 3c) (summarized in Fig. 5a, Stress+LFS). Morphine induced synaptic potentiation in naive (Fig. 1c-e) but not after chronic morphine (Fig. 3a, 2 mg · kg-1 · d-1; Fig. 3b, 20 mg · kg-1 · d-1) (summarized in Fig. 5a, Morphine). However, stress with morphine induced synaptic depression both in naive (Fig. 2a,b) and after chronic morphine (Fig. 4a,b, 20 mg · kg-1 · d-1; Fig. 4c, 2 mg · kg-1 · d-1) (summarized in Fig. 5a, Stress+Morphine). The baseline of the field EPSP amplitude (50% maximum response) was also compared between stressed and nonstressed animals. The data included the studies in the present and in the past years under the same conditions. There was no significant difference in the baseline EPSP amplitude between stressed and nonstressed animals (Fig. 5b).
Figure 5.

Multiple comparisons. a, Comparisons between naive and chronic morphine in the stress-enabled LTD induction by LFS, the morphine-induced synaptic potentiation, and the stress with morphine-induced synaptic depression. Stress-enabled LTD induction by LFS in naive but not after chronic morphine (Stress+LFS). Morphine induced synaptic potentiation in naive but not after chronic morphine (2 or 20 mg · kg-1 · d-1; Morphine). However, stress in tandem with morphine induced synaptic depression both in naive and after chronic morphine (2 or 20 mg · kg-1 · d-1; Stress+Morphine). **p < 0.01 compared between naive and chronic. b, The baseline of the field EPSP amplitude (50% maximum response) was compared between stressed and nonstressed animals. There was no significant difference in the baseline field EPSP amplitude between stressed and nonstressed animals (n = 52, 2.1 ± 0.1 mV, the field EPSP amplitude for stressed; n = 109, 2.2 ± 0.1 mV, the field EPSP amplitude for nonstressed; p > 0.05).
The present results suggest that these synaptic plasticity changes might play some role in opiate addiction. A delayed-escape paradigm of the Morris water maze was then used to assess the effect of corticosterone with morphine in the initial phase of drug use on later morphine-associated learning.
Corticosterone with morphine on a delayed-escape paradigm of the Morris water maze
Corticosterone with morphine was used in the present studies because stress raises plasma corticosterone levels and affects synaptic plasticity (Shors et al., 1989; Xu et al., 1997), and the glucocorticoid receptor plays significant roles in drug addiction (Deroche et al., 1997; Marinelli et al., 1998; Sillaber et al., 1998; Deroche-Gamonet et al., 2003; Saal et al., 2003).
The Morris water maze is widely used for the studies of hippocampal function in learning and memory (Morris, 1989; Morris et al., 1982). In the present studies, an animal was rewarded for a delay of escape (delayed-escape) in the spatial learning task of the Morris water maze. To mimic the initial phase of opiate use, we treated animals with morphine immediately after corticosterone (C-M), saline (Sal), corticosterone (Cor), morphine (Mor) during 5 d of addictive training (supplemental Table 1, available at). Each treated group was then divided into reinforced (R) or nonreinforced (N) training. In the reinforced training, if the animal avoided escape for reward or escaped to the hidden platform within 60 sec, the animal was accordingly given a reward or saline immediately. In the nonreinforced training, the animal was given a reward or saline randomly, immediately after each trial. One reinforced group and its counterpart, the nonreinforced group, were rewarded corticosterone with morphine (C-M) to assess its effect on delayed-escape behavior after previous repeated exposure. The other groups were rewarded with morphine (Mor) to assess the effect of previous repeated exposure.
With the exception of Sal_Mor/R, all reinforced groups, Cor_Mor/R, Mor_Mor/R, C-M_Mor/R, and C-M_C-M/R (Fig. 6a, black symbols), showed significant delayed-escape behavior, as indicated by longer latencies to escape, over 5 d of training, compared with their counterparts, the nonreinforced groups, Cor_Mor/N, Mor_Mor/N, C-M_Mor/N, and C-M_C-M/N (Fig. 6a, white symbols). The two nonreinforced groups, C-M_C-M/N and Cor_Mor/N, showed impairment in the spatial learning task (Fig. 6a, inverted white triangles for C-M_C-M/N; white diamonds for Cor_Mor/N) compared with the control of nonreinforced groups, Sal_Mor/N (Fig. 6a, white squares). This may be caused by the effect of corticosterone on spatial learning, in agreement with previous findings (de Quervain et al., 1998; Conrad et al., 1999; Mizoguchi et al., 2000). Remarkably, C-M_Mor/R, the reinforced group previously repeated exposure to corticosterone with morphine and showed the longest delayed-escape throughout the training days (Fig. 6a, black triangles) (p < 0.05 compared with other groups). Moreover, its counterpart, C-M_Mor/N (the nonreinforced group), performed the spatial learning task very well (Fig. 6a, white triangles) (p > 0.05 compared with Sal_Mor/N). This suggested that previous repeated exposure to corticosterone with morphine affected neither motor activity nor cognitive function.
Figure 6.

_Corticosterone with morphine on a delayed-escape paradigm of the Morris water maze. a, Delayed-escape was significantly enhanced by previous exposure to morphine, corticosterone, or corticosterone with morphine, indicated by the longer mean latencies to escape (black symbols; n = 5; p < 0.05 Mor_Mor/R compared with Mor_Mor/N; n = 6; p < 0.01 Cor_Mor/R compared with Cor_Mor/N; n = 6; p < 0.01 C-M_Mor/R compared with C-M_Mor/N; n = 6; p < 0.05 on days 9-10 C-M_C-M/R compared with C-M_C-M/N; F(9,45) = 16.1, F(9,45) = 28.6, F(9,45) = 13.0, F(9,45) = 11.0 for days 7-10, respectively), but not by previous saline exposure (n = 5; p > 0.05 Sal_Mor/R compared with Sal_Mor/N; p < 0.05 compared with other reinforced groups; F(9,45) = 16.1, F(9,45) = 28.6, F(9,45) = 13.0, F(9,45) = 11.0 for days 7-10, respectively). On the other hand, spatial learning was impaired in the group randomly rewarded with corticosterone with morphine after previous exposure to corticosterone with morphine (n = 6; p < 0.05 C-M_C-M/N compared with Sal_Mor/N; F(9,45) = 16.1, F(9,45) = 28.6, F(9,45) = 13.0 for days 7-9, respectively) and in the group randomly rewarded with morphine after previous exposure to corticosterone (n = 6; p < 0.05 Cor_Mor/N compared with Sal_Mor/N; F(9,45) = 28.6, F(9,45) = 13.0, F(9,45) = 11.0 for days 8-10, respectively). However, other nonreinforced groups performed the spatial learning task very well (white symbols; p > 0.05 cross groups compared with Sal_Mor/N). b, The reinforced groups, previously exposed to corticosterone or saline and later rewarded with morphine, and previously exposed to corticosterone with morphine and later rewarded with corticosterone with morphine, did not show morphine-seeking behavior after withdrawal for 5 d (Cor_Mor/R, Sal_Mor/R and C-M_C-M/R). However, the reinforced group, Mor_Mor/R, showed morphine-seeking behavior, as indicated by longer latencies to escape without reward, after withdrawal for 5 d but not 10 and 19 d, and a priming injection failed to reinstate on day 29 (n = 5; F(9,45) = 9.8 on day 15, p < 0.05; F(9,45) = 5.2 on day 20, F(9,45) = 14.2 on day 29; p > 0.05 compared with Mor_Mor/N; p < 0.05 compared with C-M_Mor/R). Remarkably, persistent morphine-seeking behavior was found in the reinforced group, C-M_Mor/R, over the post-training period (n = 6; p < 0.05 C-M_Mor/R compared with other groups; F(9,45) = 9.8, F(9,45) = 5.2, F(9,45) = 14.2 for days 15, 20, and 29, respectively). c, Net delay was calculated by subtracting the latencies of the nonreinforced group from that of its counterpart, the reinforced group, and then expressed as the mean of the net latencies over training days 7-10. Net delay was clearly enhanced by previous exposure to corticosterone with morphine, corticosterone, and morphine but not by previous exposure to saline (0.6 ± 2.2 sec for Sal_Mor; 17.4 ± 2.7 sec for Cor_Mor; 23.4 ± 5.9 sec for Mor_Mor; 41.0 ± 2.2 sec for C-M_Mor; 6.9 ± 2.1 sec for C-M_C-M). *p < 0.05 compared with Sal_Mor. Insets, Animals escape directly to the hidden platform in nonreinforced groups, e.g., C-M_Mor/N; animals swim slowly somewhere away from the hidden platform for a while and then rapidly escape to the hidden platform in the reinforced group, C-M_Mor/R, during the extinction test.
Extinction of the learned delayed-escape behavior was tested after withdrawal for 5, 10, and 19 d. All animals were able to escape to the hidden platform during a trial duration of 120 sec. The two reinforced groups, Mor_Mor/R and C-M_Mor/R, showed morphine-seeking behavior, as indicated by longer latencies to escape without reward, on day 15 (Fig. 6b, black circles for Mor_Mor/R; black triangles for C-M_Mor/R) (p < 0.05 compared with other groups). Meanwhile, the two nonreinforced groups, Mor_Mor/N and C-M_Mor/N, escaped to the hidden platform very fast (Fig. 6b, white circles for Mor_Mor/N; white triangles for C-M_Mor/N) (p > 0.05 compared with Sal_Mor/N). Remarkably, morphine-seeking behavior in Mor_Mor/R was extinguished (Fig. 6b, Mor_Mor/R, black circles on days 20 and 29) (p > 0.05 compared with Mor_Mor/N), but C-M_Mor/R showed persistent morphine-seeking behavior in the extinction tests on days 20 and 29 (Fig. 6b, C-M_Mor/R, black triangles on days 20 and 29) (p < 0.05 compared with other groups). Furthermore, an escape speed that was calculated using the path length in 10 sec before the animals climbed onto the hidden platform during the extinction test was the same in the nonreinforced group (C-M_Mor/N, 22.6 ± 1.3 cm/sec) as in the reinforced group (C-M_Mor/R, 21.8 ± 1.1 cm/sec; p > 0.05), although the reinforced group showed significant delayed-escape behavior. This clearly indicated that neither motor activity nor motivation to escape was affected. The learned strategy during escape, such as swimming around in the initial area or somewhere might be responsible for the delayed-escape and morphine-seeking behaviors (Fig. 6, insets).
The results on delayed-escape training during days 7-10 are summarized in Figure 6c. Net delay was calculated by subtracting the latencies of the nonreinforced group from that of its reinforced group and then expressed as mean of the net latencies over the training days 7-10. Previous exposure to corticosterone, morphine, and corticosterone with morphine significantly enhanced the net delay. Remarkably, the net delay if linearly summated that of corticosterone alone (17.4 ± 2.7 sec, Cor_Mor) with that of morphine alone (23.4 ± 5.9 sec, Mor_Mor) was the same as that of corticosterone with morphine (41.0 ± 2.2 sec, C-M_Mor), suggesting an additive effect of corticosterone with morphine on the delayed-escape behavior.
Discussion
The main finding is that acute morphine caused synaptic potentiation in naive animals, but stress in tandem with acute morphine caused synaptic depression both in naive and addicted animals. Repeated exposure to corticosterone with morphine during the initial phase of opiate use promoted remarkably later delayed-escape and morphine-seeking behaviors after withdrawal. These observations suggest possible mechanisms of corticosterone on opiate addiction.
Delayed-escape paradigm of the Morris water maze
The Morris water maze is used widely for studying the mechanisms of hippocampal function in learning and memory (Morris et al., 1982; Bliss and Collingridge, 1993; Martin et al., 2000). An animal can readily learn to escape to a hidden platform in a short time if cognitive function, motor activity, and motivation are not disturbed. In the present studies, an animal was trained to perform the spatial learning task and encouraged to learn delayed-escape by morphine reward when the animal avoided escape in 60 sec. Clearly, it is not the forced-swim test, a model of depression/anxiety induced by prolonged swimming (e.g., 30 min) in cold water and by despair of no escape. Previous exposure to corticosterone, morphine, and corticosterone with morphine later enhanced the morphine-reinforced delayed-escape behavior. Furthermore, the randomly rewarded nonreinforced group, a counterpart of the reinforced group, had the same previous exposure and later performed the spatial learning task with the same reward and had provided the information of motor activity and cognitive function for the reinforced group. Most of the nonreinforced groups performed the spatial learning task very well, under conditions such as the dose of morphine, the time of corticosterone exposure, and the stress in behavioral training. However, two nonreinforced groups showed impaired spatial learning, probably because of the combinatorial reward of corticosterone with morphine or previous exposure to corticosterone. Thus, the net delay, subtracting the latencies of the nonreinforced group from its reinforced group over the training period, made the comparison of the delayed-escape behavior between groups comparable and reliable (Fig. 6c).
Reinforcing effect and associative learning
Previous repeated exposure to saline did not cause delayed-escape by low doses of morphine reward. However, low doses of morphine effectively rewarded delayed-escape after previous repeated exposure to corticosterone, morphine, and corticosterone with morphine. From one point of view, previous exposure may enhance the reinforcing effect of morphine to cause delayed-escape, which may be consistent with the incentive-sensitization theory of addiction (Robinson and Berridge, 2000) and previous findings of the sensitization to drugs (Deroche et al., 1995; Rouge-Pont et al., 1995; Scheggi et al., 2000). On the other hand, the glucocorticoid receptor seems to be a requisite for the long-term storage of information, although it has not been fully understood why and when the effect of stress or glucocorticoids on learning and memory produce impairment, enhancement, or no effect (Shors et al., 1992; McEwen, 1994; de Kloet et al., 1999; Diamond et al., 1999; Sapolsky, 2000; Lupien and Lepage, 2001; Payne et al., 2002; Yang et al., 2003). Here, corticosterone likely enhances or impairs associative learning, depending on when it is administered, out of context during the addictive training or in context during the delayed-escape training. It is important to note the difference that the reinforced group, C-M_C-M/R, was rewarded with corticosterone with morphine to assess the combinatorial effect after previous repeated exposure, but other groups were rewarded with morphine to assess the effect of previous exposure. Corticosterone given in context may interrupt both the delayed-escape behavior and spatial learning. This is in agreement with previous findings that stress or corticosterone impairs spatial learning and memory retrieval in the Morris water maze (Diamond et al., 1996; de Quervain et al., 1998; Conrad et al., 1999; Yang et al., 2003).
Corticosterone with morphine on delayed-escape and morphine-seeking behaviors
It is very interesting that previous repeated exposure to corticosterone with morphine not only enhanced later delayed-escape behavior but also caused persistent morphine-seeking behavior after withdrawal. Mechanically, this could arise in at least two different ways. The combinatorial exposure could cause the strongest reinforcing effect on later morphine reward through an additive mechanism (Fig. 6c), leading to a slower rate of extinction. This may explain the difference of C-M_Mor/R, Cor_Mor/R, and Mor_Mor/R in delayed-escape and morphine-seeking behaviors after withdrawal, and a priming injection of morphine unable to reinstate delayed-escape behavior. Meanwhile, the previous combinatorial exposure to corticosterone with morphine could enhance the sensitivity of the animals to stress or glucocorticoids to enable withdrawal stress to trigger morphine-seeking behavior and to exacerbate the impairing effect of corticosterone on associative learning in the combinatorial reward during delayed-escape training. Alternatively, the previous combinatorial exposure was out of context and could enhance later associative learning to form the strongest memories of delayed-escape (such as the strategy to escape) (Fig. 6, insets) to preclude extinction learning. Conversely, the later combinatorial reward was in context and could impair associative learning. These mechanisms probably occur, jointly promoting the persistent delayed-escape and morphine-seeking after withdrawal.The mechanisms underlying this curious finding are not clarified, but the mechanism of hippocampal synaptic plasticity in the effects of stress or glucocorticoids on opiate addiction, such as the one described currently, may well have contributed to them.
Hippocampal synaptic plasticity in the effects of stress or glucocorticoids on opiate addiction
A wealth of evidence has revealed that stress or glucocorticoids interact with drug addiction, thus increasing drug use, drug seeking, and relapse (Erb et al., 1996; Piazza and Le Moal, 1996; Shaham et al., 2000; Stewart, 2000, 2003; Sinha, 2001; Deroche-Gamonet et al., 2003; Kauer, 2003; Saal et al., 2003; Sutton et al., 2003). The underlying mechanisms are believed to involve many brain areas, such as the ventral tegmental area and nucleus accumbens, but some may also involve the hippocampus. The hippocampus is enriched with glucocorticoid receptors and is crucially involved in regulating stress effects on synaptic plasticity and learning and memory (Shors et al., 1989; Oitzl and de Kloet, 1992; Pavlides et al., 1996; Xu et al., 1997; de Quervain et al., 1998; McEwen, 1999; Kim and Diamond, 2002). On the other hand, both synaptic plasticity (Terman et al., 1994; Little and Teyler, 1996; Mansouri et al., 1999; Pu et al., 2002) and behavioral studies (Fan et al., 1999; Lu et al., 2000; Mitchell et al., 2000) support the view that the hippocampus is involved in opiate addiction, and in other drugs addiction as suggested recently (White, 1996; Berke and Hyman, 2000; Hyman and Malenka, 2001; Nestler, 2001a; Vorel et al., 2001).
The present findings have revealed that acute morphine causes synaptic potentiation, but chronic morphine leads to the tolerance of acute morphine to induce synaptic potentiation. Stress with morphine induces synaptic depression both in naive morphine and after chronic morphine. Corticosterone with morphine during the initial phase of opiate use promotes later delayed-escape and morphine-seeking behaviors after withdrawal. Because hippocampal synaptic plasticity plays important roles in learning and memory (Bliss and Collingridge, 1993; Malenka and Nicoll, 1999; Martin et al., 2000), both the synaptic potentiation and depression may be maladaptively usurped by opiate addiction (Berke and Hyman, 2000; Hyman and Malenka, 2001; Ungless et al., 2001; Saal et al., 2003) to produce persistent delayed-escape and morphine-seeking behaviors. Stress alone does not affect basal synaptic transmission (Fig. 5b), but it may induce other changes, such as theta rhythm in the hippocampus (Simonov and Rusalova, 1980; Balleine and Curthoys, 1991; Yamamoto, 1998). A recent finding has demonstrated that theta-burst stimulation in the hippocampus may read out the memories and induce relapse (Vorel et al., 2001). Thus, the stress-related theta activity may also be involved in the initiation of the synaptic depression. The hippocampal function in memory has assumed that neuronal information is encoded and stored at a low density and in a widely distributed manner, thus increasing its storage capacity (Gluck and Myers, 1997). A redistribution of synaptic efficacy provides a learning mechanism with little net change in synaptic weight (Miller, 1996) to detect and store new information effectively (Xu et al., 1998b). Perhaps strengthening synaptic connections, and meanwhile raising stress levels of glucocorticoids (Buckingham and Cooper, 1984; Pirnik et al., 2001), and then eliciting synaptic depression during the initial phase of opiate use could endow the system with enhanced combinatorial plasticity (Dudai, 1996), usurped strongly by opiate addiction. Conversely, eliciting synaptic depression but inability to strengthen synaptic connections, presumably after stress-triggered opiate relapse, could restrain the system in consequential learning.
Footnotes
This work was supported by Chinese Academy of Sciences Grants KSCX2-SW-204-02 (N.S.) and KSCX-SW-204-04 (L.X.), National Natural Science Foundation of China Grants 30170324 and 30230130 (N.S.) and 39870280, 39925011, and 39930080 (L.X.), and National Basic Research Program Grants 2003CB515404 (N.S.) and G1999054000 (L.X.).
Correspondence should be addressed to either of the following: Dr. Lin Xu, Laboratory of Learning and Memory, Kunming Institute of Zoology, The Chinese Academy of Sciences, Kunming 650223, Yunnan, People's Republic of China, E-mail: lxu@vip.163.com; or Dr. Nan Sui, The Key Laboratory of Mental Health, Institute of Psychology, The Chinese Academy of Sciences, P.O. Box 1603, Beijing 100101, People's Republic of China, E-mail: suin@psych.ac.cn.
Copyright © 2004 Society for Neuroscience 0270-6474/04/242412-09$15.00/0
Y.Y. and X.Z. contributed equally to this work.
References
- Balleine BW, Curthoys IS (1991) Differential effects of escapable and inescapable footshock on hippocampal theta activity. Behav Neurosci 105: 202-209. [DOI] [PubMed] [Google Scholar]
- Beatty WW (1983) Opiate antagonists, morphine and spatial memory in rats. Pharmacol Biochem Behav 19: 397-401. [DOI] [PubMed] [Google Scholar]
- Berke JD, Hyman SE (2000) Addiction, dopamine, and the molecular mechanisms of memory. Neuron 25: 515-532. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31-39. [DOI] [PubMed] [Google Scholar]
- Buckingham JC, Cooper TA (1984) Differences in hypothalamo-pituitary-adrenocortical activity in the rat after acute and prolonged treatment with morphine. Neuroendocrinology 38: 411-417. [DOI] [PubMed] [Google Scholar]
- Classen W, Mondadori C (1984) Facilitation or inhibition of memory by morphine: a question of experimental parameters. Experientia 40: 506-509. [DOI] [PubMed] [Google Scholar]
- Conrad CD, Lupien SJ, McEwen BS (1999) Support for a bimodal role for type II adrenal steroid receptors in spatial memory. Neurobiol Learn Mem 72: 39-46. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Oitzl MS, Joels M (1999) Stress and cognition: are corticosteroids good or bad guys? Trends Neurosci 22: 422-426. [DOI] [PubMed] [Google Scholar]
- de Quervain DJ, Roozendaal B, McGaugh JL (1998) Stress and glucocorticoids impair retrieval of long-term spatial memory. Nature 394: 787-790. [DOI] [PubMed] [Google Scholar]
- Deroche V, Piazza PV, Casolini P, Maccari S, Le Moal M, Simon H (1992) Stress-induced sensitization to amphetamine and morphine psychomotor effects depend on stress-induced corticosterone secretion. Brain Res 598: 343-348. [DOI] [PubMed] [Google Scholar]
- Deroche V, Marinelli M, Maccari S, Le Moal M, Simon H, Piazza PV (1995) Stress-induced sensitization and glucocorticoids. I. Sensitization of dopamine-dependent locomotor effects of amphetamine and morphine depends on stress-induced corticosterone secretion. J Neurosci 15: 7181-7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroche V, Marinelli M, Le Moal M, Piazza PV (1997) Glucocorticoids and behavioral effects of psychostimulants. II. Cocaine intravenous self-administration and reinstatement depend on glucocorticoid levels. J Pharmacol Exp Ther 281: 1401-1407. [PubMed] [Google Scholar]
- Deroche-Gamonet V, Sillaber I, Aouizerate B, Izawa R, Jaber M, Ghozland S, Kellendonk C, Le Moal M, Spanagel R, Schutz G, Tronche F, Piazza PV (2003) The glucocorticoid receptor as a potential target to reduce cocaine abuse. J Neurosci 23: 4785-4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vries TJ, Schoffelmeer AN, Tjon GH, Nestby P, Mulder AH, Vanderschuren LJ (1996) Mifepristone prevents the expression of long-term behavioural sensitization to amphetamine. Eur J Pharmacol 307: R3-R4. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Fleshner M, Ingersoll N, Rose GM (1996) Psychological stress impairs spatial working memory: relevance to electrophysiological studies of hippocampal function. Behav Neurosci 110: 661-672. [DOI] [PubMed] [Google Scholar]
- Diamond DM, Park CR, Heman KL, Rose GM (1999) Exposing rats to a predator impairs spatial working memory in the radial arm water maze. Hippocampus 9: 542-552. [DOI] [PubMed] [Google Scholar]
- Dudai Y (1996) Consolidation: fragility on the road to the engram. Neuron 17: 370. [DOI] [PubMed] [Google Scholar]
- Erb S, Shaham Y, Stewart J (1996) Stress reinstates cocaine-seeking behavior after prolonged extinction and a drug-free period. Psychopharmacology (Berl) 128: 408-412. [DOI] [PubMed] [Google Scholar]
- Fan GH, Wang LZ, Qiu HC, Ma L, Pei G (1999) Inhibition of calcium/calmodulin-dependent protein kinase II in rat hippocampus attenuates morphine tolerance and dependence. Mol Pharmacol 56: 39-45. [DOI] [PubMed] [Google Scholar]
- Fratta W, Yang HY, Hong J, Costa E (1977) Stability of Met-enkephalin content in brain structures of morphine-dependent or foot shock-stressed rats. Nature 268: 452-453. [DOI] [PubMed] [Google Scholar]
- Gluck MA, Myers CE (1997) Psychobiological models of hippocampal function in learning and memory. Annu Rev Psychol 48: 481-514. [DOI] [PubMed] [Google Scholar]
- Guerra D, Sole A, Cami J, Tobena A (1987) Neuropsychological performance in opiate addicts after rapid detoxification. Drug Alcohol Depend 20: 261-270. [DOI] [PubMed] [Google Scholar]
- Holden C (2001) Drug addiction. Zapping memory center triggers drug craving. Science 292: 1039. [DOI] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC (2001) Addiction and the brain: the neurobiology of compulsion and its persistence. Nat Rev Neurosci 2: 695-703. [DOI] [PubMed] [Google Scholar]
- Kauer JA (2003) Addictive drugs and stress trigger a common change at VTA synapses. Neuron 37: 549-550. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Diamond DM (2002) The stressed hippocampus, synaptic plasticity and lost memories. Nat Rev Neurosci 3: 453-462. [DOI] [PubMed] [Google Scholar]
- Kim JJ, Foy MR, Thompson RF (1996) Behavioral stress modifies hippocampal plasticity through N-methyl-d-aspartate receptor activation. Proc Natl Acad Sci USA 93: 4750-4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Sanna PP, Bloom FE (1998) Neuroscience of addiction. Neuron 21: 467-476. [DOI] [PubMed] [Google Scholar]
- Li Z, Wu CF, Pei G, Xu NJ (2001) Reversal of morphine-induced memory impairment in mice by withdrawal in Morris water maze: possible involvement of cholinergic system. Pharmacol Biochem Behav 68: 507-513. [DOI] [PubMed] [Google Scholar]
- Little JZ, Teyler TJ (1996) Prenatal cocaine exposure leads to enhanced long-term potentiation in region CA1 of hippocampus. Brain Res Dev Brain Res 92: 117-119. [DOI] [PubMed] [Google Scholar]
- Lu L, Zeng S, Liu D, Ceng X (2000) Inhibition of the amygdala and hippocampal calcium/calmodulin-dependent protein kinase II attenuates the dependence and relapse to morphine differently in rats. Neurosci Lett 291: 191-195. [DOI] [PubMed] [Google Scholar]
- Lupien SJ, Lepage M (2001) Stress, memory, and the hippocampus: can't live with it, can't live without it. Behav Brain Res 127: 137-158. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Nicoll RA (1999) Long-term potentiation: a decade of progress? Science 285: 1870-1874. [DOI] [PubMed] [Google Scholar]
- Mansouri FA, Motamedi F, Fathollahi Y (1999) Chronic in vivo morphine administration facilitates primed-bursts-induced long-term potentiation of Schaffer collateral-CA1 synapses in hippocampal slices in vitro. Brain Res 815: 419-423. [DOI] [PubMed] [Google Scholar]
- Marinelli M, Aouizerate B, Barrot M, Le Moal M, Piazza PV (1998) Dopamine-dependent responses to morphine depend on glucocorticoid receptors. Proc Natl Acad Sci USA 95: 7742-7747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin SJ, Grimwood PD, Morris RG (2000) Synaptic plasticity and memory: an evaluation of the hypothesis. Annu Rev Neurosci 23: 649-711. [DOI] [PubMed] [Google Scholar]
- McEwen BS (1994) Corticosteroids and hippocampal plasticity. Ann NY Acad Sci 746: 134-142. [DOI] [PubMed] [Google Scholar]
- McEwen BS (1999) Stress and hippocampal plasticity. Annu Rev Neurosci 22: 105-122. [DOI] [PubMed] [Google Scholar]
- Miller K (1996) Synaptic economics: competition and cooperation in synaptic plasticity. Neuron 17: 371-374. [DOI] [PubMed] [Google Scholar]
- Mitchell JM, Basbaum AI, Fields HL (2000) A locus and mechanism of action for associative morphine tolerance. Nat Neurosci 3: 47-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi K, Yuzurihara M, Ishige A, Sasaki H, Chui DH, Tabira T (2000) Chronic stress induces impairment of spatial working memory because of prefrontal dopaminergic dysfunction. J Neurosci 20: 1568-1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG (1989) Synaptic plasticity and learning: selective impairment of learning rats and blockade of long-term potentiation in vivo by the NMDA receptor antagonist AP-5. J Neurosci 9: 3040-3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RG, Garrud P, Rawlins JN, O'Keefe J (1982) Place navigation impaired in rats with hippocampal lesions. Nature 297: 681-683. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001a) Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci 2: 119-128. [DOI] [PubMed] [Google Scholar]
- Nestler EJ (2001b) Neurobiology: total recall-the memory of addiction. Science 292: 2266-2267. [DOI] [PubMed] [Google Scholar]
- Oitzl MS, de Kloet ER (1992) Selective corticosteroid antagonists modulate specific aspects of spatial orientation learning. Behav Neurosci 106: 62-71. [DOI] [PubMed] [Google Scholar]
- Olley JE, Tiong GK, Scheer J, von Jenner NM (1990) Responses of morphine dependent opioid neurones to stressors. Prog Clin Biol Res 328: 511-514. [PubMed] [Google Scholar]
- Pavlides C, Ogawa S, Kimura A, McEwen BS (1996) Role of adrenal steroid mineralocorticoid and glucocorticoid receptors in long-term potentiation in the CA1 field of hippocampal slices. Brain Res 738: 229-235. [DOI] [PubMed] [Google Scholar]
- Payne JD, Nadel L, Allen JJ, Thomas KG, Jacobs WJ (2002) The effects of experimentally induced stress on false recognition. Memory 10: 1-6. [DOI] [PubMed] [Google Scholar]
- Pechnick P (1999) Effects of opioids on the hypothalamo-pituitary-adrenal axis. Annu Rev Pharmacol Toxicol 32: 353-383. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Le Moal ML (1996) Pathophysiological basis of vulnerability to drug abuse: role of an interaction between stress, glucocorticoids, and dopaminergic neurons. Annu Rev Pharmacol Toxicol 36: 359-378. [DOI] [PubMed] [Google Scholar]
- Pirnik Z, Schwendt M, Jezova D (2001) Single dose of morphine influences plasma corticosterone and gene expression of main NMDA receptor subunit in the adrenal gland but not in the hippocampus. Endocr Regul 35: 187-193. [PubMed] [Google Scholar]
- Pu L, Bao GB, Xu NJ, Ma L, Pei G (2002) Hippocampal long-term potentiation is reduced by chronic opiate treatment and can be restored by re-exposure to opiates. J Neurosci 22: 1914-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins TW, Everitt BJ (1999) Drug addiction: bad habits add up. Nature 398: 567-570. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC (2000) The psychology and neurobiology of addiction: an incentive-sensitization view. Addiction 95 [Suppl 2]: S91-S117. [DOI] [PubMed] [Google Scholar]
- Rocher C, Spedding M, Munoz C, Jay TM (2004) Acute stress-induced changes in hippocampal/prefrontal circuits in rats: effects of antidepressants. Cereb Cortex 14: 224-229. [DOI] [PubMed] [Google Scholar]
- Rouge-Pont F, Marinelli M, Le Moal M, Simon H, Piazza PV (1995) Stress-induced sensitization and glucocorticoids. II. Sensitization of the increase in extracellular dopamine induced by cocaine depends on stress-induced corticosterone secretion. J Neurosci 15: 7189-7195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saal D, Dong Y, Bonci A, Malenka RC (2003) Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron 37: 577-582. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM (2000) Stress hormones: good and bad. Neurobiol Dis 7: 540-542. [DOI] [PubMed] [Google Scholar]
- Scheggi S, Masi F, Tagliamonte A, Gambarana C, Tolu P, De Montis MG (2000) Rats sensitized to morphine are resistant to the behavioral effects of an unavoidable stress. Brain Res 853: 290-298. [DOI] [PubMed] [Google Scholar]
- Shaham Y, Erb S, Stewart J (2000) Stress-induced relapse to heroin and cocaine seeking in rats: a review. Brain Res Brain Res Rev 33: 13-33. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Seib TB, Levine S, Thompson RF (1989) Inescapable versus escapable shock modulates long-term potentiation in the rat hippocampus. Science 244: 224-226. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Levine S, Thompson RF (1990) Opioid antagonist eliminates the stress-induced impairment of long-term potentiation (LTP). Brain Res 506: 316-318. [DOI] [PubMed] [Google Scholar]
- Shors TJ, Weiss C, Thompson RF (1992) Stress-induced facilitation of classical conditioning. Science 257: 537-539. [DOI] [PubMed] [Google Scholar]
- Sillaber I, Montkowski A, Landgraf R, Barden N, Holsboer F, Spanagel R (1998) Enhanced morphine-induced behavioural effects and dopamine release in the nucleus accumbens in a transgenic mouse model of impaired glucocorticoid (type II) receptor function: influence of long-term treatment with the antidepressant moclobemide. Neuroscience 85: 415-425. [DOI] [PubMed] [Google Scholar]
- Simonov PV, Rusalova MN (1980) Electroencephalographic correlates of human emotional stress. Aviat Space Environ Med 51: 1109-1111. [PubMed] [Google Scholar]
- Sinha R (2001) How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 158: 343-359. [DOI] [PubMed] [Google Scholar]
- Stewart J (2000) Pathways to relapse: the neurobiology of drug- and stress-induced relapse to drug-taking. J Psychiatry Neurosci 25: 125-136. [PMC free article] [PubMed] [Google Scholar]
- Stewart J (2003) Stress and relapse to drug seeking: studies in laboratory animals shed light on mechanisms and sources of long-term vulnerability. Am J Addict 12: 1-17. [PubMed] [Google Scholar]
- Sutton MA, Schmidt EF, Choi KH, Schad CA, Whisler K, Simmons D, Karanian DA, Monteggia LM, Neve RL, Self DW (2003) Extinction-induced upregulation in AMPA receptors reduces cocaine-seeking behaviour. Nature 421: 70-75. [DOI] [PubMed] [Google Scholar]
- Terman GW, Wagner JJ, Chavkin C (1994) κ-Opioids inhibit induction of long-term potentiation in the dentate gyrus of the guinea pig hippocampus. J Neurosci 14: 4740-4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo KA, Akil H (1991) Inhibition of morphine tolerance and dependence by the NMDA-receptor antagonist MK-801. Science 251: 85-87. [DOI] [PubMed] [Google Scholar]
- Ungless MA, Whistler JL, Malenka RC, Bonci A (2001) Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature 411: 583-587. [DOI] [PubMed] [Google Scholar]
- Vorel SR, Liu X, Hayes RJ, Spector JA, Gardner EL (2001) Relapse to cocaine-seeking after hippocampal theta burst stimulation. Science 292: 1175-1178. [DOI] [PubMed] [Google Scholar]
- Wagner JJ, Etemad LR, Thompson AM (2001) Opioid-mediated facilitation of long-term depression in rat hippocampus. J Pharmacol Exp Ther 296: 776-781. [PubMed] [Google Scholar]
- Wei H, Xiong W, Yang S, Zhou Q, Liang C, Zeng BX, Xu L (2002) Propofol facilitates the development of long-term depression (LTD) and impairs the maintenance of long-term potentiation (LTP) in the CA1 region of the hippocampus of anesthetized rats. Neurosci Lett 324: 181-184. [DOI] [PubMed] [Google Scholar]
- White NM (1996) Addictive drugs as reinforcers: multiple partial actions on memory systems. Addiction 91: 921-949. [PubMed] [Google Scholar]
- Xiong W, Yang Y, Cao J, Wei H, Liang C, Yang S, Xu L (2003) The stress experience dependent long-term depression disassociated with stress effect on spatial memory task. Neurosci Res 46: 415-421. [DOI] [PubMed] [Google Scholar]
- Xu L, Anwyl R, Rowan MJ (1997) Behavioural stress facilitates the induction of long-term depression in the hippocampus. Nature 387: 497-500. [DOI] [PubMed] [Google Scholar]
- Xu L, Holscher C, Anwyl R, Rowan MJ (1998a) Glucocorticoid receptor and protein/RNA synthesis-dependent mechanisms underlie the control of synaptic plasticity by stress. Proc Natl Acad Sci USA 95: 3204-3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Anwyl R, Rowan MJ (1998b) Spatial exploration induces a persistent reversal of long-term potentiation in rat hippocampus. Nature 394: 891-894. [DOI] [PubMed] [Google Scholar]
- Yamamoto J (1998) Relationship between hippocampal theta-wave frequency and emotional behaviors in rabbits produced with stresses or psychotropic drugs. Jpn J Pharmacol 76: 125-127. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cao J, Xiong W, Zhang J, Zhou Q, Wei H, Liang C, Deng J, Li T, Yang S, Xu L (2003) Both stress experience and age determine the impairment or enhancement effect of stress on spatial memory retrieval. J Endocrinol 178: 45-54. [DOI] [PubMed] [Google Scholar]