

Abstract
Inhibitors of the isozyme cyclooxygenase-2 (COX-2) represent an important advance in pain management, although where and when these inhibitors can exert their antihyperalgesic actions are not completely understood. Here we show that unlike many peripheral tissues in which COX-2 is only expressed in physiologically significant levels after tissue injury, in the normal rat lumbar spinal cord, the majority of neurons and radial glia constitutively express high levels of COX-2 protein. Immediately after peripheral tissue injury and before any measurable upregulation of COX-2 protein in peripheral tissue or spinal cord, inhibition of constitutively expressed spinal COX-2 reduced injury-induced activation of primary afferent neurons, activation of spinal neurons, and the mechanical and thermal hyperalgesia that normally occurs after peripheral tissue injury. The present data demonstrate that constitutively expressed spinal COX-2 plays an important role in the initial hyperalgesia that follows peripheral tissue injury. These results suggest that blocking constitutive spinal COX-2 before tissue injury may reduce the initial peripheral and central sensitization that occurs after tissue injury.
Keywords: nociception, prostaglandin, spinal cord, NSAID, primary afferent, rat
Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs) are among the most commonly used drugs worldwide to treat pain and inflammation; however, just when and where NSAIDs can exert their antihyperalgesic actions have not been completely defined (Kalgutkar and Zhao, 2001). Local injury yields the release of a variety of factors that alter sensitivity of the primary afferent fibers innervating the damaged tissue, and among these factors that are important components of this peripheral sensitization are prostaglandins (Schaible and Grubb, 1993). Prostaglandins are lipidic acids that are locally synthesized by the cyclooxygenase enzymes after focal tissue injury and that sensitize peripheral nerve endings and enhance pain behavior in both animals and humans (Masferrer et al., 1994; Vane et al., 1994). Sensitization of nociceptors is initiated in part by prostaglandins binding to and activating prostanoid receptors expressed by nociceptors, which then activate adenylate cyclase and phospholipase C, leading to enhanced protein kinase C activity and increased levels of cAMP (Ahmadi et al., 2002).
In the present report, we focus on the role that constitutive spinal cyclooxygenase-2 (COX-2) may play in driving the initial hyperalgesia that occurs after peripheral tissue injury or NMDA activation of spinal neurons. Here we demonstrate that most neurons and radial glia in the rat spinal cord constitutively express high levels of COX-2 protein, and after significant tissue injury, COX-2 is involved in the early development of hyperalgesia. The mechanism by which constitutive spinal COX-2 generates this hyperalgesia appears to involve COX-2-mediated synthesis of prostaglandins, which both sensitizes the terminals of primary afferent fibers that terminate in the spinal cord and directs activation of spinal cord neurons. These results suggests that early blockade of constitutively expressed spinal COX-2 may reduce the early events in the spinal cord that have been reported to be involved in the generation and maintenance of a chronic pain state (Julius and Basbaum, 2001).
Materials and Methods
Intrathecal catheterization. For intrathecal injection, anesthetized male Holtzman rats received lumbar intrathecal catheters after an incision through the atlantooccipital membrane, and a stretched polyethene 10 tube was inserted into the intrathecal space and passed 8.5 cm so that the caudal end of the tubing localized to the lumbar enlargement. Aftera3d postoperation recovery period, rats were treated with 30 μg/10 μl COX-1 or COX-2 inhibitor (SC560 or SC58125, respectively; Amersham Biosciences, Arlington Heights, IL) or 10 μl of vehicle (DMSO; Sigma, St. Louis, MO) delivered via this catheter. Animals received this drug or vehicle at time (T) –10 min before mechanical pinch, crush, or intrathecal NMDA (0.3 μg/10 μl).
Mechanical stimulation. For paw pinch and crush, the rat was briefly anesthetized with isoflurane (2.0%) to the point of losing the withdrawal reflex. Pinch was achieved by compression of plantar skin for 30 sec with a hemostat being closed to before the first notch. In an unanesthetized rat, this compression evokes a prominent withdrawal but no evident disruption of the skin or underlying tissue. Crush was achieved by the application a hemostat across the long axis of the paw and closing the hemostat to the first notch, resulting in visible paw edema. The extent of edema was assessed using a mechanical caliper (Stoelting, Wood Dale, IL).
Tactile allodynia was assessed using a modified version of the up–down method of application of calibrated von Frey hairs (Chaplan et al., 1994). Briefly, rats were allowed to acclimatize for 15 min in a clear plastic cage with a wire mesh bottom. The 50% paw withdrawal threshold was determined with a series of von Frey filaments (Stoelting) beginning with a buckling weight of 2.0 gm. The filaments were applied to the plantar surface of the hindpaw. Paw lifting was considered a positive response and prompted the application of the next weaker filament. The next stronger filament was used when application of the filament for 5 sec did not elicit a withdrawal response. Values were calculated as described (Luo et al., 2001) and represented as mean ± SEM.
Thermal escape latencies were assessed using a radiant heat stimulation device, which has been previously described (Dirig et al., 1997). The device consists of a glass surface heated to 30°C by a focused projection bulb located immediately below the glass surface. The rats were placed on the glass surface 15 min after intrathecal NMDA (T = 0), and the stimulus was delivered separately to either hindpaw, and paw withdrawal latencies (PWLs) were assessed at T = 0, 15, 30, 60, and 90 min. These values were expressed as mean ± SEM PWL at each time point.
Tissue preparation and immunohistochemical analysis. After mechanical stimulation, anesthetized rats were perfused via the ascending aorta with 500 ml of 0.1 m PBS, pH 7.4 (22°C). The spinal cord was then quickly dissected, blocked, frozen to –20°C, cut on a cryostat at 15 μm, and mounted on gel-coated slides, all within a 2 hr period. Sections were rinsed in PBS, postfixed in 4% formalin (20 min, 22°C), exposed for 30 min (22°C) to blocking solution (1.0% normal donkey serum and 0.3% Triton X-100 in PBS), and incubated overnight with primary antibody (COX-2; Santa Cruz Biotechnology, Santa Cruz, CA; 1:250; neuron-specific nuclear protein (NeuN), Chemicon, Temecula, CA; 1:150; c-Fos, Santa Cruz Biotechnology; 1:30,000; or neurokinin-1 (NK-1), polyclonal; raised in our laboratory; 1:5000) in the blocking solution. After incubation with the primary antibody, the tissue sections were washed three times for 10 min each at 22°C in PBS, pH 7.4. Cy3-conjugated donkey anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA; 1:600) was added to the secondary incubation solution (1.0% normal donkey serum and 0.3% Triton X-100 in PBS) and applied to the tissue for 2 hr at 22°C. Finally, the tissue sections were washed three times for 10 min each at 22°C in PBS, pH 7.4, dehydrated in ethanol gradient (70, 90, and 100%), cleared in xylene, and coverslipped with DPX (Fluka, Buchs, Switzerland) mounting media.
Quantification of the immunohistochemical results was performed on 60-μm-thick spinal tissue sections from the lumbar level L4. Neurons expressing c-Fos or dendrites demonstrating NK-1 receptor internalization were counted in 10 randomly selected L4 coronal sections per animal. Dendritic NK-1 internalization was used for quantification of the effects of intrathecal NMDA because NK-1 internalization was predominantly noted on the dendrites. For both c-Fos and NK-1 internalization counts, coronal sections were viewed through a 1 cm2 eyepiece grid divided into one hundred 1 × 1 mm units. NK-1-positive dendrites that exhibited NK-1 internalization were counted and expressed as the mean number of NK-1 internalized dendrites per 900 μm2 in laminae I and II of a 60-μm-thick section. An NK-1-IR endosome was defined as an intense NK-1-IR intracellular organelle between 0.1 and 0.7 μm in diameter that was clearly not part of the external plasma membrane.
Confocal microscopy using an MRC-1024 confocal imaging system (Bio-Rad, Hercules, CA) was performed to obtain images as previously described (Mantyh et al., 1997).
Western blot. To determine COX-2 expression levels, Western blot analysis was performed on spinal cords that were harvested fresh, at the specified time points, in 50 mm Tris buffer containing 0.5% Triton X-100, 150 mm NaCl, 1 mm EDTA, and protease inhibitors. After extraction, the tissue was subjected to NuPAGE Bis-Tris (10%) gel electrophoresis and then transferred to a nitrocellulose membrane (Osmonics, Minnetonka, MN) electrophoretically. Nonspecific binding sites were blocked with 10% low-fat milk in PBS containing 0.1% Tween 20 (PBS-T) for 2 hr at 22°C. Membranes were subsequently incubated with polyclonal COX-2 antisera (Santa Cruz Biotechnology) in PBS-T buffer overnight at 4°C. The nitrocellulose membrane was then washed twice with the incubation buffer and once with a buffer containing 150 mm NaCl and 50 mm Tris-Cl, pH 7.5. The antibody–protein complexes were then blotted for 1 hr at 22°C with secondary antibodies labeled with horseradish peroxidase and detected with chemiluminescent reagents as described (Yaksh et al., 2001).
Statistical analysis. ANOVA was used to compare the behavioral effects of intrathecal drug administration compared with controls with subsequent Fisher protected least significant difference post hoc analysis. Student's t test was used to compare immunohistochemical measures between the experimental groups and their respective controls. The significance level in all analyses was set at p < 0.05. The investigator responsible for the quantification and analysis was blind as to the experimental status of each animal.
Results
To explore how NSAIDs might block COX-2 at early time points after injury, we used both immunohistochemistry and Western blot analysis to show that there is a high level of expression of COX-2 in neurons located throughout the entire gray matter and radial glia in the white matter of the rat spinal cord. Immunohistochemical localization of COX-2 analyzed with confocal microscopy of 15-μm-thick sections revealed that COX-2-IR in these neurons is perinuclear, and these COX-2-immunoreactive neurons are more prevalent in the superficial dorsal horn laminae I–III, whereas lower levels of COX-2-immunoreactive neurons are observed in the deeper laminae, including motor neurons, as shown previously (Goppelt-Struebe and Beiche 1997; Beiche et al., 1998; Spencer et al., 1998). The COX-2-IR present in this neuronal population of the L4 segment of the spinal cord (Fig. 1a) included the NK-1-expressing spinothalamic and spinoparabrachial neurons (both of which are involved in the ascending conduction of pain; Hunt, 2000; Gauriau and Bernard, 2002) because >70% of neurons that express the NK-1 receptor also expressed COX-2-IR.
Figure 1.
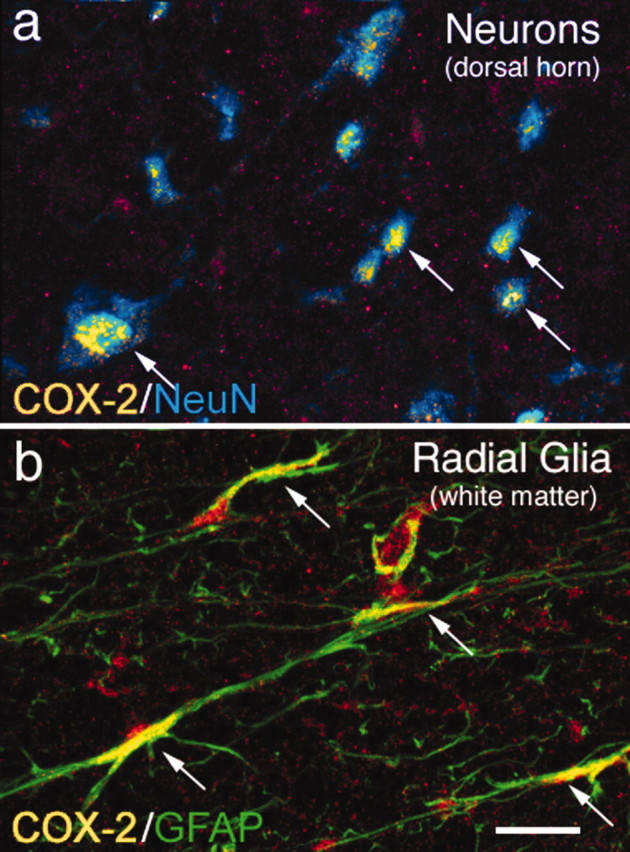
Neurons and radial glia located in the lumbar segment of the rat spinal cord constitutively express high levels of COX-2 protein. Immunohistochemical analysis reveals a high level of constitutive COX-2 expression in the majority of neurons in the rat spinal cord gray matter, as shown by colocalization of COX-2-IR (yellow) and a neuron-specific antibody, NeuN (blue; a). Confocal image analysis of 15-μm-thick sections reveals that this COX-2-IR is observed in the perinuclear region of the neuron. Radial glia in the spinal cord white matter also exhibit a high level of COX-2 expression, as shown by colocalization of COX-2-IR (yellow) and glial fibrillary acidic protein-IR (green; b) in the spinal cord white matter. Areas of colocalization appear yellow in both a and b (also indicated by arrows). Scale bar, 40 μm.
Other types of cells in the spinal cord that expressed high levels of COX-2-IR were the glial fibrillary acidic protein (GFAP)-IR radial glia located in the white matter (Fig. 1b), whereas <1% of the GFAP-IR astrocytes in the gray matter coexpress COX-2-IR.
An important consideration in assessing the role of constitutive versus inducible COX-2 is to define when spinal COX-2 levels begin to rise after peripheral tissue injury. Using Western blot analysis, we examined the expression of COX-2 protein at 0.5, 1, 2 and 4 hr after crush (Fig. 2a) or after the intrathecal administration of NMDA (Fig. 2b) and show there was no detectable upregulation of COX-2 protein until at least 2 hr after crush or intrathecal NMDA.
Figure 2.

Representative Western blot analysis showing the time course of the upregulation of COX-2 protein (top lanes) or the corresponding control β-actin (bottom lanes) in the ipsilateral lumbar spinal dorsal horn in naive animals (C) and at 0.5, 1, 2, and 4 hr after crush of the left hindpaw (a) or IT administration of 0.3μg of NMDA (b). Densitometric analysis of the Western blots confirms and extends the work of Samad et al. (2001), showing that there is significant constitutive expression of COX-2 protein in the spinal cord, and statistically significant upregulation of COX-2 does not occur until at least 2 hr after crush of the hindpaw or intrathecal administration of NMDA. Std is purified bovine COX-2 as a positive control.
To examine the degree that constitutively expressed spinal COX-1 or COX-2 contributes to nociceptive processing, pretreatment with a COX-1 or COX-2 inhibitor was performed 10 min before pinch, crush, or intrathecal administration of NMDA. In these studies, rats with indwelling intrathecal catheters were briefly anesthetized with isoflurane, and the paw was subjected to a strong but non-tissue-injurious pinch or to a significant tissue-injurious crush, with significant tissue injury as assessed by the resulting edema and erythema of the injured paw. After recovery from the anesthetic (∼15–20 min), the crush injury but not the pinch resulted in a well defined tactile allodynia that persisted for 2 hr. Thirty minutes after a unilateral paw crush, the baseline tactile threshold fell from 13.6 ± 1.9 to 6.4 ± 2.8 gm (n = 6; p < 0.05) in rats pretreated (T = –10 min) with vehicle. Rats pretreated 10 min (T = –10 min) before crush injury (T = 0) with intrathecal infusion of 30 μg/10 μl COX-2 inhibitor SC58125 did not develop significant allodynia (13.9 ± 1.1 vs 11.8 ± 0.9 gm; p > 0.10; n = 6), whereas animals treated with 30 μg/10 μl COX-1 inhibitor SC560 or vehicle (T =–10 min) showed development of significant allodynia (Fig. 3a). Previous results have suggested that spinal NMDA receptors are also involved in driving postcrush hyperalgesia (Dickenson et al., 1997), and indeed intrathecal (IT) delivery of a low dose of NMDA (0.3 μg/10 μl) results in significant thermal hyperalgesia (Fig. 3b), and this hyperalgesia was again reversed by COX-2 but not a COX-1 inhibitor or vehicle.
Figure 3.
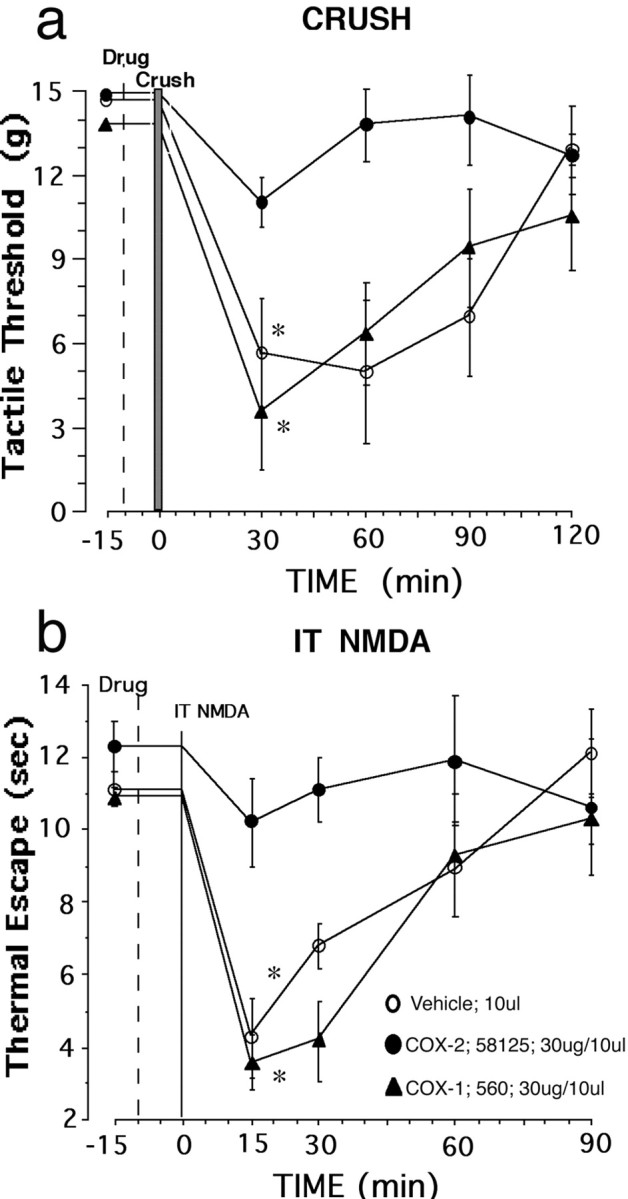
Inhibition of constitutive spinal COX-2 blocks the tactile and thermal hyperalgesia induced by peripheral tissue injury. Tactile allodynia was assessed using the up–down method with calibrated von Frey hairs, whereas thermal escape latency was assessed using a radiant heat stimulus to the undersurface of the paw through a glass plate. Tactile allodynia is significantly attenuated in animals that receive intrathecal administration of COX-2 inhibitor (30 μg/10 μl; SC58125) 10 min before unilateral crush of the hindpaw compared with pretreatment with vehicle or COX-1 inhibitor (30 μg/10 μl; SC560; a). Thermal hyperalgesia is also significantly attenuated in animals that receive intrathecal administration of COX-2 inhibitor 10 min before intrathecal administration of 0.3 μg of NMDA compared with pretreatment with vehicle or COX-1 inhibitor (b). Data are presented as mean ± SEM (*p < 0.05; n = 6 rats in each group).
To determine whether COX-2 inhibition resulted in a decrease in activation of peripheral nociceptors or neuronal activation in the spinal cord, we examined the effects that COX-2 inhibitors had on the nociceptor-induced release of substance P and the induction of c-Fos in the spinal dorsal horn after pinch or crush of the paw. Previous results have shown that nociceptive pinch or crush induces the release of dorsal horn substance P, leading to increased NK-1 receptor internalization (Mantyh et al., 1995) and an increase in the number of cells displaying c-Fos expression in the ipsilateral L3–L5 dorsal horn (Doyle and Hunt, 1999). When quantified, significantly greater NK-1 receptor internalization (Fig. 4a,b,f) and an increased incidence of c-Fos-positive neurons were observed in the ipsilateral dorsal horn after crush injury versus pinch (Fig. 4c–e) and in these rats pretreated with a COX-2 inhibitor (SC58125, 30 μg/10 μl) no effect on the c-Fos expression and NK-1 internalization generated by pinch was observed. However, after crush injury, intrathecal COX-2 resulted in a reduction in dorsal horn c-Fos and lamina I and II NK-1 internalization to levels associated with pinch activation. These observations suggest that the crush injury acutely leads to an enhanced state of nociceptive processing that can be significantly attenuated by inhibition of spinal COX-2.
Figure 4.
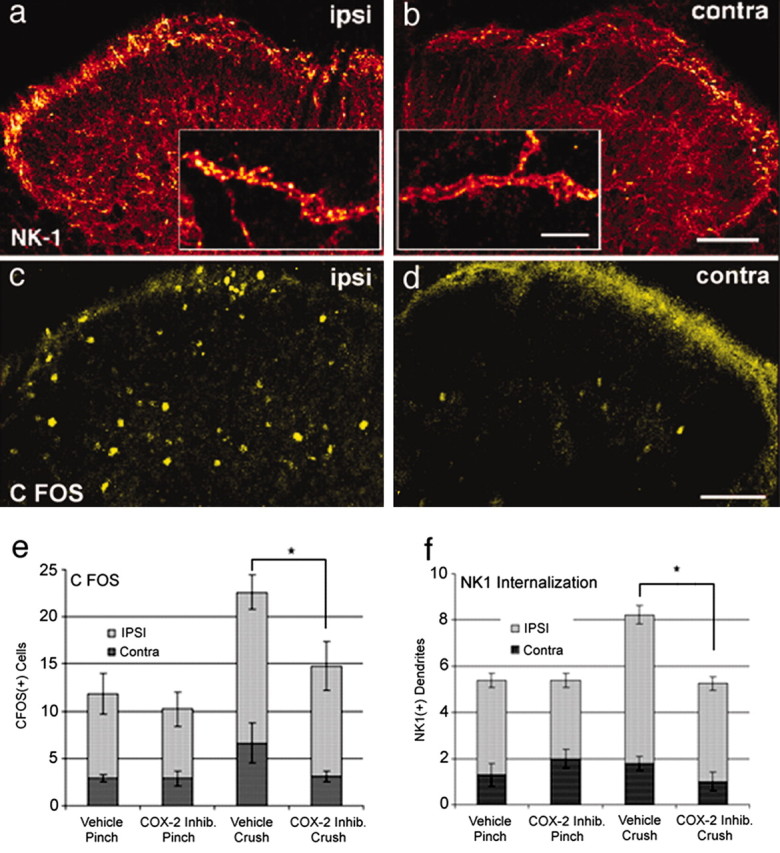
Inhibition of constitutive spinal COX-2 attenuates the release of spinal substance P and activation of spinal neurons that normally follows peripheral tissue injury. Internalization of the NK-1 receptor in dendrites located in laminae I and II of the L4 segment of the spinal dorsal horn and induction of spinal c-Fos expression in neurons located in laminae I–V are increased ipsilaterally (ipsi; a, c) after crush of the hindpaw in naive untreated animals compared with the contralateral side (contra; b, d), respectively. Significant internalization of NK-1 receptors (which is induced by spinal release of substance P from primary afferent neurons) occurs in both the cell body and the dendritic arbors (a, inset) of NK-1 receptor-expressing neurons. Note that pretreatment with an intrathecal injection of a COX-2 inhibitor (inhib.; 30 μg/10 μl SC58125 at T = – 10 min.) significantly reduced both the number of c-Fos-immunoreactive neurons in laminae I–V (e) and the number of NK-1-expressing dendrites per 900 μm2 of a 60-μm-thick section in laminae I and II that showed significant NK-1 internalization (f) in the ipsilateral dorsal horn compared with the contralateral side after unilateral crush of the hindpaw but not after a noxious but non-tissue-injuring hindpaw pinch. Data are presented as mean number of dendrites or cells ± SEM; n = 4 in each treatment group. Rats were killed 10 min after the paw stimulus. Scale bars: 100 μm; inset, 5 μm; p < 0.05.
Discussion
Classically, in most peripheral tissues, COX-1 is considered to be constitutively expressed, whereas COX-2 is inducibly expressed after tissue injury (Samad et al., 2001). In the present report, we demonstrate that unlike many peripheral tissues, the majority of neurons and radial glia in the rat spinal cord constitutively express high levels COX-2 protein. Many different types of spinal neurons express COX-2, such as the majority of lamina I NK-1-expressing neurons (Todd et al., 2002), which correspond to spinothalamic and spinoparabrachial neurons and which are involved in the ascending conduction of pain. In contrast, most glial cells such as oligodendrocytes and microglia do not express detectable levels of COX-2. Even when examining astrocytes, whereas most (>90%) radial glia located in the white matter express a very high level of COX-2, <1% of the astrocytes in the gray matter express detectable COX-2, suggesting very regulated expression of constitutive COX-2 by cells in the spinal cord.
Given the general assumption that COX-2 is primarily an inducibly rather than a constitutively expressed enzyme, an important consideration in assessing the role of constitutive versus inducible COX-2 is to define when spinal COX-2 levels begin to rise after peripheral tissue injury. We have previously shown that an acute stimulus, such as the intraplantar injection of formalin or the intrathecal injection of substance P, will lead to an immediate increase in prostaglandin (PG) release (Dirig et al., 1997). This immediate effect on extracellular PGE2 concentrations is believed to reflect the activation of phospholipase A2 secondary to increased intracellular calcium and the liberation of arachidonic acid (AA). This AA is then acted on by either COX isozyme that is constitutively present and active to result in PG release.
The current pharmacology suggests that this increased PGE2 release is mediated by COX-2 and not COX-1 pharmacology, and because the effects are observed acutely, this COX-2 must by definition be constitutively expressed. The present work indicates that the constitutively expressed COX-2 is an adequate and necessary mediator for the acute spinal facilitation induced by a small afferent input. Although it appears likely that the increase in COX-2 expression that is observed after some delay contributes to processing, its role is not known. It is important to note that COX-2 undergoes suicide inactivation, and the “inducible COX-2” may simply reflect not an increase in total functional COX-2, but an aggressive replacement motif that is necessary for an enzyme that has such a limited life span. Using Western blot analysis and immunohistochemistry, we examined the expression of COX-2 protein at 0.5, 1, 2 and 4 hr after strong crush of the hindpaw or after the intrathecal administration of NMDA. Our results show that there is no significant upregulation of COX-2 protein until at least 2 hr after crush or intrathecal injection of the excitatory NMDA.
What in many ways is unique and useful about NSAIDs is that they have little if any effect on noxious stimuli such as pinprick or pinch, which do not cause significant tissue injury but are efficacious at attenuating noxious stimuli that results from significant injury to peripheral tissue (Ehrich et al., 1999; Malmstrom et al., 1999). This clinical observation is supported by the present observations in which pretreatment with a COX-1 or COX-2 inhibitor was performed 10 min before pinch, crush, or intrathecal administration of NMDA. In these studies, rats with indwelling intrathecal catheters were briefly anesthetized with isoflurane, and the paw was subjected to a strong but non-tissue-injurious pinch or to a significant tissue-injurious crush, with significant tissue injury as assessed by the resulting edema and erythema of the injured paw. After recovery from the anesthetic (∼15–20 min), crush injury but not the pinch resulted in well defined tactile allodynia that persisted for 2 hr. Thus, rats pretreated 10 min before injury via an intrathecal infusion of a COX-2 inhibitor did not develop significant tactile allodynia, whereas animals treated with the COX-1 inhibitor or vehicle showed development of significant allodynia. Previous results have suggested that spinal NMDA receptors are also involved in driving postcrush hyperalgesia (Dickenson et al., 1997), and using IT delivery of NMDA permitted direct assessment of the spinal pharmacology of facilitated processing without having to initiate an injury state that might be altered by a systemically delivered agent. Indeed, IT delivery of a low dose of NMDA results in significant thermal hyperalgesia, and again, this hyperalgesia was again reversed by COX-2 but not a COX-1 inhibitor or vehicle.
Together, these pharmacological and behavioral data reinforce the clinical observation that what appears to be required for spinal COX-2 activation is a noxious stimulus of sufficient intensity and duration to activate the cascade necessary to form arachidonic acid and the subsequent COX-2-dependent synthesis of prostanoids. These data support the concept that the more intense the stimulus or its tendency to produce an ongoing postinjury barrage, the more likely COX-2 will be involved in initiating the prostanoid cascade (Baba et al., 2001; Southall and Vasko, 2001). In addition, these data suggest that the earlier the spinal COX-2 activation can be blocked (either preemptively or after injury), the more effectively a COX-2 inhibitor will be at blocking the hyperalgesia and allodynia that results from peripheral tissue injury such as the musculoskeletal injury used in the present study (Portenoy, 2000).
Our studies show that, unlike many peripheral tissues in which COX-2 is normally expressed at very low levels but is dramatically upregulated after tissue injury, in the spinal cord, COX-2 is constitutively expressed at high levels by both neurons and radial glia. Immediately after tissue injury, constitutive spinal COX-2 is activated and is involved in driving the initial hyperalgesia and allodynia that follows peripheral tissue injury. This evidence suggests that the early or preemptive blockade of constitutive spinal COX-2 will limit the initial hyperalgesia that accompanies peripheral tissue injury and in doing so may block early nociceptor-driven events that contribute to the development of a chronic pain state.
Footnotes
This work was supported by National Institutes of Health Grants NS23970 (P.W.M.), DA11986 (P.W.M.), and NS16541 (T.L.Y.), and by a Veterans Administration Merit Review (P.W.M.).
Correspondence should be addressed to Patrick W. Mantyh, Departments of Preventive Sciences and Neuroscience and Cancer Center, University of Minnesota, Minneapolis, MN 55455. E-mail: manty001@tc.umn.edu.
DOI:10.1523/JNEUROSCI.5054-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/242727-06$15.00/0
References
- Ahmadi S, Lippross S, Neuhuber WL, Zeilhofer HU (2002) PGE(2) selectively blocks inhibitory glycinergic neurotransmission onto rat superficial dorsal horn neurons. Nat Neurosci 5: 34–40. [DOI] [PubMed] [Google Scholar]
- Baba H, Kohno T, Moore KA, Woolf CJ (2001) Direct activation of rat spinal dorsal horn neurons by prostaglandin E2. J Neurosci 21: 1750–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beiche F, Klein T, Nusing R, Neuhuber W, Goppelt-Struebe M (1998) Localization of cyclooxygenase-2 and prostaglandin E2 receptor EP3 in the rat lumbar spinal cord. J Neuroimmunol 89: 26–34. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL (1994) Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods 53: 55–63. [DOI] [PubMed] [Google Scholar]
- Dickenson AH, Chapman V, Green GM (1997) The pharmacology of excitatory and inhibitory amino acid-mediated events in the transmission and modulation of pain in the spinal cord. Gen Pharmacol 28: 633–638. [DOI] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL (1997) Characterization of variables defining hindpaw withdrawal latency evoked by radiant thermal stimuli. J Neurosci Methods 76: 183–191. [DOI] [PubMed] [Google Scholar]
- Doyle CA, Hunt SP (1999) Substance P receptor (neurokinin-1)-expressing neurons in lamina I of the spinal cord encode for the intensity of noxious stimulation: a c-Fos study in rat. Neuroscience 89: 17–28. [DOI] [PubMed] [Google Scholar]
- Ehrich EW, Dallob A, De Lepeleire I, Van Hecken A, Riendeau D, Yuan W, Porras A, Wittreich J, Seibold JR, De Schepper P, Mehlisch DR, Gertz BJ (1999) Characterization of rofecoxib as a cyclooxygenase-2 isoform inhibitor and demonstration of analgesia in the dental pain model. Clin Pharmacol Ther 65: 336–347. [DOI] [PubMed] [Google Scholar]
- Gauriau C, Bernard J (2002) Pain pathways and parabrachial circuits in the rat. Exp Physiol 87: 251–258. [DOI] [PubMed] [Google Scholar]
- Goppelt-Struebe M, Beiche F (1997) Cyclooxygenase-2 in the spinal cord: localization and regulation after a peripheral inflammatory stimulus. Adv Exp Med Biol 433: 213–216. [DOI] [PubMed] [Google Scholar]
- Hunt S (2000) Pain control: breaking the circuit. Trends Pharmacol Sci 21: 284–286. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI (2001) Molecular mechanisms of nociception. Nature 413: 203–210. [DOI] [PubMed] [Google Scholar]
- Kalgutkar AS, Zhao Z (2001) Discovery and design of selective cyclooxygenase-2 inhibitors as non-ulcerogenic, anti-inflammatory drugs with potential utility as anti-cancer agents. Curr Drug Targets 2: 79–106. [DOI] [PubMed] [Google Scholar]
- Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL (2001) Upregulation of dorsal root ganglion (alpha)2(delta) calcium channel subunit and its correlation with allodynia in spinal nerveinjured rats. J Neurosci 21: 1868–1875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmstrom K, Daniels S, Kotey P, Seidenberg BC, Desjardins PJ (1999) Comparison of rofecoxib and celecoxib, two cyclooxygenase-2 inhibitors, in postoperative dental pain: a randomized, placebo- and active-comparator-controlled clinical trial. Clin Ther 21: 1653–1663. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, DeMaster E, Malhotra A, Ghilardi JR, Rogers SD, Mantyh CR, Liu H, Basbaum AI, Vigna SR, Maggio JE (1995) Receptor endocytosis and dendrite reshaping in spinal neurons after somatosensory stimulation. Science 268: 1629–1632. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA (1997) Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 278: 275–279. [DOI] [PubMed] [Google Scholar]
- Masferrer JL, Zweifel BS, Manning PT, Hauser SD, Leahy KM, Smith WG, Isakson PC, Seibert K (1994) Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc Natl Acad Sci USA 91: 3228–3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portenoy RK (2000) Current pharmacotherapy of chronic pain. J Pain Symptom Manage 19[Suppl 1]: S16–S20. [DOI] [PubMed] [Google Scholar]
- Samad TA, Moore KA, Sapirstein A, Billet S, Allchorne A, Poole S, Bonventre JV, Woolf CJ (2001) Interleukin-1beta-mediated induction of Cox-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature 410: 471–475. [DOI] [PubMed] [Google Scholar]
- Schaible H-G, Grubb BD (1993) Afferent and spinal mechanisms of joint pain. Pain 55: 5–54. [DOI] [PubMed] [Google Scholar]
- Southall MD, Vasko MR (2001) Prostaglandin receptor subtypes, EP3C and EP4, mediate the prostaglandin E2-induced cAMP production and sensitization of sensory neurons. J Biol Chem 276: 16083–16091. [DOI] [PubMed] [Google Scholar]
- Spencer AG, Woods JW, Arakawa T, Singer II, Smith WL (1998) Subcellular localization of prostaglandin endoperoxide H synthases-1 and -2 by immunoelectron microscopy. J Biol Chem 273: 9886–9893. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Puskar Z, Spike RC, Hughes C, Watt C, Forrest L (2002) Projection neurons in lamina I of rat spinal cord with the neurokinin 1 receptor are selectively innervated by substance p-containing afferents and respond to noxious stimulation. J Neurosci 22: 4103–4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vane JR, Mitchell JA, Appleton I, Tomlinson A, Bishop-Bailey D, Croxtall J, Willoughby DA (1994) Inducible isoforms of cyclooxygenase and nitric-oxide synthase in inflammation. Proc Natl Acad Sci USA 91: 2046–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Dirig DM, Conway CM, Svensson C, Luo ZD, Isakson PC (2001) The acute antihyperalgesic action of nonsteroidal, anti-inflammatory drugs and release of spinal prostaglandin E2 is mediated by the inhibition of constitutive spinal cyclooxygenase-2 (COX-2) but not COX-1. J Neurosci 21: 5847–5853. [DOI] [PMC free article] [PubMed] [Google Scholar]