

Abstract
Identification of the molecular mechanisms that enable synchronous firing of CA1 pyramidal neurons is central to the understanding of the functional properties of this major hippocampal output pathway. Using microfluorescence measurements of intraneuronal pH, in situ hybridization, as well as intracellular, extracellular, and K+-sensitive microelectrode recordings, we show now that the capability for synchronous gamma-frequency (20–80 Hz) firing in response to high-frequency stimulation (HFS) emerges abruptly in the rat hippocampus at approximately postnatal day 12. This was attributable to a steep developmental upregulation of intrapyramidal carbonic anhydrase isoform VII, which acts as a key molecule in the generation of HFS-induced tonic GABAergic excitation. These results point to a crucial role for the developmental expression of intrapyramidal carbonic anhydrase VII activity in shaping integrative functions, long-term plasticity and susceptibility to epileptogenesis.
Keywords: carbonic anhydrase, epilepsy, excitatory GABA, gamma oscillations, GDP, hippocampus, potassium, synchrony
Introduction
Synchronous firing of principal neurons plays a central role in neuronal plasticity (Traub et al., 1998; Linden, 1999), in integrative and representational functions (Gray, 1999; Singer, 1999), as well as in the genesis and maintenance of epileptic activity (McCormick and Contreras, 2001). High-frequency stimulation (HFS) of afferents in the CA1 area of the rat hippocampal slice has been used in a number of studies on the cellular and network mechanisms involved in the generation of synchronous firing of pyramidal neurons at gamma frequencies (20–80 Hz) (Traub et al., 1996a; Bracci et al., 1999).
The mechanisms underlying HFS-induced gamma oscillations can be divided provisionally into two categories: those that drive the oscillations by providing tonic excitation and those responsible for the temporal patterns of network activity. The latter include GABAA receptor-mediated inhibition (Whittington et al., 1995), ionotropic glutamatergic transmission (Traub et al., 1996b), gap junctions (Draguhn et al., 1998; Schmitz et al., 2001), and ephaptic effects (Bracci et al., 1999).
In the present work, the main focus is on the mechanisms underlying the generation of the HFS-induced tonic excitation in area CA1. Previous studies have suggested a role for two distinct (although not mutually exclusive) mechanisms. There is evidence that activation of interneuronal metabotropic glutamate receptors is important in the excitatory drive underlying HFS-induced gamma oscillations (Whittington et al., 1995, 1997a; Traub et al., 1996a,b). In addition, a number of studies have shown that a sustained activation of GABAA receptors leads to a long-lasting depolarization of CA1 pyramidal neurons that is often associated with pronounced spiking of the CA1 pyramidal neurons (Perreault and Avoli 1988; Grover et al., 1993; Staley et al., 1995; Kaila et al., 1997; Bracci et al., 1999; Smirnov et al., 1999). This excitatory, HFS-induced GABAergic depolarization is critically dependent on the presence of CO2/HCO3– and on functional intracellular carbonic anhydrase (Kaila et al., 1997; Voipio and Kaila, 2000). This response is caused by a fast anionic redistribution leading to a positive shift in EGABA-A, which is followed by a long-lasting increase in the extracellular K+ concentration ([K+]o) that exerts a direct depolarizing action (Kaila et al., 1997; Voipio and Kaila, 2000).
By the end of the second postnatal week, a number of qualitative changes take place at the level of network functions in the rat hippocampus, including an increased tendency for the generation of epileptiform activity (Swann et al., 1993; Gloveli et al., 1995). In addition, this is the critical postnatal time window for the developmental appearance of HFS-induced long-term potentiation (LTP) in CA1 (Harris and Teyler, 1984; Chabot et al., 1996). In this study, we demonstrate an abrupt expression of hippocampal intrapyramidal carbonic anhydrase activity at the end of the second postnatal week. This appears to be a prerequisite for the emergence of HFS-induced GABAergic excitation and for the HFS-induced synchronous gamma-frequency firing of CA1 pyramidal neurons. Furthermore, we show that among the 14 α-carbonic anhydrase isozymes (Supuran et al., 2003), intrapyramidal carbonic anhydrase activity is attributable to a specific developmental expression of the carbonic anhydrase isoform VII (CA VII).
Materials and Methods
Transverse hippocampal slices (400–600 μm) from Wistar rats younger than postnatal day 9 (P9; the day of birth is P0) were made using a McIlwain tissue chopper (Mickle Laboratory Engineering, Gomshall, Surrey, UK) or a Leica (Nussloch, Germany) VT1000 S vibrating-blade microtome. The more mature rats (and some of the younger animals) were anesthetized using an intraperitoneal injection of pentobarbital (30–40 mg/kg), and 350- to 400-μm-thick slices were made using either the tissue chopper or a Vibratome (The Vibratome Company, St. Louis, MO). Carbachol-induced oscillations were examined in horizontal slices as reported previously (Stenkamp et al., 2001).
Electrophysiological recordings on slices were done in standard physiological solution containing the following (in mm): 120 NaCl, 3 KCl, 1.1 NaH2PO4, 1.8 CaCl2, 2 MgSO4,10 d-glucose, and 25 NaHCO3 and equilibrated with 95% O2 and 5% CO2 to yield a pH of 7.4 at the experimental temperature of 32–34°C. In recordings in which tetramethylammonium (TeMA+)-sensitive microelectrodes were used, 1.5 mm TeMA chloride was added to the standard physiological solution. When recording spontaneous population oscillations, the concentrations of MgSO4 and KCl were 1.3 and 5 mm, respectively. Experiments using fluorescence imaging were performed at 20–22°C, and NaHCO3 was raised to 30 mm to keep the solution pH at 7.4. In nominally CO2/HCO3– -free HEPES-buffered solution, NaHCO3 was replaced by 20 mm HEPES plus 10 mm NaCl, pH 7.4 with NaOH, and the solution was gassed with 100% O2. The osmolarity of the standard solution and the HEPES-buffered solution was 301.3 ± 1.2 mOsm (n = 4) and 303 ± 1.3 mOsm (n = 4), respectively, measured using an Advanced Micro Osmometer (model 3300).
Unless otherwise stated, results are presented as means ± SEM. Statistical significance was tested with paired or unpaired t tests as indicated in Results.
Electrophysiological recordings. Electrophysiological experiments were performed both in an interface chamber (volume, 500 μl; solution flow, 0.8–1.0 ml/min) and in a submersion chamber (volume, 800 μl; double-sided perfusion at 3–4 ml min–1). Under interface conditions, a clear meniscus of solution was always maintained above the slice.
Extracellular field potentials were recorded using conventional NaCl-filled (150 mm) glass capillary microelectrodes (2–10 MΩ). A bipolar stimulation electrode (tungsten tips separated by 0.5 mm when using adult rats and closer with younger animals) was placed in the stratum radiatum (in adults ≤500 μm from the recording site in the CA1 stratum pyramidale, and in an anatomically corresponding location in younger animals). The stimulus intensity was adjusted to elicit a maximal population spike in response to single-pulse stimulation (8–32 V; pulse duration, 100 μsec), and trains of stimuli (40 pulses at 100 Hz) were delivered every 5 min. The HFS-induced network activity was quantified from field potential recordings by integrating off-line the apparent power of the spikes. The signal for integration was generated by first truncating the spikes at a level that was set slightly more negative (∼0.5 mV) than the negative peak of the baseline shift, followed by low-pass filtering, then subtracting the resulting signal (that accurately followed the original baseline) from the field potential and finally taking the square. The resulting plots (see Fig. 1a) represent the time course of accumulation of the apparent energy of the high-frequency stimulation-induced gamma oscillation.
Figure 1.
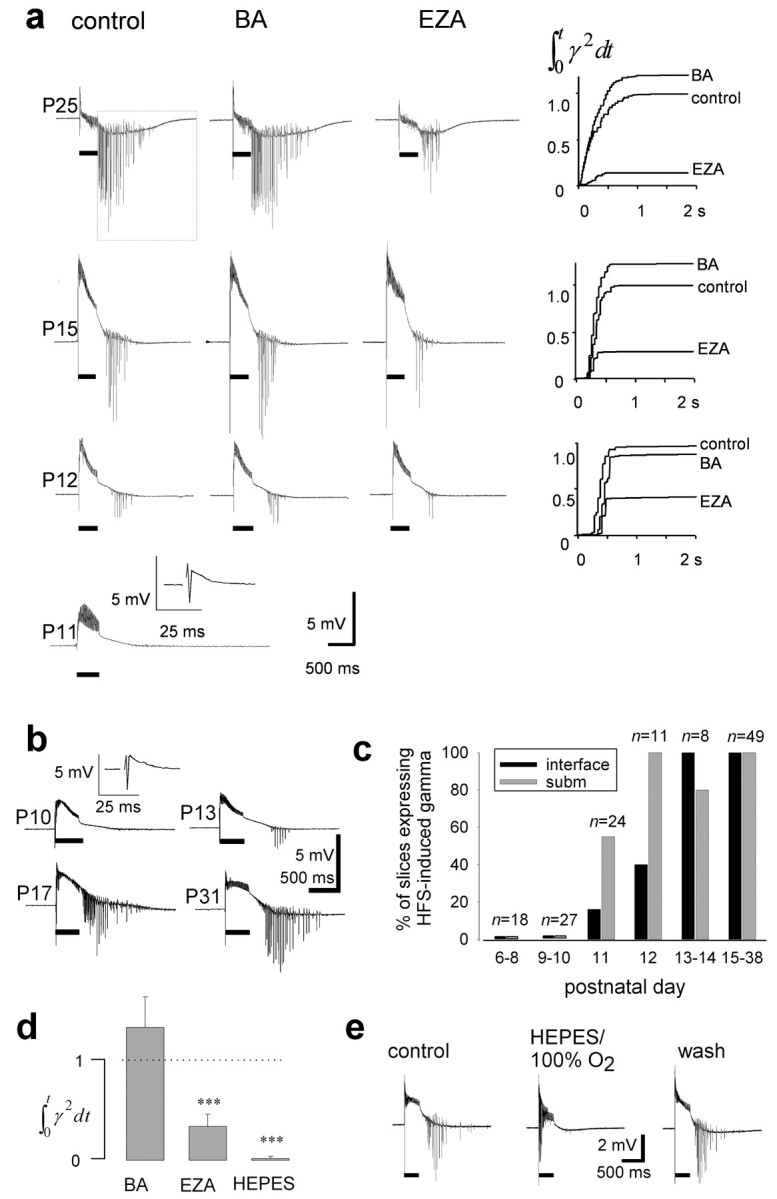
Postnatal development of HFS-induced gamma-frequency firing in CA1 stratum pyramidale and its dependence on intracellular but not interstitial carbonic anhydrase activity. a, Once HFS-induced gamma oscillation could be induced (40 stimulation pulses at 100 Hz; trains of stimuli indicated with horizontal bars), it was strongly inhibited by EZA (30–100 μm), whereas BA (10μm) had little effect. The plot showing the relative accumulation of apparent energy in the gamma-frequency range is from the time window starting 10 msec after the end of the train of stimuli (see dotted frame, P25). The field potential recordings were made in an interface chamber. b, Submerged slices show a developmental pattern of HFS-induced gamma oscillation similar to that observed in the interface chamber. Insets at P11 (a) and P10 (b) show a field response to single-pulse stimulation. c, Percentage of slices showing HFS-induced gamma oscillation plotted against postnatal day (n = 45 slices in interface and 92 in submerged chamber). d, The average of apparent energy in the gamma-frequency range in BA (n = 6), EZA (n = 6), and the nominally bicarbonate-free HEPES-buffered solution (n = 6), P23–P35, relative to that under control conditions. e, The population oscillations were abolished in a reversible manner in the absence of CO2/HCO3– (submerged slice).
Intracellular recordings from CA1 pyramidal neurons were obtained using sharp microelectrodes filled with 1.5 m K-acetate, 1.5 m K-methylsulfate, and 10 mm KCl (100–200 MΩ). Double-barreled K+-selective microelectrodes were made as described previously (Voipio et al., 1994; for response times, see Santhakumar et al., 2003) using a valinomycin-based membrane solution (Fluka, Buchs, Switzerland). In the TeMA+ electrodes (Alvarez-Leefmans et al., 1992; Nicholson, 1993) the liquid membrane solution contained 100 mg of potassium tetrakis (4-chlorophenyl) borate in 2 ml of 1,2-dimethyl-3-nitrobentzene (both chemicals from Fluka) and the backfilling solution was 150 mm TeMA chloride.
dl-2-Amino-5-phosphonopentanoic acid (dl-AP-5), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX), bicuculline methiodide, (2S)-3-{[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl)(phenylmethyl)phosphinic acid (CGP-55845), and picrotoxin were from Tocris-Cookson (Bristol, UK), and carbachol and the membrane-permeant carbonic anhydrase inhibitor ethoxyzolamide (EZA) were from Sigma (St. Louis, MO). The membrane-impermeant carbonic anhydrase inhibitor benzolamide (BA) (Maren, 1977; Saarikoski and Kaila 1992; Tong et al., 2000) was a gift from Prof. E. Swenson (Department of Medicine and Laboratory Medicine, University of Washington Medical Center, Seattle, WA).
Detection of intrapyramidal carbonic anhydrase activity. To detect intracellular carbonic anhydrase activity in hippocampal CA1 pyramidal neurons, we examined the effect of EZA (Maren, 1977) (30–100 μm) on fast intracellular pH (pHi) changes induced by transiently exposing acutely isolated cells or slices to the CO2/HCO3– -free HEPES solution. pHi was measured with the proton-sensitive fluorescent indicator 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF). The initial ΔpHi/Δt of the alkaline transient was compared in the absence and presence of EZA. In all experiments at P0–P8 and in some up to P12, EZA had little effect on the alkaline transients: the ratio of initial ΔpHi/Δt in EZA to that in control was 0.988 ± 0.041 (mean ± SD; n = 5 isolated cells and n = 64 cells in seven slices) (see Fig. 3b). This SD most likely reflects random variation and, therefore, we considered only an EZA-induced reduction in ΔpHi/Δt of at least five times higher than this SD to indicate the presence of intracellular carbonic anhydrase activity reliably.
Figure 3.

Postnatal development of intracellular carbonic anhydrase activity in CA1 pyramidal neurons. a, Specimen recording from an acutely isolated P17 neuron (steady-state pHi 7.3) shows that EZA (30 μm) attenuates the pHi shift induced by the withdrawal of CO2 in the continuous presence of BA (1 μm), indicating the presence of intracellular carbonic anhydrase. b, Data from experiments of the type shown in a quantified as the ratio of the initial ΔpHi/Δt in the presence and absence of EZA. Filled circles, Isolated CA1 pyramidal neurons; open circles, mean of 6–12 CA1 pyramidal neurons in a slice (+SEM). The inset illustrates a pair of isolated, BCECF-loaded CA1 pyramidal neurons from a P15 rat. c, Histogram showing that the percentage of pyramidal neurons expressing intracellular carbonic anhydrase increases steeply at P10–P12.
Pyramidal neurons were isolated in a region-specific manner from P6–P38 slices using the vibrodissection technique described by Pasternack et al. (1993). The cells were incubated for 5–10 min in a 1 μm solution of the acetoxy-methyl form of the indicator dye (BCECF-AM; Molecular Probes, Eugene, OR). From the BCECF-loaded cells, one to six were selected for fluorescence imaging recordings on the basis of morphological criteria, i.e., the isolated cell had a pyramidal shape, a clear apical dendrite at least two times the length of the soma and basal dendrites (see Fig. 3b, inset).
In slices from P0–P8 rats, incubation in 10 μm BCECF-AM for 40–60 min resulted in bright fluorescence of pyramidal neurons. However, after the first postnatal week, loading became poorer, and practically no fluorescing pyramidal neurons were detected in slices after P14–P16. This problem was circumvented as done previously (Stenkamp et al., 2001) by pressure injection of 1 mm BCECF-AM into the CA1 stratum pyramidale, which provided a local population of two to eight fluorescing pyramidal neurons. All fluorescence experiments were done in a submersion chamber (volume, 800 μl; one-sided perfusion at 3–4 ml/min).
The fluorescence imaging system (Photon Technology International, Lawrenceville, NJ) consisted of a 75 W xenon-arc lamp, a monochromator-chopper unit, an inverted microscope, and an intensified CCD camera (4–16 averaged frames at 0.5–1.5 Hz; excitation wavelengths, 440 and 495 nm). The pHi signal was calibrated with the high-[K+]o/nigericin technique (Thomas et al., 1979).
In situ hybridization. Rats were anesthetized with pentobarbital (30–40 mg/kg; P9 to adult) or with ketamine (20 mg/kg; P7) and then intracardially perfused with PBS, pH 7.3, followed by 4% paraformaldehyde (PFA). Their brains were removed and fixed in 4% PFA in PBS overnight at 4°C. The tissue was dehydrated through a graded ethanol an series and embedded in paraffin. Thin 7 μm sections were made using an RNase-free technique. The sections were then kept at 37°C overnight and stored at 4°C. For cRNA synthesis, an expressed sequence tag clone (BI290085) containing a specific rat CAII insert (538 bp, nucleotides 282–820; accession number NM_019291) was obtained from Invitrogen (Paisley, UK), and the sequence was verified. The forward primer: 5′CGCCAGTCACCCATCAATA3′ and reverse primer 5′AAGGATGCTTTAACCACTCG3′ were used to obtain a 700 bp fragment specific for the CAVII isoform (nucleotides 181–881; accession number XM_134293) by standard reverse transcription PCR (Li et al., 2002). The fragment was cloned into pGEM-T vector (Promega, Madison, WI), and the sequence was verified. The clones were linearized by restriction digestion and used as templates for the generation of sense and antisense 35S-labeled riboprobes for radioactive in situ hybridization or Biotin-labeled riboprobes for fluorescence in situ hybridization.
The radioactive in situ hybridization procedure using 35S-labeled sense and antisense RNA probes for CAVII was performed as described previously (Li et al., 2002). For fluorescence in situ hybridization of CAII and CAVII, we used the enzyme-labeled fluorescence mRNA in situ hybridization kit ELF97 (Molecular Probes) according to the manufacturer's protocol. The functionality of the CAII probe was verified by fluorescence in situ hybridization of CAII mRNA, which showed strong expression levels in oligodendrocyte cell bodies in the white matter of the adult rat brain (data not shown) (cf. Ghandour et al., 1980).
Quantification of hybridization signals on x-ray films was performed by computerized image analysis as described previously (Rivera et al., 2002). The region of interest for quantification of the radioactive in situ was taken to include the CA1 area from stratum oriens to the proximal stratum radiatum.
Results
Developmental expression of high-frequency stimulation-induced gamma oscillations
We first studied the capability of CA1 principal cells for synchronous gamma-frequency firing under control conditions. In experiments on rats younger than P11 (P6–P10, 40 slices in submersion and 5 in interface chamber), HFS did not induce gamma oscillations in any of the field recordings from the stratum pyramidale. This was despite the fact that single-pulse stimulation evoked a population spike of up to 3–4 mV (Fig. 1a,b). After P10, the percentage of slices exhibiting gamma oscillations increased rapidly and, after P14, HFS always induced gamma oscillations (Fig. 1c). In contrast, carbachol-induced oscillations (20 μm) (Fisahn et al., 1998) could be seen already at P7 (n = 5; data not shown) (Fisahn, 1999). This is in good agreement with all of the data provided below, which point to a specific “ontogenetic switch” at approximately P12 that is responsible for the emergence of HFS-induced synchronous spiking of CA1 pyramidal neurons at gamma frequencies.
There has been some concern (Whittington et al., 2001; Schuchmann et al., 2002) that the type of slice recording chamber has a major influence on the patterns and mechanisms of neuronal synchronization. Nevertheless, in the present study, HFS-induced gamma oscillations with comparable magnitudes, and essentially identical developmental profiles were seen under both interface and submerged (fast double-sided perfusion) conditions (Fig. 1a,b).
Local HFS of inhibitory interneurons in CA1 of slices from adult rats results in a prolonged GABAergic depolarization and spiking of pyramidal neurons (Kaila et al., 1997; Smirnov et al., 1999). This is inhibited by the membrane-permeant carbonic anhydrase inhibitor EZA, whereas the membrane-impermeant inhibitor BA has little effect (see also Burg et al., 1998; Uusisaari et al., 2000). To examine the role of intracellular carbonic anhydrase activity in the HFS-induced gamma response during development, we examined the intracellular effects of EZA (compare Fig. 3a). A quantitative comparison of the effects of BA (10 μm) and EZA (30–100 μm) was made using the maximal value attained from the cumulative plots of apparent energy of gamma activity such as those illustrated in Figure 1a (see Materials and Methods). Application of BA alone had no significant effect, whereas a subsequent addition of EZA (Fig. 1d) clearly suppressed HFS-induced gamma oscillation (p < 2 × 10–5; paired t test; n = 6). Prolonged washout (∼60 min) of EZA resulted in a near-complete recovery of the HFS-induced gamma oscillations.
The key role of intracellular carbonic anhydrase in the generation of the HFS-induced synchronous firing of principal neurons was further confirmed by recordings performed in the nominally bicarbonate-free HEPES-buffered solution. Withdrawal of CO2/HCO3– completely and reversibly blocked the gamma oscillations (P23–P35; n = 8) (Fig. 1d,e). Here, it is worth emphasizing that under the present experimental conditions, removal of CO2/HCO3– does not lead to a reduction of the efficacy of GABAergic inhibition as indicated by single pulse-evoked monosynaptic responses (Kaila et al., 1997).
We then studied the roles of glutamatergic and GABAergic transmission in the development of HFS-induced gamma oscillations. In the presence of the glutamate antagonists NBQX (10 μm) and dl-AP-5 (40 μm), HFS did not induce gamma oscillations at P8–P11 (n = 21 slices) even when HFS-evoked GABA release was enhanced using the GABAB antagonist CGP-55845A (0.5 μm; n = 9) (Bracci et al., 1999; Cobb et al., 1999) or by increasing the stimulus intensity or the number of stimuli up to 100 (n = 14). In good agreement with the data obtained with intact glutamatergic transmission, HFS resulted in gamma oscillations in all recordings after P13 (n = 31) (Fig. 2b). As under control conditions, HFS-induced gamma oscillation was strongly suppressed by EZA (P13–P37; n = 31) (Fig. 2b), whereas BA had no consistent effects. In the absence of CO2/HCO3–, the oscillations were totally abolished (n = 8; data not shown). The addition of GABAA antagonists (10 μm bicuculline or 100 μm picrotoxin; n = 3 for each drug) in the presence of the glutamate antagonists resulted in a complete block of the oscillations (data not shown). Thus, in line with a GABAergic basis of the tonic HFS-induced excitation (Kaila et al., 1997; Smirnov et al., 1999), the emergence of synchronous firing of the principal neurons requires functional intracellular carbonic anhydrase both in the presence and in the absence of fast glutamatergic transmission.
Figure 2.

Developmental expression of the GABAergic [K+]o transient and the consequent tonic depolarization. a, Intracellular recordings from CA1 pyramidal cells before and after the expression of intracellular carbonic anhydrase. At P11 (Vm = –69 mV) the pharmacologically isolated GABAA-mediated depolarizing response is small and EZA (100 μm) has little effect on it. The HFS-induced GABAergic depolarization is attenuated by EZA only after intrapyramidal carbonic anhydrase is expressed (P17; Vm = –72 mV). Spikes are truncated. b, Simultaneous GABAergic [K+]o and field potential recordings in CA1 stratum pyramidale. Both HFS-induced gamma oscillation and the [K+]o transient show a marked sensitivity to EZA only after P12. The 10 mV calibration refers to the K+ electrode differential signal (and corresponds to a 1.5 mm increase in [K+] from 3 mm), and 2 mV refers to field potential. c, The effect of EZA on the amplitude of the HFS-induced [K+]o transient plotted against postnatal age, Filled circles, Slices in an interface chamber; open circles, slices in a submersion chamber. d, Simultaneous recordings with field potential, K+- and TeMA+-sensitive microelectrodes in CA1 stratum pyramidale of a P34 slice. The 20 and 2 mV calibrations refer to the ion electrode differential signals and the field signal, respectively. All experiments were performed in the presence of NBQX, AP-5, and CGP-55845A.
Developmental expression of the GABAergic depolarization and [K+]o transient
To gain additional information on the role of intracellular carbonic anhydrase in the functional maturation of the nonsynaptic mechanisms underlying HFS-induced gamma oscillations, we performed intracellular recordings from CA1 pyramidal cells as well as measurements of [K+]o in the presence of the ionotropic glutamatergic blockers.
Before approximately P12, a prompt initial hyperpolarization was seen in the GABAergic membrane potential response on HFS (Fig. 2a), in agreement with the fact that fast inhibitory transmission is functional at this age (Swann et al., 1989; Rivera et al., 1999). The initial hyperpolarization was followed by a small and rather brief depolarization with a magnitude (11.4 ± 1.9 mV; n = 4) and a time course essentially insensitive to EZA (n = 4). As a whole, the biphasic response in the immature slices was strikingly similar to the HFS-induced response in adult slices in which the nonsynaptic depolarizing component has been selectively blocked [Smirnov et al. (1999), their Figs. 2, 3]. After P12, the GABAergic response and its sensitivity to carbonic anhydrase inhibitors were similar to that measured in adult preparations (Kaila et al., 1997), because both the amplitude and duration of the prolonged depolarization were attenuated by EZA (n = 4; P14–P35), whereas BA had no discernible effect (n = 6; P14–P35). These data clearly suggest that the prompt developmental shift seen in the HFS-induced responses is critically dependent on postnatal upregulation of intracellular carbonic anhydrase expression.
Previous work (Kaila et al., 1997) has indicated that the prolonged depolarization seen in mature slices in response to HFS is caused by a GABAergic, EZA-sensitive transient increase in [K+]o. In agreement with this, the HFS-induced [K+]o transient was very small in rats at P7–P9 even the presence of 0.5 μm CGP-55845A (0.70 ± 0.13 mm; n = 5) and was not significantly affected by EZA (paired t test; p > 0.05; n = 5) (Fig. 2b,c). At approximately P12, a marked developmental increase in the [K+]o shift was evident, and after the second postnatal week the amplitude of the [K+]o transients had reached a level (3.6 ± 0.17 mm; n = 20) similar to that measured in adult slices (Fig. 2b). In parallel with the enhancement of the [K+]o shift, there was a pronounced increase in its sensitivity to EZA (Fig. 2c), which suppressed the peak amplitude of the [K+]o response by 35 ± 3.7% (n = 9) at P30–P37. Again, BA had no detectable effect. In agreement with the complete block of HFS-induced gamma oscillations in the HEPES-buffered solution (Fig. 1d,e), withdrawal of CO2/HCO3– in the presence of EZA caused an additional decrease in the HFS-induced [K+]o shift to 47 ± 5.5% (n = 4; P14–P36) of its control value.
Extracellular volume measurements and [K+]o transients
An activity-induced increase in [K+]o can result from a cellular net release of K+, from a shrinkage of the extracellular space, or both (Dietzel et al., 1980). To find out whether, and to what extent, an activity-induced decrease in the extracellular volume fraction might have been involved in the generation of the HFS-induced [K+]o transients, we performed simultaneous recordings with field potential, K+- and TeMA+-sensitive microelectrodes (tips within 50 μm) from the CA1 stratum pyramidale of P30–P40 slices, in the presence of AP-5, NBQX, and CGP-55845A. [TeMA+]o responses were very small in amplitude (0.1 ± 0.003 mm from the baseline of 1.5 mm; n = 9) compared with [K+]o transients (Fig. 2d), indicating that changes in the extracellular volume have only a minor effect (∼7%) on the [K+]o shifts. This means that the HFS-induced [K+]o transients were mainly (∼93%) attributable to a cellular net release of potassium.
Developmental profile of intrapyramidal carbonic anhydrase expression
Although the above data indicate that an upregulation of intracellular carbonic anhydrase expression may act as a switch in the maturation of the CA1 neuronal network, it does not assign this intracellular carbonic anhydrase to any specific cell type. We next assessed the developmental expression of intrapyramidal carbonic anhydrase activity (Pasternack et al., 1993) by examining the effects of carbonic anhydrase inhibitors on fast pHi transients. Removing CO2 and HCO3– led to a rapid increase in pHi of acutely isolated pyramidal neurons, as seen in fluorescence measurements (Fig. 3a). In the presence of EZA (30–100 μm), this alkalinization was considerably slower, indicating a specific action of intracellular carbonic anhydrase, because BA (10 μm) produced only a very slight decrease in ΔpHi/Δt (Voipio, 1998). The effect of EZA on the initial pHi change was reversed during a washout period of 15–20 min.
Experiments with isolated CA1 pyramidal neurons (n = 44) indicated that the expression of intracellular carbonic anhydrase commenced at approximately P12 (Fig. 3b). The steady-state pHi before and after the expression of intrapyramidal carbonic anhydrase (7.17 ± 0.09, n = 10 and 7.20 ± 0.05, n = 16, respectively) did not differ significantly (unpaired t test; p = 0.4).
Because it turned out very difficult to isolate viable neurons in a region-specific manner from hippocampi of rats that were >1 week old, we used slice preparations to assess intracellular carbonic anhydrase activity in pyramidal neurons in P0–P8 rats (n = 64 neurons in seven slices) and compared these observations with those from P26 slices (n = 15 neurons in two slices). These experiments were performed in the continuous presence of BA because, in slices, the initial ΔpHi/Δt is rate-limited not only by the intracellular CO2/HCO3– hydration/dehydration reaction but also by the diffusion of carbonic acid buffer species within the tissue (Voipio and Ballanyi, 1997). In agreement with the data from isolated neurons, EZA had no discernible effect on the pHi response of hippocampal CA1 pyramidal neurons in the P0–P8 slices (Fig. 3b), indicating the absence of functional intracellular carbonic anhydrase. The somewhat smaller effect of intracellular carbonic anhydrase inhibition in the P26 slices compared with isolated cells of the same age (Fig. 3b) can be accounted for by the slow equilibration of the interstitial fluid after the abrupt removal of ambient CO2/HCO3–.
These data indicate that the expression of intracellular carbonic anhydrase activity is seen in all hippocampal CA1 pyramidal neurons by the end of the second postnatal week (Fig. 3c). The striking temporal correlation between the expression of intrapyramidal carbonic anhydrase and the appearance of HFS-induced gamma oscillation suggest a causal relation between these events. The time course of the upregulation of intracellular carbonic anhydrase activity in CA3 pyramidal neurons was essentially identical to that of CA1 neurons (n = 23 isolated cells; data not shown).
Identification of the intrapyramidal carbonic anhydrase isoform
A strong neuronal transcription signal for the intracellular CAVII has been reported in the mouse brain at P18 (Lakkis et al., 1997), while there is substantial evidence indicating a glial expression of another cytosolic isozyme, CAII (Agnati et al., 1995; Ridderstråle and Wistrand, 1998). However, it has been suggested recently that a widespread neuronal expression of CAII takes place in the brain, including the hippocampus (Wang et al., 2002). Other catalytic carbonic anhydrase isoforms in the parenchyma of the mammalian brain include the extracellular isoforms IV and XIV (Parkkila et al., 2001; Svichar and Chesler 2003) and the mitochondrial carbonic anhydrase CAV (Ghandour et al., 2000).
In the present work, an obvious task was to identify the neuronal, cytosolic isoform that is upregulated at approximately P12. Therefore, we focused on the expression patterns of CAII and CAVII during the first 3 weeks of postnatal life. In three series of parallel fluorescence in situ hybridization experiments (five to six time points between P7 and P22; three to five sections for each isoform and time point), there was no detectable neuronal mRNA expression of CA II (Fig. 4a). Up to P11, expression of CA VII mRNA could not be detected, whereas a steep increase in the signal was seen in the stratum pyramidale of CA1–CA3 and in the dentate gyrus at P12–P13 (Fig. 4b). A quantification of the increase in CAVII expression, based on radioactive in situ hybridization, is shown in Figure 4, c and d. These data are in excellent agreement with the idea that an upregulation of CAVII is the molecular basis of the functional developmental switch that underlies the simultaneous emergence of neuronal CA activity and of gamma oscillations as seen in the pH microfluorescence (Fig. 3) and electrophysiological experiments (Figs. 1, 2) described above.
Figure 4.

Developmental expression of carbonic anhydrase isoform VII as seen by in situ hybridization. a, CAII mRNA is not detected in fluorescence in situ hybridization (a), whereas a steep increase in CAVII mRNA is seen throughout the pyramidal layer of Ammon's horn and in the dentate gyrus by P13 (b). Scale bar, 500 μm. c, Expression pattern of CAVII mRNA demonstrated by radioactive in situ hybridization (left, sense control; right, antisense in P22 slices). Scale bar, 400μm. d, Quantification (mean + SEM) based on the radioactive in situ hybridization shows a steep upregulation of CAVII mRNA between P9 and P15.
Early spontaneous population oscillations are not dependent on CO2/HCO3–
Immature hippocampal slices are known to generate spontaneous population oscillations [SPOs; often termed giant depolarizing potentials (GDPs)] (Ben-Ari 2001) that are based on synchronous activation of both pyramidal neurons and interneurons. Such coactivation is also a characteristic of HFS-induced gamma oscillations (see Introduction). Furthermore, both the SPOs and HFS-induced oscillations are dependent on depolarizing GABAergic transmission, although the underlying ionic mechanisms are dissimilar (Kaila et al., 1997; Rivera et al., 1999).
Thus, the SPOs provide an ideal way to test whether the present results regarding the CO2/HCO3– -dependent developmental switch are specific for HFS-induced oscillations, or whether they would merely reflect a nonspecific dependence of hippocampal neuronal network activity on the presence of CO2/HCO3–. SPO activity was measured with extracellular field potential electrodes from the CA3 stratum pyramidale of P4–P6 rat hippocampal slices. In CO2/HCO3–-buffered standard solution, field SPOs were seen as negative-going shifts with an amplitude of 56.8 ± 12.7 μV (Fig. 5) (n = 6 slices; n = 10 SPOs from each experiment). In sharp contrast to HFS-induced gamma oscillations that were totally abolished (in 5 min) (Fig. 1d,e) in the nominal absence of CO2/HCO3–, there was a significant increase (53–166%; n = 6 slices; n = 10 SPOs/condition) in the amplitude of the SPOs that persisted in the HEPES buffer for up to 80 min. These results clearly demonstrate that the presence of CO2/HCO3– and carbonic anhydrase activity are not essential features of network events in the developing hippocampus. Hence, the dependence of HFS-induced gamma oscillations on bicarbonate and carbonic anhydrase activity has wide and important implications, as will be discussed below.
Figure 5.

Spontaneous population oscillations are not blocked in the absence of CO2/HCO3–. Field potential recording in the CA3 area (top traces) from a P4 rat hippocampal slice (bandpass filtered at 0.2–20 Hz) shows SPOs in the control (left) and in the CO2/HCO3–-free HEPES-buffered solution. Expanded traces of single SPOs (low-pass filtered at 500 Hz) are shown below.
Discussion
In this study, we demonstrate not only a striking temporal coincidence but also provide strong evidence suggesting a causal link between the expression of intrapyramidal carbonic anhydrase CAVII and the establishment of rhythmic synchronized pyramidal cell firing in the rat hippocampal CA1 area at approximately P12.
Previous work has shown that the long-lasting depolarizing component of the biphasic GABAA-receptor-mediated response evoked in adult CA1 pyramidal neurons on HFS is caused by a transition from synaptic to nonsynaptic K+-mediated excitatory transmission (Kaila et al., 1997; Smirnov et al., 1999; Voipio and Kaila, 2000). This mechanism of GABAergic excitation (Kaila et al., 1997) and the associated gamma oscillations (present work) are strictly dependent on the availability of bicarbonate and show a marked sensitivity to inhibitors of intracellular carbonic anhydrase. Interestingly, LeBeau et al. (2002) have shown recently that gamma activity can be evoked in the CA1 region by exogenous potassium “puffs,” which result in transient elevations of [K+]o, resembling those that are generated by the GABAergic, bicarbonate-dependent mechanism.
A key observation in the present work was that in hippocampal pyramidal neurons, HFS-induced GABAergic depolarization is hardly detectable before P12, whereas a steep upregulation of the depolarization is seen at a more mature developmental stage. This is paralleled by an increase in the effect of the intracellular carbonic anhydrase inhibitor, EZA. Because the GABAergic depolarization is caused primarily by an activity-induced [K+]o transient (Kaila et al., 1997), it is not surprising that the [K+]o transients showed a similar developmental profile with regard to their amplitude and sensitivity to EZA.
The developmental upregulation of the HFS-induced GABAergic, CO2/HCO3– -dependent depolarization was closely paralleled by an abrupt emergence of EZA- and CO2/HCO3–-sensitive HFS-induced gamma oscillations at approximately P12. Furthermore, both functional intrapyramidal carbonic anhydrase activity and the in situ hybridization signal of CAVII appeared promptly at approximately P12. The present data as a whole imply that, during postnatal development, the machinery required to generate the HFS-induced GABAergic excitation becomes functional only on expression of intrapyramidal CAVII.
A comparison of the responses of the ion electrodes sensitive to K+ and TeMA+ indicated that the [K+]o transients were attributable to a net cellular release of potassium, with a very small contribution attributable to a shrinkage of the extracellular space. GABAB-receptor-mediated potassium fluxes (Rausche et al., 1989) are not likely to provide a contribution to stimulation-induced GABAergic [K+]o shifts, because GABAB antagonists enhance, rather than suppress, the HFS-induced GABAergic depolarization (Cobb et al., 1999).
Carbonic anhydrase inhibitors have been widely used as anti-convulsant agents (Wyllie, 1997; Casini et al., 2003; Supuran et al., 2003). This is intriguing in the light of the present work, because neuronal hypersynchrony rather than hyperexcitability has repeatedly been proposed to be a hallmark of epileptic activity. It is also interesting to note that there is a sudden emergence of gamma oscillations associated with 0-Mg2+-induced epileptiform activity in rat hippocampal slices at P13 (Kohling et al., 2000). Our observations readily explain this developmental change, and point to CAVII as a novel target of carbonic anhydrase-inhibiting anticonvulsant drugs.
It is known that oscillations at gamma frequencies can result in a potentiation of excitatory connections among pyramidal neurons (Whittington et al., 1997b). Interestingly, the expression of HFS-induced LTP (Harris and Teyler, 1984; Chabot et al., 1996) takes place within the same postnatal time window as the expression of HFS-induced gamma oscillations and, indeed, [K+]o transients have been implicated in the generation of LTP (Collingridge, 1992). Because LTP in CA1 is known to require activation of NMDA receptors in the pyramidal neurons (Bliss and Collingridge, 1993), it is interesting to note that the HFS-induced GABAergic depolarization can be strong enough to remove the voltage-dependent Mg2+ block of NMDA receptors (Staley et al., 1995).
The key functional role of pyramidal CAVII in the developmental expression of HFS-induced gamma oscillations at approximately P12 is underscored by the fact that synchronous activity of interneurons and pyramidal neurons takes place in the form of SPOs (or GDPs; Ben-Ari, 2001) much earlier, with an onset at approximately P0 (Ben-Ari et al., 1989). Furthermore, as shown in the present study, SPOs are not dependent on the presence of CO2/HCO3–. The developmental time course of the qualitative changes in GABAergic transmission and of these two types of network activity seems to be based on two kinds of ontogenetic switches at the level of anion-regulatory proteins. The neuronal Cl– extruding K+-Cl– cotransporter KCC2 is upregulated from P0 to P12, which is generally thought to convert GABAA responses from depolarizing to hyperpolarizing with a consequent, progressive disappearance of SPOs during hippocampal maturation (Ben-Ari et al., 1989). However, the expression of CAVII sets the stage for another type of depolarizing GABAA response, which is caused by HFS-induced ionic shifts triggered by massive activation of pyramidal GABAA receptors.
It is also interesting to note here that interneuronal gamma activity such as that evoked by carbachol in hippocampal slices (Fisahn et al., 1998) had a much earlier onset of development than HFS-induced gamma oscillation, and could be observed already at P7 (this study; A. Fisahn, personal communication; Fisahn, 1999). These considerations point to a major difference between the cellular mechanisms underlying HFS-induced gamma oscillation (i.e., synchronous pyramidal firing at gamma frequencies) and interneuronal gamma activity (Fisahn et al., 1998). A more specific conclusion that can be drawn is that the carbachol-induced gamma oscillation with its expression already at P7 does not require intrapyramidal carbonic anhydrase activity.
In conclusion, our data point to CAVII as a major developmental switch that governs the electrophysiological behavior of CA1 pyramidal neurons, the major output pathway of the hippocampus under both physiological and pathophysiological conditions. The present study calls for additional work on changes in the expression of carbonic anhydrase isoforms during development and trauma (Agnati et al., 1995; Nógrádi et al., 1997). Such data are likely to unravel novel mechanisms underlying plasticity, epileptiform activity, and the modes of actions of CNS-targeted drugs, especially of those anticonvulsant compounds that are known to be potent carbonic anhydrase inhibitors (Perez Velazquez, 2003; Supuran et al., 2003).
Footnotes
This work was supported by grants from the Academy of Finland and from the Sigrid Juselius Foundation.
Correspondence should be addressed to Dr. Juha Voipio, Department of Biosciences, University of Helsinki, P.O. Box 65, Viikinkaari 1, 00790 Helsinki, Finland 00014. E-mail: juha.voipio@helsinki.fi.
DOI:10.1523/JNEUROSCI.5176-03.2004
Copyright © 2004 Society for Neuroscience 0270-6474/04/242699-09$15.00/0
References
- Agnati LF, Tinner B, Staines WA, Vaananen K, Fuxe K (1995) On the cellular localization and distribution of carbonic anhydrase II immunoreactivity in the rat brain. Brain Res 676: 10–24. [DOI] [PubMed] [Google Scholar]
- Alvarez-Leefmans FJ, Gamino FJ, Reuss L (1992) Cell volume changes upon sodium pump inhibition in Helix aspersa neurons. J Physiol (Lond) 458: 603–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y (2001) Developing networks play a similar melody. Trends Neurosci 24: 353–360. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL (1989) Giant synaptic potentials in immature rat CA3 hippocampal neurons. J Physiol (Lond) 416: 303–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL (1993) A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Bracci E, Vreugdenhil M, Hack SP, Jefferys JG (1999) On the synchronizing mechanisms of tetanically induced hippocampal oscillations. J Neurosci 19: 8104–8113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burg M, Heinemann U, Schmitz D (1998) Neuroactive steroids induce GABAA receptor-mediated depolarizing postsynaptic potentials in hippocampal CA1 pyramidal cells of the rat. Eur J Neurosci 10: 2880–2886. [DOI] [PubMed] [Google Scholar]
- Casini A, Antel J, Abbate F, Scozzafava A, David S, Waldeck H, Schäfer S, Supuran CT (2003) Carbonic anhydrase inhibitors: SAR and X-ray crystallographic study for the interaction of sugar sulfamates/sulfamides with isozymes I, II and IV. Bioorg Med Chem Lett 13: 841–845. [DOI] [PubMed] [Google Scholar]
- Chabot C, Bernard J, Normandin M, Ohayon M, Baudry M, Massicotte G (1996) Developmental changes in depolarization-mediated AMPA receptor modifications and potassium-induced long-term potentiation. Brain Res Dev Brain Res 93: 70–75. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Manuel NA, Morton RA, Gill CH, Collingridge GL, Davies CH (1999) Regulation of depolarizing GABA(A) receptor-mediated synaptic potentials by synaptic activation of GABA(B) autoreceptors in the rat hippocampus. Neuropharmacology 38: 1723–1732. [DOI] [PubMed] [Google Scholar]
- Collingridge GL (1992) The mechanism of induction of NMDA receptor-dependent long-term potentiation in the hippocampus. Exp Physiol 77: 771–797. [DOI] [PubMed] [Google Scholar]
- Dietzel I, Heinemann U, Hofmeier G, Lux HG (1980) Transient changes in the size of the extracellular space in the sensorimotor cortex of cats in relation to stimulus-induced changes in potassium concentration. Exp Brain Res 40: 432–439. [DOI] [PubMed] [Google Scholar]
- Draguhn A, Traub RD, Schmitz D, Jefferys JG (1998) Electrical coupling underlies high-frequency oscillations in the hippocampus in vitro. Nature 394: 189–192. [DOI] [PubMed] [Google Scholar]
- Fisahn A (1999) An investigation into cortical gamma frequency oscillations in vitro. PhD thesis, University of Oxford.
- Fisahn A, Pike FG, Buhl EH, Paulsen O (1998) Cholinergic induction of network oscillations at 40 Hz in the hippocampus in vitro. Nature 394: 186–189. [DOI] [PubMed] [Google Scholar]
- Ghandour MS, Langley OK, Vincendon G, Gombos G, Filippi D, Limozin N, Dalmasso D, Laurent G (1980) Immunochemical and immunohistochemical study of carbonic anhydrase II in adult rat cerebellum: a marker for oligodendrocytes. Neuroscience 5: 559–571. [DOI] [PubMed] [Google Scholar]
- Ghandour MS, Parkkila AK, Parkkila S, Waheed A, Sly WS (2000) Mitochondrial carbonic anhydrase in the nervous system: expression in neuronal and glial cells. J Neurochem 75: 2212–2220. [DOI] [PubMed] [Google Scholar]
- Gloveli T, Albrecht D, Heinemann U (1995) Properties of low Mg2+ induced epileptiform activity in rat hippocampal and entorhinal cortex slices during adolescence. Brain Res Dev Brain Res 87: 145–152. [DOI] [PubMed] [Google Scholar]
- Gray CM (1999) The temporal correlation hypothesis of visual feature integration: still alive and well. Neuron 24: 31–25. [DOI] [PubMed] [Google Scholar]
- Grover LM, Lambert NA, Schwartzkroin PA, Teyler TJ (1993) Role of HCO3– ions in depolarizing GABAA receptor-mediated responses in pyramidal cells of rat hippocampus. J Neurophysiol 69: 1541–1555. [DOI] [PubMed] [Google Scholar]
- Harris KM, Teyler TJ (1984) Developmental onset of long-term potentiation in area CA1 of the rat hippocampus. J Physiol (Lond) 346: 27–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaila K, Lamsa K, Smirnov S, Taira T, Voipio J (1997) Long-lasting GABA-mediated depolarization evoked by high-frequency stimulation in pyramidal neurons of rat hippocampal slice is attributable to a network-driven, bicarbonate-dependent K+ transient. J Neurosci 17: 7662–7672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohling R, Vreugdenhil M, Bracci E, Jefferys JG (2000) Ictal epileptiform activity is facilitated by hippocampal GABAA receptor-mediated oscillations. J Neurosci 20: 6820–6829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkis MM, O'Shea KS, Tashian RE (1997) Differential expression of the carbonic anhydrase genes for CA VII (Car7) and CA-RP VIII (Car8) in mouse brain. J Histochem Cytochem 45: 657–662. [DOI] [PubMed] [Google Scholar]
- LeBeau FE, Towers SK, Traub RD, Whittington MA, Buhl EH (2002) Fast network oscillations induced by potassium transients in the rat hippocampus in vitro. J Physiol (Lond) 542: 167–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Tornberg J, Kaila K, Airaksinen MS, Rivera C (2002) Patterns of cation-chloride cotransporter expression during embryonic rodent CNS development. Eur J Neurosci 16: 2358–2370. [DOI] [PubMed] [Google Scholar]
- Linden DJ (1999) The return of the spike: postsynaptic action potentials and the induction of LTP and LTD. Neuron 22: 661–666. [DOI] [PubMed] [Google Scholar]
- Maren TH (1977) Use of inhibitors in physiological studies of carbonic anhydrase. Am J Physiol (Lond) 232: F291–F297. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Contreras D (2001) On the cellular and network bases of epileptic seizures. Annu Rev Physiol 63: 815–846. [DOI] [PubMed] [Google Scholar]
- Nicholson C (1993) Ion-selective microelectrodes and diffusional measurements as tools to explore the brain cell microenvironment. J Neurosci Methods 48: 199–213. [DOI] [PubMed] [Google Scholar]
- Nógrádi A, Jonsson N, Walker R, Caddy K, Carter N, Kelly C (1997) Carbonic anhydrase II and carbonic anhydrase-related protein in the cerebellar cortex of normal and lurcher mice. Brain Res Dev Brain Res 98: 91–101. [DOI] [PubMed] [Google Scholar]
- Parkkila S, Parkkila AK, Rajaniemi H, Shah GN, Grubb JH, Waheed A, Sly WS (2001) Expression of membrane-associated carbonic anhydrase XIV on neurons and axons in mouse and human brain. Proc Natl Acad Sci USA 98: 1918–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack M, Voipio J, Kaila K (1993) Intracellular carbonic anhydrase activity and its role in GABA-induced acidosis in isolated rat hippocampal pyramidal neurones. Acta Physiol Scand 148: 229–231. [DOI] [PubMed] [Google Scholar]
- Perez Velazquez JL (2003) Bicarbonate-dependent depolarizing potentials in pyramidal cells and interneurons during epileptiform activity. Eur J Neurosci 18: 1337–1342. [DOI] [PubMed] [Google Scholar]
- Perreault P, Avoli M (1988) A depolarizing inhibitory postsynaptic potential activated by synaptically released gamma-aminobutyric acid under physiological conditions in rat hippocampal pyramidal cells. Can J Physiol Pharmacol 66: 1100–1102. [DOI] [PubMed] [Google Scholar]
- Rausche G, Sarvey JM, Heinemann U (1989) Slow synaptic inhibition in relation to frequency habituation in dentate granule cells of rat hippocampal slices. Exp Brain Res 78: 233–242. [DOI] [PubMed] [Google Scholar]
- Ridderstråle Y, Wistrand PJ (1998) Carbonic anhydrase isoforms in the mammalian nervous system. In: pH and brain function (Kaila K, Ransom B, eds), pp 21–43. New York: Wiley-Liss.
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K (1999) The K+/Cl– co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature 397: 251–255. [DOI] [PubMed] [Google Scholar]
- Rivera C, Li H, Thomas-Crusells J, Lahtinen H, Viitanen T, Nanobashvili A, Kokaia Z, Airaksinen MS, Voipio J, Kaila K, Saarma M (2002) BDNF-induced TrkB activation down-regulates the K+-Cl– cotransporter KCC2 and impairs neuronal Cl– extrusion. J Cell Biol 159: 747–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saarikoski J, Kaila K (1992) Simultaneous measurement of intracellular and extracellular carbonic anhydrase activity in intact muscle fibres. Pflügers Arch 421: 357–363. [DOI] [PubMed] [Google Scholar]
- Santhakumar V, Voipio J, Kaila K, Soltesz I (2003) Post-traumatic hyperexcitability is not caused by impaired buffering of extracellular potassium. J Neurosci 23: 5865–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Schuchmann S, Fisahn A, Draguhn A, Buhl EH, Petrasch-Parwez E, Dermietzel R, Heinemann U, Traub RD (2001) Axo-axonal coupling: a novel mechanism for ultrafast neuronal communication. Neuron 31: 831–840. [DOI] [PubMed] [Google Scholar]
- Schuchmann S, Meierkord H, Stenkamp K, Breustedt J, Windmuller O, Heinemann U, Buchheim K (2002) Synaptic and nonsynaptic ictogenesis occurs at different temperatures in submerged and interface rat brain slices. J Neurophysiol 87: 2929–2935. [DOI] [PubMed] [Google Scholar]
- Singer W (1999) Neuronal synchrony: a versatile code for the definition of relations? Neuron 24: 49–65, 111–125. [DOI] [PubMed] [Google Scholar]
- Smirnov S, Paalasmaa P, Uusisaari M, Voipio J, Kaila K (1999) Pharmacological isolation of the synaptic and nonsynaptic components of the GABA-mediated biphasic response in rat CA1 hippocampal pyramidal cells. J Neurosci 19: 9252–9260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Soldo BL, Proctor WR (1995) Ionic mechanisms of neuronal excitation by inhibitory GABAA receptors. Science 269: 977–981. [DOI] [PubMed] [Google Scholar]
- Stenkamp K, Palva JM, Uusisaari M, Schuchmann S, Schmitz D, Heinemann U, Kaila K (2001) Enhanced temporal stability of cholinergic hippocampal gamma oscillations following respiratory alkalosis in vitro. J Neurophysiol 85: 2063–2069. [DOI] [PubMed] [Google Scholar]
- Supuran CT, Scozzafava A, Casini A (2003) Carbonic anhydrase inhibitors. Med Res Rev 23: 146–189. [DOI] [PubMed] [Google Scholar]
- Svichar N, Chesler M (2003) Surface carbonic anhydrase activity on astrocytes and neurons facilitates lactate transport. Glia 41: 415–419. [DOI] [PubMed] [Google Scholar]
- Swann JW, Brady RJ, Martin DL (1989) Postnatal development of GABA-mediated synaptic inhibition in rat hippocampus. Neuroscience 28: 551–561. [DOI] [PubMed] [Google Scholar]
- Swann JW, Smith KL, Brady RJ (1993) Localized excitatory synaptic interactions mediate the sustained depolarization of electrographic seizures in developing hippocampus. J Neurosci 13: 4680–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JA, Buchsbaum RN, Zimniak A, Racker E (1979) Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18: 2210–2218. [DOI] [PubMed] [Google Scholar]
- Tong CK, Brion LP, Suarez C, Chesler M (2000) Interstitial carbonic anhydrase (CA) activity in brain is attributable to membrane-bound CA type IV. J Neurosci 20: 8247–8253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Whittington MA, Colling SB, Buzsáki G, Jefferys JG (1996a) Analysis of gamma rhythms in the rat hippocampus in vitro and in vivo. J Physiol (Lond) 493: 471–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub RD, Whittington MA, Stanford IM, Jefferys JG (1996b) A mechanism for generation of long-range synchronous fast oscillations in the cortex. Nature 383: 621–624. [DOI] [PubMed] [Google Scholar]
- Traub RD, Spruston N, Soltesz I, Konnerth A, Whittington MA, Jefferys GR (1998) Gamma-frequency oscillations: a neuronal population phenomenon, regulated by synaptic and intrinsic cellular processes, and inducing synaptic plasticity. Prog Neurobiol 55: 563–575. [DOI] [PubMed] [Google Scholar]
- Uusisaari M, Smirnov S, Voipio J, Kaila K (2000) Spontaneous epileptiform activity mediated by GABA(A) receptors and gap junctions in the rat hippocampal slice following long-term exposure to GABA(B) antagonists. Neuropharmacology 43: 563–572. [DOI] [PubMed] [Google Scholar]
- Voipio J (1998) Diffusion and buffering aspects of H+, HCO3–, and CO2 movements in brain tissue. In: pH and brain function (Kaila K, Ransom B, eds), pp 45–66. New York: Wiley-Liss.
- Voipio J, Ballanyi K (1997) Interstitial PCO2 and pH, and their role as chemostimulants in the isolated respiratory network of neonatal rats. J Physiol (Lond) 499: 527–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voipio J, Kaila K (2000) GABAergic excitation and K+-mediated volume transmission in the hippocampus. Prog Brain Res 125: 329–338. [DOI] [PubMed] [Google Scholar]
- Voipio J, Pasternack M, MacLeod KT (1994) Ion-sensitive microelectrodes: microelectrode techniques. In: The Plymouth workshop handbook (Ogden D, ed), pp 275–316. Cambridge, UK: The Company of Biologist.
- Wang W, Bradley SR, Richerson GB (2002) Quantification of the response of rat medullary raphe neurones to independent changes in pHo and PCO2 J Physiol (Lond) 540: 951–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Traub RD, Jefferys JG (1995) Synchronized oscillations in interneuron networks driven by metabotropic glutamate receptor activation. Nature 373: 612–615. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Stanford IM, Colling SB, Jefferys JG, Traub RD (1997a) Spatiotemporal patterns of gamma frequency oscillations tetanically induced in the rat hippocampal slice. J Physiol (Lond) 502: 591–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Traub RD, Faulkner HJ, Stanford IM, Jefferys JG (1997b) Recurrent excitatory postsynaptic potentials induced by synchronized fast cortical oscillations. Proc Natl Acad Sci USA 94: 12198–12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittington MA, Doheny HC, Traub RD, LeBeau FE, Buhl EH (2001) Differential expression of synaptic and nonsynaptic mechanisms underlying stimulus-induced gamma oscillations in vitro J Neurosci 21: 1727–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyllie E (1997) The treatment of epilepsy: principles and practice. Baltimore: Williams and Wilkins.