

Abstract
GABAA receptor function can be conceptually divided into interactions between ligand and receptor (binding) and the opening and closing of the ligand-bound channel (gating). The relationship between binding, gating, and receptor structure remains unclear. Studies of mutations have identified many amino acid residues that contribute to the GABAbinding site. Most of these studies assayed changes in GABA dose–response curves, which are macroscopic measures that depend on the interplay of many processes and cannot resolve individual microscopic transitions. Understanding the microscopic basis of binding and gating is critical, because kinetic transition rates predict how receptors will behave at synapses. Furthermore, microscopic rates are directly related to the molecular interactions underlying receptor function.
Here, we focused on a residue (β2-R207) previously identified as lining the GABA-binding site that, when mutated to cysteine, greatly reduces apparent GABA affinity and was predicted to affect both binding and gating. To better understand the role of β2-R207, we expressed α1β2 and α1β2-R207C receptors in human embryonic kidney 293 cells and studied receptor kinetics using fast solution applications. The mutation accelerated deactivation by 10-fold, without altering desensitization in the presence of saturating GABA. Maximum open probability and single-channel open times were also unaltered by the mutation, but the GABA-binding rate was reduced eightfold. Therefore, the effects of this mutation in a predicted binding site residue are solely attributable to changes in GABA-binding and unbinding kinetics, with no changes in channel gating. Because β2-R207 stabilizes GABA in the binding pocket, it may directly contact the GABA molecule.
Keywords: kinetics, HEK293 cells, mutation, single channel, agonist, antagonist
Introduction
During fast synaptic transmission, neurotransmitter activates receptors in the postsynaptic membrane. The amplitude and time course of the subsequent postsynaptic conductance depend on the two interrelated processes of binding (the interaction of receptor and ligand) and gating (transitions of the bound receptor between open, closed, and desensitized states). Binding is mediated by molecular interactions between the ligand and amino acid residues that line the ligand-binding pocket, that then trigger gating.
A description of binding-pocket structure is rapidly emerging for the superfamily of ligand-gated ion channels, which includes the nicotinic acetylcholine, glycine, serotonin type 3, and GABAA receptors. Site-directed mutagenesis, cysteine-scanning mutagenesis, and photoaffinity labeling have shown that the ligand-binding pocket is at the interface between two subunits, with each subunit contributing multiple discontinuous “loops” (for review, see Galzi, 1994). For example, in the GABAA receptor, the α and β subunits each contribute three regions to the binding pocket (Sigel et al., 1992; Amin and Weiss, 1993; Smith and Olsen, 1994; Westh-Hansen et al., 1997, 1999; Boileau et al., 1999, 2002; Wagner and Czajkowski, 2001; Holden and Czajkowski, 2002; Newell and Czajkowski, 2003).
To evaluate the role of individual residues in binding or gating, most previous studies have examined the effects of mutations on ligand dose–response curves. This approach measures macroscopic parameters such as the concentration of ligand that gives the half-maximal response (EC50), which is itself determined by the more fundamental microscopic processes of ligand binding– unbinding, channel opening– closing, and desensitization–resensitization. Dose–response curves cannot directly reveal these microscopic processes (Colquhoun, 1998). For wild-type GABAA receptors, significant progress toward understanding microscopic kinetics has been made using single-channel recording and rapid ligand application techniques to develop kinetic models of receptor function and to estimate individual rate constants (Twyman et al., 1990; Celentano and Wong, 1994; Jones and Westbrook, 1995; Lavoie and Twyman, 1996; Jones et al., 1998; Haas and Macdonald, 1999; Burkat et al., 2001). Although the combination of kinetic approaches with mutagenesis would obviously be more powerful than either method alone, only a few studies have examined the effects of mutations on microscopic kinetics (Chang and Weiss, 1999; Bianchi and Macdonald, 2001; Scheller and Forman, 2002).
In this study, we applied the techniques of rapid ligand application, single-channel recording, and kinetic modeling to clarify the biophysical role that the arginine at position 207 of the β2 subunit of the rat GABAA receptor plays in channel function. This residue is likely to face into the GABA-binding pocket, and its mutation to cysteine causes a 70-fold increase in the EC50 for GABA (Wagner and Czajkowski, 2001). The present experiments demonstrate that the mutation, β2-R207C, causes a 20-fold increase in the ligand-unbinding rate, an eightfold decrease in the ligand-binding rate, and has no effect on any of the rates associated with gating. Therefore, β2-R207 is important for stabilizing the agonist–receptor complex, and may enjoy a direct interaction with the GABA molecule.
Materials and Methods
Cell culture and transfection. Human embryonic kidney (HEK-293) cells were cultured in Minimum Essential Medium with Earle's salts (Invitrogen, Gaithersburg, MD) containing 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO) in a 37°C incubator under a 5% CO2 atmosphere. When the cells reached 60–90% confluence in 60 mm culture dishes, they were transfected with 1 μg each of α1-FLAG and β2 (wild type or R207C) rat cDNAs subcloned into the pCEP4 vector (Invitrogen) using the LipofectAMINE2000 reagent (Invitrogen) according to the supplied protocol. At 20–28 hr after transfection, the cells were replated on 12 mm sterile glass coverslips, transferred to a 30°C incubator, and recorded at 24–96 hr after replating. Transfected cells were visualized by incubation with anti-FLAG antibody-coated beads (Sigma-Aldrich), which recognize the flag epitope sequence (DYKDDDDK) inserted between the sixth and seventh amino acid of the mature α1 subunit. The flag epitope insertion has been shown previously not to alter function (Connolly et al., 1996).
Patch-clamp electrophysiology. Recordings from outside-out patches excised from HEK cells were made using borosilicate glass pipettes filled with (in mm): 140 KCl, 10 EGTA, 2 MgATP, 20 phosphocreatine, and 10 HEPES, pH 7.3. For macroscopic current recording, patches were voltage clamped at –60 mV and placed in the stream of a multibarreled flowpipe array (Vitrodynamics, Rockaway, NJ) mounted on a piezoelectric bimorph (Vernitron, Bedford, OH). A computer-controlled constant current source drove the bimorph to move solution interfaces over the patch with 10–90% exchange times of ∼200 μsec, as measured by the liquid junction current at the open pipette tip after each experiment. GABA and SR-95531 were dissolved in the perfusion solution, which contained (in mm): 145 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 4 mm Glucose, pH 7.4, 315 mOsm. For extracellular solutions that contained >10 mm GABA, the concentration of NaCl was reduced to 95 mm, and a combination of sucrose and GABA was added to compensate for the reduced osmolarity. When using low NaCl extracellular solution, the concentration of KCl in the pipette solution was also reduced to 90 mm, and 50 mm K-gluconate was added to maintain a constant Cl– driving force. GABA and SR-95531 were obtained from Sigma-Aldrich Chemicals. Currents were low-pass filtered at 2–5 kHz with a four-pole Bessel filter and digitized at a rate no less than twice the filter frequency. Data were collected using an Axopatch 200B amplifier and Digidata 1320A digitizer, controlled by AxoGraph software (Axon Instruments, Foster City, CA) running on a Macintosh G4 (Apple Computers, Cupertino, CA). Curve fitting was also performed using AxoGraph software.
Single-channel currents were recorded from patches held at –80 mV, sampled at 20 kHz, and filtered at 2 kHz. Before analysis, the current records were digitally refiltered using a 1 kHz Gaussian filter. Events were detected using amplitude windows on the basis of a 15 pS conductance and using a 200 μsec minimum event width, with homewritten routines in Matlab 5.2 (Mathworks, Natick, MA). Events that included multiple openings were discarded. Open time distributions were fit by the maximum likelihood method, with corrections for missed events, and the number of open times was determined using the log-likelihood ratio test (Colquhoun and Sigworth, 1995). Only events lasting ≥200 μsec were accepted for analysis.
Peak open probability analysis. Nonstationary variance analysis (Sigworth, 1980) was performed on currents recorded from a series of saturating GABA pulses (2 msec, 10 sec intervals) from which ensemble mean current (I) and variance (σ2) were calculated for each data point. The mean current was divided into 100 equally sized bins, and the corresponding variances were averaged. Plots of binned variance versus current were fit with the equation: σ2 = iI – I2N–1, where i is the single-channel current, and N is the number of channels. Variance resulting from slow drift (i.e., rundown or run-up) was corrected by local linear fitting of the drift, calculating the variance attributable to this trend at each point, and subtracting the drift variance (scaled by the square of normalized current amplitude) from the calculated variance before fitting. This method yields accurate estimates of i and N when tested on simulated data with drift.
Kinetic modeling. Kinetic modeling was performed with homewritten software using the Q-matrix method (Colquhoun and Hawkes, 1995a,b). During optimization, the rate constants kon, α2, α1, kon-ant, and koff-ant were constrained to values obtained from experiments presented in this study. The unconstrained rate constants were optimized by a simultaneous fit to current responses from wild-type receptors to 2 and 500 msec saturating (10 mm) GABA pulses and a paired pulse protocol (2 msec, 10 mm pulses separated by 0.1, 0.3 and 1 sec). After simultaneous optimization, all of the rates except for koff, d2, r2, and p were fixed, and the model was optimized to fit individual responses to 2 msec (six patches) and 500 msec (five patches) pulses of 10 mm GABA. The final values for koff, d2, r2, and p are the means of the values collected from these 11 individual optimizations (Fig. 6). Optimization used a simplex algorithm to minimize the amplitude-weighted sum of squared errors between actual and simulated currents. In all cases, significant differences were tested using a two-tailed unpaired Student's t test at a significance level of p ≤ 0.05.
Figure 6.

Kinetic modeling demonstrates that solely altering GABA-binding and -unbinding rates can completely account for the changes caused by the β2-R207C mutation. A, The Markov model used to simulate GABAA responses (previously described by Jones and Westbrook, 1995, 1998). B, Rate constants used to simulate wild-type and mutant responses. The values for koff, d2, r2, and p are reported as mean ± SEM, because they were allowed to vary while the model was optimized to current responses from individual patches to account for patch-to-patch variability. koff was the only nonconstrained rate constant that was significantly different when comparing mutant and wild-type models (Student's t test). C, Current responses evoked by application of 10 mm GABA to patches containing α1β2 receptors (black) overlaid with simulated responses (red). The GABA application protocols are (from right to left) 2 msec step, 500 msec, and paired 2 msec pulses with intervals of 100, 300, and 1000 msec. D, Currents from α1β2-R207C receptors evoked by the same application protocols used inC using 100 mm GABA (black) are overlaid with simulated responses using the parameters for α1β2-R207C receptors.
Results
Mutation of β2-R207 alters GABAA receptor kinetics
We initially characterized the effect of the β2-R207C mutation on receptor kinetics by observing macroscopic deactivation (relaxation after removal of agonist) and desensitization (relaxation in the presence of saturating agonist). After a brief (2 msec) pulse of saturating GABA, currents from α1β2-R207C receptors deactivated 10 times faster than currents from wild-type receptors (Fig. 1A, Table 1). Because the time course of deactivation depends on the interaction of multiple transition rates such as channel opening, channel closing, desensitization, resensitization, and agonist unbinding (Jones and Westbrook, 1995), a change in deactivation does not immediately inform us as to which rates are altered by the mutation. However, the mutation had no effect on desensitization during a long (1 sec) pulse of saturating GABA (Fig. 1B, Table 1). Like deactivation, desensitization is shaped by multiple transition rates. In fact, desensitization depends on all of the same transitions that shape deactivation, except for the agonist-unbinding rate (i.e., the contribution of the unbound state becomes so small that it is as if the channel never unbinds). Therefore, the result that the mutation alters deactivation but not desensitization suggests that the β2-R207C mutation accelerates agonist unbinding but has no effect on channel gating.
Figure 1.
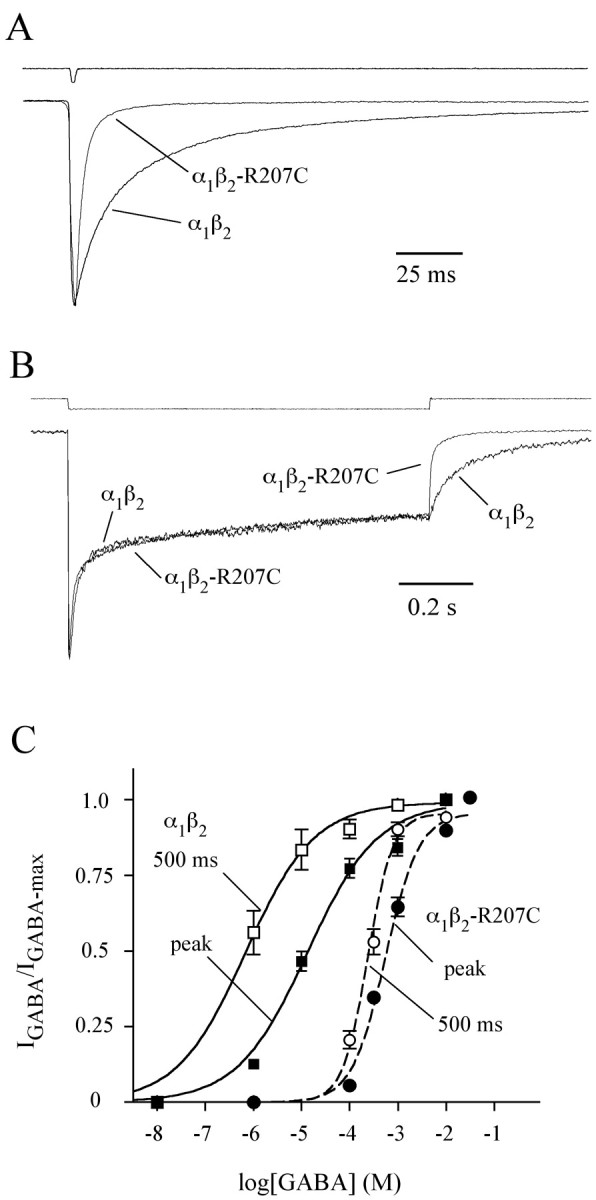
The mutationβ2-R207C increases both the rate of deactivation and GABA EC50 but does not affect desensitization. A, B, α1β2 or α1β2-R207C receptors were challenged with 2 msec (A) or 1 sec(B) pulses of GABA. All traces are peak normalized. Peak currents of traces shown varied between 65 and 150 pA. Because of differences in EC50, GABAconcentrations used on wild-type and R207C-containing receptors were 10 and 100 mm, respectively. Each trace is the average of between 25 and 200 sweeps, recorded while the patches were held at –60 mV. The top traces in both panels are recordings from open electrode tips made at the end of each experiment to evaluate the speed of solution exchange. C, GABA concentration–response curves for wild-type (solid lines, squares) and mutant (dashed lines, circles) receptors for 1 sec GABA pulses. The filled symbols show data generated by measuring peak GABA currents, and the open symbols show data from currents measured 500 msec after GABA application. The curves were generated by fitting the data with the equation IGABA/IGABA-max = Ymax/(1 + 10^(logEC50 – log[GABA])^N), where N is the Hill coefficient, using Prism software (GraphPad Software, San Diego, CA). EC50 and Hill coefficient values were: 14μm, 0.6 (wild-type peak); 0.7 μm, 0.6 (wild type, 500 msec); 561 μm, 1.2 (mutant peak); and 240 μm, 1.6 (mutant, 500 msec). All EC50 values were significantly different from each other except for the mutant at peak and mutant at 500 msec, which had overlapping 95% confidence intervals (generated by fitting software). All Hill coefficients had overlapping 95% confidence intervals.
Table 1.
Parameters from exponential fits to deactivation and desensitization and paired pulse recovery
|
Deactivation (2 msec pulse) |
Percentage of τ1 |
τ1 (msec) |
Percentage of τ2 |
τ2 (msec) |
Percentage of τ3 |
τ3 (msec) |
τw (msec) |
n |
| α1β2 | 53 ± 3 | 9.1 ± 0.5 | 35 ± 3 | 54 ± 5 | 12 ± 1 | 283 ± 28 | 53 ± 3 | 22 |
| α1β2(R207C) |
89 ± 2** |
2.8 ± 0.3** |
12 ± 3** |
45 ± 12 |
n/a |
n/a |
4.9 ± 0.5** |
15 |
| Desensitization (1 sec pulse) |
Percentage of τ1
|
τ1 (msec) |
Percentage of τ2
|
τ2 (msec) |
|
Extent (%) |
τw (msec) |
n
|
| α1β2 | 46 ± 3 | 15 ± 2 | 27 ± 3 | 630 ± 90 | 27 ± 3 | 260 ± 50 | 10 | |
| α1β2(R207C) |
52 ± 2 |
15 ± 1 |
25 ± 3 |
480 ± 70 |
|
24 ± 2 |
160 ± 25 |
14 |
| Paired pulse recovery |
Percentage of τ1
|
τ1 (msec) |
Percentage of τ2
|
τ2 (msec) |
|
Intercept (%) |
τw (msec) |
n
|
| α1β2 | 43 ± 3 | 133 ± 25 | 13 ± 5 | 3564 ± 545 | 43 ± 6 | 700 ± 78 | 7 | |
| α1β2(R207C) |
25 ± 4* |
43 ± 24** |
6 ± 2 |
1486 ± 662* |
|
69 ± 3** |
147 ± 32** |
5 |
All data are given as mean ± SEM. Significant differences between wild-type and mutant parameters were calculated using Student's t test (*p < 0.05; **p < 0.01). τw is the weighted time constant computed as Σai·τi/Σai where ai and τi are the amplitude and time constant of component i. Multi-exponential curve fits were performed with Axograph (deactivation and desensitization) or Prism (paired-pulse recovery).
Accelerated deactivation could be caused by more rapid unbinding of GABA or by reduction in mean channel open time, both of which would reduce the fraction of time spent in bound states and thus predict a rightward shift in the macroscopic dose–response curve. Alternatively, faster deactivation could be attributable to increased desensitization, which would increase the fraction of time in bound states and thus predict a leftward shift. Figure 1C illustrates that the β2-R207C mutation caused a 40-fold rightward shift in the dose–response curve for peak current and a 340-fold rightward shift for current measured at 500 msec. Therefore, the mutation reduces the fraction of time spent in bound states, consistent with an accelerated GABA-unbinding rate.
Another phenomenon that depends on the interplay between gating and binding is paired-pulse depression. When pairs of brief (2–3 msec) saturating GABA pulses were given to α1β2 receptors at variable intervals, the second pulse evoked a smaller peak response that recovered as the inter-pulse interval grew longer (Fig. 2A). In patches containing α1β2-R207C receptors, the depression of the second pulse was significantly reduced, and the receptors recovered more quickly than wild type (Fig. 2B,C). This result could imply that the desensitized state in the mutant receptor has a slower entry rate or a shorter lifetime. However, it is difficult to reconcile this interpretation with the fact that the mutation does not affect desensitization during long pulses (Fig. 1). Furthermore, it has been shown that paired-pulse depression of neuronal GABAA receptors is similarly attenuated when using β-alanine, an agonist that has a faster unbinding rate than GABA but does not affect gating (Jones and Westbrook, 1995; Jones et al., 1998). Therefore, reduced paired-pulse depression in α1β2-R207C receptors is also likely to be caused by an increase in the GABA-unbinding rate.
Figure 2.
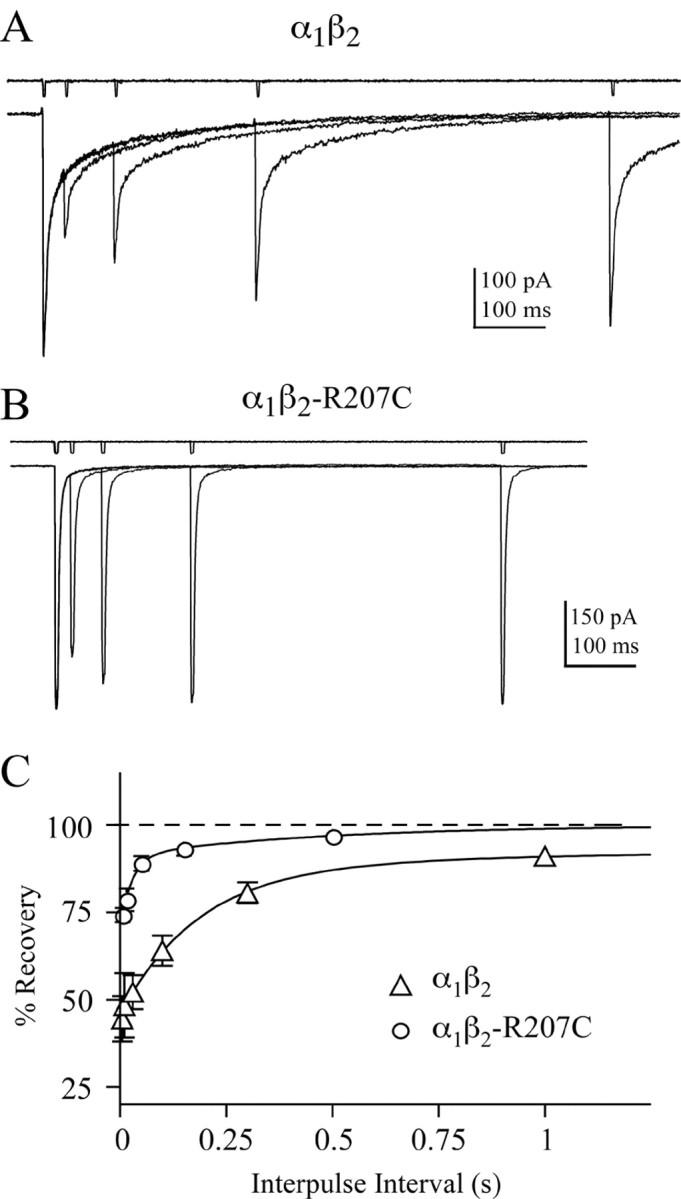
α1β2-R207C receptors recover more rapidly than wild-type after a short pulse of GABA. A, Pairs of GABA pulses (10 mm; 2–3 msec) with inter-pulse intervals (IPIs) that varied from 30 to 3000 msec were given to patches containing wild-type receptors. Five trials separated by 15 sec were interleaved, averaged, and normalized to the first response. B, Paired pulses of 100 mm GABA on α1β2-R207C receptors with inter-pulse intervals that varied from 5 to 1500 msec (5 trials separated by 10 sec). C, The percentage recovery [(Ipeak2 – Ionset)/(Ipeak1 – Ionset) × 100] is plotted as a function of inter-pulse interval. Ipeak1 and Ipeak2 are the amplitudes of the first and second responses in a pair, and Ionset is the current just before the start of the second response. Results for wild-type (open triangles) and β2-R207C (open circles)-containing receptors are plotted as mean ± SEM. The solid lines are fit to the equation; percentage recovery = 100 – 100 * [a1 exp (–IPI/τ1) + a2 exp (–IPI/τ2)], where a1 and τ1 are the amplitude and time constant of component i. The best fit values given as mean ± SEM are: α1β2, τ1(a1) = 0.13 ± 0.03 sec (0.43), τ2(a2) = 3.6 ± 0.5 sec (0.13), n = 7; α1β2-R207C, τ1(a1) = 0.043 ± 0.024 sec (0.25), τ2(a2) = 1.5 ± 0.6 sec (0.06), n = 5.
The mutation does not alter peak open probability or single-channel open times
The hypothesis that the β2-R207C mutation does not affect gating is supported by the fact that desensitization during long GABA pulses is indistinguishable between mutant and wild-type receptors. Alternatively, it is possible that the β2-R207C mutation causes precisely balanced shifts in multiple gating rates so that there is no obvious effect on macroscopic desensitization. We tested whether this might be the case by measuring the peak open probability in response to a saturating GABA pulse (Po-max). Like macroscopic desensitization, Po-max depends on the interplay of several gating processes. We measured Po-max by nonstationary variance analysis (Sigworth, 1980) and found no significant difference between mutant and wild-type receptors (Fig. 3), further supporting the idea that the β2-R207C mutation does not affect gating.
Figure 3.

Nonstationary variance analysis demonstrates that the mutation does not affect the peak probability of opening (Po-max). A, B, Superposition of 10 consecutive current responses from wild-type (A) or mutant (B) receptors to a 2 msec application of saturating GABA aligned with plots of the variance of the current at each point (>100 current responses were used for calculation of variance). Piezo-stimulus artifacts have been removed. C, Plots of current versus variance for wild-type (open circles; Po-max = 0.40 ± 0.08; n = 7) and mutant receptors (closed circles; Po-max = 0.46 ± 0.09; n = 10; mean ± SD). These values were not significantly different (Student's t test).
To further test whether the β2-R207C mutation affects gating, the channel closing rates (αn) were measured by single-channel recording. Open time distributions from single patches containing either α1β2 receptors (10 mm GABA) or α1β2-R207C receptors (100 mm GABA) are shown in Figure 4. There was no difference between open time histograms from wild-type and mutant receptors. In every case, open time distributions collected from a single patch were best fit with three exponential components consisting of two equally dominant open states with lifetimes of ≈ 0.4 and 1.3 msec and one rarely visited open state with a lifetime of ≈ 3.5 msec. Mean open times and relative areas were: for α1β2 receptors, 0.43 ± 0.15 msec, 0.5; 1.3 ± 0.2 msec, 0.46; and 3.2 ± 0.6 msec, 0.05 (six patches); and for α1β2-R207C receptors, 0.40 ± 0.09 msec, 0.54; 1.4 ± 0.2 msec, 0.40; and 3.8 ± 0.8 msec, 0.06 (six patches; mean ± SD). Wild-type and α1β2-R207C receptors both displayed two conductance states. Wild-type conductances were 14.6 ± 0.6 pS (20%) and 10.5 ± 0.8 pS (six patches). R207C conductances were 12.9 ± 0.5 pS (23%) and 9.3 ± 0.4 pS (five patches; mean ± SD). We do not believe that these minor, but significant (p < 0.01), differences are caused by the mutation. Rather, they most likely occur because of the lower chloride concentration in the mutant recording solutions ([Cl]wt, 90 mm; [Cl]mut, 40mm), which was reduced to accommodate the addition of 100 mm GABA (see Materials and Methods).
Figure 4.

The mutation does not alter single-channel open-time distributions. A, Single-channel recordings for wild-type and mutant receptors made from outside-out patches held at –80 mV in the presence of 10 mm (α1β2) or 100 mm (α1β2-R207C) GABA. Traces are sequential segments of a continuous record. B, C, Open-time histograms from one patch each for wild-type and mutant receptors fitted with the sum of three exponential functions by the maximum likelihood method. For theα1β2 histogram shown, the individual components had time constants (and relative areas) of 0.5 msec (0.47), 1.6 msec (0.50), and 4.2 msec (0.03). For theα1β2-R207C histogram shown, the individual components had time constants (and relative areas) of 0.6 msec (0.53), 1.8 msec (0.44), and 3.9 msec (0.03).
Mutation of β2-R207 reduces the binding rate of GABA
When comparing α1β2 and α1β2-R207C receptors, we could not detect any change in gating by examining desensitization, Po-max, or mean open times. Because the mutation does not affect gating, it appears that the observed increase in deactivation rate and reduction in paired-pulse inhibition must be attributable to an increase in the GABA-unbinding rate (koff-GABA). One might expect a binding site mutation that effects the unbinding rate to simultaneously alter the binding rate (kon-GABA) (Jones et al., 1998). However, none of the data presented thus far can provide a measure of kon-GABA.
To measure kon-GABA, we performed experiments in which the competitive antagonist SR-95531 and GABA were simultaneously applied and allowed to compete with each other to occupy the receptor. In this “race” experiment, the percentage of channels occupied by each ligand at the peak of current depends on their relative binding rates. Therefore, if the SR-95531-binding rate (kon-SR) is known, the ratio of the peak current in GABA alone to the peak current in GABA plus SR-95531 (Irace) can be used to calculate kon-GABA (Clements et al., 1992; Jones et al., 1998). We determined kon-SR using the equation kon-SR = KD-SR/koff-SR, where KD-SR and koff-SR (the dissociation constant and microscopic unbinding rate for SR-95531) were directly measured using the deconvolution method developed by Jones et al. (2001). This method exploits the fact that, after equilibration in competitive antagonist, the current evoked by a jump into GABA is shaped by the convolution of the time course of antagonist unbinding and the waveform of the control current (evoked with no pre-equilibration in antagonist). Therefore, the time course of antagonist unbinding can be obtained by deconvolving the control current from the current recorded after antagonist equilibration (Fig. 5A–C). The time course of antagonist unbinding is fit with an exponential function giving koff-SR and the percentage of receptors occupied by antagonist at equilibrium (see Fig. 5 legend). KD-SR is obtained by repeating this experiment with different concentrations of SR-95531 and plotting the antagonist occupancy versus concentration. We found that the mutation had no effect on the binding rate, unbinding rate, or dissociation constant for SR-95531 [α1β2: koff-SR = 11 ± 3 sec–1, KD-SR = 210 nm, kon-SR = (5.5 ± 1.7) × 107 M–1s–1, n = 11; α1β2-R207C: koff-SR = 16 ± 6 sec–1, KD-SR = 180 nm, kon-SR = (8.9 ± 3.4) × 107 M–1s–1, n = 11; mean ± SD].
Figure 5.

β2-R207C reduces the GABA-binding rate. A,B, Competitive antagonist unbinding slows the rise of agonist activated current. Currents were evoked by 10 mm (wild type; A) or 100 mm (mutant; B) GABA alone in outside-out patches with and without pre-equilibration in 1μm SR-95531. The unbinding rate of the competitive antagonist can be extracted from the data shown in A and B by deconvolving the control and experimental responses. C, Plots showing the deconvolution of currents with antagonist pre-equilibration from those without pre-equilibration. The data shown is the mean ± SEM of multiple deconvolutions (wild type, n = 5, mutant, n = 4). The solid lines are curves fit to the data resulting from the deconvolutions with the equation A(t) = [P∞ – (P∞ – P0)e – t/τunbind]N, where A(t) is the fraction of receptors not occupied by SR-95531, P0 and P∞ are the probabilities of being available initially (at t = 0) and at steady state (t =∞), τunbind is the time constant of antagonist unbinding at each site, and N is the number of binding sites. Best fits were always obtained with N = 1. D, A race experiment (see Results) on a patch containing wild-type receptors in which the current evoked by simultaneous application of 1 mm GABA and 100 μm SR-95531 is compared with the current evoked by 1 mm GABA alone. E, A race experiment on mutant receptors using 10 mm GABA and 100μm SR-95531. The current ratios from D and E were used to compute kon-GABA, using Equation 12 from Jones et al. (1998). Inset traces in D and E are the result of simulations of the race experiments shown using the model and rate constants from Figure 6.
However, during race experiments that measure the ratio of current activated by coapplication of GABA and SR-95531 to that activated by GABA alone, α1β2-R207C receptors required ∼10-fold higher concentrations of GABA to achieve the same current ratio (Irace) observed in α1β2 receptors (Fig. 5D,E). This difference indicates that the binding rate of GABA, relative to that of SR-95531, is much slower in mutant than in wild-type receptors. From these Irace values and the SR-95531-binding rates measured above, we computed kon-GABA using Equation 12 from Jones et al. (1998) and found that the mutation caused an eightfold decrease in the GABA-binding rate [α β = (6.85 ± 2.8) × 106 M–1s–1, n = 14; α1β2-R207C = (0.8 ± 0.3) × 106 M–1s–1, n = 13; mean ± SD; < 0.001).
An interesting result observed during these experiments is that when GABA and SR-95531 were coapplied, there was an apparent increase in the rate of macroscopic desensitization that was more pronounced in the mutant receptor. It is possible that SR-95531 causes this effect by altering receptor gating. However, a simpler explanation is that as the system goes to equilibrium, some receptors unbind GABA, allowing SR-95531 to occupy a larger percentage of receptors than it did during the peak current. To test this idea, we simulated race experiments for both wild-type and mutant receptors using a model in which SR-95531 acts as a pure competitive antagonist and has no effect on GABA-mediated gating (see model and rates in Fig. 6). As seen in actual currents, the presence of antagonist increased apparent desensitization of simulated currents (Fig. 5D,E, insets), demonstrating that this phenomenon occurs even if SR-95531 has no effect on channel gating.
The effects of the mutation can be explained solely by altering binding and unbinding rates
If, as the data suggest, mutation of β2-R207 affects only agonist binding and unbinding, then a kinetic model that describes α1β2 receptors should be convertible into one that describes α1β2-R207C receptors simply by adjusting kon-GABA and koff-GABA while leaving all other rates unchanged. A kinetic model that accurately describes the behavior of α1β2 receptors was developed using the model shown in Figure 6A (Jones et al., 1998). Before optimization, kon was fixed to its measured rate, α1 was set to the fastest measured closing rate, and α2 was set to the intermediate closing rate. The longest open state, which accounts for ∼5% of all openings, was ignored. The remaining rate constants were allowed to vary while the model was optimized to simultaneously fit responses to short, long, and paired pulses of saturating GABA that were scaled to the measured value of Po-max (see Materials and Methods). The optimized rate constants are shown in Figure 6B, and simulated currents generated with this model are overlaid on actual patch responses in Figure 6C. The model was then fit to currents recorded from α1β2-R207C with kon-GABA constrained to the value measured for mutant receptors. After optimization, the only unconstrained rate constant that was significantly different between the mutant and wild-type models was koff-GABA, which shifts from 19 sec–1 in wild-type receptors to 400 sec–1 in mutant receptors. As an independent check on the quality of the models, we simulated dose–response curves, which had not been used during optimization. The resulting EC50 values were: α1β2, 12 μm at peak and 0.8 μm at 500 msec; α1β2-R207C, 620 μm at peak and 180 μm at 500 msec. In all cases, these numbers are within 25% of the actual values (10% on average), verifying that these models are good predictors of receptor behavior. In summary, all kinetic effects of the β2-R207C mutation can be explained solely by an eightfold decrease in kon-GABA coupled with a 20-fold increase in koff-GABA.
Discussion
Determining whether an amino acid residue is involved in ligand binding or channel gating through mutational analysis has proven to be a nontrivial problem. Mutational effects are commonly quantified by shifts in EC50 or KD. However, because these measures depend on both binding and gating, it is often impossible to distinguish between alterations in binding or gating using them (Colquhoun, 1998). Changes in maximum open probability are potentially useful for diagnosing gating changes. Unfortunately, the notoriously variable expression in heterologous systems makes it difficult to control for the number of receptors, so that only very large (>10-fold) shifts in peak current are reliably detectable. Despite these difficulties, techniques have been developed that can potentially identify binding or gating changes using EC50 and peak current measurements. For example, the relative efficacy of a partial agonist has been used as an internal control for receptor expression, and changes in relative efficacy have thus been interpreted as being caused by gating changes (Maksay et al., 2000; O'Shea and Harrison, 2000; Wagner and Czajkowski, 2001). In contrast, evidence for binding changes has included: (1) a change in the potency of GABA that is not accompanied by a change in the potency of a modulator that opens the channel but does not bind at the GABA-binding site, and (2) large changes in GABA potency in the absence of large changes in maximal current (Amin and Weiss, 1993). Although these methods can suggest that a change in binding or gating has occurred, they cannot rule out whether both have changed or reveal which microscopic transitions were affected.
The limitations inherent in using dose–response curves can be overcome by adopting methods that explicitly relate a structural change to changes in elementary functional properties, namely the conformational states of the receptor and the transition rates between them. To this end, we applied single-channel recording, rapid ligand application, and kinetic modeling to quantify the effects of mutating a predicted binding site residue on individual binding and gating processes. Our results lead to three conclusions: the β2-R207C mutation (1) has no effect on gating, (2) greatly increases the likelihood that the GABA molecule will leave the binding site, and (3) greatly reduces the likelihood that GABA will bind in the first place.
Mutation of β2-R207 does not affect gating
Multiple lines of evidence indicate that the mutation does not effect gating. Macroscopic desensitization and Po-max, both of which depend on the interplay of multiple gating processes, are not affected by the β2-R207C mutation. It is possible that a mutation could induce a balanced shift in multiple microscopic gating rates so that neither of these macroscopic parameters appears to be affected. However, because individual transition rates influence desensitization and Po-max to different degrees, it seems unlikely that alterations in multiple gating rates would exactly balance out so that both of these processes appear unchanged. In addition, mutant and wild-type receptors have indistinguishable single-channel open time distributions, demonstrating that the channel-closing rate is unaffected. This conclusion, of course, assumes that the measured events are actual openings and not bursts that appear to be openings because their closures are too brief to measure. Because the small conductance of these channels necessitated filtering at 1–2 kHz, the distributions in Figure 4 probably do contain at least some bursts. Ironically, this potential source of error further strengthens our conclusion that gating is not affected by the mutation. If the distributions are contaminated by bursts, yet are unaffected by the mutation, then the burst durations must be unaffected. Because burst duration depends on multiple gating steps (e.g., opening, closing, and desensitization) rather than merely channel closure, unchanged burst distributions imply that none of the intraburst-gating steps are affected.
We previously reported that this mutation affects gating as well as binding (Wagner and Czajkowski, 2001). This conclusion was reached by observation, in oocytes, that the mutation altered the relative efficacy of the partial agonist piperidine-4-sulfonic acid (P4S; α1β2: Ipeak-P4S/Ipeak-GABA = 0.5; α1β2-R207C: Ipeak-P4S/Ipeak-GABA = 0.21). This result conflicts with the present finding that the mutation has no effect on gating. It could be that the mutation only alters P4S-mediated gating but not GABA-mediated gating. However, a simpler explanation is that the apparent efficacy change in oocytes is actually an artifact of slow solution exchange. When agonist is rapidly applied, the resulting current peaks within the first few milliseconds. However, in a typical oocyte experiment, the GABA concentration probably requires more than 1 sec to reach peak [Wagner and Czajkowski (2001) used a flow rate of 70 μl/sec and a bath volume of 100 μl, which predicts an exchange time constant of 1.4 sec, assuming good mixing]. The simulations in Figure 7, A and B, show that slow application results in severe underestimation of peak current and falsely indicates a reduction in peak open probability for α1β2-R207C receptors, effects not observed in rapid application experiments. In addition, simulations of slow application using the increased unbinding rate, but no gating changes as observed in the present study, showed a false change in the relative efficacy of P4S (Fig. 7C) (α1β2: Ipeak-P4S/Ipeak-GABA = 0.5; α1β2-R207C: Ipeak-P4S/Ipeak-GABA = 0.33). In other words, a change in the relative efficacy of a partial agonist can lead to erroneous conclusions about gating if solution exchange is not sufficiently rapid (i.e., milliseconds or less for fast ligand-gated channels). Therefore, our previous prediction that β2-R207C affected gating was an error, probably because of the fact that slow ligand application precludes accurate measurement of efficacy or even relative efficacy.
Figure 7.

Slow agonist application can result in erroneous conclusions about the effects of mutations on gating. A, The model and rate constants from Figure 6 were used to simulate current responses to a fast step of saturating GABA (α1β2, 1mm; α1β2-R207C, 30 mm), as used in the present study. As expected for identical gating between wild type and mutant, the currents are essentially identical. B, The same model and rates were used to simulate a slow exponentially rising application of GABA, as used in oocyte experiments, with a time constant of 1 sec and a final concentration of 1 mm forα1β2 and 30 mm forα1β2-R207C. The mutant yields a smaller peak than wild type if the agonist is not applied rapidly, erroneously suggesting that gating has been altered. C, Simulations of current responses to GABA and P4S using slow agonist applications as shown in B. The partial agonism of P4S was modeled as a reduction in mean channel open time, but wild-type and mutant gating in P4S were identical. However, the simulated P4S/GABA current ratio is smaller for the mutant, again falsely suggesting that the mutant alters gating. The concentrations of P4S used were 1 mm for α1β2 receptors and 3 mm forα1β2R207C receptors. The agonist concentrations used here are the same as those used by Wagner and Czajkowski (2001) and are at least 20-fold greater than the EC50 value measured in oocytes for each receptor–agonist concentration.
β2-R207C alters both GABA binding and unbinding
Because deactivation is accelerated in α1β2-R207C receptors while gating is unaltered, the mutation must increase the microscopic unbinding rate. An increased unbinding rate is also supported by the result that mutant receptors are less susceptible to, and recover more quickly from, paired-pulse depression. An increase in unbinding rate would have this effect by reducing the opportunity for the receptor to enter a desensitized state after the channel closes. Finally, simulations demonstrate that an increase in koff-GABA paired with a decrease in kon-GABA are the only changes necessary to account for the kinetic effects of the mutation.
The case for a mutation-induced decrease in the GABA-binding rate is even more straightforward, because this rate was measured in race experiments. However, there are some assumptions that must be true in order for the Irace method of determining kon-GABA to give accurate values. The original equation used to calculate kon-GABA from Irace and kon-SR (Jones et al., 1998) was derived under the assumption that there is only one binding site for SR-95531, and that SR-95531 binding precludes GABA binding at that site only. Under that scheme, if there are two GABA-binding sites, one molecule of GABA and one of SR-95531 could be bound at the same time, and the channel would be prevented from opening. The assumption of a single SR-95531-binding site was supported by Jones et al. (1998, 2001). In contrast, it remains unknown whether or not SR-95531 and GABA can co-occupy the receptor. In fact, the kinetic model used here does not allow co-occupancy (Fig. 6A). When the race equation is rederived under this assumption, the resulting kon-GABA is half of the value obtained from the original equation, because GABA has two chances to bind for every chance had by SR-95531. The resulting simulations produced EC50 values that agreed with the experimentally determined values to within 10% on average (see Results). In contrast, the alternate assumption, that SR-95531 and GABA can co-occupy the receptor, resulted in a five-fold larger average discrepancy. These findings imply that binding of one SR-95531 molecule precludes GABA binding at both sites, not only to the site occupied by the antagonist. Because the two GABA-binding sites probably do not overlap physically, this mechanism requires some sort of allosteric exclusion between the two sites. However, such allosteric exclusion in no way contradicts the idea that SR-95531 is competitive, because competition is formally defined as mutually exclusive binding of agonist and antagonist, regardless of the physical arrangement of their binding sites (Segel, 1976).
Another assumption that must hold in order for the race equation to accurately estimate kon-GABA is that any given receptor only binds agonist or antagonist once before the current peaks. This holds in wild-type receptors for which GABA and SR-95531 both have relatively slow unbinding rates. However, GABA unbinds from α1β2-R207C receptors much more quickly, which may give SR-95531 extra opportunities to compete with the agonist before the current peaks. In this case, the race equation yields a lower limit on kon-GABA. Fortunately, only ∼5% of singly bound GABA receptors will unbind rather than binding a second GABA or entering gating steps [i.e., koff/(koff + [GABA]kon + β1 + d1)], and of those, only ∼30% will unbind before the peak of current (τoff, 2.4 msec; time of peak, ≈1 msec), approximately one-third of which will then bind SR-95531. Therefore, we expect rapid unbinding in the mutant receptor to cause an insignificant (<1%) underestimate of kon-GABA.
In neuronal GABAA receptors, decreasing the length of full agonists decreases affinity by simultaneously speeding unbinding and slowing binding (Jones et al., 1998). This correlation between length and affinity was explained by a simple physical model, in which the binding site must undergo an energy-requiring motion during binding. The affinity reduction of β2-R207C receptors is also attributable to a simultaneous speeding of unbinding and slowing of binding, exactly as would be predicted by that model if removing the arginine side chain effectively expands the binding site making GABA appear smaller in comparison. The model predicts a similar effect if, instead of expanding the binding site, removing the side chain reduced the depth of the energy well associated with components of the binding site that directly interact with the GABA molecule. In contrast, antagonist binding and unbinding kinetics are not closely correlated with each other, suggesting that separate forces (hence separate structures) influence agonist and antagonist affinity (Jones et al., 2001). Consistent with this idea, the β2-R207C mutation had no effect on SR-95531 binding or unbinding kinetics.
β2-R207 may directly interact with the GABA molecule
The mutation β2-R207C affects binding and unbinding rates but does not affect gating. One might expect this very profile when mutating a residue that directly stabilizes GABA in the binding pocket. If there is a direct interaction between GABA and β2-R207, it likely occurs as a favorable electrostatic interaction between the positively charged guanido group at the end of the arginine side chain and the negatively charged carboxylate group on the GABA molecule. However, if β2-R207 were solely responsible for stabilizing the carboxylate group, we would then expect the cysteine mutation to completely abolish GABA binding. Therefore, other residues, and possibly other arginines, must also be involved. Indeed, such an interaction has already been postulated to occur between the arginine at position 66 of the α1 subunit and the GABA carboxylate group (Holden and Czajkowski, 2002). Like β2-R207, α1R66 faces into the binding pocket, and its mutation to cysteine causes a 300-fold rightward shift in GABA-EC50. There is also a third arginine, at position 131 of the α1 subunit, that faces into the binding pocket and the mutation to cysteine of which causes a 70-fold shift in GABA-EC50 (Holden and Czajkowski, 2004). Because mutation of each of these arginines similarly reduces GABA affinity, although no single mutant completely abolishes binding, all three of these residues may collaborate to stabilize the GABA carboxylate group. It might seem surprising that side chains from different subunits would share an interaction with the same end of the GABA molecule. Nonetheless, this hypothesis appears to be supported by a molecular model of the extracellular domain of the GABAA receptor derived from the crystal structure of the homologous acetylcholine-binding protein (Brejc et al., 2001; Cromer et al., 2002; Holden and Czajkowski, 2002). In this model, β2-R207, α1-R66, and α1-R131 can come together to form a triangle of positive charge that spans a narrow part of the agonist-binding pocket.
We propose that this “crown of arginines” forms a region of positive charge that accommodates the negatively charged carboxylate group of the GABA molecule. Such an interaction would explain why single mutations among these three arginines reduce affinity but do not abolish binding altogether. This arrangement would imply that the GABA molecule is oriented vertically within the binding site (i.e., perpendicular to the plane of the plasma membrane), with the amino group facing away from the arginine crown (Holden and Czajkowski, 2002). In addition, antagonists such as SR-95531 have bulky aromatic groups attached to the amino end of the GABA-like moiety, which would also face away from the arginine crown, allowing these bulky groups to interact with structures outside the core of the binding site. This could explain why mutations in the arginine crown have little effect on SR-95531 interactions, whereas some mutations above (e.g., α1-R119C) or below (e.g., α1-D62C) the core of the binding site can be protected from interacting with sulfhydryl-specific reagents by SR-95531 but not by GABA (Holden and Czajkowski, 2001, 2004). In contrast, kinetic studies suggest that competitive antagonist affinity is dominated by hydrophobic interactions (Jones et al., 2001), consistent with the large increases in SR-95531 EC50 caused by mutation of aromatic residues α1-F64, β2-Y97, or β2-Y205 (Wagner and Czajkowski, 2001; Boileau et al., 2002; Holden and Czajkowski, 2002). These aromatic residues may help to stabilize the crown against electrostatic repulsion between arginines, possibly through cation-pi interactions. Similar bonds with the amino group of GABA and antagonists, and additional hydrophobic bonds with antagonists, might contribute to binding of these ligands. Additional studies on the microscopic kinetics of binding site mutations will be a valuable complement to existing structural methods in piecing together the interactions between ligands and the GABAA receptor.
Footnotes
This work was supported by Epilepsy Foundation Award (M.J.), Fellowship (D.A.W.), and Grant NS34727 (C.C.).
Correspondence should be addressed to David A. Wagner, Department of Biological Sciences, Marquette University, P.O. Box 1881, Milwaukee, WI 53201-1881. E-mail: david.wagner@marquette.edu.
DOI:10.1523/JNEUROSCI.4316-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/242733-09$15.00/0
References
- Amin J, Weiss DS (1993) GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature 366: 565–569. [DOI] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL (2001) Mutation of the 9′ leucine in the GABA(A) receptor gamma2L subunit produces an apparent decrease in desensitization by stabilizing open states without altering desensitized states. Neuropharmacology 41: 737–744. [DOI] [PubMed] [Google Scholar]
- Boileau AJ, Evers AR, Davis AF, Czajkowski C (1999) Mapping the agonist binding site of the GABAA receptor: evidence for a β-strand. J Neurosci 19: 4847–4854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boileau AJ, Newell JG, Czajkowski C (2002) GABA(A) receptor beta 2 Tyr97 and Leu99 line the GABA-binding site. Insights into mechanisms of agonist and antagonist actions. J Biol Chem 277: 2931–2937. [DOI] [PubMed] [Google Scholar]
- Brejc K, van Dijk WJ, Klaassen RV, Schuurmans M, van Der Oost J, Smit AB, Sixma TK (2001) Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature 411: 269–276. [DOI] [PubMed] [Google Scholar]
- Burkat PM, Yang J, Gingrich KJ (2001) Dominant gating governing transient GABA(A) receptor activity: a first latency and Po/o analysis. J Neurosci 21: 7026–7036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celentano JJ, Wong RKS (1994) Multiphasic desensitization of the GABAA receptor in outside-out patches. Biophys J 66: 1039–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y, Weiss D (1999) Channel opening locks agonist onto the GABAC receptor. Nat Neurosci 2: 219–225. [DOI] [PubMed] [Google Scholar]
- Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL (1992) The time course of glutamate in the synaptic cleft. Science 258: 1498–1501. [DOI] [PubMed] [Google Scholar]
- Colquhoun D (1998) Binding, gating, affinity and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol 125: 924–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG (1995a) The principles of the stochastic interpretation of ion-channel mechanisms. In: Single-channel recording (Sakmann B, Neher E, eds), pp 397–482. New York: Plenum.
- Colquhoun D, Hawkes AG (1995b) A Q-matrix cookbook. How to write only one program to calculate the single-channel and macroscopic predictions for any kinetic mechanism. In: Single-channel recording (Sakmann B, Neher E, eds), pp 589–633. New York: Plenum.
- Colquhoun D, Sigworth FJ (1995) Fitting and statistical analysis of single-channel records. In: Single-channel recording, Ed 2 (Sakmann B, Neher E, eds), pp 483–587. New York: Plenum.
- Connolly CN, Krishek BJ, McDonald BJ, Smart TG, Moss SJ (1996) Assembly and cell surface expression of heteromeric and homomeric gammaaminobutyric acid type A receptors. J Biol Chem 271: 89–96. [DOI] [PubMed] [Google Scholar]
- Cromer BA, Morton CJ, Parker MW (2002) Anxiety over GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci 27: 280–287. [DOI] [PubMed] [Google Scholar]
- Galzi J-LC, Changeux J-P (1994) Neurotransmitter-gated ion channels as unconventional allosteric protiens. Curr Opin Struct Biol 4: 554–565. [Google Scholar]
- Haas KF, Macdonald RL (1999) GABAA receptor subunit gamma2 and delta subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol (Lond) 514: 27–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holden JH, Czajkowski C (2001) Mapping agonist and antagonist binding site subdomains in the α1M113-α1T125 region of the GABAA receptor. Soc Neurosci Abstracts 31: 35.37. [Google Scholar]
- Holden JH, Czajkowski C (2002) Different residues in the GABA(A) receptor alpha 1T60-alpha 1K70 region mediate GABA and SR-95531 actions. J Biol Chem 277: 18785–18792. [DOI] [PubMed] [Google Scholar]
- Holden JH, Czajkowski C (2004) α1Gly124-α1Leu132: a novel binding site region on the GABAA receptor that undergoes distinct conformational rearrangements during ligand binding and allosteric modulation. Soc Neurosci Abstr 34: 50.1. [Google Scholar]
- Jones MV, Westbrook GL (1995) Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 15: 181–191. [DOI] [PubMed] [Google Scholar]
- Jones MV, Sahara Y, Dzubay JA, Westbrook GL (1998) Defining affinity with the GABAA receptor. J Neurosci 18: 8590–8604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MV, Jonas P, Sahara Y, Westbrook GL (2001) Microscopic kinetics and energetics distinguish GABA(A) receptor agonists from antagonists. Biophys J 81: 2660–2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie AM, Twyman RE (1996) Direct evidence for diazepam modulation of GABAA receptor microscopic affinity. Neuropharmacology 35: 1383–1392. [DOI] [PubMed] [Google Scholar]
- Maksay G, Thompson SA, Wafford KA (2000) Allosteric modulators affect the efficacy of partial agonists for recombinant GABA(A) receptors. Br J Pharmacol 129: 1794–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell JG, Czajkowski C (2003) The GABAA receptor alpha 1 subunit pro174-Asp191 segment is involved in GABA binding and channel gating. J Biol Chem 278: 13166–13172. [DOI] [PubMed] [Google Scholar]
- O'Shea SM, Harrison NL (2000) Arg-274 and Leu-277 of the gamma-aminobutyric acid type A receptor alpha 2 subunit define agonist efficacy and potency. J Biol Chem 275: 22764–22768. [DOI] [PubMed] [Google Scholar]
- Scheller M, Forman SA (2002) Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABAA receptors. J Neurosci 22: 8411–8421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segel I (1976) Biochemical calculations, Ed 2, pp 246–252. New York: Wiley.
- Sigel E, Baur R, Kellenberger S, Malherbe P (1992) Point mutations affecting antagonist affinity and agonist dependent gating of GABAA receptor channels. EMBO J 11: 2017–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ (1980) The variance of sodium current fluctuations at the node of Ranvier. J Physiol (Lond) 307: 97–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GB, Olsen RW (1994) Identification of a [3H]muscimol photoaffinity substrate in the bovine gamma-aminobutyric acidA receptor alpha subunit. J Biol Chem 269: 20380–20387. [PubMed] [Google Scholar]
- Twyman RE, Rogers CJ, Macdonald RL (1990) Intraburst kinetic properties of the GABAA receptor main conductance state of mouse spinal chord neurons in culture. J Physiol (Lond) 423: 193–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner DA, Czajkowski C (2001) Structure and dynamics of the GABA binding pocket: a narrowing cleft that constricts during activation. J Neurosci 21: 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westh-Hansen SE, Rasmussen PB, Hastrup S, Nabekura J, Noguchi K, Akaike N, Witt MR, Nielsen M (1997) Decreased agonist sensitivity of human GABA(A) receptors by an amino acid variant, isoleucine to valine, in the alpha1 subunit. Eur J Pharmacol 329: 253–257. [PubMed] [Google Scholar]
- Westh-Hansen SE, Witt MR, Dekermendjian K, Liljefors T, Rasmussen PB, Nielsen M (1999) Arginine residue 120 of the human GABAA receptor alpha 1, subunit is essential for GABA binding and chloride ion current gating. NeuroReport 10: 2417–2421. [DOI] [PubMed] [Google Scholar]