

Abstract
The lobster heart is synaptically driven by the cardiac ganglion, a spontaneously bursting neural network residing within the cardiac lumen. Here, we present evidence that nitric oxide (NO) plays an inhibitory role in lobster cardiac physiology. (1) NO decreases heartbeat frequency and amplitude. Decreased frequency is a direct consequence of a decreased ganglionic burst rate. Decreased amplitude is an indirect consequence of decreased burst frequency, attributable to the highly facilitating nature of the synapses between cardiac ganglion neurons and muscle fibers (although, during prolonged exposure to NO, amplitude recovers to the original level by a frequency-independent adaptation mechanism). NO does not alter burst duration, spikes per burst, heart muscle contractility, or amplitudes of synaptic potentials evoked by stimulating postganglionic motor nerves. Thus, NO acts on the ganglion, but not on heart muscle. (2) Two observations suggest that NO is produced within the lobster heart. First, immunoblot analysis shows that nitric oxide synthase (NOS) is strongly expressed in heart muscle relative to other muscles. Second, l-nitroarginine (l-NA), an NOS inhibitor, increases the rate of the heartbeat (opposite to the effects of NO). In contrast, the isolated ganglion is insensitive to l-NA, suggesting that heart muscle (but not the ganglion) produces endogenous NO. Basal heart rate varies from animal to animal, and l-NA has the greatest effect on the slowest hearts, presumably because these hearts are producing the most NO. Thus, because the musculature is a site of NOS expression, whereas the ganglion is the only intracardiac target of NO, we hypothesize that NO serves as an inhibitory retrograde transmitter.
Keywords: crustacean, heart, neuromodulation, central pattern generator, negative inotropy, negative chronotropy, nitric oxide
Introduction
Nitric oxide (NO) is a ubiquitous signaling molecule used by most eukaryotic and prokaryotic organisms (Hanafy et al., 2001; Torreilles, 2001; Adak et al., 2002; Neill et al., 2002). Because of its rapid membrane-permeating characteristics and short half-life in biological tissues, NO is typically used to transmit information between closely juxtaposed cells. For example, NO serves as a local modulator of blood pressure (Shesely et al., 1996; Hanafy et al., 2001), cardiac function (Balligand et al., 1993; Keaney et al., 1996; Zahradnikova et al., 1997; Gyurko et al., 2000), neuronal development (Cramer et al., 1998; Scholz et al., 1998; Champlin and Truman, 2000), synaptic transmission (Schuman and Madison, 1994; Huang, 1997), and oscillatory behavior in pattern-generating networks of neurons (Pape and Mager, 1992; Moroz et al., 1993; Gelperin, 1994; Elphick et al., 1995; Hedrick and Morales, 1999; Scholz et al., 2001; Newcomb and Watson, 2002; McLean and Sillar, 2000).
Extracts of crustacean heart muscle contain high levels of a calcium-sensitive form of nitric oxide synthase (NOS) (Scholz et al., 2002), suggesting the possibility that contraction of the crustacean heart may be accompanied by NO production. A likely target for NO produced by cardiac muscle would be the cardiac ganglion, situated within the lumen of the heart and well within the diffusional radius of the short-lived NO molecule. Indeed, NO is known to trigger a biochemical response (increased cGMP metabolism) when applied to the isolated cardiac ganglion of the crab (Scholz et al., 2002). Because the function of the cardiac ganglion is to provide the synaptic input that drives the contractions of the heart (Alexandrowicz, 1932; Cooke, 2002), it is attractive to speculate that NO may provide feedback information about the status of cardiac muscle that could be used to modulate the activity of the ganglion. A conceptually similar retrograde signaling role has been described for NO at a number of synapses in the vertebrate CNS (Daniel et al., 1993; Boulton et al., 1995; Arancio et al., 1996; Boxall and Garthwaite, 1996; Lev-Ram et al., 1997; Prast and Philippu, 2001).
To test the hypothesis that NO plays a signaling role in crustacean cardiac physiology, we have examined the effects of exogenously applied NO on the intact lobster heart and on an isolated, spontaneously bursting cardiac ganglion preparation. In addition, we have analyzed the effects of NO on nerve-evoked contractions (elicited after ablating the ganglion) and on intact hearts exposed to elevated potassium (which evokes contractures while shutting down the rhythmic output of the ganglion), to determine whether NO can affect the muscle in the absence of ganglionic input. To investigate whether endogenously produced NO might play a role in cardiac physiology, we have confirmed the presence of NOS immunoreactivity in cardiac extracts, and evaluated the physiological effects of the NOS inhibitor l-nitroarginine.
Materials and Methods
Animals. Lobsters were purchased from Commercial Lobster (Boston MA) and Harris-Teeter (Chapel Hill, NC) and kept in aerated artificial seawater tanks at 11°C. All experiments were performed in chilled (9–11°C) lobster saline. The saline solution contained the following (in mm): 479 NaCl, 12.7 KCl, 13. 7 CaCl2, 20 MgSO4, 3.9 Na2SO4, 5 HEPES, and 10 glucose, pH 7.45.
Immunoblotting. Lobsters were anesthetized on ice. Freshly dissected muscle tissues were rapidly frozen on dry ice, and then stored temporarily at –80°C. Frozen tissues were thawed, then homogenized in 10 mm HEPES, pH 7.0, containing 1 mm EDTA and a 1:100 dilution of a protease inhibitor mixture [2.5 mm 4-(2-aminoethyl)-benzenesulfonyl fluoride, 38 μm pepstatin A, 35 μm trans-epoxysuccinyl-l-leucylamido(4-guanidino)butane, 100 μm bestatin, 55 μm leupeptin, and 2 μm aprotinin, obtained from the Sigma, St. Louis MO]. Homogenates were centrifuged for 30 min at 100,000 × g, and the supernates were fractionated on 8% SDS-PAGE gels, transferred to Protran nitrocellulose blotting membrane (Schleicher and Schuell BioScience, Keene NH), and analyzed by Western blotting with a universal NOS (uNOS) antibody (diluted 1:100; Affinity Bioreagents, Golden, CO). Immunoreactive proteins were detected by enhanced chemiluminescence (Renaissance Reagent, NEN Life Science Products, Boston, MA), using an HRP-coupled goat-anti-rabbit secondary antibody (diluted 1:5000; Jackson ImmunoResearch, West Grove, PA).
Intact heart preparation. The tail, the anterior part of the carapace, the stomach, and the hepatopancreas were removed after anesthetizing the animals, and the sternal artery was cut near the ventral nerve cord. Cuts were then made laterally along the carapace on either side of the heart, and a small piece of dorsal carapace (with heart attached) was separated from the rest of the thorax. For studies of the intact cardiac preparation, the noncardiac muscles around the heart were excised and the hypodermis with the attached heart was removed from the carapace by blunt dissection and pinned to a dish partially filled with clear Sylgard (Dow Corning, Midland, MI). To measure displacement during contraction, the sternal artery (which is normally under tension in vivo) was attached with a silk thread (0.4 mm; Genzyme, Fall River, MA) to a Grass FT03 force transducer (Astro-Med, West Warwick, RI) and stretched at an angle ∼30° off of vertical, closely paralleling the normal trajectory taken by the artery in the intact animal. The lumen of the heart was cannulated by creating an opening in the anterior wall of the heart just beneath the five anterior arteries. Chilled saline (10–14°C) was perfused into the lumen at ∼5 ml/min. The external surface of the heart was also superfused with saline at ∼1 ml/min. When the effects of NO donors were analyzed, the drugs were simultaneously added to (and removed from) both the external and internal solutions.
Isolated cardiac ganglion preparation. A V-shaped cut was made from each of the two ventral ostia toward the sternal artery on the ventral wall of an isolated heart to expose the Y-shaped cardiac ganglion, which sits directly on the dorsal inner wall of the heart muscle and gives rise to a set of extensively arborizing motor nerve roots that penetrate and innervate the musculature. The ganglion was freed from the muscle by transecting each of these roots at the distalmost point at which the root penetrates the muscle, and the isolated ganglion with attached roots was pinned dorsal side up in a Sylgard-coated dish. This enabled us to record the electrical activity of motor axons in the nerve roots using stainless steel monopolar extracellular electrodes isolated in petroleum jelly wells, and intracellular activity in the motor neuron cell bodies using 20–40 MΩ microelectrodes filled with 0.6 m K2SO4 and 20 mm KCl connected to an Axoclamp 2A amplifier (Axon Instruments, Union City, CA). Isolated ganglia were superfused with chilled saline (in the absence or presence of NO donors) at 10–14°C and ∼3 ml/min.
Stimulus-driven synaptic preparation. For these experiments, we opened the heart from the ventral side and pinned it flat in a Sylgard-coated dish (internal surface facing upward). We then ablated the entire ganglionic trunk containing the neuronal cell bodies, and used a suction electrode to stimulate the remaining stump of one of the postganglionic motor nerve roots that innervates the cardiac musculature. Stimulation was delivered with an A-M Systems (Everett, WA) isolated pulse stimulator, either in the form of single 0.5 msec rectangular pulses or as trains of such pulses organized into bursts of four (or, in some experiments, five) pulses with an interpulse interval of 70 msec. Muscle contraction was minimal in response to such short bursts, allowing us to obtain stable intracellular recordings of muscle membrane potential. The preparation was continuously superfused as described above for the intact heart.
Stimulus-driven contractile preparation. For these experiments, a small hole was cut in the ventral heart muscle wall and the ganglionic trunk was cut at the level of the posterior lateral nerves (Cooke, 2002). A suction electrode was introduced through this hole and the anterior portion of the ganglion, containing most of the motor neurons, was sucked up and stimulated with bursts. Each individual burst, consisting of 0.5 msec pulses given at 80 Hz for 200 msec, was designed to mimic the naturally occurring bursts that are typically generated by the cardiac ganglion. If such bursts are applied at a regular frequency, the bursting of the anterior motor neurons is quickly entrained, and the frequency at which the heart muscle contracts can then be manually controlled by controlling the frequency of the stimulus. In the experiments reported below, the burst frequency was varied between 0.8 and 0.2 Hz, and the resulting contractions were measured using a displacement sensor attached to the sternal artery as described above.
The absolute values of contraction amplitudes recorded in these cases were lower than those of the intact heart, because the hole in the ventral muscle wall decreased the strength of contraction. In all preparations in which cardiac ganglion activity was evoked with a suction electrode (n = 9) contraction amplitude also declined over the 2–3 hr time course of the experiment, likely because of damage caused by repeated wear and tear across the suction electrode positioned within the contracting heart.
Data acquisition and analysis. Data were digitized at 5 kHz for extracellular and intracellular recordings and at 1 kHz for tension recordings using a Digidata 1200 data acquisition board (Axon Instruments) and analyzed using Clampfit 8.0 (Axon Instruments), Igor Pro (WaveMetrics, Lake Oswego, OR), Spike2 (Cambridge Electronics Design, Cambridge, England), and Excel (Microsoft, Bellevue, WA). Intracellular records from muscle fibers were filtered at 0.1 kHz and smoothed to remove stimulus artifacts. Tension recordings were filtered at 10 Hz during acquisition. Instantaneous beat frequency was calculated as the reciprocal of the time interval between the current beat and the beat that immediately preceded it (in Hertz). Moving bin analysis was performed with a custom macro developed in Igor Pro, which allowed us to determine the means of all events occurring within successive, nonoverlapping time windows of defined length (typically 60 or 120 sec). Data were plotted and statistical tests performed using Sigma Plot and Sigma Stat (SPSS, San Rafael, CA) or Excel (Microsoft). The reported p values were obtained either from a two-tailed t test (paired or unpaired, as appropriate) or from a one-way repeated-measures ANOVA followed by Student–Newman–Keuls pairwise comparisons. Data are presented as means ± SE.
Chemicals. The NO donor S-nitroso-N-acetyl-penicillamine (SNAP) and the NOS inhibitor l-nitroarginine (l-NA) were obtained from Alexis Biochemicals (Lausanne, Switzerland). The NO donor 3-[2-hydroxy-2-nitroso-1-propylhydrazino]-1-propanamine (NOC-15) was obtained from Sigma-Aldrich (St. Louis, MO).
Results
Lobster heart muscle is enriched for NOS-like immunoreactivity
In a recent study of NOS expression in a crab (Cancer productus), Scholz et al. (2002) demonstrated that, compared with other tissues, crab heart muscle extracts are enriched for a calcium-activated form of NOS. The high levels of enzyme activity observed in this previous study correlated well with the presence of an immunoreactive protein that was detected on Western blots of crab heart muscle cytosol, using a well characterized anti-NOS antibody directed against a strongly conserved core catalytic region of the enzyme. With this same antibody, we now show that extracts of lobster heart (generated from isolated muscle after removal of the ganglion) also express a 132 kDa putative NOS, whereas a comparable protein was not observed in other lobster muscles (Fig. 1). Thus, in crustaceans, NOS expression appears to be enriched in the heart relative to other contractile tissues.
Figure 1.

Immunoblot analysis of NOS expression in lobster contractile tissues. Each lane contains 60 μg of cytosolic protein obtained, as indicated, from heart (H), walking leg dactyl opener muscle (DO), walking leg dactyl closer muscle (DC), superficial abdominal extensor muscle (SE), and dorsal thoracic body wall muscle (DM). To the left is an immunoblot generated with a uNOS antibody that recognizes a core domain conserved across a variety of vertebrate and invertebrate species. The black arrowhead denotes the position of the NOS-like protein. To the right is the Ponceau S stain of the same blot generated just before immunostaining, showing that equivalent protein loads were applied to each lane. Positions of molecular weight standards (in kilodaltons) are as indicated.
NO decreases the amplitude and frequency of the lobster heartbeat
The observation that high levels of NOS are present in both crab and lobster cardiac tissue suggests that NO might play a local role in controlling aspects of crustacean cardiac physiology. To investigate this hypothesis, we measured the effects of exogenously applied NO on the frequency, amplitude, rise time, and fall time of the lobster heartbeat, as measured with a displacement sensor attached to the spontaneously active in vitro preparation. Under control conditions, after an initial stabilization period, the isolated hearts beat steadily at a mean frequency of 0.84 ± 0.07 Hz and with a mean contraction amplitude of 0.29 ± 0.03 mm (n = 9 typical experiments, chosen at random). For each individual heartbeat, the contraction phase was approximately two times faster than the relaxation phase: the average rise time (required to reach half of the maximal amplitude) was 121 ± 11 msec, and the average fall time (to half maximal amplitude) was 225 ± 16 msec.
The NO donor SNAP (at 10–5 m) decreased both the rate and amplitude of the heartbeat, as seen in Figure 2A (examples of tension recordings obtained before and after application of the drug). Although the control frequencies and amplitudes varied somewhat from preparation to preparation, the effects of SNAP were consistent (Fig. 2B): on average, at the peak of the response, contraction frequency decreased by 39 ± 6% (to a mean value of 0.53 ± 0.08 Hz; n = 9; p < 0.001 relative to control), and amplitude by 31 ± 8% (to a mean value of 0.21 ± 0.04 mm; n = 9; p < 0.001). SNAP also slowed the fall time of each contraction without significantly affecting its rise time. This is most easily appreciated by scaling control and SNAP-treated heartbeats to the same amplitude (Fig. 2C). The average rise time in the presence of SNAP was 124 ± 12 msec (n = 9; p = 0.32 relative to control), whereas the average fall time was 292 ± 29 msec (n = 9; p = 0.002 relative to control).
Figure 2.
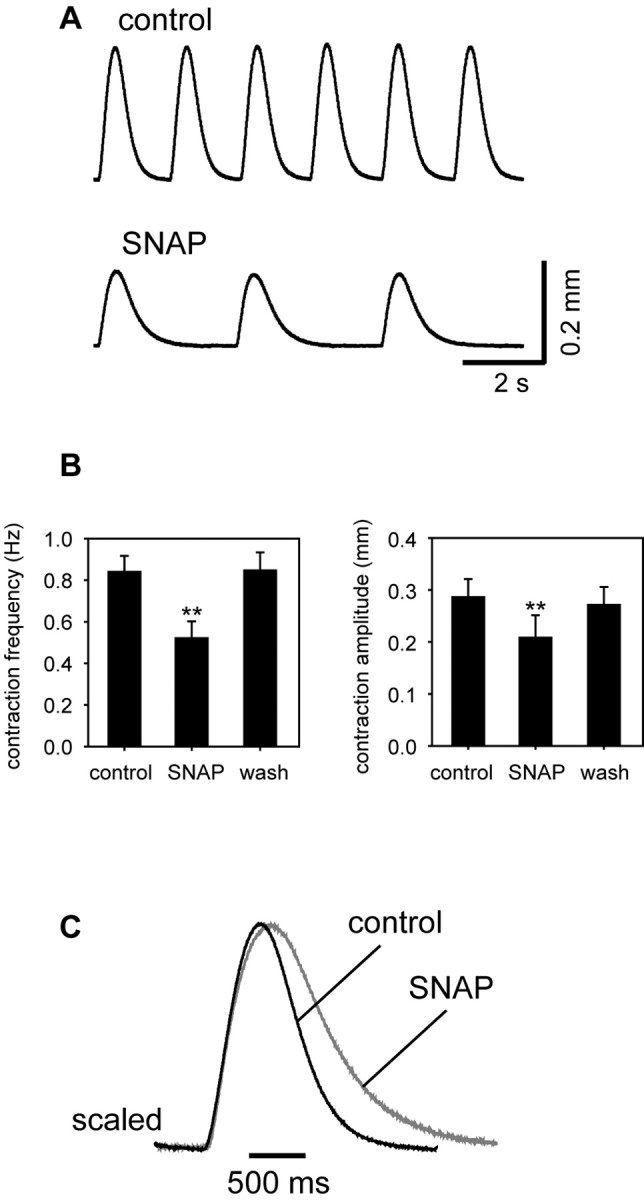
Negative inotropic and chronotropic effects of NO on the lobster heart. A, Representative traces showing displacement over time measured under control conditions (top trace) and at the peak of the response to SNAP (bottom trace). B, Mean responses of nine preparations before, during, and after washout of NO. **p < 0.001 relative to control, paired two-tailed t test. C, Two superimposed heart beats (each the average of 6 consecutive contractions) taken before (black; control) and at the peak response to 10–5 m SNAP (gray; SNAP). The traces are normalized to peak amplitude.
The time course of the change in amplitude is very similar to that of the change in frequency. Figure 3A shows data from an individual preparation, whereas Figure 3B shows the normalized average of the responses of nine independent preparations. The inotropic and chronotropic effects occur with comparable latencies, and the rate of change of amplitude parallels the rate of change of frequency throughout the response (Fig. 3B). Furthermore, as an individual preparation responds to SNAP, its beat-to-beat changes in amplitude and frequency are essentially linearly related to one another, as shown in Figure 4A. The graph plots the amplitude of each heartbeat as a function of its instantaneous frequency (i.e., the reciprocal of the time interval between the current beat and the beat that preceded it). The values enclosed by the circle in the upper right represent beats that occurred under control conditions; the values falling on the diagonal line from upper right to lower left represent beats that occurred as the preparation was beginning to respond to 10–5 m SNAP; and the values circled in the lower left represent beats that occurred after the response had reached its maximum value.
Figure 3.

Time course of the effects of SNAP on the isolated, spontaneously beating heart. A, Amplitude (white symbols) and instantaneous contraction frequency (black symbols) from a representative experiment plotted as a function of time before, during (black bar), and after washout of 10–5 m SNAP, followed by the application of 10–5 m SNAP solution that had been allowed to decay for more than eight half-lives (degassed SNAP). B, The initial phase of the response to 10–5 m SNAP (applied at the arrow) as a function of time (data represent means ± SE of 9 preparations). To combine data from different preparations, we scaled the control amplitude and frequency to 1 for each experiment and normalized subsequent heartbeats relative to control. Data were subdivided into consecutive 30 sec time bins, and the mean amplitude and frequency were determined in each bin (moving bin analysis). The average of these means (and the SE) were then calculated for each time bin across the nine experiments. After an initial lag (attributable in large part to the dead space in the perfusion line) the amplitude and frequency decrease in parallel. The rates of change of each parameter with respect to time are given by the solid and dashed lines, respectively.
Figure 4.
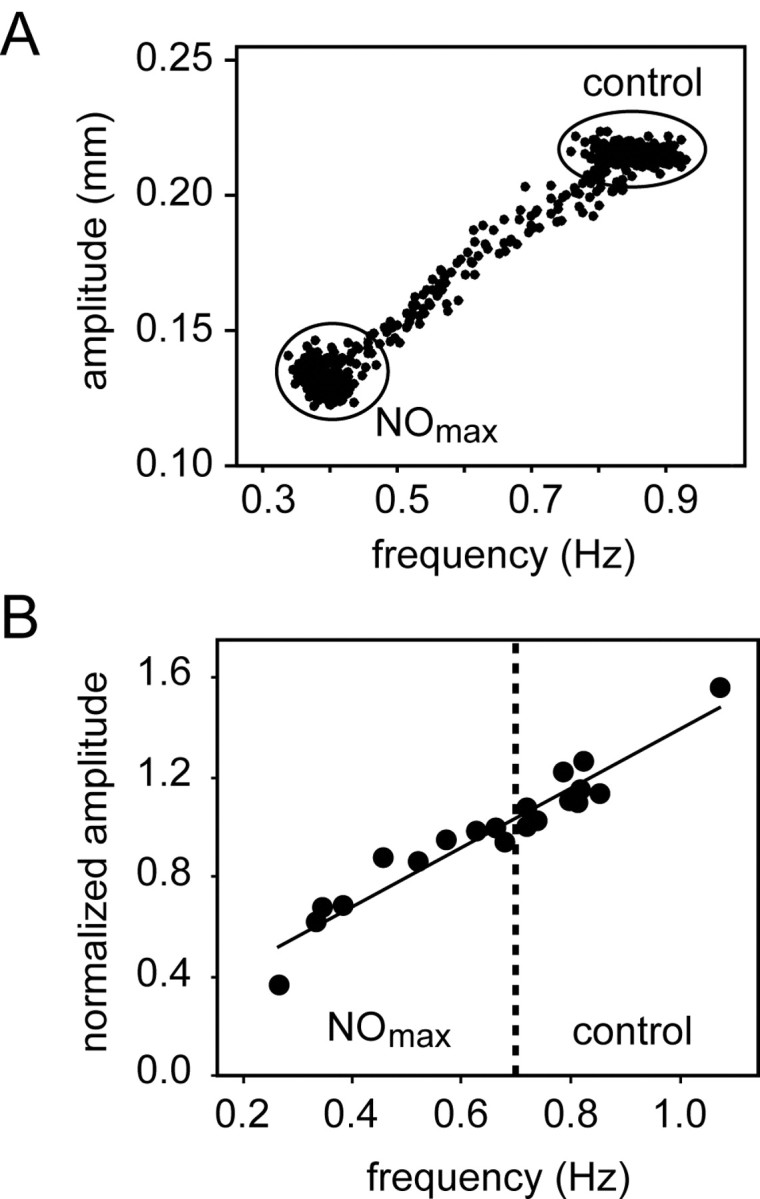
The frequency and amplitude of the heartbeat are linearly correlated over a range of frequencies. A, Data from a single preparation before and during its response to 10–5 m SNAP, with the amplitude of each heartbeat plotted as a function of the instantaneous frequency of that beat. (See Results for details.) B, Amplitude versus frequency data from nine preparations. Contraction amplitudes were first normalized to 1 at 0.7 Hz contraction frequency for each preparation. Mean normalized control amplitudes (all at frequencies >0.7 Hz) and mean normalized amplitudes at the peak of the SNAP response (all at frequencies <0.7 Hz) were plotted against the respective mean contraction frequencies in the absence (control) and presence (NOmax) of SNAP. The solid line represents a linear regression fit (r2 = 0.92; p < 0.001).
This relationship between frequency and amplitude holds for multiple preparations over a large range of frequencies, as shown in Figure 4B. For ease of presentation, this figure focuses only on the starting (control) and final maximal (NOmax) values for each of 10 preparations stimulated with SNAP. To permit comparison across animals, amplitudes are normalized to the amplitude at 0.7 Hz. Thus, each animal is represented by two values: its normalized control value (amplitude as a function of frequency), plotted to the right of the dashed line at 0.7 Hz, and its normalized NO-stimulated value (amplitude as a function of frequency), plotted to the left of the dashed line. These aggregate data are fit well by a straight line (n = 10; r2 = 0.92).
Adaptation is observed during prolonged exposure to NO
The marked temporal correlation and linear relationship between frequency and amplitude during the onset of the response to SNAP suggests that a common underlying mechanism regulates both parameters, a hypothesis that will be developed further below. The close correspondence between frequency and amplitude is not maintained, however, during prolonged (>10 min) exposures to SNAP. Under these conditions, an adaptive process begins to reverse the effects of the stimulus, and the rate and extent of adaptation is typically much greater in the amplitude domain than in the frequency domain.
In contrast with the reproducible nature of the onset of the response, adaptation actually varies widely from preparation to preparation. Nevertheless, certain general principles can be observed in Figure 5A (which shows the behavior of an individual preparation during 60 min of continuous exposure to SNAP) and Figure 5B (the average of six such responses, normalized as in Fig. 3B to correct for differences in the control frequency and control amplitude of each preparation). Most commonly, complete adaptation was observed in the amplitude domain during our studies, whereas partial adaptation was more characteristic in the frequency domain. In addition, amplitude responses began to adapt sooner than frequency responses, and amplitude usually showed a small “off response” (increased contractility) when the SNAP was washed out. However, we have also observed preparations in which adaptation failed to occur altogether, or in which frequency adapted more completely than amplitude. The reasons for this variability are not evident. Still, it is clear from the data presented in Figures 3,4,5 that amplitude and frequency are typically tightly coupled during the onset phase of the response, but are usually uncoupled during the adaptation phase.
Figure 5.

Nonequivalent adaptation of amplitude and frequency during prolonged exposure to SNAP. A, Time course of changes in heartbeat amplitude (white symbols) and instantaneous frequency (black symbols) of an individual preparation before, during (black bar), and after a 60 min exposure to 10–5 m SNAP. B, Averaged data from six such experiments, normalized as in Figure 3B with 2 min time bins.
Verification that NO is responsible for the physiological actions of SNAP
SNAP spontaneously breaks down in solution, producing two molecules of NO and one molecule of N-acetyl-d,l-penicillamine disulfide per molecule of SNAP (t1/2 ≅ 5 hr at room temperature, pH 7; Roy et al., 1994) The NO is also unstable, decomposing rapidly to form a mixture of nitrate and nitrite (Ignarro, 1990). To determine whether the effects described above are caused by NO or other SNAP breakdown products, we investigated the physiological actions of a SNAP solution that had been allowed to incubate at room temperature until all of the SNAP and all of the NO had decayed (more than eight half-lives). Such “degassed” SNAP solutions had no effect on the preparation (Fig. 3A).
Figure 6 compares concentration response curves for SNAP and degassed SNAP. We applied each test concentration for 15 min, measured the effect (if any) at the peak of the response, and interposed a 45 min washout period between exposures. The order of application of high and low concentrations was varied from experiment to experiment. Using this protocol we saw no obvious anomalies in heartbeat frequency that could be attributed to long-term desensitization or adaptation, whereas the effects of SNAP on amplitude were rather more variable from preparation to preparation. For this reason, we have not plotted the amplitude data. Experiments with freshly prepared SNAP revealed a concentration-dependent inhibitory effect on frequency that is well described by the standard Michaelis–Menten equation using an IC50 for SNAP of 5 × 10–6 m. Note, however, that the concentration of NO must have been well below that of the SNAP that was being used to generate it. In contrast, the degassed solution was inactive even at the highest concentration tested.
Figure 6.

The effects of SNAP are concentration dependent. Heart rate plotted as a function of SNAP concentration (white symbols; n = 8 animals), or degassed SNAP (black symbols; n = 3). Each point represents the mean ± SE response to a 15 min application. Data were fitted to a standard single binding site isotherm with an IC50 of 5 × 10–6 m.
As an additional control, we exposed intact hearts to NOC-15, another NO donor that is chemically distinct from SNAP. NOC-15 generates NO more rapidly than does SNAP, producing one molecule of NO per molecule of NOC-15 with a t1/2 ≅ 77 min at 22°C, pH 7 (Keefer et al., 1996). At 10–4 m, the inhibitory effects of NOC-15 on the frequency and amplitude of the heartbeat were similar to those of SNAP (n = 5), whereas degassed NOC-15 was physiologically inert (n = 2) (data not shown). The consistent effects of two independent NO donors (and their degassed derivatives) argues that the observed inhibition is caused by NO rather than by any alternative degradation products or by the precursors themselves.
NO slows burst frequency of the isolated cardiac ganglion
The cardiac ganglion generates the rhythmic motor pattern that drives lobster heart contractions, and lobster cardiac muscle has no myogenic contraction (Cooke, 2002). Therefore, any SNAP-induced change in contraction frequency of the heart must be caused by changes in the firing patterns of the motor neurons found in the cardiac ganglion. This could occur either through a direct effect of NO on the ganglion itself or through an action of NO on some component of the musculature, which then transmits a secondary signal to the ganglion. To determine whether NO specifically affects the burst properties of the ganglion, we applied NO donors directly to isolated cardiac ganglia.
Under control conditions, isolated ganglia burst spontaneously at a mean basal frequency of 0.63 ± 0.09 Hz (n = 6). This is slightly slower than the mean beat frequency of the intact heart, a difference noted previously that is thought to be caused by the elimination of stretch-receptor-mediated feedback when the ganglion is separated from the musculature (Cooke, 2002). Figure 7, A and B, shows that bath application of 10–5 M SNAP decreased the burst frequency of the isolated ganglion, with a rate of onset and washout similar to the actions of SNAP on the intact heart. The average burst frequency after a 15 min exposure to 10–5 m SNAP was 0.5 ± 0.1 Hz (n = 6; p = 0.002 relative to control; paired Student's t test). The percentage decrease in burst frequency induced by SNAP in the isolated ganglion (21%) was less than the percentage decrease in heart rate observed in the intact heart (39%), perhaps because the isolated ganglion is already operating at a lower basal frequency, as noted above.
Figure 7.
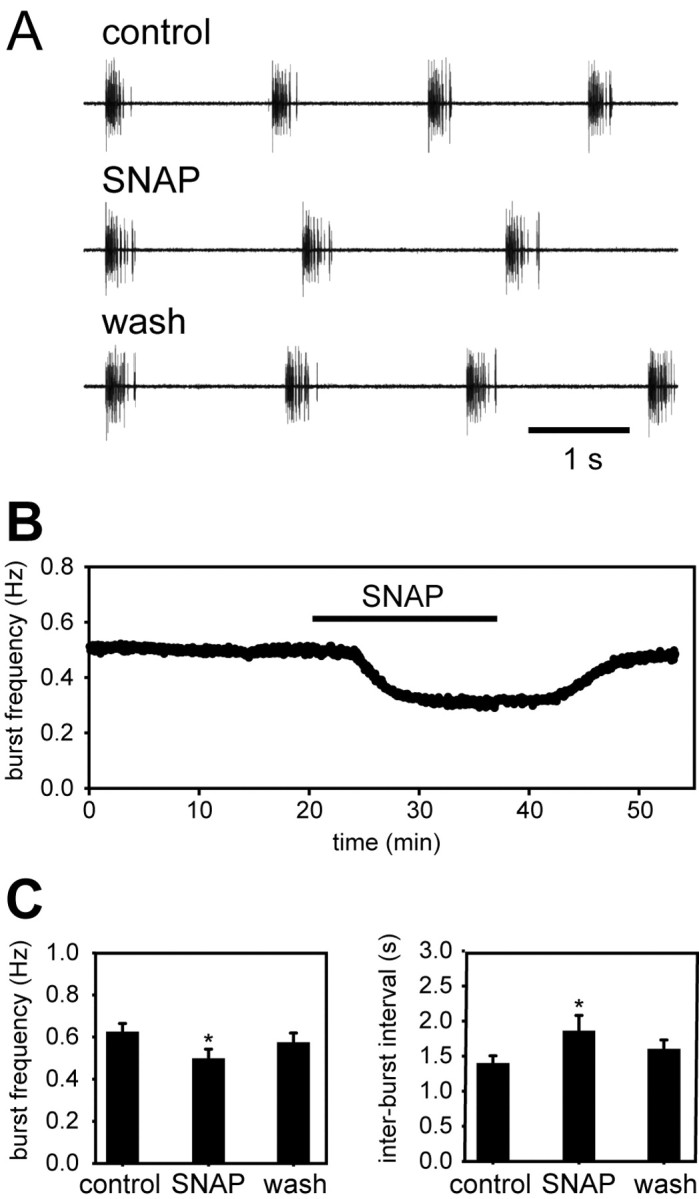
NO decreases the burst frequency of the isolated cardiac ganglion in vitro. A, Representative extracellular recordings from an isolated lobster cardiac ganglion before (top), during (middle), and after (bottom) application of 10–5 m SNAP. B, Time course of the response of a single ganglion to SNAP (delivered during the period denoted by the horizontal bar). C, Mean responses of six independent preparations. *p < 0.05, paired two-tailed t test.
Figure 7C shows that the SNAP-induced decrease in burst frequency was caused by an increase in the interburst interval (from 1.38 ± 0.2 to 1.84 ± 0.5 sec; n = 6; p = 0.011), with no significant change in the burst duration (0.24 ± 0.1 sec in control and 0.25 ± 0.1 sec in SNAP), intraburst spike frequency (66.9 ± 11.5 Hz in control and 68.4 ± 11.6 Hz in SNAP), and spike number per burst (15.7 ± 5.8 in control and 17.0 ± 7.1 in SNAP). Again, these effects of SNAP were similar to those of NOC-15 at 10–4 m (n = 7) (data not shown). Degassed SNAP and NOC-15 solutions of the appropriate concentrations, tested after a minimum of eight half-lives in each case, showed no effect on burst frequency or any other burst parameter (n = 3) (data not shown).
The NO-induced decrease in ganglionic burst frequency provides a straightforward mechanism to explain the decrease in heartbeat frequency observed when NO is applied to the intact heart. Furthermore, because muscle fibers and stretch receptor endings have been removed from the isolated cardiac ganglion preparation, it is evident that the negative chronotropic response to NO represents a direct action on the ganglion. However, because the structure of each burst remained unchanged in the presence of NO (i.e., each burst contained the same number of spikes delivered at the same frequency), this leaves open the question of how NO is affecting heartbeat amplitude. Mechanisms that could be responsible for such a negative inotropic effect fall into two general classes: those that decrease the ability of muscle fibers to contract in response to depolarization (by altering some aspect of excitation/contraction coupling) and those that decrease the strength of the synaptic input to the muscle fibers (either by reducing presynaptic release of the transmitter or by postsynaptic response to the transmitter). The experiments described below explore each of these possibilities.
NO does not affect the contractility of heart muscle fibers
To determine whether NO directly inhibits the ability of heart muscle to contract, we induced a submaximal contracture in a spontaneously beating heart by elevating K+ in the bathing medium, and then applied SNAP in the continued presence of elevated K+ (Fig. 8). In response to the K+-induced depolarization (perfusion time indicated by the black bars below the trace), the cardiac ganglion stopped bursting and the muscle showed a prolonged contracture, whose amplitude was lower than those of the neurally driven heartbeats. Adding a very high concentration of SNAP (2 × 10–4 m; perfusion time indicated by the white bars above the trace) to the high K+ saline had no effect on the K+-induced contracture (n = 3) (Fig. 8), although, when the high K+ saline was washed out and the cardiac ganglion resumed bursting, the same concentration of SNAP now markedly slowed the rate and decreased the amplitude of the ganglion-mediated heartbeat, as expected (Fig. 8, inset). Thus, NO did not significantly alter excitation/contraction coupling in cardiac muscle fibers induced to contract by a tonic depolarizing stimulus.
Figure 8.

NO does not directly affect muscle contraction. Main trace, tension recording of rhythmic heart contractions before, during, and after application of 2 × 10–4 m SNAP (white bars above trace), delivered in normal saline (right half of trace) or in saline containing five times the normal amount of KCl (left half of trace, as indicated by the black bar). Inset above the main trace shows the effect of SNAP on an expanded time scale, after washout of high KCl saline. Delay represents the dead volume in the perfusion line.
NO does not directly modulate neuromuscular transmission
In the beating heart, a critical determinant of contraction amplitude is the total synaptic depolarization delivered to the muscle fiber during a burst. Because the number and timing of spikes in the burst was not affected by NO, we investigated the possibility that NO might decrease the amplitude of the depolarization (synaptic potential) evoked by a single spike or a burst of spikes. To perform these experiments, we first removed the cardiac ganglion (to abolish endogenous rhythmic activity) and then used a suction electrode to stimulate one of the remaining motor nerves. This generated excitatory junctional potentials (EJPs) in the innervated muscles, which were recorded using sharp intracellular microelectrodes. Trains of stimulus-evoked EJPs showed facilitation, depression, and/or summation, depending on the frequency of the stimulation (Fig. 9A). At 0.8 Hz, a firing rate comparable with the normal burst frequency of the unstimulated cardiac ganglion, single EJPs in the cardiac muscle facilitated over the time course of several seconds, and this facilitation was unaffected by NO (Fig. 9B).
Figure 9.

NO does not affect facilitation, summation, or depression at the cardiac neuromuscular synapse. EJPs were elicited in cardiac muscle fibers of a deganglionated heart by stimulating the motor nerves with a suction electrode. A, EJPs evoked by 1 or 6 Hz stimulation (0.5 msec pulses), as marked, showing facilitation at 1 Hz and a combination of depression (arrow), summation, and facilitation at 6 Hz. B, Facilitation index (the ratio of the nth EJP amplitude to the first EJP amplitude) for muscle fibers stimulated every 3 min with a train of nine pulses (interpulse interval, 1.25 sec) during a control period (squares), after a 15 min exposure to 10–5 m SNAP (circles), and after washout of the SNAP (n = 3 preparations). C, Overlaid recordings of EJP bursts (average of 10 individual traces each) in control (inner envelope) and SNAP (outer envelope), produced by stimuli consisting of five pulses/burst at an interpulse interval of 70 msec and an interburst interval of 1.25 sec. Quantifying the first (D) and fourth (E) EJP amplitudes obtained from experiments like those in C (n = four animals) shows that NO does not significantly change either EJP amplitude directly (first EJP) or the combined effects of depression and summation in the burst (the value of the peak of the fourth EJP relative to resting muscle membrane potential).
To evaluate the effect of NO on a more complex EJP waveform, we stimulated the motor nerves with short bursts consisting of five pulses (at 70 msec intervals), using a burst frequency of 0.8 Hz. This stimulus paradigm revealed the effects of NO on depression and summation within each burst, while keeping the duration of the burst short enough to avoid significant contraction. Bath application of 10–5 m SNAP did not cause any significant change in the amplitude or shape of the complex wave of depolarization induced by single bursts (Fig. 9C, overlaid traces). To confirm this quantitatively, we measured the amplitude of the first EJP and the level of depolarization at the fourth EJP relative to the membrane resting potential. NO did not directly affect the steady-state amplitude of the first EJP in a burst (Fig. 9D) (n = 4; p = 0.50) nor did it change shortterm depression or summation kinetics, both of which are integrated in our measure of membrane depolarization at the fourth EJP (Fig. 9E) (n = 4; p = 0.13).
NO indirectly modulates synaptic transmission: the NO-induced decrease in burst frequency reduces both synaptic depolarization and contraction amplitude.
As shown in the preceding sections, the negative inotropic effect of NO is not caused by direct changes in muscle contractility or excitability. Indeed, of all the parameters that we have measured, only one (ganglionic burst frequency) was significantly altered by NO. We have also noted (Fig. 9) that the amplitudes of cardiac muscle EJPs are strongly frequency dependent. If there is facilitation of the compound EJPs that are evoked by bursts, then slowing the burst frequency (chronotropic effect) should decrease facilitation, and this alone would result in a weaker contraction (inotropic effect). To test this, we first looked to see whether interburst facilitation occurs. Figure 10A shows compound EJPs induced by a train consisting of bursts of four pulses (separated by 70 msec) delivered at the standard control burst frequency of 0.8 Hz. Within each burst of EJPs depression was observed, attributable to the high frequency of spikes within the burst, but the bursts themselves facilitated robustly, and climbed to a steady state value over ∼10 sec. When the burst frequency was then decreased from 0.8 to 0.45 Hz (the average shift observed after exposure to 10–5 m SNAP), the total EJP depolarization decreased, although none of the other burst parameters changed.
Figure 10.

Synaptic depolarization of cardiac muscle is linearly related to burst frequency. A, EJP bursts (4 pulses/burst; interpulse interval, 70 msec) delivered at 0.8 Hz to a quiescent preparation. At the break in the trace, the stimulation frequency was changed to 0.45 Hz. B, The amplitude of the first EJP in the burst is plotted as a function of time, during application of stimuli at different frequencies, as noted below the data points. Stimulus parameters are as in A. C, Amplitude of the first EJP in each burst (normalized to 1 at 0.8 Hz for each preparation) as a function of stimulation frequency. Data were pooled from eight independent preparations, each stimulated at multiple frequencies, as in A and B; the solid line represents a linear regression fit (r2 = 0.88; p < 0.001).
Such frequency-dependent changes in burst amplitude were fully reversible and could be evoked repeatedly (Fig. 10B). They also occurred linearly over a range of burst frequencies from 0.8 to 0.3 Hz, as shown in Figure 10C (n = 8; r2 = 0.89; p < 0.001). This figure plots the amplitude of the first EJP in each burst as a function of burst frequency (normalized to 1 for bursts at 0.7 Hz, to enable comparison across animals). Thus, over the range of frequencies observed in control and NO-stimulated preparations, a decrease in burst frequency leads directly to a decrease in the total depolarization in the heart muscle fibers, and this should lead to a weaker heartbeat.
To confirm directly that the frequency-dependent changes in synaptic strength are sufficient to account for the negative inotropic effect of NO, we used a suction electrode to externally drive contractions of the heart muscle, and changed the frequency to see whether this would provoke appropriate changes in contraction amplitude. Figure 11A shows evoked contractions recorded at a number of different burst frequencies in a single preparation (each trace in the figure is the mean of four to five individual contractions, taken shortly after the frequency shift, when the amplitude had come to its new level). From this, and other such experiments (Fig. 11B) (n = 2), it is clear that decreasing burst frequency in and of itself decreases contraction amplitude. Indeed, there was good quantitative correlation between the magnitudes of the frequency-evoked shift in contraction amplitude and the NO-evoked shift in contraction amplitude: shifting the frequency from 0.8 to 0.44 Hz (a frequency change that mimics the effects of 10–5 m SNAP) caused a change in contraction amplitude from 0.21 to 0.15 mm, a 29% decrease similar to the 31% decrease in contraction amplitude observed when the intact heart was exposed to 10–5 m SNAP (Fig. 2B). This similarity was maintained over the entire range of frequencies tested: Figure 11C replots the data obtained when we externally controlled the frequency of the deganglionated preparation (taken from Fig. 11B) along with the data obtained when we used NO to shift the frequency of the intact preparation (taken from Fig. 4B).
Figure 11.
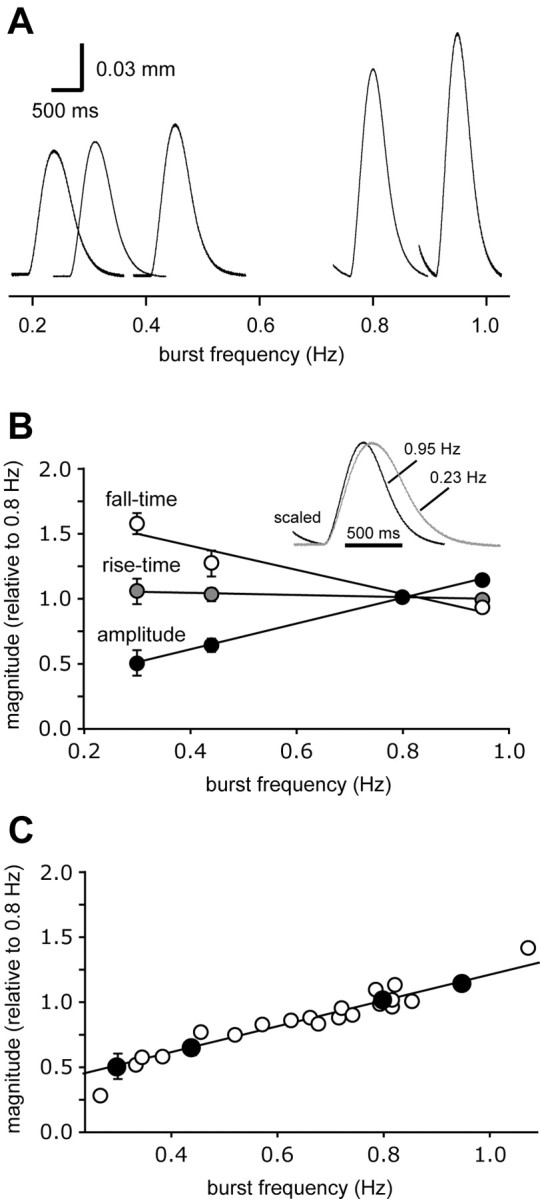
Contractile responses of cardiac muscle are linearly related to burst frequency. When bursts of stimuli (16 pulses at an interpulse interval of 12.5 msec) are delivered through a suction electrode to a cardiac ganglion exposed within the lumen of the heart, the burst output of the ganglion (and therefore the contraction of the heart) becomes entrained to the input stimulus, allowing external control over the frequency of cardiac contractions. A, Contractions evoked over a physiologically relevant range of burst frequencies. Each waveform is the average of six consecutive contractions collected shortly after a shift from 0.8 Hz to a new frequency, as indicated by the scale below the figure. B, Mean contraction amplitude (black), rise time (gray), and fall time (white) plotted as a function of stimulation frequency (data combined from two experiments performed as in A). Inset, Averaged waveforms from A, superimposed and scaled to the same amplitude. C, Data from B, normalized to a value of 1 at 0.7 Hz (black symbols) and replotted with the data presented in Figure 4B (white symbols), to show that the dependency of amplitude on frequency is similar whether frequency is shifted manually or by the application of NO.
Not only did decreasing the burst frequency decrease the amplitude of the evoked contraction amplitude, but it also increased the fall time [from 208 ± 53 to 261 ± 90 msec (n = 2 animals; mean ± extremes) for a frequency shift from 0.8 to 0.44 Hz], again mimicking the actions of SNAP (compare Fig. 11B, inset, with Fig. 2C). Thus, the change in half-fall time, described above for intact preparations exposed to NO (Fig. 2C), is not caused by a direct effect of NO on the contraction/relaxation apparatus of the cardiac muscle, but rather is again an indirect result of the change in the frequency of the cardiac ganglion bursts. Hence, changes in contraction frequency per se are sufficient to account for the changes in contraction amplitude and fall-time induced by NO donors.
As a final test of this idea, we used a suction electrode to drive the cardiac ganglion for prolonged periods with manually generated bursts at 0.8 Hz (thereby producing cardiac contractions at a defined and invariant frequency) and measured the effect of NO on the amplitudes of the evoked contractions (Fig. 12). Although there was a clear decline in the vitality of the preparation over time (evidenced by a slow, essentially linear decrease in contraction amplitude), there was no discernible effect of NO on contractility under these conditions (i.e., when heartbeat frequency was held constant, heartbeat amplitude remained constant despite the presence of NO). These data provide independent confirmation of the conclusions drawn from Figures 8 (NO does not affect EC coupling within muscle fibers) and 9 (NO does not affect synaptic transmission between motor neuron terminals and muscle fibers).
Figure 12.

NO does not affect the amplitudes of contractions when they are evoked at a fixed frequency. A suction electrode was used to entrain the ganglion, as described in the legend to Figure 11, and contractions were evoked continuously at 0.8 Hz. Twitch amplitudes were measured during a control period, and then for 15 min in the presence of SNAP (at either 10–5 or 3 × 10–5 m). To combine data from multiple preparations, the twitch amplitudes for each experiment were binned together in 60 sec intervals, a mean value was determined for each bin, and the data were normalized to fix the amplitude at 1 in the bin just preceding the time of SNAP application (time 0 on the ordinate). Data points represent the means ± SE obtained from five independent experiments. The error increases progressively on either side of 0 because different preparations showed different rates of run-down, as described in Results. The data points have been fit to a straight line by linear regression (r2 = 0.97).
Evidence for endogenous release of NO from the cardiac musculature
The data presented above demonstrate that the lobster cardiac ganglion is able to respond to exogenous NO, and that cardiac tissue potentially has the capacity to generate NO (as suggested by the presence of NOS-like immunoreactivity), but they do not bear on the question of whether the tissue does generate NO. To examine this possibility, we exposed isolated, beating hearts to l-NA, an NOS inhibitor. If the cardiac preparation does spontaneously generate NO, then the simplest a priori hypothesis would be that, by inhibiting endogenous NO production, l-NA should elicit a response that is opposite in polarity to the inhibitory responses triggered by exogenous NO. This prediction is complicated, however, by the adaptation phenomenon described above; specifically, if a tissue has adapted to its own basal level of NO, then l-NA may have little or no discernible stimulatory effect on amplitude [because NO-induced changes in amplitude usually adapt completely and show only a small off response when NO is removed (Fig. 5)], whereas l-NA should have a greater stimulatory effect on frequency (because NO-induced changes in frequency usually show only partial adaptation).
When we tested the effects of l-NA on the intact heart, these predictions were confirmed (Fig. 13A). When averaged over a number of preparations (n = 16), 300 μm l-NA caused a statistically significant increase in heartbeat frequency with no detectable effect on amplitude. However, l-NA was not equally effective on all preparations. An example of a responsive preparation is shown in Figure 13B, left, whereas an unresponsive preparation is shown in Figure 13B, right. When a heart failed to respond to l-NA this did not reflect an underlying insensitivity to NO (a consequence, for example, of complete adaptation to the presence of endogenous NO), because such preparations could still respond robustly to exogenously added NO (Fig. 13B). This suggests that the variability was attributable to animal-to-animal differences in basal NO production. The intensity of the response to l-NA was strongly related to the basal heartbeat frequency of an individual preparation: the stimulatory effect of the inhibitor was much more evident if the basal frequency was low (Fig. 13C). This is, perhaps, not surprising, because the effects of l-NA should be most pronounced in the animals that are producing the most NO, and (because NO decreases beat frequency) these are the animals that should have had the slowest basal heart rates. The mean frequency of the control heartbeats in Figure 13A is lower than the mean frequency reported for randomly chosen hearts in Figure 2A, because we focused on the lower end of the frequency spectrum to evaluate the effects of l-NA on slower hearts more effectively.
Figure 13.

l-NA affects the beat frequency of the intact heart when the basal heart rate is low, but does not affect the burst frequency of the isolated cardiac ganglion. A, Heartbeat amplitude (white bars) and heartbeat frequency (black bars) from 16 independent hearts, measured either just before (–) or after 30 min continuous application (+) of 300 μm l-NA (means ± SE). B, Heartbeat frequencies plotted as a function of time before, during (black bar), and after washout of 300 μm l-NA for two representative intact preparations, one of which had a basal heart rate of ∼0.3 Hz (left) and the other of ∼0.73 Hz (right). The slower heart sped up in response to l-NA, whereas the faster heart did not. The faster heart was also exposed to 10–5 m SNAP immediately after exposure to the l-NA, as indicated. The robust response to SNAP indicates that the preparation was not desensitized to NO. Data were smoothed using a moving bin average with a bin size of 1 min. C, l-NA-induced change in heart rate (percentage change from the basal rate), plotted as a function of basal frequency. Each data point is from an independent experiment. D, Instantaneous burst frequencies plotted as a function of time before, during (black bar), and after washout of 300 μm l-NA for two representative isolated cardiac ganglia, one of which had a basal burst rate of ∼0.25 Hz (left) and the other of ∼0.95 Hz (right). Neither preparation responded to l-NA. Data were smoothed using a moving bin average with a bin size of 1 min.
There was no statistically significant effect of l-NA on the isolated cardiac ganglion, regardless of frequency (Fig. 13D). We tested 13 ganglia, whose basal burst frequency spanned the range from 0.25 to 0.9 Hz (selecting sufficient slowly bursting preparations to ensure that the entire frequency range was well represented). No parameter that we measured was affected by l-NA, including burst frequency (0.51 ± 0.06 Hz in control and 0.54 ± 0.06 Hz in l-NA; p = 0.33), interburst interval (and 1.76 ± 0.2 sec in control and 1.62 ± 0.2 sec in l-NA; p = 0.23), burst duration (0.58 ± 0.1 sec in control and 0.58 ± 0.1 sec in l-NA; p = 0.98), intraburst spike frequency (128 ± 8 Hz in control and 129 ± 7 Hz in l-NA; p = 0.65), and spike number per burst (75 ± 14 in control and 75 ± 12 in l-NA; p = 0.96). This lack of sensitivity to l-NA indicates that the ganglion in isolation does not generate NO. Therefore, the NO produced by the intact heart must be generated by some component of the wall of the heart. In conjunction with our finding that the ganglion is the only NO-sensitive component of the intact cardiac preparation, this leads us to conclude that NO serves as a retrograde inhibitory signaling molecule, carrying information from the wall of the heart back to the ganglion that innervates it.
Discussion
The heart is a dynamic organ whose output is regulated by a number of feedback mechanisms. In crustaceans, short-term control over cardiac performance is provided by excitatory and inhibitory nerves that modulate heart rate and contractility for periods of up to several seconds (Florey, 1960; Field and Larimer, 1975). Longer-term excitation is provided by a variety of circulating hormones that increase the rate and strength of the heartbeat for periods lasting from minutes to hours (Cooke and Hartline, 1975; Benson, 1984; Miller et al., 1984; Sullivan and Miller, 1984; Worden et al., 1995; Berlind, 1998). This raises the question of whether crustaceans might also employ long-term inhibitory mechanisms to counteract these long-lasting excitatory mechanisms.
Our data reveal that NO is a good candidate for such an inhibitory modulator. First, the lobster heart reacts to exogenously applied NO with strong negative inotropic and chronotropic responses. Second, using l-NA, we have been able to demonstrate the endogenous production of NO in ∼50% of the cardiac preparations that we have tested. Third, additional confirmation for the intracardiac expression of NOS is provided by our Western blot data. However, it is apparent from the variable effects of l-NA that either NOS protein expression or NOS enzyme activity must be differentially regulated in individual animals. We do not yet understand the basis for this variability. Many possibilities could be considered, including variables such as gender, stress level before the experiment, volume status, diet, hypoxia, or social dominance. What is clear, however, is that basal heart rate varies considerably from animal to animal, and that the level of endogenous NO production appears to be one determinant of this variability.
In animals that do express active NOS, the presence of this calcium-activated enzyme in the cardiac musculature provides a mechanism by which local NO production could be indexed to cardiac performance, because any input (neural or hormonal) that increases the rate and strength of the heartbeat will also increase the rate and extent of calcium entry, and thus the rate and extent of NO production. Increased NO will, in turn, inhibit the cardiac ganglion and counter, at least in part, the effects of the excitatory stimulus. Thus, NO may serve as the mediator of a negative feedback mechanism capable of limiting the level of excitation delivered by the cardiac ganglion to the muscle. It might be useful for an animal to be able to turn such a mechanism on or off, as dictated by environmental or behavioral imperatives.
In addition to this postulated local mechanism, NO might also be delivered to the heart from an extracardiac source. Although NO is normally considered to be quite labile in biological fluids, serum proteins that form stable complexes with NO have been identified in both mammals (Jai et al., 1996) and arthropods (Ribeiro et al., 1993). Although such NO-binding proteins have not yet been identified in crustaceans, it remains an intriguing possibility that other NOS-expressing organs, such as the hepatopancreas (Scholz et al., 2002), might be able to regulate cardiac performance by delivering NO to the heart through the circulation. In addition, it is also possible that NO produced by the heart could be delivered to other downstream, NO-sensitive target organs (such as the stomatogastric ganglion; Scholz et al., 2001).
As a gaseous signaling molecule able to diffuse across cell membranes, NO has unique properties that allow it to affect multiple target cells. Despite the diffuse nature of its delivery, it is clear that NO activates and modulates particular neural network targets with stereotypical functional consequences (Moroz et al., 1993; Gelperin, 1994; Elphick et al., 1995; Scholz et al., 2001). This suggests that particular distributions of NO receptors (and/or other downstream targets) may enable a diffuse NO signal to sculpt the responses of specific cells within a population. The physiological network under study in this paper is composed of two anatomically and spatially distinct cellular compartments: (1) the cardiac ganglion, comprised of four interneuron pacemaker cells and five motor neurons situated within the lumen of the heart; and (2) the wall of the heart, which contains the striated muscle fibers and fibroblasts that form the cardiac chamber, as well as the terminations of stretch receptors and motor axons, both of which originate from the ganglion and are embedded within the heart musculature. Our data show distinct functional differences between these compartments: the wall of the heart produces NO but is not affected by it, whereas the ganglion is affected by NO but does not produce it. Thus, NO does not alter the electrical excitability of the muscle in response to ganglionic input, the contractility of the muscle in response to electrical excitation, or the input/output characteristics (facilitation, depression, summation, or excitation/secretion coupling) of the synaptic terminals that innervate the muscle. Indeed, the sole physiological parameter that we have found to be affected by NO is the burst frequency of the ganglion. This single action of NO leads directly to a decrease in heart rate, which, as a consequence of facilitation at the motor neuron-to-muscle synapse, leads to a reduction in heartbeat amplitude. A similar dependency of contraction amplitude on burst frequency has been observed in other rhythmically contracting crustacean muscles (Mercier and Wilkins, 1984).
In addition to the direct link between frequency and amplitude, we have also noted an adaptation process that uncouples these two parameters after prolonged exposure to NO. The mechanism that underlies this adaptation process is unknown, although the transient increase in contraction amplitude (the off response) that occurs when NO is washed out of the preparation suggests that adaptation may involve reversible changes in the contractility of the muscle fibers themselves. The idea that the crustacean heart may be programmed to adjust its contractility to a pre-established “set point” is an intriguing possibility.
Our current understanding of the inhibitory effects of NO also leads to the prediction that an increase in burst frequency will by itself lead to an increase in contraction amplitude. This raises the possibility that some (or all) of the many known cardioexcitatory modulators may exert their effects on heart contractility solely by increasing the burst rate of the cardiac ganglion. Alternatively, cardioactive agents like serotonin, octopamine, and proctolin, which have been shown to enhance contractility at other types of neuromuscular junctions (Kravitz et al., 1985), may augment their chronotropic effects on the cardiac ganglion with a direct inotropic action on heart muscle.
To selectively modulate the physiological properties of the cardiac ganglion, it is perhaps self-evident that NO must activate a signaling cascade that is selectively expressed within the ganglion, although the molecular components of this cascade are currently undefined. The most widely recognized downstream effector for NO is the soluble, heme-containing form of guanylate cyclase, whose activity is strongly enhanced by NO (Hanafy et al., 2001). Consistent with this idea, NO has been shown to elevate cGMP levels when applied to isolated cardiac ganglia (Scholz et al., 2002), an effect whose potential significance is underscored by the observation that the NO-sensitive guanylate cyclase enzyme is not, in general, very abundant in the crustacean nervous system (Scholz et al., 1996; Prabhakar et al., 1997). Linkage between NO and cGMP in the lobster heart would also be consistent with the intertwined inhibitory roles for these signaling molecules in a variety of contexts in the mammalian cardiovascular system (Ignarro et al., 1999; Kone, 2001). However, NO can also stimulate other heme-containing enzymes (e.g., cyclooxygenase or cytochrome P-450), can activate ADP-ribosyltransferases, and can induce the nonenzymatic nitration and nitrosylation of tyrosine and cysteine residues, respectively (Hanafy et al., 2001). Thus, the role of cGMP in the inhibitory actions of NO cannot be predicted, and must be evaluated in future studies.
Modulation of the cardiac system in Homarus americanus has direct functional consequences because it affects the flow of hemolymph through arteries to multiple organs, including the brain and the ventral ganglia (McMahon, 1995). In this paper, we have presented evidence that supports the hypothesis that the lobster heart regulates itself via a physiological feedback loop involving the cardiac ganglion and its target, the cardiac musculature, using the diffuse gaseous neuromodulator NO. This role for NO is consistent with its known functions as a cardiovascular regulator in vertebrate systems, as well as its documented role as a retrograde trans-synaptic signaling molecule in the nervous system, and extends our understanding of how NO functionally regulates discrete neural networks despite being a short-lived diffuse neuromodulator.
Footnotes
This work was supported by National Science Foundation Grant 0236320 (M.G.), National Institutes of Health Grant NS 17813 (E.M.), an American Psychological Association Predoctoral Fellowship in Neuroscience (N.D.C.-B.), and a Howard Hughes Medical Institute Predoctoral Fellowship (A.M.). We thank Drs. Dan Cox and Kathleen Dunlap for writing the Igor Pro procedure file used to analyze muscle tension recordings, Dr. Dirk Bucher for writing the Spike2 scripts used to analyze extracellular recordings, and Kathleen Dunlap for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Michael F. Goy, University of North Carolina, Department of Cell and Molecular Physiology, 5309B Medical Biomolecular Research Building, CB#7545, 103 Mason Farm Road, Chapel Hill, NC 27599. E-mail: mgoy@med.unc.edu.
DOI:10.1523/JNEUROSCI.3779-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/242813-12$15.00/0
References
- Adak S, Aulak KS, Stuehr DJ (2002) Direct evidence for nitric oxide production by a nitric-oxide synthase-like protein from Bacillus subtilis J Biol Chem 277: 16167–16171. [DOI] [PubMed] [Google Scholar]
- Alexandrowicz JS (1932) The innervation of the heart of the crustacea I. decapoda Q J Microsc Sci 75: 181–255. [Google Scholar]
- Arancio O, Lev-Ram V, Tsien RY, Kandel ER, Hawkins RD (1996) Nitric oxide acts as a retrograde messenger during long-term potentiation in cultured hippocampal neurons. J Physiol (Paris) 90: 321–322. [DOI] [PubMed] [Google Scholar]
- Balligand JL, Kelly RA, Marsden PA, Smith TW, Michel T (1993) Control of cardiac muscle cell function by an endogenous nitric oxide signaling system. Proc Natl Acad Sci USA 90: 347–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson JA (1984) Octopamine alters rhythmic activity in the isolated cardiac ganglion of the crab, Portunus sanguinolentus Neurosci Lett 44: 59–64. [DOI] [PubMed] [Google Scholar]
- Berlind A (1998) Dopamine and 5-hydroxytryptamine actions on the cardiac ganglion of the lobster Homarus americanus J Comp Physiol [A] 182: 363–376. [Google Scholar]
- Boulton CL, Southam E, Garthwaite J (1995) Nitric oxide-dependent long-term potentiation is blocked by a specific inhibitor of soluble guanylyl cyclase. Neuroscience 69: 699–703. [DOI] [PubMed] [Google Scholar]
- Boxall AR, Garthwaite J (1996) Long-term depression in rat cerebellum requires both NO synthase and NO-sensitive guanylyl cyclase. Eur J Neurosci 8: 2209–2212. [DOI] [PubMed] [Google Scholar]
- Champlin DT, Truman JW (2000) Ecdysteroid coordinates optic lobe neurogenesis via a nitric oxide signaling pathway. Development 12: 3543–3551. [DOI] [PubMed] [Google Scholar]
- Cooke IM (2002) Reliable, responsive pacemaking and pattern generation with minimal cell numbers: the crustacean cardiac ganglion. Biol Bull 202: 108–136. [DOI] [PubMed] [Google Scholar]
- Cooke IM, Hartline DK (1975) Neurohormonal alteration of integrative properties of the cardiac ganglion of the lobster Homarus americanus J Exp Biol 63: 33–52. [DOI] [PubMed] [Google Scholar]
- Cramer KS, Leamey CA, Sur M (1998) Nitric oxide as a signaling molecule in visual system development. Prog Brain Res 118: 101–114. [DOI] [PubMed] [Google Scholar]
- Daniel H, Hemart N, Jaillard D, Crepel F (1993) Long-term depression requires nitric oxide and guanosine 3′:5′ cyclic monophosphate production in rat cerebellar Purkinje cells. Eur J Neurosci 5: 1079–1082. [DOI] [PubMed] [Google Scholar]
- Elphick MR, Kemenes G, Staras K, O'Shea M (1995) Behavioral role for nitric oxide in chemosensory activation of feeding in a mollusc. J Neurosci 15: 7653–7664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field LH, Larimer JL (1975) The cardioregulatory system of crayfish: neuroanatomy and physiology. J Exp Biol 62: 519–530. [DOI] [PubMed] [Google Scholar]
- Florey E (1960) Studies on the nervous regulation of the heartbeat in decapod crustaceans. J Exp Biol 43: 1061–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelperin A (1994) Nitric Oxide mediates network oscillations of olfactory interneurons in a terrestrial mollusc. Nature 369: 61–63. [DOI] [PubMed] [Google Scholar]
- Gyurko R, Kuhlencordt P, Fishman MC, Huang PL (2000) Modulation of mouse cardiac function in vivo by eNOS and ANP. Am J Physiol 278: H971–H981. [DOI] [PubMed] [Google Scholar]
- Hanafy KA, Krumenacker JS, Murad F (2001) NO, nitrotyrosine, and cyclic GMP in signal transduction. Med Sci Monit 7: 801–819. [PubMed] [Google Scholar]
- Hedrick MS, Morales RD (1999) Nitric oxide as a modulator of central respiratory rhythm in the isolated brainstem of the bullfrog (Rana catesbeiana). Comp Biochem Physiol A Mol Integr Physiol 124: 243–251. [DOI] [PubMed] [Google Scholar]
- Huang EP (1997) Synaptic plasticity: a role for nitric oxide in LTP. Curr Biol 7: R141–143. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ (1990) Biosynthesis and metabolism of endothelium derived nitric oxide. Annu Rev Pharmacol Toxicol 30: 535–560. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Cirino G, Casini A, Napoli C (1999) Nitric oxide as a signaling molecule in the vascular system: an overview. J Cardiovasc Pharmacol 34: 879–886. [DOI] [PubMed] [Google Scholar]
- Jai L, Bonaventura C, Bonaventura J, Stamler J (1996) S-nitrosohaemoglobin: a dynamic activity of blood involved in vascular control. Nature 380: 221–226. [DOI] [PubMed] [Google Scholar]
- Keaney Jr JF, Hare JM, Balligand JL, Loscalzo J, Smith TW, Colucci WS (1996) Inhibition of nitric oxide synthase augments myocardial contractile responses to beta-adrenergic stimulation. Am J Physiol 271: H2646–H2652. [DOI] [PubMed] [Google Scholar]
- Keefer LK, Nims RW, Davies KM, Wink DA (1996) “NONOates” (1-substituted diazen-1-ium-1,2-diolates) as nitric oxide donors: convenient nitric oxide dosage forms. Methods Enzymol 268: 281–293. [DOI] [PubMed] [Google Scholar]
- Kone BC (2001) Molecular biology of natriuretic peptides and nitric oxide synthases. Cardiovasc Res 51: 429–441. [DOI] [PubMed] [Google Scholar]
- Kravitz EA, Beltz BS, Glusman S, Goy MF, Harris-Warrick RM, Johnston MF, Livingstone MS, Schwarz TL (1985) The well-modulated lobster: the roles of serotonin, octopamine and proctolin in the lobster nervous system. In: Model neural networks and behaviour (Selverston AI, ed), pp 339–360. New York: Plenum.
- Lev-Ram V, Jiang T, Wood J, Lawrence DS, Tsien RY (1997) Synergies and coincidence requirements between NO, cGMP, and Ca2+ in the induction of cerebellar long-term depression. Neuron 18: 1025–1038. [DOI] [PubMed] [Google Scholar]
- McLean DL, Sillar KT (2000) The distribution of NADPH-diaphoraselabelled interneurons and the role of nitric oxide in the swimming system of Xenopus laevis larvae. J Exp Biol 203: 705–713. [DOI] [PubMed] [Google Scholar]
- McMahon BR (1995) The physiology of gas exchange, circulation, ion regulation and nitrogenous excretion: an integrative approach. In: Biology of the lobster Homarus americanus (Factor JR, ed), pp 497–517. San Diego: Academic.
- Mercier AJ, Wilkins JL (1984) Analysis of the scaphognathite ventilatory pump in the shore crab Carcinus maenas III. Neuromuscular mechanisms. J Exp Biol 113: 83–99. [Google Scholar]
- Miller MW, Benson JA, Berlind A (1984) Excitatory effects of dopamine on the cardiac ganglia of the crabs Portunus sanguinolentus and Podophthalmus vigil J Exp Biol 108: 97–118. [DOI] [PubMed] [Google Scholar]
- Moroz LL, Park JH, Winlow W (1993) Nitric oxide activates buccal motor patterns in Lymnaea stagnalis NeuroReport 4: 643–646. [DOI] [PubMed] [Google Scholar]
- Neill SJ, Desikan R, Clarke A, Hurst RD, Hancock JT (2002) Hydrogen peroxide and nitric oxide as signalling molecules in plants. J Exp Bot 53: 1237–1247. [PubMed] [Google Scholar]
- Newcomb JM, Watson WH 3rd (2002) Modulation of swimming in the gastropod Melibe leonina by nitric oxide. J Exp Biol 205: 397–403. [DOI] [PubMed] [Google Scholar]
- Pape HC, Mager R (1992) Nitric oxide controls oscillatory activity in thalamocortical neurons. Neuron 9: 441–448. [DOI] [PubMed] [Google Scholar]
- Prabhakar S, Short DB, Scholz NL, Goy MF (1997) Identification of nitric oxide-sensitive and -insensitive forms of cytoplasmic guanylate cyclase. J Neurochem 69: 1650–1660. [DOI] [PubMed] [Google Scholar]
- Prast H, Philippu A (2001) Nitric oxide as modulator of neuronal function. Prog Neurobiol 64: 51–68. [DOI] [PubMed] [Google Scholar]
- Ribeiro JMC, Hazzard JMH, Nussenzveig RH, Champagne DE, Walker FA (1993) Reversible binding of nitric oxide by a salivary heme protein from a bloodsucking insect. Science 260: 539–541. [DOI] [PubMed] [Google Scholar]
- Roy B, du Moulinet d'Hardemare A, Fontecave M (1994) New thionitrites: synthesis, stability and nitric oxide generation. J Org Chem 59: 7019–7026. [Google Scholar]
- Scholz NL, Goy MF, Truman JW, Graubard K (1996) Nitric oxide and peptide neurohormones activate cGMP synthesis in the crab stomatogastric nervous system. J Neurosci 16: 1614–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz NL, Chang ES, Graubard K, Truman JW (1998) The NO/cGMP pathway and the development of neural networks in postembryonic lobsters. J Neurobiol 34: 208–226. [DOI] [PubMed] [Google Scholar]
- Scholz NL, de Vente J, Truman JW, Graubard K (2001) Neural network partitioning by NO and cGMP. J Neurosci 21: 1610–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz NL, Labenia JS, DeVente J, Graubard K, Goy MF (2002) Expression of nitric oxide synthase and nitric oxide-sensitive guanylate cyclase in the crustacean cardiac ganglion. J Comp Neurol 454: 158–167. [DOI] [PubMed] [Google Scholar]
- Schuman EM, Madison DV (1994) Nitric oxide and synaptic function. Annu Rev Neurosci 17: 153–183. [DOI] [PubMed] [Google Scholar]
- Shesely EG, Maeda N, Kim HS, Desai KM, Krege JH, Laubach VE, Sherman PA, Sessa WC, Smithies O (1996) Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc Natl Acad Sci USA 93: 13176–13181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RE, Miller MW (1984) Dual effects of proctolin on the rhythmic burst activity of the cardiac ganglion. J Neurobiol 15: 173–196. [DOI] [PubMed] [Google Scholar]
- Torreilles J (2001) Nitric oxide: one of the more conserved and widespread signaling molecules. Front Biosci 6: D1161–D1172. [DOI] [PubMed] [Google Scholar]
- Worden MK, Kravitz EA, Goy MF (1995) Peptide F1, an N-terminally extended analog of FMRFamide, enhances contractile activity in multiple target tissues in lobster. J Exp Biol 198: 97–108. [DOI] [PubMed] [Google Scholar]
- Zahradnikova A, Minarovic I, Venema RC, Meszaros LG (1997) Inactivation of the cardiac ryanodine receptor calcium release channel by nitric oxide. Cell Calcium 22: 447–454. [DOI] [PubMed] [Google Scholar]