

Abstract
Hippocampal long-term depression (LTD) is a long-lasting decrease in synaptic strength that is most commonly studied at glutamatergic inputs to pyramidal cells in hippocampal area CA1. Activation of G-protein-coupled group I (including types 1 and 5) metabotropic glutamate receptors (mGluRs) by the pharmacological agonist (RS)-3,5-dihydroxyphenylglycine (DHPG) elicits LTD in area CA1 of the hippocampus. Recent reports have shown that de novo protein synthesis is necessary for DHPG-induced LTD. However, relatively little is known about the signaling pathways that couple mGluRs to translation initiation. In this study, we investigated whether the activation of the phosphoinositide 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR) pathway, which has been shown to regulate translation initiation, is necessary for mGluR-LTD induced by DHPG. We found that brief incubations of mouse hippocampal slices with DHPG resulted in increased phosphorylation of Akt and mTOR in hippocampal area CA1. Two structurally unrelated PI3K inhibitors, LY294002 and wortmannin, blocked the DHPG-induced increases in phosphorylation of Akt and mTOR. Biochemical fractionation studies showed that the DHPG-induced increase in the phosphorylation of Akt and mTOR could be detected in synaptoneurosome preparations, and immunohistochemical analysis revealed that similar increases could be detected in both stratum pyramidale and stratum radiatum in area CA1. Finally, we observed that both PI3K inhibitors and rapamycin, an mTOR inhibitor, prevented mGluR-LTD induced by DHPG. Together, our findings indicate that activation of the PI3K-Akt-mTOR signaling cascade is required for mGluR-LTD and suggest that this pathway may couple group I mGluRs to translation initiation in hippocampal area CA1.
Keywords: CA1, hippocampus, phosphorylation, protein kinase, translation initiation, synaptic plasticity
Introduction
Long-term depression (LTD) in the hippocampus may provide a cellular substrate for certain forms of learning and memory (Braunewell and Manahan-Vaughan, 2001). Two mechanistically distinct forms of LTD coexist at Schaffer collateral synapses in area CA1 of hippocampus (Oliet et al., 1997), an NMDA receptor-dependent form (Mulkey and Malenka, 1992) and a metabotropic glutamate receptor (mGluR)-dependent form (Bolshakov and Siegelbaum, 1994; Oliet et al., 1997). The mGluR-dependent form of LTD (mGluR-LTD) is particularly interesting, because it requires rapid translation of preexisting dendritic mRNA (Huber et al., 2000).
mGluR-LTD can be selectively induced by (RS)-3,5-dihydroxyphenylglycine (DHPG), an agonist that activates the group I mGluRs, including mGluR1 and mGluR5 (Palmer et al., 1997; Fitzjohn et al., 1999; Schnabel et al., 1999; Huber et al., 2000). Several signaling molecules have been shown to be required for mGluR-LTD in area CA1, including Gq-type G-proteins (Kleppisch et al., 2001), p38 mitogen-activated protein (MAP) kinase (Rush et al., 2002), and tyrosine phosphatases (Moult et al., 2002). However, little is known concerning the signaling pathways that couple mGluRs to translation initiation during mGluR-LTD.
Phosphoinositide (PI) lipid signaling is involved in both synaptic plasticity (Kelly and Lynch, 2000; Daw et al., 2002; Raymond et al., 2002; Sanna et al., 2002; Opazo et al., 2003) and translation initiation. After phosphorylation by phosphoinositide 3-kinase (PI3K), lipids such as phosphatidylinositol-3,4,5-trisphosphate (PIP3) act as second messengers to recruit and activate protein kinases such as phosphoinositide-dependent kinase 1 (PDK1) and Akt (Franke et al., 1997; Chan et al., 1999). Akt affects numerous downstream targets, including proteins that regulate translation. These include a protein kinase termed mammalian target of rapamycin (mTOR) and its downstream targets S6 kinase (S6K) (Dufner and Thomas, 1999) and the eukaryotic initiation factor 4E (eIF4E)-binding protein (4E-BP) (Sonenberg and Gingras, 1998; Raught and Gingras, 1999). S6K is activated via mTOR-dependent phosphorylation and can phosphorylate ribosomal protein S6, an event that results in enhanced translation of 5′ terminal oligopyrimidine tract-containing (5′ TOP) mRNAs (Terada et al., 1995; Jefferies et al., 1997). The mTOR-dependent phosphorylation of 4E-BP (Brunn et al., 1997b) causes dissociation of 4E-BP from eIF4E (Lin et al., 1994; Pause et al., 1994). This dissociation permits eIF4E to recruit an initiation complex of multiple translation factors to the mRNA cap structure m7GpppN, an essential step necessary for translation initiation (Lawrence and Abraham, 1997). Thus, mTOR is a protein kinase that is critical for regulation of translation initiation.
In this study, we investigated the role of the PI3K-Akt-mTOR signaling pathway in mGluR-LTD in area CA1 of mouse hippocampal slices using biochemical, immunohistochemical, and electrophysiological approaches. We found that treatment of hippocampal slices with DHPG increases the phosphorylation of PDK1, Akt, and mTOR in a PI3K-dependent manner in area CA1. We also found that inhibition of either PI3K or mTOR resulted in the blockade of mGluR-LTD induced by DHPG. Together, our findings indicate that activation of the PI3K-Akt-mTOR signaling pathway is required for mGluR-LTD in hippocampal area CA1 and suggest that this cascade couples mGluRs to the protein translation machinery during mGluR-LTD.
Materials and Methods
Materials. All primary antibodies used were purchased from Cell Signaling Technology (Beverly, MA). The horseradish peroxidase-linked goat anti-rabbit IgG was obtained from Promega (Madison, WI). Indocarbocyanine (Cy3)-conjugated AffiniPure goat anti-rabbit IgG was purchased from Jackson ImmunoResearch (West Grove, PA). DHPG, 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and LY367385 were obtained from Tocris Cookson (Ellisville, MO). LY294002, LY353011, and wortmannin were purchased from Sigma (St. Louis, MO). Rapamycin was obtained from Cell Signaling Technology. Ascomycin was purchased from Calbiochem (San Diego, CA). Enhanced chemiluminescence (ECL) Western blotting detection reagents were obtained from Amersham Biosciences (Piscataway, NJ).
Hippocampal slice preparations. Hippocampal slices from male C57BL/6 mice 6-8 weeks of age were removed, and 400 μm slices were prepared. Slices were placed in saline solution containing (in mm) 124 NaCl, 4.4 KCl, 26 NaHCO3, 10 d-glucose, 2 CaCl2, and 2 MgCl2, gassed with 95% O2/5% CO2, pH 7.4, for 1 hr at room temperature and then transferred to a 32°C artificial CSF (ACSF) containing (in mm) 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 2 CaCl2, 1 MgCl2, 25 d-glucose saturated 95% O2/5% CO2, pH 7.4, for 1 hr. Slices then were exposed to different compounds of interest for the indicated times and snap frozen over dry ice. The CA1 regions were microdissected and sonicated in ice-cold homogenization buffer (HB) containing phosphatase and protease inhibitors (200 nm calyculin, 10 μg/ml leupeptin, 2 μg/ml aprotinin, 1 mm sodium orthovanadate, and 1 μm microcystin-LR). Synaptoneurosome fractions were prepared as described previously (Johnson et al., 1997) by passing the sonicate through membranes of decreasing pore size (100 to 5 μm). The final filtrate was centrifuged at 10,000 × g (20 min; 4°C), and the pellet containing the synaptoneurosomes was resuspended in HB. The protein concentration was measured by the method of Bradford (1976) using bovine serum albumin as the standard.
Western blot analysis. Equivalent amounts of protein for each sample were resolved in 10% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The PVDF membranes were blocked in 5% nonfat dry milk for 1 hr in Tris-buffered saline containing Tween 20 and then incubated with the phospho-specific antibody of interest [phospho-Akt (Ser473) antibody, 1:1000; phospho-mTOR (Ser2448) antibody, 1:1000; phospho-PDK1 (Ser 241) antibody, 1:2000] for 1 hr at room temperature followed by incubation with horseradish peroxidase-linked goat anti-rabbit IgG (1:2500 dilution) and developed using ECL. The blots then were incubated in stripping buffer (62 mm Tris-HCl, pH 6.8, 2% SDS, and 100 mm β-mercaptoethanol) for 1 hr at 55-60°C followed by incubation in Tris-buffered saline with Tween 20 for 30 min. The stripped blots were incubated with an antibody directed against total levels of the respective protein (Akt antibody, 1:1000; mTOR antibody, 1:1000; PDK1 antibody, 1:2000). Densitometric analysis of phospho-immunoreactivity and total immunoreactivity for each protein was conducted using Scion image software (Scion, Frederick, MD). Phosphorylated immunoreactivity was normalized to total immunoreactivity for each kinase. The statistical analysis described in the figure legends was performed on non-normalized data.
Immunohistochemistry. Control slices and slices treated with either DHPG or DHPG plus LY294002 were immediately put in ice-cold 4% paraformaldehyde/0.1% glutaraldehyde in PBS, pH 7.4, and fixed overnight. The slices then were put in 30% sucrose overnight at 4°C and embedded with optimal cutting temperature compound. The slices were sectioned into 20 μm sections using a sliding microtome. Free-floating sections were blocked with 10% normal goat serum in PBS/0.7% Triton X-100 (PBS-TX) overnight at 4°C. Sections were then incubated overnight at 4°C with primary antibodies [phospho-Akt antibody (ser 473), 1:100; phospho-mTOR antibody (ser 2448), 1:50; MAP-2 antibody, 1:100]. After washing three times with PBS-TX, sections were incubated for 2 hr at room temperature with Cy3-conjugated AffiniPure goat anti-rabbit IgG and FITC-conjugated goat anti-mouse IgG and diluted 1:500 in blocking solution. Sections were then washed and mounted onto poly-l-lysine-coated slides. Sections were analyzed and imaged using a Zeiss LSM 510 meta confocal microscope system (Zeiss, Oberkochen, Germany).
Electrophysiological recordings. Extracellular recordings were obtained from area CA1 of C57/BL6 mouse hippocampal slices. The 400-μm-thick slices were prepared as described previously. The slices were placed in an interface-recording chamber and equilibrated with oxygenated ACSF at a flow rate of 1 ml/min at 30°C for at least 1 hr before recording. Extracellular recordings of field EPSPs (fEPSPs) were obtained from the stratum radiatum using microelectrodes filled with ACSF (resistance, 1-4 MΩ). A bipolar Teflon-coated platinum electrode was placed in stratum radiatum to activate Schaffer collateral-commissural afferents at 0.05 Hz. The stimulation strength was set to elicit a response equivalent to 50% of the maximal fEPSPs. In all experiments, baseline synaptic transmission was monitored for a minimum of 10 min before drug administration. The slope of the fEPSP was expressed as a percentage of the baseline average before drug application. Normalized data were averaged and expressed as the mean ± SEM. Significant differences between groups were determined using either an independent Student's t test or ANOVA performed on a 10 min average taken 50 min after DHPG application.
Results
Activation of group I mGluRs with DHPG increases the phosphorylation of PDK1, Akt, and mTOR in hippocampal area CA1
To examine whether activation of group I mGluRs could trigger the activation of the PI3K-Akt-mTOR pathway in area CA1, we first used phospho-specific antibodies to measure the relative levels of the phosphorylated, active forms of PDK1 and Akt after hippocampal slices were treated with DHPG. For PDK1, we used an antibody specific for phosphorylated serine 241, which is on the activation loop of PDK1 and is necessary for kinase activity (Casamayor et al., 1999). For Akt, we used an antibody specific for phosphorylated serine 473, which is essential for maximal activation of Akt (Alessi et al., 1996). Incubation of hippocampal slices with 50 μm DHPG for 5 min increased the phosphorylation of PDK1 and Akt as measured on Western blots of homogenates from area CA1 (Fig. 1A,B). The DHPG-induced increase in the phosphorylation of PDK1 and Akt was transient, returning to control levels 20 min after washout of DHPG (Fig. 1A,B). PI3K and Akt can phosphorylate and activate mTOR (Gingras et al., 1998; Nave et al., 1999; Sekulic et al., 2000), a protein kinase that has been shown to regulate translation initiation (Gingras et al., 2001). Therefore, we also examined whether activation of group I mGluRs with DHPG could result in an increase in the levels of phosphorylated, active mTOR in area CA1. We used an antibody specific for phosphorylated serine 2448, which has been reported to be important in the control of mTOR activity (Brunn et al., 1997a; Scott et al., 1998). Incubation of hippocampal slices with DHPG for 5 min resulted in an increase in the phosphorylation of mTOR (Fig. 1C,D). The DHPG-induced increase in the phosphorylation of mTOR was transient, returning to control levels by 5 min after washout of DHPG (Fig. 1C,D). Interestingly, the DHPG-induced increase in mTOR phosphorylation was more transient than the DHPG-induced increases in PDK1 and Akt phosphorylation, suggesting that DHPG may also activate an mTOR-sensitive protein phosphatase. Together, these findings indicate that activation of group I mGluRs with DHPG is associated with transient increases in the phosphorylation of PDK1, Akt, and mTOR in hippocampal area CA1.
Figure 1.

DHPG induces increases in the phosphorylation of PDK1, Akt, and mTOR in hippocampal area CA1. Hippocampal slices were treated with DHPG (50 μm) for 5 min. Slices then were incubated with ACSF in the absence of DHPG for the indicated times, and the level of phosphorylation in PDK1, Akt, and mTOR was assessed via Western blot analysis. A, Representative Western blots for phoshorylated PDK1 (p-PDK1), total PDK1, phosphorylated Akt (p-Akt), and total Akt. Numbers indicate molecular weight markers. Con, Control. B, p-PDK1 and p-Akt immunoreactivity were normalized to total PDK1 and total Akt immunoreactivity. Values are means ± SEM. p-PDK1 values: 0 min, 154 ± 20% of control, n = 6; 5 min, 210 ± 57% of control, n = 4; 10 min, 161 ± 18% of control, n = 4; 20 min, 120 ± 11% of control, n = 4. p-Akt values: 0 min, 146 ± 19% of control, n = 13; 5 min, 199 ± 52% of control, n = 4; 10 min, 152 ± 28% of control, n = 4; 20 min, 90 ± 8% of control, n = 4. C, Representative Western blots for phosphorylated mTOR (p-mTOR) and total mTOR. Numbers indicate molecular weight markers. D, p-mTOR immunoreactivity was normalized to total mTOR immunoreactivity. Values are means ± SEM. p-mTOR values: 0 min, 149 ± 19% of control, n = 10; 5 min, 92 ± 25% of control, n = 4; 10 min, 108 ± 22% of control, n = 4; 20 min, 104 ± 36% of control, n = 4. * denotes statistical significance compared with control with a Student's t test (p < 0.05).
DHPG-induced increases in PDK1, Akt, and mTOR phosphorylation are PI3K dependent
It is well known that PDK1, Akt, and mTOR are downstream effectors of PI3K. Therefore, we determined whether either LY294002 or wortmannin, two structurally distinct PI3K inhibitors, could block the DHPG-induced increases in the phosphorylation in PDK1, Akt, and mTOR. We found that both LY294002 (50 μm) and wortmannin (100 nm) completely blocked the increases in PDK1, Akt, and mTOR phosphorylation induced by DHPG (Fig. 2). Neither LY294002 nor wortmannin alone altered the basal levels of PDK1, Akt, and mTOR phosphorylation (data not shown). These results indicate that activation of PI3K is required for the activation of PDK1, Akt, and mTOR triggered by activation of group I mGluRs in hippocampal area CA1.
Figure 2.

DHPG-induced increases in the phosphorylation of PDK1, Akt, and mTOR are PI3K dependent. Hippocampal slices were preincubated with either normal ACSF, LY294002 (50 μm), or wortmannin (100 nm) for 30 min and then exposed to DHPG (50 μm) for 5 min in the presence of either normal ACSF or the inhibitors. A, Representative Western blots for phosphorylated PDK1 (p-PDK1), total PDK1, phosphorylated Akt (p-Akt), total Akt, phosphorylated mTOR (p-mTOR), and total mTOR. B, Phosphorylated kinase immunoreactivity was normalized to total kinase immunoreactivity for each kinase. Values are means ± SEM. p-PDK1 values: DHPG, 154 ± 20% of control, n = 6; DHPG plus LY294002, 91 ± 6% of control, n = 3; DHPG plus wortmannin, 103 ± 11% of control, n = 3. p-Akt values: DHPG, 146 ± 19% of control, n = 13; DHPG plus LY294002, 98 ± 18% of control, n = 6; DHPG plus wortmannin, 57 ± 8% of control, n = 6. p-mTOR values: DHPG, 149 ± 20% of control, n = 10; DHPG plus LY294002, 88 ± 27% of control, n = 6; DHPG plus wortmannin, 76 ± 13% of control, n = 6. * denotes statistical significance compared with control with a Student's t test (p < 0.05).
The effects of mGluR1 and mGluR5 antagonists on DHPG-induced increases in Akt and mTOR phosphorylation
Group I mGluRs include the mGluR1 and mGluR5 subtypes (Anwyl, 1999), both of which are activated by DHPG. We were interested in determining the relative contributions of these two group I mGluR subtypes in the DHPG-induced increases in the phosphorylation of Akt and mTOR. To address this issue, we determined whether either LY367385 (100 μm), an mGluR1 antogonist, or MPEP (10 μm), an mGluR5 antagonist, could block the DHPG-induced increases in the phosphorylation of Akt and mTOR. Incubation of hippocampal slices with either LY367385 or MPEP alone significantly reduced the DHPG-induced increase in the phosphorylation of Akt and mTOR (Fig. 3A,B). Incubation of the slices with LY367385 and MPEP together completely blocked the DHPG-induced increase in the phosphorylation of Akt and mTOR (Fig. 3A,B). Incubation of slices with either LY367385 or MPEP alone did not alter basal levels of phosphorylated Akt and mTOR (data not shown). In addition, incubation of slices with 50 μm 2-amino-5-phosphonopentanoic acid, an NMDA receptor antagonist, did not affect the DHPG-induced increases in the phosphorylation of Akt and mTOR (data not shown). Overall, these results suggest that the activation of both mGluR1 and mGluR5 subtypes of group I mGluRs are required for the DHPG-induced increase in the phosphorylation of Akt and mTOR in hippocampal area CA1.
Figure 3.
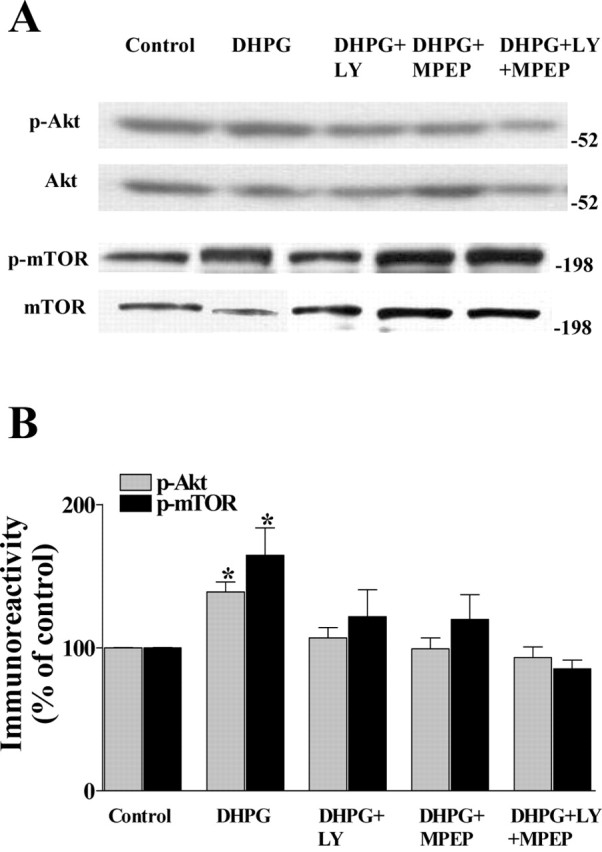
The effects of mGluR1 and mGluR5 antagonists on DHPG-induced increases in Akt and mTOR phosphorylation. Hippocampal slices were preincubated with either normal ACSF, LY367385 (LY; 100 μm), a selective mGluR1 antagonist, MPEP (10 μm), a selective mGluR5 antagonist, or LY367385 plus MPEP for 30 min and then exposed to DHPG (50 μm) for 5 min in the presence of either normal ACSF or the antagonists. A, Representative Western blots for phosphorylated Akt (p-Akt), total Akt, phosphorylated mTOR (p-mTOR), and total mTOR. B, Phosphorylated kinase immunoreactivity was normalized to total kinase immunoreactivity for each kinase. Values are means ± SEM. p-Akt values: DHPG, 137 ± 10% of control, n = 12; DHPG plus LY367385, 105 ± 5% of control, n = 9; DHPG plus MPEP, 99 ± 6% of control, n = 9; DHPG plus LY367385 plus MPEP, 93 ± 7% of control. p-mTOR values: DHPG, 164 ± 19% of control, n = 13; DHPG plus LY367385, 121 ± 19% of control, n = 12; DHPG plus MPEP, 119 ± 17% of control, n = 12; DHPG plus LY367385 plus MPEP, 85 ± 6% of control. * denotes statistical significance compared with control with a Student's t test (p < 0.05).
DHPG-induced increases in the phosphorylation of Akt and mTOR in synaptoneurosomes from hippocampal area CA1
To determine whether the DHPG-induced increases in Akt and mTOR phosphorylation occurred in specific subcellular compartments, we treated hippocampal slices with DHPG, isolated synaptoneurosomes from area CA1, and probed the synaptoneurosome preparations for phosphorylated Akt and phosphorylated mTOR. We observed that slices treated with DHPG significantly increased the phosphorylation of both Akt and mTOR in synaptoneurosome preparations (Fig. 4). Similar to our findings with hippocampal homogenates, we found that the PI3K inhibitor LY294002 blocked DHPG-induced increases in phosphorylation of Akt and mTOR in synaptoneurosomes (Fig. 4). Together, these data indicate that group I mGluR activation can activate the PI3K-Akt-mTOR pathway in synaptic compartments, consistent with the idea that this signaling pathway is involved in mGluR-LTD.
Figure 4.

DHPG-induced increases in the phosphorylation of Akt and mTOR in hippocampal synaptoneurosomes are PI3K dependent. Hippocampal slices were preincubated with either normal ACSF or LY294002 (50 μm) for 30 min and then exposed to DHPG (50 μm) for 5 min in the presence of either normal ACSF or the inhibitor. After the exposure to DHPG, the slices were frozen, and synaptoneurosomes were prepared as described in Materials and Methods. A, Representative Western blots for phosphorylated Akt (p-Akt), total Akt, phosphorylated mTOR (p-mTOR), and total mTOR. B, Phosphorylated kinase immunoreactivity was normalized to total kinase immunoreactivity for each kinase. Values are means ± SEM. p-Akt values: DHPG, 140 ± 11% of control, n = 3; DHPG plus LY294002, 81 ± 17% of control, n = 3. p-mTOR values: DHPG, 123 ± 6% of control, n = 3; DHPG plus LY294002, 80 ± 9% of control, n = 3. * denotes statistical significance compared with control with a Student's t test (p < 0.05).
DHPG-induced increases the phosphorylation of Akt and mTOR in both the soma and dendrites of pyramidal neurons in hippocampal area CA1
To further investigate the subcellular localization of phosphorylated Akt and phosphorylated mTOR in hippocampal slices after DHPG stimulation, we used immunohistochemical techniques coupled with confocal microscopy. Treatment of hippocampal slices with DHPG dramatically increased phospho-Akt and phospho-mTOR immunoreactivity in both the soma of the stratum pyramidale and dendritic processes of stratum radiatum in area CA1 (Fig. 5). Preincubation of the slices with the PI3K inhibitor LY294002 significantly reduced the DHPG-induced increases in phosphorylation of Akt and mTOR in cell bodies and dendrites of pyramidal cells (Fig. 5). These results indicate that group I mGluR activation triggers the activation of the PI3K-Akt-mTOR signaling pathway in both somatic and dendritic compartments in pyramidal cells in hippocampal area CA1.
Figure 5.

DHPG-induced increases in the phosphorylation of Akt and mTOR are present in both soma and dendrites of pyramidal cells in hippocampal area CA1. Representative confocal images were obtained from slices that were double labeled using antibodies specific for phospho-Akt, phospho-mTOR, and the dendritic marker MAP2. Phosphorylated Akt (p-Akt) and phosphorylated mTOR (p-mTOR) labeling is indicated by red, MAP2 labeling is indicated by green, and dual labeling is indicated by yellow. A, D, Sham-treated slices. B, E, Slices treated with 50 μm DHPG for 5 min. C, F, Slices that were preincubated with 50 μm LY294002 for 30 min and then incubated with 50 μm DHPG for 5 min in the presence of the inhibitor. Stratum pyramidale (s.p.) and stratum radiatum (s.r.) are indicated. Images were obtained with a 20× objective (numerical aperture, 0.75) at zoom 2.0.
DHPG-induced mGluR-LTD in hippocampal area CA1 is dependent on PI3K
To examine whether PI3K activity was required for mGluR-LTD, we induced mGluR-LTD by incubating hippocampal slices with DHPG (50 μm) for 5 min in the presence of either LY294002 or wortmannin. In agreement with previous studies, DHPG caused an acute depression of synaptic transmission that was followed after washout of DHPG by a long-lasting depression of synaptic transmission (Fig. 6). Treatment of the slices with either LY294002 (50 μm) or wortmannin (100 nm) had no effect on either baseline synaptic transmission or the acute depression induced by DHPG (Fig. 6). In contrast, both LY294002 (Fig. 6A,B) and wortmannin (Fig. 6C,D) blocked mGluR-LTD. Treatment of the slices with LY303511 (50 μm), an inactive structural analog of LY294002, had no effect on mGluR-LTD induced by DHPG (Fig. 6E,F). Together with our biochemical and immunohistochemical findings, these data suggest that activation of the PI3K signaling pathway is required for mGluR-LTD.
Figure 6.

DHPG-induced mGluR-LTD is dependent on PI3K in hippocampal area CA1. A-F, mGluR-LTD was induced by incubation of slices with DHPG (50 μm) for 5 min in the presence of either vehicle, LY294002 (A, B), wortmannin (C, D), or LY303511 (E, F). Either the vehicle or the inhibitors were present in the perfusing solution 20 min before and during the treatment with DHPG, as indicated by the bars by each panel. A, Plotted are ensemble averages for the initial slope of the fEPSP for 30 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or LY294002 (50 μm). LTD was significantly inhibited by LY294002 as determined by a Student's t test (p < 0.01) at the final time point (vehicle: closed squares; fEPSP slope, 76 ± 4% of baseline; n = 8; LY294002: open squares; fEPSP slope, 98 ± 4% of baseline; n = 7). B, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or LY294002. C, Plotted are ensemble averages for the initial slope of the fEPSP for 30 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or wortmannin (100 nm). LTD was significantly inhibited by wortmannin as determined by a Student's t test (p < 0.05) at the final time point (vehicle: closed squares; fEPSP slope, 76 ± 2% of baseline; n = 8; wortmannin: open squares; fEPSP slope, 90 ± 6% of baseline; n = 8). D, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or wortmannin. E, Plotted are ensemble averages for the initial slope of the fEPSP for 30 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or LY303511 (50 μm). LY303511 had no effect on LTD at the 50 min time point (vehicle: closed squares; fEPSP slope, 75 ± 5% of baseline; n = 8; LY303511: open squares; fEPSP slope, 68 ± 10% of baseline; n = 5; p > 0.05 with a Student's t test). F, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or LY303511. Calibration: (B, D, F) 5 mV, 2 msec.
DHPG-induced mGluR-LTD in hippocampal area CA1 is dependent on mTOR
Previous studies have shown that mGluR activation can induce synaptic protein synthesis (Weiler and Greenough, 1993; Angenstein et al., 1998) and that mGluR-LTD requires de novo protein synthesis (Huber et al., 2000). It is well known that mTOR is a regulator of several aspects of translation initiation. To determine whether mTOR is required for mGluR-LTD, we treated hippocampal slices with DHPG in the presence of the mTOR inhibitor rapamycin. Treatment of hippocampal slices with rapamycin (20 nm) abolished mGluR-LTD induced by DHPG (Fig. 7A,B). The effect of rapamycin was specific to mGluR-LTD, because rapamycin altered neither baseline synaptic transmission nor the acute depression induced by DHPG (Fig. 7A). It has been shown that rapamycin forms a complex with the immunophilin FK506-binding protein 12 (FKBP12), which then inhibits the protein kinase activity of mTOR (Brown et al., 1995; Sabers et al., 1995). To confirm the specificity of the action of rapamycin on mTOR, we used ascomycin, an analog of FK506, which binds to FKBP12 but does not inhibit mTOR activity (Petros et al., 1992; Kawai et al., 1993). Preincubation of slices with ascomycin (20 nm) did not affect DHPG-induced mGluR-LTD (Fig. 7C,D). Together with the results and our biochemical and immunohistochemical studies, these results indicate that activation of mTOR is required for mGluR-LTD induced by DHPG in hippocampal area CA1.
Figure 7.

DHPG-induced mGluR-LTD is dependent on mTOR in hippocampal area CA1. A-D, mGluR-LTD was induced by incubation of slices with DHPG (50 μm) for 5 min in the presence of either vehicle, rapamycin (A, B), or ascomycin (C, D). Either vehicle or the inhibitors were present in the perfusing solution 10 min before, during, and 10 min after the treatment with DHPG, as indicated by the bars in each panel. A, Plotted are ensemble averages for the initial slope of the fEPSP for 20 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or rapamycin (20 nm). LTD was significantly inhibited by rapamycin as determined with a Student's t test (p < 0.0001) at the final time point (vehicle: closed squares; fEPSP slope, 76 ± 3% of baseline; n = 12; rapamycin: open squares; fEPSP slope, 92 ± 1% of baseline; n = 8). B, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or rapamycin. C, Plotted are ensemble averages for the initial slope of the fEPSP for 20 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or ascomycin (20 nm). Ascomycin had no effect on LTD at the 50 min time point (vehicle: closed squares; fEPSP slope, 75 ± 4% of baseline; n = 8; ascomycin: open squares; fEPSP slope, 79 ± 6% of baseline; n = 4; p > 0.05 with a Student's t test). F, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or ascomycin. Calibration: (B, D) 5 mV, 2 msec.
The role of mGluR1 and mGluR5 in DHPG-induced LTD in hippocampal area CA1
In a previous study of mGluR-LTD in rat hippocampal slices, it was shown that mGluR1 was required for the acute depression and that mGluR5 was required for the long-lasting depression induced by DHPG (Faas et al., 2002). Because we found that both LY367385, an mGluR1 antagonist, and MPEP, an mGluR5 antagonist, blocked DHPG-induced increases in the phosphorylation of Akt and mTOR (Fig. 3), we examined the contributions of mGluR1 and mGluR5 to mGluR-LTD in mouse hippocampal slices. We found that MPEP (10 μm) significantly attenuated mGluR-LTD but did not reduce the acute depression-induced DHPG (Fig. 8A,B), a finding consistent with the previous study in rat hippocampal slices (Faas et al., 2002). In contrast, LY367385 (100 μm) significantly attenuated the acute depression induced by DHPG, but alone did not alter mGluR-LTD (Fig. 8C,D). Treatment of hippocampal slices with both MPEP and LY367385 resulted in an attenuation of the acute depression and a complete blockade of mGluR-LTD induced by DHPG (Fig. 8E,F). Our findings indicate that in mouse hippocampal area CA1, both mGluR1 and mGluR5 activation are essential for the full expression of mGluR-LTD, whereas only mGluR1 activation is involved in the acute depression caused by DHPG.
Figure 8.

The role of mGluR1 and mGluR5 in DHPG-induced LTD in mouse hippocampal area CA1. A-F, mGluR-LTD was induced by incubation of slices with DHPG (50 μm) for 5 min in the presence of either vehicle, MPEP (A, B), LY367385 (C, D), or LY367385 plus MPEP (E, F). Either vehicle or the antagonists were present in the perfusing solution 20 min before and during the treatment with DHPG, as indicated by the bars in each panel. A, Plotted are ensemble averages for the initial slope of the fEPSP for 30 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or MPEP (10 μm). LTD was significantly inhibited by MPEP as determined with a Student's t test (p < 0.01) at the final time point (vehicle: closed squares; fEPSP slope, 74 ± 2% of baseline; n = 14; MPEP: open squares; fEPSP slope, 89 ± 3% of baseline; n = 8). B, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or MPEP. C, Plotted are ensemble averages for the initial slope of the fEPSP for 30 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or LY367385 (100 μm). LTD was not significantly inhibited by LY367385 as determined with a Student's t test (p > 0.05) at the final time point (vehicle: closed squares; fEPSP slope, 76 ± 4% of baseline; n = 8; wortmannin: open squares; fEPSP slope, 79 ± 11% of baseline; n = 14). D, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or LY367385. E, Plotted are ensemble averages for the initial slope of the fEPSP for 30 min before, during, and 50 min after treatment of DHPG in the presence of either vehicle or LY367385 plus MPEP. LTD was significantly inhibited by LY367385 plus MPEP as determined with a Student's t test (p < 0.001) at the final time point (vehicle: closed squares; fEPSP slope, 74 ± 2% of baseline; n = 14; LY367385 plus MPEP: open squares; fEPSP slope, 98 ± 2% of baseline; n = 5). F, Representative fEPSPs before and 50 min after treatment with DHPG in the presence of either vehicle or LY367385 plus MPEP. Calibration: (B, D, F) 5 mV, 2 msec.
Discussion
The PI3K-Akt-mTOR signaling pathway is required for mGluR-LTD and may couple group I mGluRs to translation initiation in hippocampal area CA1
Previous studies have shown that activation of mGluRs increases synaptic protein synthesis (Weiler and Greenough, 1993; Angenstein et al., 1998) and that DHPG-induced mGluR-LTD is dependent on dendritic protein synthesis (Huber et al., 2000). However, the signal transduction pathways that couple group I mGluR to translation initiation is poorly understood. In this report, we have shown that activation of group I mGluRs with DHPG can trigger the activation of the PI3K-Akt-mTOR signaling pathway in mouse hippocampal area CA1. We detected the activation of this pathway in whole homogenates and synaptoneurosomes as well as in the dendrites of hippocampal area CA1. The activation of this pathway is required for mGluR-LTD because two structurally unrelated PI3K inhibitors, LY294002 and wortmannin, as well as an mTOR inhibitor, rapamycin, blocked the long-lasting depression induced by DHPG. These findings strongly suggest that the PI3K-Akt-mTOR pathway is a signaling pathway that couples group I mGluRs to the translation initiation in hippocampal area CA1.
mTOR is required for protein synthesis-dependent forms of synaptic plasticity
One way that the activation of mTOR can contribute to translation initiation is by phosphorylating 4E-BP, which is bound to eIF4E. The phosphorylated 4E-BP is replaced by eIF4G, a scaffolding translation factor that assembles several critical components of the cap-dependent translation initiation complex on the mRNA via its interaction with cap-bound eIF4E (Lin and Lawrence, 1996; Brunn et al., 1997b; Gingras et al., 1998, 1999). It has been shown that mGluR-LTD is blocked by postsynaptic injection of the cap analog m7GpppG (Huber et al., 2000), consistent with the possibility that mTOR phosphorylates 4E-BP during mGluR-LTD. mTOR can also regulate translation via either direct or indirect phosphorylation and activation of S6K (Gingras et al., 2001). S6K then phosphorylates ribosomal protein S6 (Fumagalli and Thomas, 2000), which can result in enhanced translation of 5′TOP mRNAs that encode numerous components of the translation machinery. It remains to be determined whether mTOR is required for the phosphorylation of either 4E-BP, to regulate cap-dependent translation, or S6K, to regulate S6-directed translation during mGluR-LTD.
Several forms of protein synthesis-dependent synaptic plasticity have been shown to require mTOR. In Aplysia sensory neurons, rapamycin inhibits stabilization of long-term facilitation (LTF) induced by serotonin (Casadio et al., 1999). Similarly, it was shown that rapamycin inhibits LTF at the crayfish neuro-muscular junction (Beaumont et al., 2001). More recently, it was shown that late-phase long-term potentiation (L-LTP), potentiation induced by brain-derived neurotrophic factor (BDNF), and DHPG-induced depotentiation of LTP in the hippocampus are blocked by rapamycin (Tang et al., 2002; Zho et al., 2002). In this study, we found that rapamycin blocked mGluR-LTD (Fig. 7A,B). Thus, mGluR-LTD induced by DHPG should be added to the list of protein synthesis-dependent forms of synaptic plasticity that require mTOR activity.
The upstream signaling pathways that trigger activation of mTOR during protein synthesis-dependent forms of synaptic plasticity, including mGluR-LTD, are not well understood but likely involve PI3K and Akt (Gingras et al., 1998). Inhibitors of PI3K have been shown to block crayfish LTF (Beaumont et al., 2001) and various forms of LTP (Kelly and Lynch, 2000; Sanna et al., 2002; Opazo et al., 2003). In addition, BDNF-dependent phosphorylation and activation of mTOR in cultured neurons is blocked by PI3K inhibitors (Takei et al., 2001). We found that inhibition of PI3K blocked the activation of both Akt and mTOR induced by activation of group I mGluRs by DHPG (Figs. 2, 5). The precise delineation of the signaling cascades that couple glutamate, serotonin, and neurotrophin receptors to activation of mTOR via PI3K and Akt during synaptic plasticity is an important issue that remains to be addressed. With respect to group I mGluRs, recently a cytoplasmic PI3K enhancer (PIKE) termed PIKE-L has been reported to couple group I mGluRs to PI3K via Homer (Rong et al., 2003). Whether PIKE-L is required for the activation of PI3K during mGluR-LTD is a possibility that remains to be examined.
Localization of Akt and mTOR activation during mGluR-LTD
The locus of mGluR-LTD expression is unclear. Several studies have reported that mGluR-LTD is associated with increases in paired-pulse facilitation (Fitzjohn et al., 2001; Faas et al., 2002) and changes in miniature EPSC frequency (Oliet et al., 1997; Fitzjohn et al., 2001), suggesting that the expression of mGluR-LTD is preynaptic. In addition, it was reported that the presynaptic vesicle release and cycling are altered during mGluR-LTD (Zakharenko et al., 2002). However, these findings do not eliminate the possibility of a postsynaptic contribution to mGluR-LTD. For example, it has been shown that mGluR-LTD is dependent on postsynaptic protein synthesis (Huber et al., 2000, 2001). In this study, we have shown that activation of group I mGluRs triggers the PI3K-Akt-mTOR signaling cascade and that increases in Akt and mTOR phosphorylation occur in dendrites in area CA1 (Fig. 5). Although we cannot exclude the possibility of an additional presynaptic locus, our findings are consistent with the idea that the PI3K-Akt-mTOR signaling cascade couples group I mGluRs to translation initiation to enhance dendritic protein synthesis that is required for the expression of mGluR-LTD.
mGluR1 and mGluR5 are differentially involved in DHPG-induced mGluR-LTD in mouse hippocampal area CA1
We found that the mGluR5 antagonist MPEP attenuated DHPG-induced mGluR-LTD (Fig. 8A,B). In contrast, the mGluR1 antagonist LY367385 suppressed acute depression of transmission induced by DHPG but did not block DHPG-induced mGluR-LTD (Fig. 8C,D). Consistent with our findings, it has been shown that DHPG-induced mGluR-LTD is absent in mGluR5 mutant mice (Huber et al., 2001). In addition, it has been shown that MPEP blocked DHPG-induced mGluR-LTD in rat hippocampal slices (Faas et al., 2002), whereas the mGluR1 antagonist LY367385 reduced the acute depression induced by DHPG, but it did not reduce mGluR-LTD (Fitzjohn et al., 1999; Fass et al., 2002). Previous studies have demonstrated that mGluR5 and mGluR1 have different distribution in CA1 pyramidal cells, with mGluR5 being the most abundant of the group I mGluRs in this region (Romano et al., 1995; Ferraguti et al., 1998). Interestingly, we found that either MPEP or LY367385 alone reduced the DHPG-induced increases in the phosphorylation of Akt and mTOR; when MPEP and LY367385 together were incubated with slices, the DHPG-induced increases in the phosphorylation of Akt and mTOR were completely blocked (Fig. 3). In addition, we found that incubation of slices with MPEP and LY367385 together completely blocked DHPG-induced mGluR-LTD (Fig. 8E,F). Overall, our findings indicate that both mGluR1 and mGluR5 are required for the maximum activation of Akt and mTOR induced by DHPG and that both mGluR1 and mGluR5 are necessary for the full expression of mGluR-LTD in mouse hippocampal slices.
A possible mechanism to identify newly synthesized proteins during mGluR-LTD
An important question that has not been investigated to date is the identity of newly synthesized dendritic proteins during mGluR-LTD. One likely candidate is the fragile X mental retardation protein (FMRP). FMRP is found predominantly in the cytoplasm where it is thought to dimerize and form a complex with mRNAs, mRNA-binding proteins, FMRP-associated proteins, and ribosomes, tonically inhibiting the translation of the mRNAs in the complex (Jin and Warren, 2003). FMRP and Fmr1 mRNA have been shown to be present in dendrites (Feng et al., 1997; Weiler et al., 1997), and stimulation of mGluRs has been shown to result in a rapid translation of FMRP in synaptoneurosomes (Weiler et al., 1997). Recently, Huber et al. (2002) showed that hippocampal mGluR-LTD is enhanced in slices from Fmr1 knock-out mice. These investigators proposed a model in which mGluR-LTD normally enhances synthesis of FMRP, which serves to limit expression of LTD by inhibiting translation of other synaptic mRNAs, that encode proteins important for synaptic function. Examination of the role of these proteins in mGluR-LTD should provide insight into how specific mRNAs are translated in response to synaptic stimulation that ultimately results in synaptic plasticity.
In conclusion, our findings demonstrate that activation of the PI3K-Akt-mTOR pathway is required for mGluR-LTD, a form of synaptic plasticity that is known to require dendritic protein synthesis. We observed that the DHPG induced a PI3K-dependent activation of both Akt and mTOR in dendrites in hippocampal area CA1, indicating that this signaling pathway could couple group I mGluRs to translation initiation during mGluR-LTD. Determination of downstream effectors of mTOR during mGluR-LTD will be necessary to begin to elucidate the identities of the proteins that are synthesized in dendrites during this type of synaptic plasticity.
Footnotes
This work was supported by National Institutes of Health Grant NS34007 and the FRAXA Research Foundation. We thank Marcia Antion and Jessica Banko for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Eric Klann, Department of Molecular Physiology and Biophysics, Baylor College of Medicine, 1 Baylor Plaza, Houston, TX 77030. E-mail: eklann@bcm.tmc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246352-10$15.00/0
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541-6551. [PMC free article] [PubMed] [Google Scholar]
- Angenstein F, Greenough WT, Weiler IJ (1998) Metabotropic glutamate receptor-initiated translocation of protein kinase p90rsk to polyribosomes: a possible factor regulating synaptic protein synthesis. Proc Natl Acad Sci USA 95: 15078-15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwyl R (1999) Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev 29: 83-120. [DOI] [PubMed] [Google Scholar]
- Beaumont V, Zhong N, Fletcher R, Froemke RC, Zucker RS (2001) Phosphorylation and local presynaptic protein synthesis in calcium- and calcineurin-dependent induction of crayfish long-term facilitation. Neuron 32: 489-501. [DOI] [PubMed] [Google Scholar]
- Bolshakov VY, Siegelbaum SA (1994) Postsynaptic induction and presynaptic expression of hippocampal long-term depression. Science 264: 1148-1152. [DOI] [PubMed] [Google Scholar]
- Bradford MM (1976) Rapid and sensitive method for quantification of microgram quantities of protein using the principle of protein-dye binding. Anal Biochem 72: 248-252. [DOI] [PubMed] [Google Scholar]
- Braunewell KH, Manahan-Vaughan D (2001) Long-term depression: a cellular basis for learning? Rev Neurosci 12: 121-140. [DOI] [PubMed] [Google Scholar]
- Brown EJ, Beal PA, Keith CT, Chen J, Shin TB, Schreiber SL (1995) Control of p70S6 kinase by kinase activity of FRAP in vivo Nature 377: 441-446. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Fadden P, Haystead TA, Lawrence Jr JC (1997a) The mammalian target of rapamycin phosphorylates sites having a (Ser/Thr)-Pro motif and is activated by antibodies to a region near its COOH terminus. J Biol Chem 272: 32547-32550. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Hudson CC, Sekulic A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC, Abraham RT (1997b) Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 227: 99-101. [DOI] [PubMed] [Google Scholar]
- Casadio A, Martin KC, Giustetto M, Zhu H, Chen M, Bartsch D, Bailey CH, Kandel ER (1999) A transient, neuron-wide form of CREB-mediated long-term facilitation can be stabilized at specific synapses by local protein synthesis. Cell 99: 221-237. [DOI] [PubMed] [Google Scholar]
- Casamayor A, Morrice NA, Alessi DR (1999) Phosphorylation of Ser-241 is essential for the activity of 3-phosphoinositide-dependent protein kinase-1: identification of five sites of phosphorylation in vivo Biochem J 342: 287-292. [PMC free article] [PubMed] [Google Scholar]
- Chan TO, Rittenhouse SE, Tsichlis PN (1999) AKT/PKB and other D3 phosphoinositide-regulated kinases: kinase activation by phosphoinositide-dependent phosphorylation. Annu Rev Biochem 68: 965-1014. [DOI] [PubMed] [Google Scholar]
- Daw MI, Bortolotto ZA, Saulle E, Zaman S, Collingridge GL, Isaac JT (2002) Phosphatidylinositol 3 kinase regulates synapse specificity of hippocampal long-term depression. Nat Neurosci 5: 835-836. [DOI] [PubMed] [Google Scholar]
- Dufner A, Thomas G (1999) Ribosomal S6 kinase signaling and the control of translation. Exp Cell Res 253: 100-109. [DOI] [PubMed] [Google Scholar]
- Faas GC, Adwanikar H, Gereau IV RW, Saggau P (2002) Modulation of presynaptic calcium transients by metabotropic glutamate receptor activation: a differential role in acute depression of synaptic transmission and long-term depression. J Neurosci 22: 6885-6890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Gutekunst CA, Eberhart DE, Yi H, Warren ST, Hersch SM (1997) Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J Neurosci 17: 1539-1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraguti F, Conquet F, Corti C, Grandes P, Kuhn R, Knopfel T (1998) Immunohistochemical localization of the mGluR1beta metabotropic glutamate receptor in the adult rodent forebrain: evidence for a differential distribution of mGluR1 splice variants. J Comp Neurol 400: 391-407. [PubMed] [Google Scholar]
- Fitzjohn SM, Kingston AE, Lodge D, Collingridge GL (1999) DHPG-induced LTD in area CA1 of juvenile rat hippocampus; characterisation and sensitivity to novel mGlu receptor antagonists. Neuropharmacology 38: 1577-1583. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Palmer MJ, May JE, Neeson A, Morris SA, Collingridge GL (2001) A characterisation of long-term depression induced by metabotropic glutamate receptor activation in the rat hippocampus in vitro J Physiol (Lond) 537: 421-430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC (1997) PI3K: downstream AKTion blocks apoptosis. Cell 88: 435-437. [DOI] [PubMed] [Google Scholar]
- Fumagalli S, Thomas G (2000) S6 phosphorylation and signal transduction. In: Translational control of gene expression (Sonenberg N, Hershey JWB, Mathews MB, eds), pp 685-717. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Gingras AC, Kennedy SG, O'Leary MA, Sonenberg N, Hay N (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev 12: 502-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N (1999) Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes Dev 13: 1422-1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N (2001) Regulation of translation initiation by FRAP/mTOR. Genes Dev 15: 807-826. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF (2000) Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science 288: 1254-1257. [DOI] [PubMed] [Google Scholar]
- Huber KM, Roder JC, Bear MF (2001) Chemical induction of mGluR5- and protein synthesis-dependent long-term depression in hippocampal area CA1. J Neurophysiol 86: 321-325. [DOI] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF (2002) Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci USA 99: 7746-7750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jefferies HB, Fumagalli S, Dennis PB, Reinhard C, Pearson RB, Thomas G (1997) Rapamycin suppresses 5′TOP mRNA translation through inhibition of p70S6K. EMBO J 16: 3693-3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Warren ST (2003) New insights into fragile X syndrome: from molecules to neurobehaviors. Trends Biochem Sci 28: 152-158. [DOI] [PubMed] [Google Scholar]
- Johnson MW, Chotiner JK, Watson JB (1997) Isolation and characterization of synaptoneurosomes from single rat hippocampal slices. J Neurosci Methods 77: 151-156. [DOI] [PubMed] [Google Scholar]
- Kawai M, Lane BC, Hsieh GC, Mollison KW, Carter GW, Luly JR (1993) Structure-activity profiles of macrolactam immunosuppressant FK-506 analogues. FEBS Lett 316: 107-113. [DOI] [PubMed] [Google Scholar]
- Kelly A, Lynch MA (2000) Long-term potentiation in dentate gyrus of the rat is inhibited by the phosphoinositide 3-kinase inhibitor, wortmannin. Neuropharmacology 39: 643-651. [DOI] [PubMed] [Google Scholar]
- Kleppisch T, Voigt V, Allmann R, Offermanns S (2001) G(alpha)q-deficient mice lack metabotropic glutamate receptor-dependent long-term depression but show normal long-term potentiation in the hippocampal CA1 region. J Neurosci 21: 4943-4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence Jr JC, Abraham RT (1997) PHAS/4E-BPs as regulators of mRNA translation and cell proliferation. Trends Biochem Sci 22: 345-349. [DOI] [PubMed] [Google Scholar]
- Lin TA, Lawrence Jr JC (1996) Control of the translational regulators PHAS-I and PHAS-II by insulin and cAMP in 3T3-L1 adipocytes. J Biol Chem 271: 30199-30204. [DOI] [PubMed] [Google Scholar]
- Lin TA, Kong X, Haystead TA, Pause A, Belsham G, Sonenberg N, Lawrence Jr JC (1994) PHAS-I as a link between mitogen-activated protein kinase and translation initiation. Science 266: 653-656. [DOI] [PubMed] [Google Scholar]
- Moult PR, Schnabel R, Kilpatrick IC, Bashir ZI, Collingridge GL (2002) Tyrosine dephosphorylation underlies DHPG-induced LTD. Neuropharmacology 43: 175-180. [DOI] [PubMed] [Google Scholar]
- Mulkey RM, Malenka RC (1992) Mechanisms underlying induction of homosynaptic long-term depression in area CA1 of the hippocampus. Neuron 9: 967-975. [DOI] [PubMed] [Google Scholar]
- Nave BT, Ouwens DM, Withers DJ, Aless DR, Shepherd PR (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and aminoacid deficiency on protein translation. Biochem J 344: 427-431. [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Malenka RC, Nicoll RA (1997) Two distinct forms of long-term depression coexist in CA1 hippocampal pyramidal cells. Neuron 18: 969-982. [DOI] [PubMed] [Google Scholar]
- Opazo P, Watabe AM, Grant SG, O'Dell TJ (2003) Phosphatidylinositol 3-kinase regulates the induction of long-term potentiation through extracellular signal-related kinase-independent mechanisms. J Neurosci 23: 3679-3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer MJ, Irving AJ, Seabrook GR, Jane DE, Collingridge GL (1997) The group I mGluR receptor agonist DHPG induces a novel form of LTD in the CA1 region of the hippocampus. Neuropharmacology 36: 1517-1532. [DOI] [PubMed] [Google Scholar]
- Pause A, Belsham GJ, Gingras AC, Donze O, Lin TA, Lawrence Jr JC, Sonenberg N (1994) Insulin-dependent stimulation of protein synthesis by phosphorylation of a regulator of 5′-cap function. Nature 371: 762-767. [DOI] [PubMed] [Google Scholar]
- Petros AM, Gemmecker G, Neri P, Olejniczak ET, Nettesheim D, Xu RX, Gubbins EG, Smith H, Fesik SW (1992) NMR studies of an FK-506 analog, [U-13C]ascomycin, bound to FK-506-binding protein. J Med Chem 35: 2467-2473. [DOI] [PubMed] [Google Scholar]
- Raught B, Gingras AC (1999) eIF4E activity is regulated at multiple levels. Int J Biochem Cell Biol 31: 43-57. [DOI] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ, Crouch MF (2002) The phosphoinositide 3-kinase and p70 S6 kinase regulate long-term potentiation in hippocampal neurons. Neuroscience 109: 531-536. [DOI] [PubMed] [Google Scholar]
- Romano C, Sesma MA, McDonald CT, O'Malley K, Van den Pol AN, Olney JW (1995) Distribution of metabotropic glutamate receptor mGluR5 immunoreactivity in rat brain. J Comp Neurol 355: 455-469. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, Tu J, Worley PF, Snyder SH, Ye K (2003) PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci 6: 1153-1161. [DOI] [PubMed] [Google Scholar]
- Rush AM, Wu J, Rowan MJ, Anwyl R (2002) Group I metabotropic glutamate receptor (mGluR)-dependent long-term depression mediated via p38 mitogen-activated protein kinase is inhibited by previous high-frequency stimulation and activation of mGluRs and protein kinase C in the rat dentate gyrus in vitro J Neurosci 22: 6121-6128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT (1995) Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem 270: 815-822. [DOI] [PubMed] [Google Scholar]
- Sanna PP, Cammalleri M, Berton F, Simpson C, Lutjens R, Bloom FE, Francesconi W (2002) Phosphatidylinositol 3-kinase is required for the expression but not for the induction or the maintenance of long-term potentiation in the hippocampal CA1 region. J Neurosci 22: 3359-3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabel R, Kilpatrick IC, Collingridge GL (1999) An investigation into signal transduction mechanisms involved in DHPG-induced LTD in the CA1 region of the hippocampus. Neuropharmacology 38: 1585-1596. [DOI] [PubMed] [Google Scholar]
- Scott PH, Brunn GJ, Kohn AD, Roth RA, Lawrence Jr JC (1998) Evidence of insulin-stimulated phosphorylation and activation of the mammalian target of rapamycin mediated by a protein kinase B signaling pathway. Proc Natl Acad Sci USA 95: 7772-7777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekulic A, Hudson CC, Homme JL, Yin P, Otterness DM, Karnitz LM, Abraham RT (2000) A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res 60: 3504-3513. [PubMed] [Google Scholar]
- Sonenberg N, Gingras AC (1998) The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol 10: 268-275. [DOI] [PubMed] [Google Scholar]
- Takei N, Kawamura M, Hara K, Yonezawa K, Nawa H (2001) Brain-derived neurotrophic factor enhances neuronal translation by activating multiple initiation processes: comparison with the effects of insulin. J Biol Chem 276: 42818-42825. [DOI] [PubMed] [Google Scholar]
- Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci USA 99: 467-472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada N, Takase K, Papst P, Nairn AC, Gelfand EW (1995) Rapamycin inhibits ribosomal protein synthesis and induces G1 prolongation in mitogen-activated T lymphocytes. J Immunol 155: 3418-3426. [PubMed] [Google Scholar]
- Weiler IJ, Greenough WT (1993) Metabotropic glutamate receptors trigger postsynaptic protein synthesis. Proc Natl Acad Sci USA 90: 7168-7171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiler IJ, Irwin SA, Klintsova AY, Spencer CM, Brazelton AD, Miyashiro K, Comery TA, Patel B, Eberwine J, Greenough WT (1997) Fragile X mental retardation protein is translated near synapses in response to neuro-transmitter activation. Proc Natl Acad Sci USA 94: 5395-5400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharenko SS, Zablow L, Siegelbaum SA (2002) Altered presynaptic vesicle release and cycling during mGluR-dependent LTD. Neuron 35: 1099-1110. [DOI] [PubMed] [Google Scholar]
- Zho WM, You JL, Huang CC, Hsu KS (2002) The group I metabotropic glutamate receptor agonist (S)-3,5-dihydroxyphenylglycine induces a novel form of depotentiation in the CA1 region of the hippocampus. J Neurosci 22: 8838-8849. [DOI] [PMC free article] [PubMed] [Google Scholar]