

Abstract
Torsion dystonia-1 (DYT1) dystonia, the most common inherited form of dystonia, is caused by a three base pair deletion that eliminates a single amino acid from the disease protein, torsinA. TorsinA is an “AAA” protein thought to reside in the endoplasmic reticulum (ER), yet both its cellular function and the basis for neuronal dysfunction in DYT1 remain unknown. A clue to disease pathogenesis is the fact that mutant, but not wild-type, torsinA forms membranous inclusions in cell culture. To explore the pathobiology of DYT1 dystonia, we generated PC12 neural cell lines that inducibly express wild-type or mutant torsinA. Although in this model torsinA displays some properties consistent with ER localization, mutant torsinA also accumulates in the nuclear envelope (NE), a structure contiguous with cytoplasmic ER. Consistent with this, membranous inclusions formed by mutant torsinA are shown to derive not from the ER, as thought previously, but from the NE. We demonstrate further that torsinA forms different disulfide-linked complexes that may be linked functionally to subcellular localization in the NE versus cytoplasmic ER. Despite mutant TA accumulation in NE structures, nucleocytoplasmic transport of a reporter protein was unaffected. These findings, together with parallel studies failing to demonstrate perturbation of ER function, implicate the NE as a primary site of dysfunction in DYT1. DYT1 dystonia can be added to the growing list of inherited neurological disorders involving the NE.
Keywords: dystonia, torsinA, DYT1, nuclear envelope, karmellas, endoplasmic reticulum
Introduction
Torsion dystonia-1 (DYT1) is the most common inherited dystonia, a disabling autosomal dominant neurological disorder characterized by involuntary muscle contractions that lead to abnormal postures (Fahn et al., 1998). The gene responsible for DYT1, TOR1A, encodes torsinA (TA), an endoplasmic reticulum (ER) glycoprotein belonging to the AAA family of proteins (ATPases associated with various cellular activities) (Ozelius et al., 1997). DYT1 is caused by a common GAG deletion in TOR1A that eliminates one of a pair of glutamic acids near the C terminus of TA. Although several reports suggest a role for TA in protein quality control (McLean et al., 2002; Caldwell et al., 2003), relatively little is known about disease pathogenesis, including the neurobiological function of TA and its dysfunction in the disease state.
Immunohistochemical staining for TA indicates that the protein is primarily cytoplasmic in neurons throughout the brain (Shashidharan et al., 2000; Konakova and Pulst, 2001; Konakova et al., 2001). Postmortem analysis of DYT1 brain has not identified specific neuropathological findings or changes in TA expression or subcellular distribution (Augood et al., 2002; Walker et al., 2002; Rostasy et al., 2003). Studies in cell-based models of DYT1, however, have revealed aberrant localization of mutant TA (TAmut). In transfected cells, wild-type TA (TAwt) shows diffuse cytoplasmic staining consistent with ER localization, whereas TAmut is found primarily within cytoplasmic membranous inclusions (Hewett et al., 2000; Kustedjo et al., 2000).
Further understanding of the mechanisms underlying inclusion formation by TAmut, including their subcellular point of origin, may provide insight into the normal function of TA and its dysfunction in the disease state. Our studies described here implicate the nuclear envelope (NE) as the site of origin and not the ER, as has been believed. The NE functions as a gated barrier that regulates transport between these two cellular compartments. It is composed of the outer nuclear membrane (ONM), inner nuclear membrane (INM), nuclear pore complexes (NPCs), and the nuclear lamina (Burke and Stewart, 2002). The ONM, a subdomain of the ER, is contiguous with the INM, which contains membrane proteins that interact with the nuclear lamina and nucleoplasm. Recently, a proteomics screen revealed unexpectedly complex protein composition to the NE, suggesting that it functions as much more than a simple barrier (Schirmer et al., 2003). Consistent with this view, an increasing number of neurological disorders are linked to dysfunction of this organelle (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003; Ostlund and Worman, 2003; Schirmer et al., 2003).
In this study, we have generated an inducible PC12 neural cell model of DYT1 to study the pathobiology of DYT1. Our initial focus has been to define the origin and significance of TAmut-derived inclusions. Using complementary cell biological and biochemical approaches, we show that TAmut, unlike TAwt, preferentially resides within the NE and forms membranous inclusions derived from this structure. We also study the potential impact of TAmut on cellular processes linked to the NE and ER, including nucleocytoplasmic transport and the secretory pathway.
Materials and Methods
Generation of inducible cell lines. To create stably transfected, TA-expressing neural cell lines, we elected to use PC6-3 cells, a well studied subclone of PC12 cells that develops into sympathetic neuron-like cells on treatment with nerve growth factor (NGF) (Pittman et al., 1993). A PC6-3 clonal line stably expressing the tet-repressor (PC6-3/TR) was generously provided by Stefan Strack (University of Iowa, Iowa City). cDNAs encoding TAwt or TAmut were excised from pcDNA3-TAwt and pcDNA3-TAmut (kindly provided by Xandra Breakefield, Massachusetts General Hospital, Boston) with BamHI and XbaI and cloned into the tet-regulatable expression vector pcDNA5TO (Invitrogen, Carlsbad, CA). After sequence verification, pcDNA5TO-TAwt and pcDNA5TO-TAmut were transfected separately into PC6-3/TR cells using Lipofectamine Plus (Invitrogen, Bethesda, MD) according to the manufacturer's instructions. Transfected cells were cultured as described previously (Chai et al., 1999b) in medium containing blasticidine (5 μg/ml) and hygromycin (100 μg/ml), and 48 clones were selected for each transfected plasmid. Clones were screened by adding 1.5 μg/ml doxycycline to the culture media for 24 hr and assessing inducible TA expression by Western blot analysis (WB) of lysates. Clones showing the tightest, inducible expression were then screened by indirect immunofluorescence microscopy (IF) to identify those clones with the most homogeneous expression of TA across the entire cell population. These clones, TAmut 9, 33, and 39 and TAwt 25, 27, and 28, were selected for the further characterization described in this paper. To achieve a differentiated neural phenotype, PC6-3 clones were cultured in low serum, NGF-containing media (2% equine serum and 1% fetal bovine serum; 100 ng/ml NGF) in collagen-coated dishes or slides.
Expression plasmids and antibodies. Additional plasmids used for transfection included green fluorescent protein (GFP)-emerin and laminB receptor fused to GFP at either the N- or C-terminal end (GFP-LBR and LBR-GFP; a kind gift of Tokuko Hagaruchi, Kansai Advanced Research Center, Kobe, Japan) (Haraguchi et al., 2000), GFP-laminC (generously provided by Jos Broers, University of Maastricht, The Netherlands), GFP-laminA, GFP-sec61β, GFP-p62, GFP-histoneH1, and GFP-signal peptidase 25 kDa (kindly provided by Tom Rapoport, Harvard Medical School, Boston) (Rolls et al., 1999), GFP-glucocorticoid receptor (GFP-GR; a kind gift of Mario Galigniana, University of Michigan, Ann Arbor) (Galigniana et al., 2001), GFP-human dopamine transporter (GFP-hDAT; kindly provided by Ulrik Gether, University of Copenhagen, Denmark) (Granas et al., 2003), and yellow fluorescent protein-cystic fibrosis transmembrane regulator protein (YFP-CFTR) (kindly provided by Michael Welsh, University of Iowa, Iowa City).
Anti-TA antibodies, generously provided by Xandra Breakefield (Massachusetts General Hospital), included polyclonal rabbit antiserum generated against a TA-maltose binding protein fusion protein (MBP), rabbit polyclonal TAB1, and the mouse monoclonal antibody D-MG10 (Hewett et al., 2000). Additional rabbit polyclonal antibodies used were anti-calnexin (Stressgen Biotechnologies, Victoria, Canada), anti-calreticulin (Affinity Bioreagents, Golden, CO), anti-NFB42/Fbx2p (a kind gift of Randall Pittman, University of Pennsylvania, Philadelphia) (Erhardt et al., 1998), and anti-emerin, anti-laminA, goat polyclonal anti-laminB, and anti-nucleoporin NUP98 (all from Santa Cruz Biotechnology, Santa Cruz, CA). Mouse monoclonal antibodies included anti-α-tubulin, anti-SC35 (both from Sigma, St. Louis, MO), anti-laminA/C, anti-karyopherin β3 (both from Santa Cruz Biotechnology), anti-KDEL (Stressgen), and anti-ubiquitin (Zymed, South San Francisco, CA).
Treatments. To induce the unfolded protein response (UPR), we treated cells with either 2.5 μg/ml tunicamycin for 2.5 hr or 400 nm thapsigargin for 4 hr (both from Sigma). For assessment of the N-glycosylation state of TA, protein lysates were incubated with PNGase F or EndoH (New England Biolabs, Beverly, MA) following the manufacturer's recommendations. We inhibited ER to Golgi transport by incubation with 5 μg/ml brefeldin A for 2 hr, and disrupted ER-associated degradation (ERAD) with 10 μm lactacystin (both from Calbiochem, San Diego, CA) for 8 hr. For microtubule disruption, cells were treated with 20 μm nocodazole (Sigma) for 2 hr before fixation, and to inhibit translation in cells transfected with GFP-DAT, 10μm cycloheximide (Sigma) was added to the culture media 6 hr before fixation.
Western blot analysis. Unless noted otherwise, cells were harvested for WB analysis 48 hr after doxycycline induction as described previously (Chai et al., 1999a). To assess the level of induction, we quantified WB signal as described previously (Gonzalez-Alegre et al., 2003). For nuclear extraction experiments, we followed a previously described protocol with minor modifications (Perez et al., 1999). Briefly, cells were washed and scraped in ice-cold PBS with protease inhibitors and pelleted at 800 rpm for 5 min at 4°C. The pellet was resuspended in hypotonic buffer, and cell membranes were disrupted by multiple passes through a 27 gauge needle. Cell lysis was confirmed by light microscopy, and nuclei were then pelleted at 500 × g for 10 min at 4°C. The supernatant was saved as the cytosolic fraction, and the nuclear pellet was rinsed in hypotonic buffer, resuspended in high salt buffer, and placed on ice for 10 min. The nuclear fraction was then centrifuged at 14,000 rpm for 5 min at 4°C. The pellet was resuspended in IMP buffer containing 1% NP-40 and briefly sonicated. When indicated, we performed the experiments in the absence of reducing agent (DTT) but added 5 mm N-ethylmaleimide to avoid aberrant disulfide link formation. We used laminA/C and calreticulin as fractionation controls. As opposed to other cellular components or ER lumenal proteins, the separation of the membranous “cytoplasmic” ER and the “nuclear” ER (or NE) by subfractionation techniques is very difficult because they are physically contiguous organelles (Schirmer et al., 2003). Cytoplasmic ER membrane proteins can be found in the nuclear fraction at higher concentrations than expected, and it is not possible to ascertain whether they were present in the cytoplasmic or nuclear ER membrane. For that reason, we used calnexin (an ER-membrane protein) as a partitioning control for the fractionation of the NE–ER-membrane components.
Indirect immunofluorescence. For IF, cells were fixed in 4% paraformaldehyde in PBS and processed as described previously (Chai et al., 1999a). In experiments assessing internalization of GFP-GR, cells were fixed by immersion in ice-cold methanol for 15 min. Fluorescence visualization of fixed cells was performed with a Zeiss (Thornwood, NY) Axioplan fluorescence microscope. Digital images were collected on separate red, green, and blue fluorescence channels using either a Diagnostics SPOT (Sterling Heights, MI) or Axiocam HRm (Zeiss) digital cameras. Live cell images of cells expressing GFP-fusion proteins were collected with a Kodak MDS 290 digital camera mounted on an Olympus CK40 inverted microscope equipped for GFP fluorescence and phase-contrast microscopy. Digitized images were assembled using Adobe Photoshop 6.0.
Transmission electron microscopy. For transmission electron microscopy (TEM), induced and noninduced TAmut cells were processed as described previously (Ashbourne Excoffon et al., 2003). In summary, cells were submerged in 2.5% glutaraldehyde and postfixed in osmium tetroxide followed by 2.5% aqueous uranyl acetate. Samples were then dehydrated in a graded series of ethanol and embedded in Eponate-12 resin (Ted Pella Inc., Redding, CA). Ultrathin sections (80 nm) were counterstained with uranyl acetate and lead citrate before being viewed on a Hitachi TEM H-7000 (Pleasanton, CA).
Glucocorticoid receptor translocation. To assess the impact of TA on nuclear import of the GR, cells were transfected with GFP-GR (Galigniana et al., 2001). We incubated transfected cells for 48 hr with medium containing 10% charcoal–dextran-stripped fetal bovine serum (HyClone, Logan, UT) with or without doxycycline and then added 1 μm dexamethasone (Sigma) to induce nuclear transport of GFP-GR. Images of single cells were obtained immediately before dexamethasone treatment and 1, 3, 5, 8, 10, 15, 20, 25, and 30 min after treatment. Initial experiments established the time frame during which the most prominent changes in GFP-GR location occurred, which was 10–20 min after dexamethasone addition. Accordingly, we then transfected NGF-differentiated cells with GFP-GR and fixed parallel samples immediately before and 10, 15, and 20 min after dexamethasone treatment. Internalization of GFP-GR was quantified using a previously reported method (Galigniana et al., 2001) that assigns a translocational score of 4 for nuclear fluorescence much greater than cytoplasmic fluorescence, 3 for nuclear fluorescence greater than cytoplasmic fluorescence, 2 for nuclear fluorescence equal to cytoplasmic fluorescence, 1 for nuclear fluorescence less than cytoplasmic fluorescence, and 0 for nuclear fluorescence much less than cytoplasmic fluorescence. The mean translocational score was calculated for 100 GFP-GR-expressing cells in each condition.
Results
Generation of inducible neural cell lines expressing torsinA
We successfully generated stably transfected PC6-3 clonal cell lines that inducibly express TAwt or TAmut and undergo neural differentiation in the presence of NGF. We obtained multiple clones that robustly induced TAwt or TAmut synthesis on exposure to doxycycline, detected both by WB (with an average ∼20-fold induction over endogenous levels) and by IF (Fig. 1A,B). IF of TAwt-expressing cells revealed a diffuse cytoplasmic distribution for TAwt. In cells expressing TAmut, however, the diffuse cytoplasmic pattern was lost, and instead we observed numerous cytoplasmic TA-containing inclusions (Fig. 1B). In NGF-differentiated cells, the cytoplasmic inclusions were equally abundant, as shown in Figure 1C, and were also rarely found in neuritic processes (Fig. 1D). These results were observed with several different anti-TA antibodies. The antibody MBP recognized only overexpressed TA by WB when used at a 1:1000 dilution, but weakly detected endogenous TA at 1:250 dilution. In IF experiments, this antibody did not detect endogenous TA, or did so only faintly, and thus was a useful antibody for most of our studies. All IF figures shown here were obtained with MBP unless noted otherwise. Antibodies TAB1 and D-MG10, which were used primarily in confirmatory experiments, detected both endogenous and induced TA.
Figure 1.

An inducible neural cell model of DYT1. Shown are stably transfected PC6-3 clonal cells displaying doxycycline-inducible expression of TAwt or TAmut by WB (A) [anti-TA (MBP) 1:1000], where calnexin is shown as a loading control, and by IF (B), where TA is seen in red and nuclear DAPI staining is in blue. TAwt distributes as a diffuse cytoplasmic pattern, whereas TAmut forms multiple cytoplasmic inclusions. C, A similar IF pattern is seen after TAmut induction in differentiated cells, where TA is seen in red, α-tubulin is in green, and DAPI nuclear staining is in blue. D, Inclusions are sometimes found in neuritic processes.
Electron microscopy of TAmut-expressing cells showed multiple, cytoplasmic ring-shaped structures formed by concentric, bimembranous layers (Fig. 2), similar to those described previously (Hewett et al., 2000). These structures were never found in noninduced cells, indicating that they correspond to the inclusions seen by IF. These results were replicated in three different clones each for TAwt and TAmut and confirmed with multiple antibodies specific for TA (data not shown). The contrasting cellular behavior of TAwt and TAmut in our inducible neural clones recapitulates the results described previously in transiently transfected cell lines (Hewett et al., 2000; Kustedjo et al., 2000) and validates them as cell models in which to study aberrant properties of TAmut.
Figure 2.

TAmut inclusions are multilamellar bimembrane structures. Shown are representative transmission electron microscopy views of TAmut-expressing cells. The left panel shows a single cell with three different inclusions (arrows). Inset is a higher magnification of one of the inclusions showing loosely arranged concentric membranes. The inclusion also contains a darker, smaller inclusion formed by tightly arranged concentric membranes (arrowhead). The top right panel shows another typical cytoplasmic inclusion, and the right bottom picture shows a possible intranuclear lamellar inclusion (arrow). Scale bars, 1 μm.
Wild-type and mutant torsinA preferentially localize to distinct ER subdomains
Previous studies have shown that TAwt colocalizes with various ER markers in cultured cells (Hewett et al., 2000; Kustedjo et al., 2000). We confirmed this, observing diffuse colocalization of TAwt with ER luminal [binding protein (BiP) (Fig. 3A); calreticulin (data not shown)] and membrane [calnexin (Fig. 3B)] proteins. As noted by others (Hewett et al., 2000; Kustedjo et al., 2000), TAmut did not colocalize with these ER markers, and the inclusions formed by TAmut did not show enhanced immunostaining for ER resident proteins (Figs. 3A,B). This led us to examine whether the inclusion formation reflected altered handling of TAmut by ER-linked pathways. The major routes controlling ER protein homeostasis are (1) translocation of new proteins into the lumen, (2) transit from the ER to the Golgi, and (3) retrotranslocation to the cytosol of proteins destined for destruction by the ubiquitin-dependent process known as ERAD. Perturbations in any of these three might result in inclusion formation by TAmut. To address this, we blocked ER to Golgi transport with brefeldin A and ERAD with lactacystin in our TA-expressing cell lines. These treatments did not induce inclusion formation in cells expressing TAwt, suggesting that neither ER retention nor inefficient destruction of TA is responsible for inclusion formation. Moreover, treatment of TAmut-expressing cells with these same agents did not affect inclusion formation, suggesting that traditional routes of exit from the ER are not required for their genesis (Fig. 3C). WB analysis of lysates did not confirm increased TA levels after proteasome inhibition (see Fig. 8), but in some cells lactacystin treatment seemed to enhance TAmut perinuclear signal. It is possible that proteasome inhibition causes a redistribution of TA, leading to an increase in perinuclear signal without an actual change in total TA levels. We also confirmed that both TAwt and TAmut contain similar high-mannose, asparagine-linked glycans, a biochemical feature of proteins that reside in the ER (Fig. 3D), consistent with previous reports (Hewett et al., 2000; Kustedjo et al., 2000). These results establish that, despite their clear difference in subcellular localization, both forms of TA reside in a pre-Golgi compartment.
Figure 3.

TAwt and TAmut are pre-Golgi proteins. IF images show colocalization of TAwt, but not TAmut, with BiP (A) and calnexin (B). C, Blocking ER-exit pathways by brefeldinA and lactacystin neither induces inclusion formation by TAwt nor prevents formation of inclusions by TAmut. D, Both forms of TA, detected by anti-TA (MBP) 1:1000, are deglycosylated equally by EndoH and PNGase F, indicating high-mannose, N-linked glycosylation. The lower intensity of deglycosylated bands could result from decreased antibody recognition of TA when deglycosylated or partial degradation of the protein. E, Microtubule disruption by nocodazole alters neuritic processes but not the presence of inclusions.
Figure 8.

Expression of TAmut does not induce the unfolded protein response. WB analysis of cells expressing TAwt or TAmut showed no changes in BiP or calnexin levels when compared with uninduced cells. In contrast, treatment with lactacystin, a proteasome inhibitor that activates the UPR, significantly increased BiP expression. Note that after inhibition of proteasomal degradation there are no significant changes in levels of TA. α-tubulin is shown as a loading control.
We incubated TAmut-expressing cells with nocodazole to assess whether inclusion formation depends on microtubule integrity. The inclusions did not disappear with nocodazole treatment as has been described for both aggresomes and a type of ER-derived inclusions (Kamhi-Nesher et al., 2001); however, microtubule disruption caused the inclusions to concentrate in a more “perinuclear” location, probably as a result of the dramatic structural changes induced by nocodazole (Fig. 3E). We conclude that TAwt and TAmut are both pre-Golgi glycoproteins, yet display striking differences in subcellular location that reflect divergent distribution of the two proteins within distinct ER subdomains. The evidence suggests that TAwt and TAmut, when overexpressed, localize to spatially distinct pre-Golgi, membrane-limited structures. Thus, pathogenesis in DYT1 may reflect perturbed homeostasis of a membrane-bound organelle.
Mutant torsinA inclusions originate from the nuclear envelope
By IF microscopy, cells expressing TAmut showed a thin, intensely immunostained, perinuclear rim suggesting the presence of TAmut in the NE (Fig. 4A). In contrast, prominent nuclear rim staining was not seen in cells expressing TAwt (Fig. 4A). Because the NE is a subdomain of the ER, physically connected via the ONM (Voeltz et al., 2002), we asked whether TAmut preferentially localized to this nuclear ER subdomain. Such a location would be consistent with the pre-Golgi, high-mannose glycosylation state of TAmut. We observed colocalization of this rim staining with the NE resident proteins laminA (Fig. 4B), laminA/C, laminB, and emerin (Fig. 5), supporting this hypothesis. Interestingly, on incubation of TA-expressing cells with 10 μm H2O2 for 30 min, both TAwt and TAmut accumulated in the perinuclear rim (data not shown), as described recently by Hewett et al. (2003). To provide additional experimental support for localization to the nuclear ER or NE, we performed subcellular fractionation experiments. We detected both forms of TA in the nuclear and cytoplasmic fractions (Fig. 4C), supporting the presence of TA in the NE. TAmut localizing to the cytoplasmic fraction probably corresponds to the inclusions rather than the ER itself.
Figure 4.

TAmut accumulates in the nuclear envelope. A, Representative immunofluorescence images show a TA-immunoreactive perinuclear rim (green) surrounding the nucleus (DAPI staining; blue) in a cell expressing TAmut, but not in a cell expressing TAwt. TA was stained using D-MG10 antibody. Higher magnification of the nuclear area without DAPI staining is also shown. B, Double immunofluorescence shows colocalization of TAmut in the perinuclear rim with laminA. C, Subcellular fractionation experiments show a clear separation of ER lumenal (calreticulin) and nuclear lamina (laminA/C) proteins in the cytoplasmic and nuclear fractions. ER-membrane proteins (calnexin) are found in both compartments, indicating that they are present in cytoplasmic ER and ONM of the NE. TorsinA, whether wild type or mutant, is also found in both fractions. The increased calnexin signal seen in cells expressing TA in this blot was not observed consistently.
Figure 5.
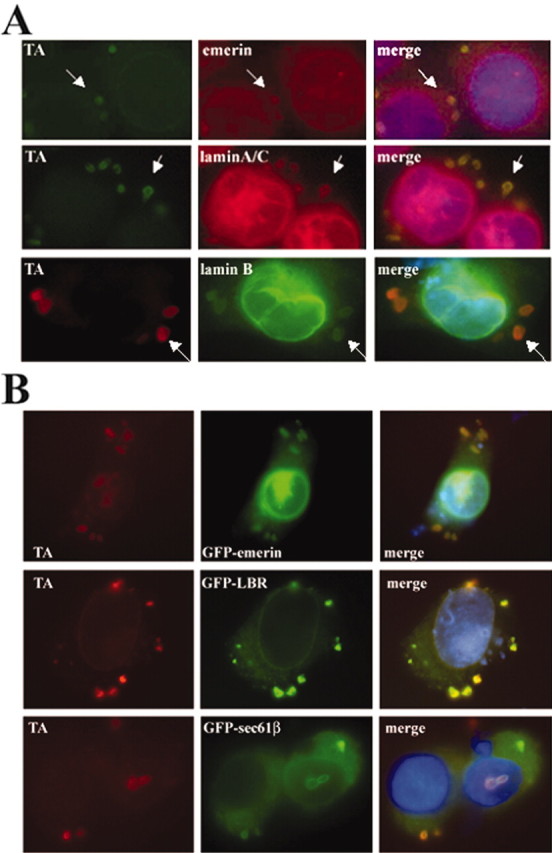
TAmut inclusions contain nuclear envelope proteins. A, Double IF images showing colocalization of endogenous emerin, laminA/C, and laminB with TAmut-derived inclusions (arrows). B, Transfected GFP-fusion NE proteins were also targeted to inclusions.
These results suggested that, as TAmut accumulates in the NE, inclusions originate from this ER subdomain. To test this, we performed IF with antibodies directed against components of the NE. In cells expressing TAmut, perinuclear inclusions were consistently and intensely stained with antibodies directed either against endogenous laminA/C (against the N-terminal part of the protein, which is shared by laminA-type isoforms) or against emerin (Fig. 5A); these proteins reside in the nuclear lamina and the INM, respectively. The inclusions were not immunostained with an antibody that recognizes only the A isoform of laminA/C (Fig. 4B). It is possible that the laminA/C antibody detects a different isoform of type-A lamins (other than laminA or laminC) generated by alternative splicing of the same gene, or that it cross-reacts with another NE protein. Consistent with this hypothesis, GFP-laminA and GFP-laminC fusion proteins did not localize to the inclusions (data not shown). Another possibility is that the fusion of GFP to lamins alters their normal physiology.
Another component of the nuclear lamina, laminB, was also detected in the inclusions (Fig. 5A), although less intensely than when using the laminA/C antibody. As discussed later, we hypothesize that TAmut interacts directly with INM proteins, whereas the presence of lamins in the inclusions may result from indirect TAmut–lamin interactions through INM proteins, explaining why lamins are detected less consistently in inclusions. In TAmut-expressing cells, but never in noninduced cells, we detected cytoplasmic inclusions coimmunoreactive for both emerin and laminA/C (data not shown), supporting their formation from NE-derived membranes. Proteins from other subcellular structures linked directly or indirectly to the NE, including the NPCs, the nuclear transport machinery, and the nucleoplasm, were not found in the inclusions by IF analysis (Table 1).
Table 1.
Analysis of the colocalization of endogenous and transfected GFP-fusion proteins to TAmut inclusions
|
Subcellular structure |
Endogenous proteins |
|
GFP-fusion proteins |
|
|---|---|---|---|---|
| Nuclear envelope—ER membrane | LaminA/C (nuclear lamina) | + | GFP-laminA (nuclear lamina) | - |
| LaminA (nuclear lamina) | - | GFP-laminC | - | |
| LaminB (nuclear lamina) | +/- | GFP-emerin (INM) | ++ | |
| Emerin (INM) | + | GFP-LBR (INM) | ++ | |
| Nup98(NPC) | - | GFP-p92 (NPC) | - | |
| Calnexin (ONM-ER) | - | GFP-sp25 (ONM-ER) | + | |
| GFP-sec61β (ONM-ER) | + | |||
| Nucleoplasm | SC35 | - | GFP-histoneH1 | - |
| ER-lumenal | Calreticulin | - | ||
| Plasma membrane | GFP-DAT | + | ||
| YFP-CFTR | + | |||
| Other | Karyopherin-β3 (nuclear transport) | - | GFP | - |
| Fbx2 (cytoplasmic) | - | GFP-GR | - | |
| α-Tubulin (cytoplasmic) | - | GFP-Q19 | - | |
|
|
Ubiquitin (diffuse) |
- |
|
|
Results are expressed as - (no signal), +/- (weak signal), + (moderate signal), and ++ (strong signal) in TAmut inclusions.
To gain further experimental evidence for an NE origin of inclusions, we transfected TA-expressing cell lines with GFP-fusion proteins known to localize to various NE and ER subcompartments. As summarized in Table 1 and shown in Figure 5B, GFP-fusion proteins that localize to the INM or ONM of the NE were also targeted to inclusions. Localization to inclusions was observed in fixed and unfixed cells, indicating that inclusions are not a fixation artifact. Two controls, GFP vector alone or GFP fused to a 19 polyglutamine tract, did not colocalize to inclusions (data not shown). Together, our IF and GFP-fusion experiments indicate that the deletion of a single glutamic acid residue causes TAmut to accumulate in the NE, resulting in the formation of inclusions derived from the NE.
To further assess differences in TAwt and TAmut proteins, we performed WB analysis of cell lysates in the presence or absence of reducing agent. These studies revealed differences both in the subcellular distribution of endogenous and overexpressed TA and in the formation of higher molecular weight (MW) complexes by TAwt and TAmut. In the absence of DTT, lysates from TAmut-expressing cells contained a higher MW band (∼85 kDa) immunostained with anti-TA antibodies. A second, even higher MW band was present equally in induced and noninduced cells (Fig. 6A). To determine whether these higher MW species localized to specific subcellular compartments, we studied their distribution in cell fractionation experiments. With TAmut, the ∼85 kDa band was weakly but consistently detected in the nuclear fraction and occasionally in the cytoplasmic fraction (data not shown). When high concentrations of protein were loaded, we also detected this band in the nuclear but not the cytoplasmic fraction of cells expressing TAwt, although less intensely seen than with TAmut. This band was absent in DTT-treated lysates, suggesting that it corresponds to a disulfide-linked TA complex, either a homodimer or a heterodimer of TA with another protein.
Figure 6.

WB analysis of TA under nonreducing conditions suggests the presence of TA oligomers. A, Denatured protein lysates in the absence of DTT show a large (>220 kDa) TA-immunoreactive band [anti-TA (MBP) 1:250] in induced and noninduced cells (black arrowhead) and a ∼85 kDa TA-immunoreactive band seen only in TAmut-expressing cells (empty arrowhead). B, Subcellular fractionation showed that the >220 kDa TA-immunoreactive band was found almost exclusively in the cytoplasmic fraction of induced and noninduced cells (black arrowhead). After the addition of DTT to the protein lysates, this band disappeared, and a faint band appeared at the expected MW for TA in the noninduced cytoplasmic lanes, suggesting that it corresponds to endogenous TA or another torsin-related protein detected by this antibody [anti-TA (MBP) 1:250].
The second higher MW band, which was seen equally in induced and uninduced cells, localized almost exclusively to the cytoplasmic compartment. In the presence of DTT, this high MW species reduced to the expected MW for TA (Fig. 6B), suggesting that it corresponds to endogenous TA existing in a larger, disulfide-linked protein complex. These results are consistent with previous sedimentation analyses showing oligomeric complexes of TAwt and TAmut (Kustedjo et al., 2000) and with the prediction that TA, as an AAA protein, functions as an oligomeric complex (Breakefield et al., 2001). The results further suggest that endogenous TA is present primarily in the cytoplasmic ER and not the nuclear ER. It is also possible that the antibody used in these experiments cross-reacts with torsinB or other torsin isoforms of similar MW, resulting in the detection of a torsin-related, but not torsinA-specific, protein.
TAmut expression does not alter nuclear import
The accumulation of TAmut in the NE and resultant inclusion formation could compromise nucleocytoplasmic transport by altering the number, location, or function of NPCs. To address this possibility, we tested whether TAmut expression impaired nuclear import of the GR, a well studied model protein for hormone-regulated nuclear import. GR is an appropriate model protein because it is expressed in central dopaminergic neurons, which are TA-expressing neurons implicated in DYT1 pathophysiology (Augood et al., 2002; Walker and Shashidharan, 2003). Live cell imaging of cells transfected with GFP-GR showed the onset of GFP-GR nuclear import 3–5 min after the addition of dexamethasone and completed translocation within 30 min (Fig. 7A). Because the bulk of import occurred 10–20 min after adding dexamethasone, we focused on this time frame for quantitative measures of transport rate. TAwt- or TAmut-expressing cells were transfected with GFP-GR expression plasmid, incubated for 48 hr in charcoal–dextran-stripped medium with or without doxycycline to induce TA expression, and then fixed either immediately before or 10, 15, and 20 min after adding dexamethasone. The rate of nuclear translocation of GFP-GR did not differ in the presence or absence of TA expression, whether TAwt or TAmut (Fig. 7B). This finding, together with the lack of sequestration of NPC proteins in TAmut inclusions, suggests that TAmut expression in the NE does not perturb active nucleocytoplasmic transport.
Figure 7.

TAmut does not alter the nuclear import of the GR. A, Live cell analysis of nuclear import of GFP-GR in a cell expressing TAmut, before and after dexamethasone treatment. B, Quantification of nuclear import of GFP-GR in fixed cells expressing TAwt, TAmut, and noninduced cells at different times after dexamethasone treatment. Results are the mean (±SD) of 100 cells for each group.
Influence of TAmut on ER function
Several features of TA, including its subcellular localization, similarity to the HSP 100/Clp (heat shock protein 100/CLP proteases) protein family, and putative role in protein quality control (McLean et al., 2002; Caldwell et al., 2003), suggest that TAmut could act by altering ER function. One possibility is that TAmut could activate ER stress responses such as the UPR. To address this hypothesis, we tested whether overexpression of TAwt or TAmut caused an increase in BiP levels, a marker of UPR activation. BiP levels did not change, suggesting that the presence of TAmut does not lead to significant ER stress (Fig. 8). Moreover, treatment of the parent PC6-3 cell line with the ER stressors thapsigargin and tunycamycin did not alter levels of endogenous TA (data not shown), in agreement with a previous report (Hewett et al., 2003). This suggests that TA is not upregulated during general ER stress responses.
Alternatively, TAmut expression might impair protein processing or maturation in the ER secretory pathway without causing detectable ER stress. To address this possibility, we tested whether TAmut expression impairs normal ER protein processing and surface transport of a candidate protein. We chose the hDAT as a candidate protein because it is a cell-surface, integral membrane glycoprotein present in dopaminergic cells. In TAmut cells coexpressing GFP-hDAT, the GFP signal was not highly enriched in inclusions, although many TAmut-derived inclusions contained small amounts of GFP signal (Fig. 9A,B). This is perfectly consistent with our proposed NE origin of inclusions, because ongoing protein synthesis and translocation are known to occur at the ONM of the NE. Indeed when GFP-hDAT was expressed in the absence of TAmut, some GFP-hDAT signal localized to a perinuclear rim, consistent with NE localization (Fig. 9C). When protein translation was blocked with cycloheximide, GFP-hDAT signal disappeared from the NE and became detectable only at the cell surface. This occurred in both TAmut-expressing and noninduced control cells. Thus, even in the presence of TAmut, GFP-hDAT was transported effectively to the cell surface. We obtained similar results with a second glycoprotein, YFP-CFTR (data not shown).
Figure 9.

The human dopamine transporter weakly colocalizes to TAmut-derived inclusions. A, A differentiated TAmut-expressing cell transfected with GFP-hDAT, showing both GFP and TAmut signal (red) in the inclusions and the NE. Nuclear DAPI staining is shown in blue. A higher-magnification view of the indicated areas highlights the colocalization of TA and hDAT in the inclusions (B) and in the nuclear envelope (C).
Despite the successful surface transport of fusion proteins in the presence of TAmut inclusions, we cannot rule out minor effects of TAmut on ER protein synthesis and trafficking. It should be noted that the intensity of the GFP-DAT and YFP-CFTR signal detected in inclusions was much less than that seen for proteins of the INM such as GFP-emerin, GFP-LBR, or LBR-GFP. Indeed, in TAmut cells expressing these NE GFP-fusion proteins, GFP-containing inclusions were easily seen by live-cell microscopy, clearly distinguishable from the pattern found in noninduced cells. This distinction was much less obvious for GFP-hDAT or YFP-CFTR, both of which required IF for us to detect the GFP signal colocalizing with TAmut inclusions.
Discussion
In this study, we created TA-expressing neural cell lines to study the pathobiology of DYT1 dystonia. Using this cell model, we obtained several lines of evidence indicating that TAmut preferentially accumulates in the NE, eventually leading to the formation of inclusions derived from this organelle. First, TAwt and TAmut displayed divergent subcellular distributions even before inclusions were formed, with TAmut preferentially localizing to a perinuclear rim pattern consistent with the NE. Second, TAmut colocalized with endogenous components of the NE, both in the perinuclear rim and in inclusions. Third, when coexpressed as fusion proteins, resident proteins of the NE colocalized with TAmut to the perinuclear rim and inclusions. Fourth, subcellular fractionation revealed the presence of TAmut in the nuclear fraction, presumably within the NE. Finally, all ER membrane proteins that did colocalize with TAmut to inclusions were ones also known to reside in the ONM, consistent with a NE origin; conversely, some proteins found in inclusions were not ER resident proteins. Taken together, these findings suggest that the common DYT1 mutation, which eliminates a single glutamic acid residue from TAmut, causes this disease protein to preferentially localize to the NE and form NE-derived inclusions. The significance of our results is bolstered by a recent report, published while this manuscript was under review, that describes similar findings, including the accumulation of TAmut in the NE in human disease tissue (Goodchild and Dauer, 2004). For these reasons, we believe that our studies of TA in neural lines provide valuable insight into potential disease mechanisms in DYT1 dystonia.
TorsinA and the nuclear envelope
Our finding that TAmut accumulates in the NE is also supported by previous reports that showed immunostaining for TAwt in a perinuclear rim in transfected cells (Kustedjo et al., 2000), the accumulation of TA in the NE after treatment with H2O2 (Hewett et al., 2003), and the presence of OOC-5 (oocyte formation-5), a torsin-related protein in Caenorhabditis elegans, in a perinuclear rim (Basham and Rose, 2001). Why does TAmut accumulate in the NE? At least two hypotheses could explain this phenomenon. The “diffusion-retention” model posits that INM proteins localize to the NE by interacting with NPCs, lamins, or other NE proteins (Rolls et al., 1999; Holmer and Worman, 2001). It is unlikely that TAmut interacts with NPCs because we did not find NPC proteins in the inclusions, and although INM proteins that interact with lamins typically have a long domain extending into the nucleoplasm, TA begins with an N-terminal transmembrane domain and is otherwise an entirely lumenal protein (Kustedjo et al., 2000; Hewett et al., 2003; Liu et al., 2003). In the case of TAmut, the glutamic acid deletion may promote aberrant interactions with one or more INM proteins, causing TA to be retained in the NE. Consistent with this hypothesis, TAwt lacking its transmembrane domain is secreted into the culture media, whereas TAmut is not (Liu et al., 2003).
A second possibility is that the mutation could impair the ATPase activity of TA. In this scenario, TAmut could still bind putative NE substrates or adaptor proteins yet be unable to hydrolyze ATP and so fail to act on and then release the interacting substrates. Such “enzyme trapping” could sequester TAmut in the NE. Keys to addressing this possibility will be (1) the identification of TA-binding partners in the NE, (2) a comparison of the ATPase activities of TAwt versus TAmut, and (3) a cellular analysis of TA that has been mutated in its ATP binding domains. Two recent publications have addressed these possibilities. Kustedjo et al. (2003) did not find differences in ATP hydrolysis activity in an assay using recombinant TA proteins. Conversely, Goodchild and Dauer (2004) showed that in cultured cells, TA with impaired ATP hydrolyzing ability also accumulated in the NE. These divergent, but not necessarily incompatible, results could reflect the different experimental systems used.
Our data also suggest that once TAmut accumulates in the NE, inclusions arise from this organelle. It has been proposed that the inclusions originate from the ER because they contain protein disulfide isomerase (PDI) (Hewett et al., 2000). This finding, however, is also consistent with a NE origin. PDI has been demonstrated to be present in noncytoplasmic ER locations, including the NE (Turano et al., 2002). As mentioned earlier, the translation–translocation machinery is present in the ONM, and PDI is part of this machinery, assisting in the proper folding and disulfide formation of newly synthesized proteins. Moreover, the inclusions were immunoreactive for some proteins of the nuclear lamina, and they are not synthesized in the ER.
Why does TAmut induce the formation of membranous inclusions? The simplest explanation is that it occurs because TAmut has accumulated in the NE. Overexpression of certain membrane proteins is known to increase membrane synthesis to compensate for this “protein overload,” resulting in the formation of anomalous membranous structures such as karmellas or crystalloid ER. We suggest that a similar process occurs in the NE when TAmut is overexpressed, and that the membranous inclusions may exist as a “protective” mechanism to clear the NE of abnormally accumulated protein.
The membranous inclusions induced by TAmut appear to be a novel type of inclusion. The absence of proteins of the NPC shows that they are not annulate lamellas, i.e., stacked ER membranes highly enriched in nuclear pores (Walther et al., 2003). As discussed in Results, they also are not derived from the recently described “ER quality control compartment” (Kamhi-Nesher et al., 2001). They do share, however, some similarities with karmellas, which are characterized by a concentric accumulation of paired membranes around the NE (Wright et al., 1988). Similar but predominantly nuclear membranous inclusions have been described in tellurium-treated Schwann cells (Berciano et al., 2000). TAmut-inclusions may correspond to a novel karmella-like structure derived from the NE.
It is important to mention that TAmut inclusions have not been seen in DYT1 disease brain (Walker et al., 2002; Rostasy et al., 2003). The mutation may cause TA to localize preferentially to the NE, which results in inclusion formation when the mutant protein is overexpressed. In DYT1 patients, lower levels of TAmut could accumulate in the NE yet not lead to inclusion formation, especially if inclusions are a byproduct of TA overexpression. Careful immunohistological analysis of the NE in human DYT1 brain is needed to answer this question.
An intriguing observation is the difference in subcellular distribution and electrophoretic behavior that we observed for endogenous versus overexpressed TA. By cellular fractionation, endogenous TA proved to be predominantly cytoplasmic. In contrast, overexpressed TA also partitioned into the nuclear fraction, more so with TAmut than with TAwt. Moreover, endogenous TA ran on nonreducing gels as a discrete disulfide-linked complex of >220 kDa, whereas overexpressed TA electrophoresed both as monomer and as an ∼85 kDa disulfide-linked complex, perhaps representing TA dimers. One possible explanation for these differences is a saturable mechanism that either retains TA in the cytoplasmic ER or exports TA from the NE. Whatever the underlying mechanism, our results suggest that TA undergoes dynamic flux between these two cellular compartments and that this flux may be tied to the oligomeric or redox status of TA. With respect to this last point, TA accumulated in the NE after H2O2 treatment in our cell model, confirming a previous report (Hewett et al., 2003). The potential role of disulfide linkages in regulating TA function and localization will be important to study in the future.
TorsinA and protein quality control
Because evidence has suggested a role for TA in protein quality control (McLean et al., 2002; Caldwell et al., 2003), we studied the potential relationship between TAmut expression and ER stress. In our cellular model, overexpression of TAmut did not activate the UPR, nor were TA levels upregulated after ER stress. TA therefore does not seem to be a stress-response protein (Hewett et al., 2003). These findings, together with the lack of misfolded protein aggregates and neurodegeneration in brains of DYT1 patients, suggest that a general failure in ER protein quality control does not play a crucial role in the disease state.
TAmut expression in neurons could impair aspects of the ER secretory pathway, however, altering the intracellular maturation and transport of membrane or secretory proteins. Although we detected a small amount of overexpressed GFP-hDAT and YFP-CFTR in inclusions, we suspect that this reflects partial localization to the ONM during biosynthesis and may not relate to disease mechanisms. Indeed, both proteins were able to reach the plasma membrane even in the presence of inclusions. Further study of dopaminergic neurons in DYT1 animal models should address this important question in vivo.
The potential pathogenic role of TAmut in the NE is intriguing because it adds to the growing list of neurological diseases linked to dysfunction in this organelle, including inherited neuropathies, muscular dystrophy, and premature aging (De Sandre-Giovannoli et al., 2003; Eriksson et al., 2003; Ostlund and Worman, 2003; Schirmer et al., 2003). In addition to playing a structural role, the NE is involved in various important cellular processes that might be altered in these disorders, including control of nucleocytoplasmic transport, signal transduction pathways, and regulation of gene expression (Burke and Stewart, 2002; Greber and Carafoli, 2002; Hutchison, 2002). Further understanding of the function of TA within the NE may help define the underlying pathogenic mechanism and lead toward rational therapies for this incurable disorder.
Footnotes
This work was supported by a Menkes/Markham Fellowship of the Dystonia Medical Research Foundation to P.G.-A. We are grateful to Drs. Xandra Breakefield, Jos Broers, Mario Galigniana, Ulrik Gether, Tokuko Hagaruchi, Randall Pittman, Tom Rappoport, Stefan Strack, and Michael Welsh for providing plasmids, cell lines, and antibodies. We thank Kenneth Moore and Jian Shao from the Central Microscopy Research Facility (University of Iowa) for their assistance with electron microscopy and Dr. Kevin Glenn and Victor Miller for their critical review of this manuscript.
Correspondence should be addressed to Dr. Pedro Gonzalez-Alegre, Department of Neurology, University of Iowa Hospitals and Clinics, 200 Hawkins Drive, Iowa City, IA 52242. E-mail: gonzalezp@mail.medicine.uiowa.edu.
DOI:10.1523/JNEUROSCI.4461-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/242593-09$15.00/0
References
- Ashbourne Excoffon KJ, Moninger T, Zabner J (2003) The coxsackie B virus and adenovirus receptor resides in a distinct membrane microdomain. J Virol 77: 2559–2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augood SJ, Hollingsworth Z, Albers DS, Yang L, Leung JC, Muller B, Klein C, Breakefield XO, Standaert DG (2002) Dopamine transmission in DYT1 dystonia: a biochemical and autoradiographical study. Neurology 59: 445–448. [DOI] [PubMed] [Google Scholar]
- Basham SE, Rose LS (2001) The Caenorhabditis elegans polarity gene ooc-5 encodes a Torsin-related protein of the AAA ATPase superfamily. Development 128: 4645–4656. [DOI] [PubMed] [Google Scholar]
- Berciano MT, Fernandez R, Pena E, Calle E, Villagra NT, Rodriguez-Rey JC, Lafarga M (2000) Formation of intranuclear crystalloids and proliferation of the smooth endoplasmic reticulum in Schwann cells induced by tellurium treatment: association with overexpression of HMG CoA reductase and HMG CoA synthase mRNA. Glia 29: 246–259. [DOI] [PubMed] [Google Scholar]
- Breakefield XO, Kamm C, Hanson PI (2001) TorsinA: movement at many levels. Neuron 31: 9–12. [DOI] [PubMed] [Google Scholar]
- Burke B, Stewart CL (2002) Life at the edge: the nuclear envelope and human disease. Nat Rev Mol Cell Biol 3: 575–585. [DOI] [PubMed] [Google Scholar]
- Caldwell GA, Cao S, Sexton EG, Gelwix CC, Bevel JP, Caldwell KA (2003) Suppression of polyglutamine-induced protein aggregation in Caenorhabditis elegans by torsin proteins. Hum Mol Genet 12: 307–319. [DOI] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL (1999a) Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet 8: 673–682. [DOI] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Bonini NM, Paulson HL (1999b) Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci 19: 10338–10347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N (2003) Lamin A truncation in Hutchinson-Gilford progeria. Science 300: 2055. [DOI] [PubMed] [Google Scholar]
- Erhardt JA, Hynicka W, DiBenedetto A, Shen N, Stone N, Paulson H, Pittman RN (1998) A novel F box protein, NFB42, is highly enriched in neurons and induces growth arrest. J Biol Chem 273: 35222–35227. [DOI] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS (2003) Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423: 293–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fahn S, Bressman SB, Marsden CD (1998) Classification of dystonia. Adv Neurol 78: 1–10. [PubMed] [Google Scholar]
- Galigniana MD, Radanyi C, Renoir JM, Housley PR, Pratt WB (2001) Evidence that the peptidylprolyl isomerase domain of the hsp90-binding immunophilin FKBP52 is involved in both dynein interaction and glucocorticoid receptor movement to the nucleus. J Biol Chem 276: 14884–14889. [DOI] [PubMed] [Google Scholar]
- Granas C, Ferrer J, Loland CJ, Javitch JA, Gether U (2003) N-terminal truncation of the dopamine transporter abolishes phorbol ester- and substance P receptor-stimulated phosphorylation without impairing transporter internalization. J Biol Chem 278: 4990–5000. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Alegre P, Miller VM, Davidson BL, Paulson HL (2003) Toward therapy for DYT1 dystonia: allele-specific silencing of mutant TorsinA. Ann Neurol 53: 781–787. [DOI] [PubMed] [Google Scholar]
- Goodchild RE, Dauer WT (2004) Mislocalization to the nuclear envelope: an effect of the dystonia-causing torsinA mutation. Proc Natl Acad Sci USA (published on-line Jan. 7). [DOI] [PMC free article] [PubMed]
- Greber UF, Carafoli E (2002) Signaling takes control of nucleo-cytoplasmic trafficking. Workshop on signal-regulated nuclear transport. EMBO Rep 3: 410–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haraguchi T, Koujin T, Hayakawa T, Kaneda T, Tsutsumi C, Imamoto N, Akazawa C, Sukegawa J, Yoneda Y, Hiraoka Y (2000) Live fluorescence imaging reveals early recruitment of emerin, LBR, RanBP2, and Nup153 to reforming functional nuclear envelopes. J Cell Sci 113: 779–794. [DOI] [PubMed] [Google Scholar]
- Hewett J, Gonzalez-Agosti C, Slater D, Ziefer P, Li S, Bergeron D, Jacoby DJ, Ozelius LJ, Ramesh V, Breakefield XO (2000) Mutant torsinA, responsible for early-onset torsion dystonia, forms membrane inclusions in cultured neural cells. Hum Mol Gen 9: 1403–1413. [DOI] [PubMed] [Google Scholar]
- Hewett J, Ziefer P, Bergeron D, Naismith T, Boston H, Slater D, Wilbur J, Schuback D, Kamm C, Smith N, Camp S, Ozelius LJ, Ramesh V, Hanson PI, Breakefield XO (2003) TorsinA in PC12 cells: localization in the endoplasmic reticulum and response to stress. J Neurosci Res 72: 158–168. [DOI] [PubMed] [Google Scholar]
- Holmer L, Worman HJ (2001) Inner nuclear membrane proteins: functions and targeting. Cell Mol Life Sci 58: 1741–1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CJ (2002) Lamins: building blocks or regulators of gene expression? Nat Rev Mol Cell Biol 3: 848–858. [DOI] [PubMed] [Google Scholar]
- Kamhi-Nesher S, Shenkman M, Tolchinsky S, Fromm SV, Ehrlich R, Lederkremer GZ (2001) A novel quality control compartment derived from the endoplasmic reticulum. Mol Biol Cell 12: 1711–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konakova M, Pulst SM (2001) Immunocytochemical characterization of torsin proteins in mouse brain. Brain Res 922: 1–8. [DOI] [PubMed] [Google Scholar]
- Konakova M, Huynh DP, Young W, Pulst SM (2001) Cellular distribution of Torsin A and Torsin B in normal human brain. Arch Neurol 58: 921–927. [DOI] [PubMed] [Google Scholar]
- Kustedjo K, Bracey MH, Cravatt BF (2000) Torsin A and its torsion dystonia-associated mutant form are lumenal glycoproteins that exhibit distinct subcellular localizations. J Biol Chem 275: 27933–27939. [DOI] [PubMed] [Google Scholar]
- Kustedjo K, Deechongkit S, Kelly JW, Cravatt BF (2003) Recombinant expression, purification, and comparative characterization of torsinA and its torsion dystonia-associated variant deltaE-torsinA. Biochemistry 42: 15333–15341. [DOI] [PubMed] [Google Scholar]
- Liu Z, Zolkiewska A, Zolkiewski M (2003) Characterization of human torsinA and its dystonia-associated mutant form. Biochem J 374: 117–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean PJ, Kawamata H, Shariff S, Hewett J, Sharma N, Ueda K, Breakefield XO, Hyman BT (2002) TorsinA and heat shock proteins act as molecular chaperones: suppression of alpha-synuclein aggregation. J Neurochem 83: 846–854. [DOI] [PubMed] [Google Scholar]
- Ostlund C, Worman HJ (2003) Nuclear envelope proteins and neuromuscular diseases. Muscle Nerve 27: 393–406. [DOI] [PubMed] [Google Scholar]
- Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, Fahn S, Risch NJ, Buckler AJ, Gusella JF, Breakefield XO (1997) The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat Genet 17: 40–48. [DOI] [PubMed] [Google Scholar]
- Perez MK, Paulson HL, Pittman RN (1999) Ataxin-3 with an altered conformation that exposes the polyglutamine domain is associated with the nuclear matrix. Hum Mol Genet 8: 2377–2385. [DOI] [PubMed] [Google Scholar]
- Pittman RN, Wang S, DiBenedetto AJ, Mills J (1993) A system for characterizing cellular and molecular events in programmed neuronal cell death. J Neurosci 64: 566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls MM, Stein PA, Taylor SS, Ha E, McKeon F, Rapoport TA (1999) A visual screen of a GFP-fusion library identifies a new type of nuclear envelope membrane protein. J Cell Biol 146: 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rostasy K, Augood SJ, Hewett JW, Leung JC, Sasaki H, Ozelius LJ, Ramesh V, Standaert DG, Breakefield XO, Hedreen JC (2003) TorsinA protein and neuropathology in early onset generalized dystonia with GAG deletion. Neurobiol Dis 12: 11–24. [DOI] [PubMed] [Google Scholar]
- Schirmer EC, Florens L, Guan T, Yates III JR, Gerace L (2003) Nuclear membrane proteins with potential disease links found by subtractive proteomics. Science 301: 1380–1382. [DOI] [PubMed] [Google Scholar]
- Shashidharan P, Kramer BC, Walker RH, Olanow CW, Brin MF (2000) Immunohistochemical localization and distribution of torsinA in normal human and rat brain. Brain Res 853: 197–206. [DOI] [PubMed] [Google Scholar]
- Turano C, Coppari S, Altieri F, Ferraro A (2002) Proteins of the PDI family: unpredicted non-ER locations and functions. J Cell Physiol 193: 154–163. [DOI] [PubMed] [Google Scholar]
- Voeltz GK, Rolls MM, Rapoport TA (2002) Structural organization of the endoplasmic reticulum. EMBO Rep 3: 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker RH, Brin MF, Sandu D, Good PF, Shashidharan P (2002) TorsinA immunoreactivity in brains of patients with DYT1 and non-DYT1 dystonia. Neurology 58: 120–124. [DOI] [PubMed] [Google Scholar]
- Walker RH, Shashidharan P (2003) Developments in the molecular biology of DYT1 dystonia. Mov Disord 18: 1102–1107. [DOI] [PubMed] [Google Scholar]
- Walther TC, Askjaer P, Gentzel M, Habermann A, Griffiths G, Wilm M, Mattaj IW, Hetzer M (2003) RanGTP mediates nuclear pore complex assembly. Nature 424: 689–694. [DOI] [PubMed] [Google Scholar]
- Wright R, Basson M, D'Ari L, Rine J (1988) Increased amounts of HMG-CoA reductase induce “karmellae”: a proliferation of stacked membrane pairs surrounding the yeast nucleus. J Cell Biol 107: 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]