

Abstract
Immunization with amyloid-β (Aβ) 1-42 has been shown to reduce amyloid burden and improve cognition in Alzheimer's disease (AD) model mice. In a human trial, possible cognitive benefit was found but in association with significant toxicity in a minority of patients. We proposed that immunization with nonfibrillogenic Aβ derivatives is much less likely to produce toxicity and have previously shown that one such derivative (K6Aβ1-30) can reduce amyloid burden in mice to a similar extent as Aβ1-42. Here, we immunized AD model mice (Tg2576) with Aβ1-30[E18E19] or with K6Aβ1-30[E18E19]. These peptides were designed to be nontoxic and to produce less T-cell response, which has been linked to toxicity. K6Aβ1-30[E18E19] induced primarily an IgM response, whereas Aβ1-30[E18E19] induced an IgG titer that was lower than previously seen with K6Aβ1-30 or Aβ1-42. However, both treated animal groups performed better than Tg controls in the radial arm maze. Amyloid burden was similar in Aβ1-30[E18E19]-vaccinated mice and their Tg controls, whereas the number of medium and small sized plaques was reduced (29-34%) in K6Aβ1-30[E18E19]-immunized mice compared with Tg controls. Amyloid burden in these mice correlated inversely with plasma IgM levels. The cognitive benefit and amyloid reduction in the K6Aβ1-30[E18E19]-vaccinated mice are likely to be related to peripheral clearance of Aβ, because IgM does not cross the blood-brain barrier because of its large size. Our results indicate that these nontoxic Aβ derivatives produce an attenuated antibody response, which is less likely to be associated with negative side effects while having cognitive benefits.
Keywords: Mamyloid-β, immunization, transgenic mice, behavior, immunoglobulin Ms, Alzheimer
Introduction
Alzheimer's disease (AD) is characterized in part by extensive deposition of amyloid β (Aβ) in the brain. An emerging therapeutic approach for AD is immune modulation to enhance clearance of Aβ, primarily on the basis of a report showing that immunization with fibrillar Aβ1-42 reduces Aβ plaque burden and associated pathology in mouse brains (Schenk et al., 1999). Previous and subsequent studies indicated that this effect was likely to be antibody mediated (Bard et al., 2000) and suggested that the clearance of plaques was attributable to: (1) microglial activation (Bard et al., 2000), (2) direct antibody-mediated disassembly of plaques (Solomon et al., 1997; Bacskai et al., 2002; Das et al., 2003), and (3) drawing of Aβ from the brain via peripheral clearance of soluble Aβ (DeMattos et al., 2001; Sigurdsson et al., 2001; Lemere et al., 2003). After these promising mouse studies, clinical trials were initiated using fibrillar Aβ1-42 along with QS-21 adjuvant, which were subsequently halted because of encephalitis observed in a small subset of patients (Schenk, 2002). These adverse effects may be related to a cytotoxic T-cell response (Schenk, 2002).
Various algorithms can predict antibody binding sites on peptides as well as potential T-cell epitopes. The major antibody epitopes of Aβ are located within the first 30 amino acids (Jameson and Wolf, 1988). Predicted T-cell epitopes depend on the haplotype of the major histocompatibility complex (MHC) but are located throughout the peptide (Singh and Raghava, 2001; Singh and Raghava, 2003). As shown by these computer algorithms, common cytotoxic (MHC I; CD8+) and T-helper (MHC II; CD4+) epitopes include the C terminus and the middle hydrophobic region of Aβ. Given the side effects observed with Aβ1-42 vaccination, we suggest that the future of immune therapy for AD is in using Aβ derivatives that have a less intrinsic neurotoxicity as well as elicit reduced overall T-cell responses. We reported previously that K6Aβ1-30 elicits a similar antibody response as Aβ1-42 in mice, and these antigens have comparable therapeutic efficacy (Schenk et al., 1999; Sigurdsson et al., 2001). We now have characterized other Aβ derivatives that, like K6Aβ1-30, are designed to have no intrinsic toxicity while also containing internal substitutions to diminish the T-cell response.
Materials and Methods
Peptides and other chemicals. K6Aβ1-30-NH2[E18E19], Aβ1-30-NH2[E18E19], K6Aβ1-30-NH2, and Aβ1-42 were synthesized at the Keck Foundation at Yale University, as described previously (Sigurdsson et al., 2001). The Aβ derivatives used for immunizations maintain the two major immunogenic sites of Aβ peptides, which are residues 1-11 and 22-28 of Aβ1-42 based on an antigenic index (Jameson and Wolf, 1988). These peptides are amidated on the C terminus to maintain the immunogenicity of that epitope and are referred to as K6Aβ1-30[E18E19], Aβ1-30[E18E19], and K6Aβ1-30. The glutamate substitutions in positions 18 and 19 reduce the β-sheet content of the peptide, which reduces fibrillogenicity (Hilbich et al., 1992) and neurotoxicity (Pike et al., 1993) and also eliminates major T-cell epitopes (Singh and Raghava, 2001, 2003). The six lysyl residues on the N terminus were added to enhance immunogenicity and further reduce β-sheet content. All other chemicals were from Sigma (St. Louis, MO) unless otherwise stated.
Study of amyloid fibril formation in vitro. Aliquots of the peptides prepared in 0.1 mol/l Tris, pH 7.4, were incubated for different times, and their fibril formation was compared with that of Aβ1-42. In vitro fibrillogenesis was evaluated by a thioflavin T assay, as we described previously (Soto et al., 1998; Sigurdsson et al., 2001). Thioflavin T binds specifically to amyloid. This binding shifts its emission spectrum, producing a fluorescent enhancement proportional to the amount of amyloid formed.
Neurotoxicity. The potential neurotoxicity of K6Aβ1-30[E18E19] and Aβ1-30[E18E19] (10 μmol/l) was evaluated at 6 d in a human neuroblastoma cell line (SK-N-SH) using the MTT assay as described previously (Sigurdsson et al., 2001), with Aβ1-42 as control. Briefly, cells were plated at 10,000 cells per 100 μl of culture medium per well in flat-bottom, 96-well microtiter plates. After cell attachment to the plate overnight in an incubator (37°C; 5.0% CO2), 10 μl of freshly prepared peptide solution (in sterile H2O) was added. Subsequent steps were as described previously (Sigurdsson et al., 2001).
Animals. The vaccination was performed in heterozygous Tg2576 amyloid precursor protein (APP) mouse model (Hsiao et al., 1996). These mice develop Aβ plaques as early as 11-13 months of age. The animals were maintained on a 12 hr light/dark cycle, and animal care was in accordance with institutional guidelines.
Vaccine administration. Aβ1-30[E18E19] and K6Aβ1-30[E18E19] were administered as described previously (Sigurdsson et al., 2001). Briefly, the peptide was dissolved in PBS at a concentration of 2 mg/ml and then mixed 1:1 (v/v) with the adjuvant or PBS. Complete Freund's adjuvant was used for the first injection, incomplete Freund's adjuvant for the next three injections, and PBS from the fifth injection forward. The mice received a subcutaneous injection of 100 μl followed by a second injection 2 weeks later and then monthly thereafter. Vaccination using the Aβ1-30[E18E19] peptide started when the mice were 6-8 months of age, and after 14 immunizations, the mice were killed at 19-21 months of age (n = 6-8 per group). The K6Aβ1-30[E18E19] peptide was first administered when the mice were 11-13 months of age, and after nine immunizations, the mice were killed at 19-21 months of age (n = 14-18 per group). For comparison of IgG/IgM profile, plasma was analyzed from Tg2576 mice immunized with antigens (K6Aβ1-30, Aβ1-42; n = 6 per group) that we and others have shown to result in a robust IgG response (Schenk et al., 1999; Sigurdsson et al., 2001). These control mice were bled at 18 months after eight immunizations over 7 months. As additional controls for immune response and subsequent acid unmasking, wild-type littermates (n = 13) of the Aβ1-30[E18E19] group were bled in addition to wild-type mice (3-4 months of age; n = 6 per group) that were immunized with K6Aβ1-30, K6Aβ1-30[E18E19], or Aβ1-30[E18E19] and bled 6 months later after five injections.
Behavior. Spatial learning was evaluated using an eight-arm radial maze with a food well at the end of each arm. Clear Plexiglas guillotine doors, operated by a remote pulley system, controlled access to the arms from a central area from which the animals entered and exited the apparatus. After 2 d of adaptation, food-restricted mice (3-4 hr daily access to food; mice maintained at 10% body weight loss) were given one training session per day for nine consecutive days. For each session, all arms were baited with fruit loop cereal, and animals were permitted to enter all arms until the eight rewards had been consumed. The number of errors (entries to previously visited arms) and time to complete each session were recorded.
Antibody levels. Antibody levels were determined by 1:200 and 1:500 dilutions of plasma using ELISA as described previously (Sigurdsson et al., 2001), in which Aβ or its derivative is coated onto microtiter wells. The antibodies were detected by a goat anti-mouse IgG linked to a horse-radish peroxidase (Amersham Biosciences, Piscataway, NJ) or a goat anti-mouse IgM peroxidase conjugate (Sigma; A8786), and tetramethyl benzidine (TMB; Pierce, Rockford, IL) was the substrate.
For antibody unmasking, plasma was diluted 1:500 with acid dissociation buffer (1.5% bovine serum albumin and 0.2 m glycine-acetate in PBS, pH 2.5) and subsequently incubated at room temperature for 20 min (Li et al., 2004). To remove dissociated Aβ, the solution was then centrifuged in a Microcon filter device (10,000 molecular weight cutoff; Millipore, Bedford, MA) at 8000 × g for 20 min at room temperature. The solution containing the antibody was then collected according to the instructions of the manufacturer and its pH adjusted to 7.0 with 1 m Tris buffer, pH 9.0, and brought to the initial volume with ELISA dilution buffer. Similar antigen-antibody unmasking protocols have been reported previously (Lillo et al., 1993; Quinn et al., 1993).
Histology. Mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.), perfused transaortically with phosphate buffer, and the brains processed as described previously (Sigurdsson et al., 1996). The right hemisphere was immersion-fixed in periodate-lysine-paraformaldehyde, whereas the left hemisphere was snap-frozen for measurements of Aβ levels using established ELISA methods (Janus et al., 2000; Mehta et al., 2000). Serial coronal sections (40 μm) were cut, and every fifth section was stained with 6E10, a monoclonal antibody that recognizes Aβ and stains both pre-amyloid and Aβ plaques (Signet, Dedham, MA) (Kim et al., 1990). Staining was performed as described previously (Sigurdsson et al., 1996; Soto et al., 1998). Briefly, sections were incubated in 6E10 at a 1:1000 dilution. A mouse-on-mouse immunodetection kit (Vector Laboratories, Burlingame, CA) was used, with the anti-mouse IgG secondary antibody at a 1:2000 dilution. The sections were reacted in 3,3-diaminobenzidine tetrahydrochloride with nickel ammonium sulfate (Ni; Mallinckrodt, Paris, KY) intensification.
Image analysis. Immunohistochemistry of tissue sections was quantified with a Bioquant image analysis system (BIOQUANT Image Analysis Corporation, Nashville, TN), and unbiased sampling was used (West, 1999). All procedures were performed by an individual blinded to the experimental condition of the study. The cortical area analyzed was dorsomedially from the cingulate cortex and extended ventrolaterally to the rhinal fissure within the right hemisphere. The area of the grid was 800 × 800 μm2, and amyloid load was measured in 20 cortical frames per mouse (640 × 480 μm2 each) chosen randomly. The Aβ burden is defined as the percentage of area in the measurement field occupied by reaction product. The number of plaques was also counted, and the plaques were divided into three groups on the basis of their size (small, 0.01-50 μm2; medium, 50.01-1000 μm2; large, >1000 μm2).
Tissue homogenization and sandwich ELISA assay for soluble Aβ levels. Brain homogenates, 10% (w/v), were prepared in 20 mmol/l Tris, pH 7.4, 250 mmol/l sucrose, 1 mmol/l EDTA, and 1 mmol/l EGTA. Immediately before use, 1:100 volume of 100 mmol/l PMSF solution (in ethanol) and 1:1000 volume of LAP (5 mg each of leupeptin, antipain, and pepstatin A per milliliter of N-N-dimethylformamide) were added to the homogenization buffer. After mixing with an equal volume of 0.4% diethylamine/100 mmol/l NaCl and centrifugation at 135,000 × g for 1 hr at 4°C, the samples were neutralized with 1:10 volume 0.5 mol/l Tris, pH 6.8, and then aliquoted, flash-frozen on dry ice, and stored at -80°C until analyzed. The sandwich ELISA incorporates 6E10 as the capture antibody and rabbit anti-Aβ1-40 (Chemicon, Temecula, CA) or anti-Aβ1-42 (Biosource, Camarillo, CA) for detection antibody. Secondary antibody was peroxidase-linked anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA), and TMB was the chromogen. Resulting optical density values (450 nm) were compared with standard curves. Assay sensitivity was ∼30-50 pg/ml.
Data analysis. The cell culture data were analyzed by one-way ANOVA, followed by a Neuman-Keuls test for post hoc analysis (Statistica 6.1). Amyloid burden and the levels of soluble Aβ within the brain were analyzed by a Student's t test, one-tailed. The radial arm maze data were analyzed by two-way ANOVA repeated measures and a Neuman-Keuls post hoc test. Correlation was determined by calculating the Pearson r correlation coefficient.
Results
Fibril formation and neurotoxicity
Aβ1-30[E18E19] and K6Aβ1-30[E18E19] were not fibrillogenic, whereas Aβ1-42 contained fibrils throughout the incubation period (Fig. 1A). Aβ1-42 was also neurotoxic in human neuroblastoma (SK-N-SH) cell culture but not the two Aβ derivatives (Fig. 1B) (one-way ANOVA; p < 0.001). After a 6 d incubation, 10 μm Aβ1-42 reduced cell viability by 27% (Neuman-Keuls post hoc test; p < 0.001). As with these two Aβ derivatives, we have demonstrated previously that K6Aβ1-30 does not make fibrils and is not toxic in cell culture under these conditions (Sigurdsson et al., 2001).
Figure 1.

A, Thioflavin T fluorometric assay. Fibril formation of Aβ1-42, Aβ1-30[E18E19], and K6Aβ1-30[E18E19] was measured in vitro in triplicates (37°C; 0-18 d). The Aβ derivatives were not fibrillogenic compared with Aβ1-42, which readily formed fibrils. This particular lot of Aβ1-42 already formed fibrils at t=0, and the amount of fibrils did not increase substantially over time. B, MTT assay. Aβ1-42 was toxic to human neuroblastoma cells (SK-N-SH) in culture, whereas the Aβ derivatives had no effect compared with the control group. ***p < 0.001, compared with control group (one-way ANOVA; Neuman-Keuls post hoc test).
Behavior
The mice immunized with Aβ1-30[E18E19] performed much better in the radial arm maze than vehicle-treated mice, and their performance approached that of their wild-type littermates (Fig. 2A) (ANOVA, repeated measures; treatment effect, p < 0.0001; days effect, p = 0.03; Neuman-Keuls post hoc test: vehicle vs peptide, p = 0.02; vehicle vs wild type, p < 0.001). Wild-type littermates were immunized with the peptide in adjuvant or received adjuvant alone with peptide vehicle (PBS). No difference was observed between these two groups, which were then combined for subsequent analysis. Likewise, mice injected with K6Aβ1-30[E18E19] showed improved performance in the radial arm maze (Fig. 2B) (ANOVA, repeated measures; treatment effect, p < 0.05; days effect, p = 0.02).
Figure 2.

A, Group differences were observed in the radial arm maze (two-way ANOVA, repeated measures; treatment, p < 0.0001; days, p = 0.03). Vehicle-treated (n = 8) transgenic mice performed significantly worse in the maze compared with Aβ1-30[E18E19]-treated transgenic mice (p = 0.02; n = 6) and their wild-type littermates (p < 0.001; n = 17). B, K6Aβ1-30[E18E19]-treated transgenic mice (n = 13) had significantly fewer errors in the radial arm maze compared with their vehicle-treated (n = 19) controls (two-way ANOVA, repeated measures; treatment, p < 0.05; days, p = 0.02).
Immune response
In contrast to our previous findings with K6Aβ1-30, Aβ1-30[E18E19] elicited a more modest IgG reponse (Fig. 3A). IgM response was also low overall, although a few mice had a moderate IgM level against the antigen (data not shown). The other Aβ derivative, K6Aβ1-30[E18E19], also elicited a modest IgG response (Fig. 3B), but this peptide produced a robust IgM response (Fig. 3C). The IgM antibodies from the K6Aβ1-30[E18E19] mice cross-reacted with Aβ1-40, but minimal reactivity was observed with plasma from mice treated with other Aβ derivatives, Aβ1-42 or adjuvant alone (Fig. 3D). Although mice immunized with K6Aβ1-30 or Aβ1-42 had low IgM levels, the plasma from these animals provided a strong IgG signal toward the immunogen or Aβ1-40 (K6Aβ1-30, 1.2 ± 0.7; Aβ1-42, 0.9 ± 0.5, against Aβ1-40 at 1:500 dilution; n = 6 per group), as we and others have reported previously (Schenk et al., 1999; Sigurdsson et al., 2001).
Figure 3.

A, Immunization with Aβ1-30[E18E19] resulted in a modest increase in IgG recognizing the antigen (black), Aβ1-40 (dark gray), and Aβ1-42 (light gray) on ELISA plates as detected in plasma samples diluted 1:200 obtained at the end of the study (n = 6-8 per group). Controls are plasma samples from mice injected with adjuvant and peptide vehicle incubated on the same peptide-coated ELISA plates as plasma from Aβ1-30[E18E19]. The peptides used for coating the plates are listed above the respective bars. B, Likewise, immunization with K6Aβ1-30[E18E19] elicited a modest IgG response against the antigens Aβ1-40 and Aβ1-42 at the same plasma dilution (n = 14-18 per group). The controls are as described in A, and the peptides used for coating the ELISA plates are listed above the bars. C, K6Aβ1-30[E18E19] induced a substantially more pronounced IgM response against itself compared with IgG response as detected in plasma at 1:500 dilution. The controls are as described in A. D, The IgM antibodies generated after K6Aβ1-30[E18E19] immunization cross-reacted with Aβ1-40 (n = 12). Plasma from mice immunized with other Aβ derivatives, Aβ1-42 (n = 6-9 per group) or control mice that received adjuvant with peptide vehicle (n = 16), had minimal IgM reactivity toward Aβ1-40.
Antibody unmasking with acid treatment of the plasma increased IgG detection of the immunogen and Aβ1-40 both in vehicle-treated and immunized Tg2576 and wild-type mice, indicating that this enhancement was nonspecific under our conditions.
Plaques and Aβ levels
Although cognitive improvement was observed in the animals immunized with Aβ1-30[E18E19], there was not a significant difference in histological amyloid plaque burden or Aβ ELISA levels between the groups, and these measurements did not correlate with behavioral outcome (data not shown).
In contrast, there was a strong trend for a reduction in overall amyloid burden in the K6Aβ1-30[E18E19]-treated group (26% reduction in total plaque area, p = 0.10). Additional analysis indicated that small and medium sized plaques were preferentially affected (Fig. 4A-C; A, small plaques: 34% reduction in plaque number, p = 0.02; B, medium plaques: 29% reduction, p = 0.04). It should be more difficult to clear larger plaques that are more encapsulated by astroglia than smaller plaques. Aβ ELISA levels did not differ significantly between the groups. Although cognitive performance did not correlate with plaque burden or levels of soluble Aβ (data not shown), high IgM titer correlated with low amyloid burden (Fig. 4D-F), suggesting that IgM was mediating a peripheral clearance of Aβ. The subsequent shift in equilibrium of unbound Aβ within and outside the brain then resulted in an expected efflux of Aβ from the brain, thereby diminishing plaque burden. To verify that the reduction in brain amyloid burden was not attributable to binding of IgM to plaques, sections from several animals were stained with anti-IgM antibodies under various conditions, and no immunoreactive material associated with the plaques was detected.
Figure 4.
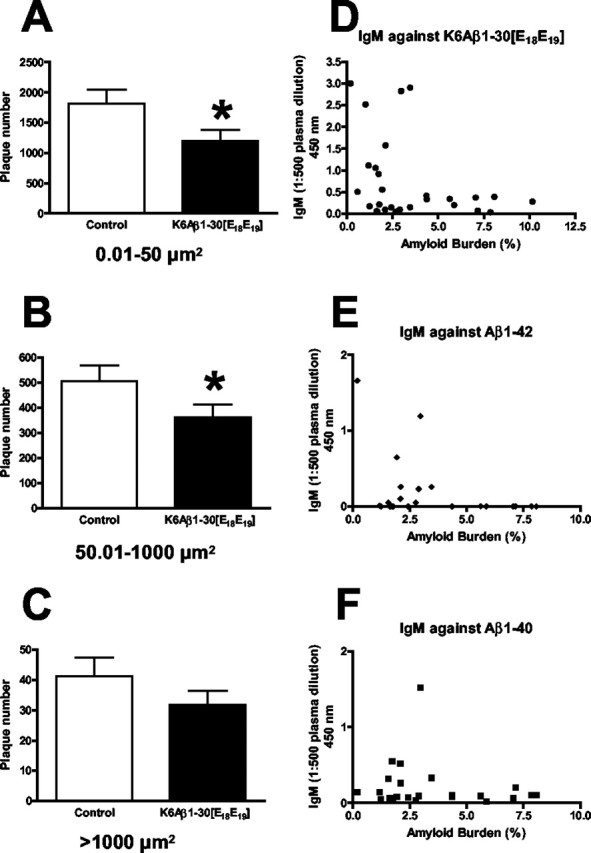
Immunization with K6Aβ1-30[E18E19] preferentially reduced small (A) (34% reduction; p = 0.02) and medium-sized (B) (29% reduction; p = 0.04) plaques. C, Large plaques were not significantly affected (n = 18 per group). D, E, Low amyloid plaque burden correlated with high IgM levels against the immunogen (D) (p < 0.05) and Aβ1-42 (E) (p = 0.05). F, A trend for correlation was seen for IgM recognizing Aβ1-40.
Discussion
We show that treatment of AD model mice with both of our Aβ derivatives leads to cognitive improvements. With Aβ1-30[E18E19], this effect was not associated with a reduction in amyloid burden or levels of soluble brain Aβ. Immunization with our Aβ derivatives may result in the clearance of a subpopulation of Aβ, such as oligomers that may be more directly linked to behavioral outcome than total Aβ levels or amount of plaques. Cognitive improvements have been observed in immunized mice without obvious relationship to certain Aβ measurements (Janus et al., 2000; Morgan et al., 2000; Dodart et al., 2002; Kotilinek et al., 2002). Our other Aβ derivative, K6Aβ1-30[E18E19], elicited a different and more pronounced immune response, which was associated with cognitive improvement and a clearance of small and medium sized plaques (29-34% reduction in size). Furthermore, plasma levels of IgM correlated inversely with the amyloid plaque burden, suggesting that IgM was mediating a peripheral clearance of Aβ, thereby drawing Aβ from the brain because of a shift in equilibrium of Aβ within and outside the brain. The observation that this derivative elicited primarily an IgM response instead of an IgG response indicates a T-cell-independent immune response (Baumgarth, 2000), which is commonly seen for antigens with a repetitive structure such as polysaccharides but can also be observed for proteins (Szomolanyi-Tsuda et al., 2001). In contrast, K6Aβ1-30 and Aβ1-42 elicit a T-cell-dependent response, in which recruitment of T-cells facilitates isotype switching from IgM to IgG and results in a robust IgG response after the second injection of the immunogen (Schenk et al., 1999; Sigurdsson et al., 2001). This kind of modulation of the vaccine response is likely related to a loss of a helper T-cell epitope within the mutated region (amino acids 18 and 19). Because of the lysine residues, K6Aβ1-30[E18E19] is likely to be more immunogenic than Aβ1-30[E18E19] and, therefore, elicits a more pronounced humoral response. That the attenuated immune response produced by our Aβ derivatives is nonetheless associated with cognitive improvement has important implications for AD vaccine therapy.
Over the years, we have evaluated various behavioral tests to detect cognitive differences in the Tg APP model (Tg2576) compared with wild-type mice. The radial arm maze is the test that consistently shows us cognitive impairments in these Tg2576 mice compared with their wild-type littermates. This test has been used for many years by numerous laboratories, and there is a consensus in the field of behavioral neuroscience that it measures working memory in mice with a strong spatial component. Confinement to the central arena by doors discourages the use of nonspatial search strategies. Another popular spatial learning task is the water maze, but this particular test is more suitable for rats. In addition, it is a very stressful task, and the Tg2576 strain appears to have a lower stress tolerance than their wild-type counterparts. In the water maze, it is also easier for the animal to use different search strategies to finish the task, and spatial deficits detected in one of these tests are often not seen in the other (Hodges, 1996). For these reasons, we relied on the radial arm maze and consistently observed cognitive improvements with our Aβ derivatives.
To determine whether we were potentially underestimating the amount of anti-Aβ antibodies, we dissociated endogenous Aβ bound to the serum antibodies with mild acid denaturation. Although this unmasking protocol increased antibody titer substantially in mice with low titer but not in animals with high titer, this effect was also observed in nonimmunized Tg2576 and wild-type mice. Hence, under our conditions, this procedure does not add to our understanding of the relationship between antibody titer, amyloid burden, and behavior.
The serious side effects observed in a subset of patients in the phase II clinical trial of Aβ1-42 vaccination, which included encephalitis leading to death, have raised the bar for approval of related approaches. Evidence suggests that this toxicity was related to the Th-1, cytotoxic T-cell response (Schenk, 2002), which may be elicited by both the full length Aβ and the QS-21 adjuvant that promotes a cell-mediated Th-1 immune response (Kensil et al., 1995). More recently, Aβ-reactive T-cells have been detected in Alzheimer's patients and controls (Giubilei et al., 2003; Monsonego et al., 2003), and T-cell responses toward Aβ fragments have been modulated with amino acid substitutions (Monsonego et al., 2003), supporting our approach (Sigurdsson et al., 2001; Knudsen et al., 2003).
There is a predicted overlap of Aβ derived MHC I and MHC II epitopes (Singh and Raghava, 2001, 2003). Given the variety of individual haplotypes in the general population, it is unlikely that cytotoxic T-cell epitopes can be eliminated without affecting T-helper epitopes in most individuals. A safer vaccine should be generated by reducing the occurrence of both types of promiscuous T-cell binding sites while still effectively enhancing cognition, as our findings indicate. When T-cells are not recruited, immunological memory is not generated, and antibody production is transient. This type of response resembles passive immunization but should have fewer complications. The multiple antibody injections required to treat a chronic disease like AD are likely to generate anti-idiotypic antibodies. The resulting serum immune complexes can subsequently lead to vasculitis and glomerulonephritis. Considering these potential side effects, we suggest that vaccination with a T-cell-independent Aβ derivative will be preferable and safer as a chronic treatment. IgM is also six times larger than IgG; therefore, it is less prone to enter the brain (Nerenberg and Prasad, 1975; Vermes, 1983). Antibodies within the brain may result in inflammatory side effects, whereas peripheral antibodies drawing out cerebral Aβ should be less toxic.
The development of immune-based AD therapy is critically dependent on limiting toxicity. This depends on a better understanding of how immunization improves cognition in AD model mice and ultimately in humans, as well as by defining the type of immune response directly linked to the encephalitis that occurred in a subset of patients. Our results show that a cognitive benefit can be seen with a modest humoral response, and that an IgM-mediated clearance of Aβ has a promising therapeutic potential while being potentially safer than other approaches.
Footnotes
This research was supported by National Institutes of Health Grants AG20197, AG20245, AG05891, and AR02594 and by the Alzheimer's Association and Mindset Biopharmaceuticals. We thank Thorir Sigmundsson for technical assistance.
Correspondence should be addressed to either of the following: Einar M. Sigurdsson, Departments of Psychiatry and Pathology, New York University School of Medicine, Millhauser Laboratories, Room HN418, 560 First Avenue, New York, NY 10016, E-mail: einar.sigurdsson@med.nyu.edu; or Thomas Wisniewski, Departments of Neurology, Pathology, and Psychiatry, Millhauser Laboratories, Room HN419, 560 First Avenue, New York, NY 10016, E-mail: thomas.wisniewski@med.nyu.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246277-06$15.00/0
References
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT (2002) Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci 22: 7873-7878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T (2000) Peripherally administered antibodies against amyloid β-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6: 916-919. [DOI] [PubMed] [Google Scholar]
- Baumgarth N (2000) A two-phase model of B-cell activation. Immunol Rev 176: 171-180. [DOI] [PubMed] [Google Scholar]
- Das P, Howard V, Loosbrock N, Dickson D, Murphy MP, Golde TE (2003) Amyloid-β immunization effectively reduces amyloid deposition in FcRγ-/- knock-out mice. J Neurosci 23: 8532-8538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM (2001) Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA 98: 8850-8855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, De-Long CA, Wu S, Wu X, Holtzman DM, Paul SM (2002) Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nat Neurosci 5: 452-457. [DOI] [PubMed] [Google Scholar]
- Giubilei F, Antonini G, Montesperelli C, Sepe-Monti M, Cannoni S, Pichi A, Tisei P, Casini AR, Buttinelli C, Prencipe M, Salvetti M, Ristori G (2003) T cell response to amyloid-β and to mitochondrial antigens in Alzheimer's disease. Dement Geriatr Cogn Disord 16: 35-38. [DOI] [PubMed] [Google Scholar]
- Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K (1992) Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer's disease βA4 peptides. J Mol Biol 228: 460-473. [DOI] [PubMed] [Google Scholar]
- Hodges H (1996) Maze procedures: the radial-arm and water maze compared. Brain Res Cogn Brain Res 3: 167-181. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Jameson BA, Wolf H (1988) The antigenic index: a novel algorithm for predicting antigenic determinants. Comput Appl Biosci 4: 181-186. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D (2000) Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408: 979-982. [DOI] [PubMed] [Google Scholar]
- Kensil CR, Wu JY, Soltysik S (1995) Structural and immunological characterization of the vaccine adjuvant QS-21. Pharm Biotechnol 6: 525-541. [DOI] [PubMed] [Google Scholar]
- Kim KS, Wen GY, Bancher C, Chen CMJ, Sapienza V, Hong H, Wisniewski HM (1990) Detection and quantification of amyloid β-peptide with 2 monoclonal antibodies. Neurosci Res Commun 7: 113-122. [Google Scholar]
- Knudsen EL, Wisniewski T, Quartermain D, Sage D, Scholtzova H, Frangione B, Sigurdsson EM (2003) Immunization with amyloid-β derivatives improves cognition while provoking a weak antibody response. Soc Neurosci Abstr 133.10.
- Kotilinek LA, Bacskai B, Westerman M, Kawarabayashi T, Younkin L, Hyman BT, Younkin S, Ashe KH (2002) Reversible memory loss in a mouse transgenic model of Alzheimer's disease. J Neurosci 22: 6331-6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, Matsuoka Y, Taylor JW, DeMattos RB, Holtzman DM, Clements JD, Selkoe DJ, Duff KE (2003) Evidence for peripheral clearance of cerebral Aβ protein following chronic, active Aβ immunization in PSAPP mice. Neurobiol Dis 14: 10-18. [DOI] [PubMed] [Google Scholar]
- Li Q, Cao C, Chackerian B, Schiller J, Gordon M, Ugen KE, Morgan D (2004) Overcoming antigen masking of anti-Aβ antibodies reveals breaking of B cell tolerance by virus-like particles in Aβ immunized amyloid precursor protein transgenic mice. BMC Neurosci, in press. [DOI] [PMC free article] [PubMed]
- Lillo FB, Cao Y, Concedi DR, Varnier OE (1993) Improved detection of serum HIV p24 antigen after acid dissociation of immune complexes. AIDS 7: 1331-1336. [DOI] [PubMed] [Google Scholar]
- Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM (2000) Plasma and cerebrospinal fluid levels of amyloid β proteins 1-40 and 1-42 in Alzheimer disease. Arch Neurol 57: 100-105. [DOI] [PubMed] [Google Scholar]
- Monsonego A, Zota V, Karni A, Krieger JI, Bar-Or A, Bitan G, Budson AE, Sperling R, Selkoe DJ, Weiner HL (2003) Increased T cell reactivity to amyloid β protein in older humans and patients with Alzheimer disease. J Clin Invest 112: 415-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Duff K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW (2000) Aβ peptide vaccination prevents memory loss in an animal model of Alzheimer's disease. Nature 408: 982-985. [DOI] [PubMed] [Google Scholar]
- Nerenberg ST, Prasad R (1975) Radioimmunoassays for Ig classes G, A, M, D, and E in spinal fluids: normal values of different age groups. J Lab Clin Med 86: 887-898. [PubMed] [Google Scholar]
- Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW (1993) Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci 13: 1676-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn TC, Kline R, Moss MW, Livingston RA, Hutton N (1993) Acid dissociation of immune complexes improves diagnostic utility of p24 antigen detection in perinatally acquired human immunodeficiency virus infection. J Infect Dis 167: 1193-1196. [DOI] [PubMed] [Google Scholar]
- Schenk D (2002) Amyloid-β immunotherapy for Alzheimer's disease: the end of the beginning. Nat Rev Neurosci 3: 824-828. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P (1999) Immunization with amyloid-β attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400: 173-177. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Lorens SA, Hejna MJ, Dong XW, Lee JM (1996) Local and distant histopathological effects of unilateral amyloid-β 25-35 injections into the amygdala of young F344 rats. Neurobiol Aging 17: 893-901. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Scholtzova H, Mehta PD, Frangione B, Wisniewski T (2001) Immunization with a non-toxic/non-fibrillar amyloid-β homologous peptide reduces Alzheimer's disease associated pathology in transgenic mice. Am J Pathol 159: 439-447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Raghava GP (2001) ProPred: prediction of HLA-DR binding sites. Bioinformatics 17: 1236-1237. [DOI] [PubMed] [Google Scholar]
- Singh H, Raghava GP (2003) ProPred1: prediction of promiscuous MHC Class-I binding sites. Bioinformatics 19: 1009-1014. [DOI] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frankel D, Hanan-Aharon E (1997) Disaggregation of Alzheimer β-amyloid by site-directed mAb. Proc Natl Acad Sci USA 94: 4109-4112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C, Sigurdsson EM, Morelli L, Kumar RA, Castano EM, Frangione B (1998) β-sheet breaker peptides inhibit fibrillogenesis in a rat brain model of amyloidosis: implications for Alzheimer's therapy. Nat Med 4: 822-826. [DOI] [PubMed] [Google Scholar]
- Szomolanyi-Tsuda E, Brien JD, Dorgan JE, Garcea RL, Woodland RT, Welsh RM (2001) Antiviral T-cell-independent type 2 antibody responses induced in vivo in the absence of T and NK cells. Virology 280: 160-168. [DOI] [PubMed] [Google Scholar]
- Vermes LM (1983) Cerebrospinal fluid proteins: III. Normal values of immunoglobulins G, A and M (variations related to race, sex and age). Ar Qneuropsiquiatr 41: 25-49. [DOI] [PubMed] [Google Scholar]
- West MJ (1999) Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 22: 51-61. [DOI] [PubMed] [Google Scholar]