

Abstract
To determine the nature of dopamine modulation of dendritic Ca2+ signaling in layers V-VI prefrontal cortex (PFC) neurons, whole-cell Ca2+ potentials were evoked after blockade of Na+ and K+ channels. Soma-dendritic Ca2+ spikes evoked by suprathreshold depolarizing pulses, which could be terminated by superimposed brief intrasomatic hyperpolarizing pulses, are blocked by the L-type Ca2+ channel antagonist nimodipine (1 μm). The D1/D5 receptor agonist dihydrexidine (DHX) (0.01-10 μm; 5 min) or R-(+)SKF81291 (10 μm) induced a prolonged (>30 min) dose-dependent peak suppression of these Ca2+ spikes. This effect was dependent on [Ca2+]i- and protein kinase C (PKC)-dependent mechanisms because [Ca2+]i chelation by BAPTA or inhibition of PKC by bisindolymaleimide (BiM1), but not inhibition of [Ca2+]i release with heparin or Xestospongin C, prevented the D1-mediated suppression of Ca2+ spikes. Depolarizing pulses subthreshold to activating a Ca2+ spike evoked a nimodipine-sensitive Ca2+ “hump” potential. D1/D5 stimulation induced an N-[2-((o-bromocinamyl)amino)ethyl]-5-isoquinolinesulfonamide (H-89)- or internal PKA inhibitory peptide[5-24]-sensitive (PKA-dependent) transient (∼7 min) potentiation of the hump potential to full Ca2+ spike firing. Furthermore, application of DHX in the presence of the PKC inhibitor BiM1 or internal PKC inhibitory peptide[19-36] resulted in persistent firing of full Ca2+ spike bursts, suggesting that a D1/D5-PKA mechanism switches subthreshold Ca2+ hump potential to fire full Ca2+ spikes, which are eventually turned off by a D1/D5-Ca2+-dependent PKC mechanism. This depolarizing state-dependent, D1/D5-activated, bi-directional switching of soma-dendritic L-type Ca2+ channels via PKA-dependent potentiation and PKC-dependent suppression may provide spatiotemporal regulation of synaptic integration and plasticity in PFC.
Keywords: synaptic plasticity, schizophrenia, dendrites, synaptic integration, amplification, PKA, PKC
Introduction
The mesocortical dopamine (DA) input to the deep layers V-VI of the prefrontal cortex (PFC) (Berger et al., 1991; Krimer et al., 1997) is crucial for processing short-term working memory that is used to guide forthcoming behaviors and attentional control (Goldman-Rakic, 1998; Miller and Cohen, 2001; Tzschentke, 2001). An optimal level of D1 receptor (D1R) activation in the PFC is required to facilitate processing of short-term memory-related activity in PFC (Zahrt et al., 1997; Goldman-Rakic et al., 2000).
D1Rs are located exclusively on dendritic spines and shafts (Smiley et al., 1994) of the extensive dendritic arbor of pyramidal PFC neurons, in which dendritic Ca2+ channels also reside (Westenbroek et al., 1992; Hell et al., 1993; Smiley and Goldman-Rakic, 1993; Sabatini et al., 2001). Cortical neurons possess all three major subtypes (L, P, and N) of high voltage-activated (HVA) Ca2+ channels (Brown et al., 1993; Ye and Akaike, 1993; Lorenzon and Foehring, 1995). L-type Ca2+ channel immunoreactivity is distributed primarily in the soma-basal dendrite-proximal apical dendrite compartments, whereas N- and P-type immunoreactivity is distributed throughout the apical dendrites of pyramidal neurons (Hillman et al., 1991; Usowicz et al., 1992; Westenbroek et al., 1992; Mills et al., 1994). The importance of DA modulation of dendritic functions is exemplified by the findings that dysfunction of the mesocortical DA inputs to the dendrites, dendritic degeneration, and spine loss in PFC neurons are implicated in schizophrenia (Garey et al., 1998; Selemon and Goldman-Rakic, 1999; Glantz and Lewis, 2000; Kulisevsky, 2000; Levy and Farrow, 2001; Weinberger et al., 2001).
Ca2+ spikes can be activated at the distal apical dendrites of cortical pyramidal neurons by strong synaptic stimulation or by back-propagated action potentials (Yuste and Tank, 1996; Stuart et al., 1997; Larkum et al., 1999a; Hausser et al., 2000; Larkum and Zhu, 2002). In PFC neurons, a Ca2+ “hump” potential activated in the proximal dendrites by subthreshold depolarizing pulses has also been characterized functionally (Seamans et al., 1997). Both Ca2+ spikes in distal apical dendrites and Ca2+ hump potentials in proximal-basal dendritic compartments may contribute to spatiotemporal synaptic signal amplification, integration, and plasticity (Magee et al., 1998; Yang et al., 1999).
Although D1R may optimally modulate incoming synaptic inputs by attenuating dendritic Ca2+ spikes that normally amplify propagating distal synaptic signals en route to the soma in PFC neurons (Yang and Seamans, 1996; Yang et al., 1999), the functional roles of a possible DA modulation of L-type Ca2+ channels activated near soma-proximal-basal dendritic compartments are less understood. In this study, we examined the extent to which D1R activation functionally modulates soma-dendritic Ca2+ signaling and determined how D1R modulates nimodipine-sensitive dendritic Ca2+ spikes and hump potentials that were evoked by suprathreshold and subthreshold depolarizing current pulses in PFC pyramidal neurons. We showed that D1/D5R activation dynamically switches bi-directionally between dendritic L-type Ca2+ spike suppression or hump potential activation to Ca2+ spikes in a distinct temporal- and voltage-dependent manner via differential PKC or PKA activation, respectively.
Preliminary results have been published previously in abstract form (Young and Yang, 2002).
Materials and Methods
Animals. Young adult [postnatal day (P) 20-24] male rats were obtained from Harlan Bioproducts for Science, Inc. (Indianapolis, IN) during October to February. The euthanasia method was approved by the Lilly Animal Use Committee, the policies of which adhere closely to the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals (Public Health Service policy) and the National Institute of Health's Guide for the Care and Use of Laboratory Animals. The rats were maintained on a 12 hr light/dark cycle, with the light phase being 7:00 A.M. to 7:00 P.M. Food and water were available ad libitum. After decapitation by a guillotine (using a plastic Decapicone rat restrainer; Braintree Scientific, Braintree, MA), the brain was dissected rapidly and immersed for 1 min in ice-cold low-Ca2+ artificial CSF (ACSF) containing (in mm): 124 NaCl, 3 KCl, 26 NaHCO3, 4 MgCl2, 0.5 CaCl2, 0.4 ascorbic acid, 0.8 thiourea, and 10 glucose. The solution was oxygenated continuously with carbogen (95% O2, 5% CO2).
A block of tissue containing the forebrain was glued onto the cutting stage of a Vibratome (Vibroslice; World Precision Instruments, Sarasota, FL) by cyanoacrylic-based glue and sliced in the coronal plane (350 μm) in ice-cold low-Ca2+ ACSF solution. The cut PFC slices [corresponding to anteroposterior = +2-3 mm from bregma; lateromedial = 0.5-1 mm; dorsoventral = 2-4 mm from the cortical surface, according to Paxinos and Watson (1998)] were immediately transferred to an incubation solution containing (in mm): 124 NaCl, 3 KCl, 26 NaHCO3, 1,3 MgCl2, 2.3 CaCl2, 0.4 ascorbic acid, 0.8 thiourea, and 10 glucose and incubated at 31-34°C for 0.5 hr on a hot plate before they are cooled to room temperature for the rest of the day. After 1 hr of cooling, a single slice was transferred to a submersion-type recording chamber (RC-26G; Warner Instruments, Hamden, CT), and the slice was perfused with gravity-fed ACSF (maintained at 30-31°C) at 3-4 ml/min. The temperature of the ACSF was heated immediately to the preset temperature using an in-line heater connected to an automatic feedback controller (SH-27B; Warner Instruments). All solutions were gassed continuously with carbogen (95% O2 + 5% CO2).
Whole-cell patch-clamp recordings. Layers V-VI PFC pyramidal neurons from the prelimbic area of the PFC were visualized using an upright Olympus BX50WI microscope equipped with differential interference contrast optics and an infrared high performance vidicon camera (C2400-07ER; Hamamatsu, Bridgewater, NJ). PFC pyramidal neurons were easily recognizable via a 40× water-immersion lens by the pyramidal shape of their cell bodies and by the presence of long apical dendrites that extend toward superficial layers and the pia. Biocytin (3 mg/ml)-filled patch pipettes were used to fill some neurons followed by streptavidin staining according to the method used by Yang et al. (1996) for morphological examination.
In the recording chamber, the brain slice was held by a nylon net (RC-22C; Warner Instruments). For whole-cell recordings, standard patch electrodes were made using thick-walled borosilicate micropipettes (1.5 mm outer diameter, 1.1 mm inner diameter; Sutter Instrument, Novato, CA) and were pulled by a horizontal micropipette puller (Flaming-Brown P-97; Sutter Instrument). Microelectrodes contained (in mm): 100 KMeSO4, 10 HEPES, 1 EGTA, 2 MgCl2·H2O, 2 Na2ATP, 0.5 Tris-GTP, 10 Na2-phosphocreatine, and 60 sucrose. The electrodes had a resistance of 4-6 MΩ in ACSF. The microelectrode was connected to the headstage of an Axoclamp-200B amplifier (Axon Instruments, Foster City, CA) with Ag/AgCl wire. The recorded voltage signals were amplified in current-clamp bridge mode, digitized by a 12 bit analog-to-digital board (Digidata 1322A; Axon instruments), and sampled on-line using a PC-based computer. The captured data were analyzed off-line using ClampFit software (Version 8.0, Axon Instruments).
Isolation of a suprathreshold Ca2+ spike and subthreshold Ca2+ hump potentials. To isolate the high-threshold Ca2+ spikes, tetrodotoxin (TTX; 1 μm) and tetraethylammonium hydrochloride (TEA; 20 mm) were first used to block Na+ and K+ channels, respectively. Depolarizing pulses with incremental pulse amplitude and duration were used to characterize the firing properties of the Ca2+ spike (Fig. 1). Step-wise increases in depolarizing current pulses eventually evoked all-or-none Ca2+ spikes. A “suprathreshold” current was determined by using the current amplitude that was two to three incremental steps above that which caused the first Ca2+ spike. This suprathreshold current step was rechecked before D1 agonist application to ensure that the current needed to evoke suprathreshold Ca2+ had not changed over time. Incremental pulse durations (50-450 msec) in these cells resulted in an initial single short-lasting spike that was followed by a plateau (∼75% of the cells) (Fig. 1C). In other cells this stepwise increment of depolarizing pulse duration evoked a burst of several repetitive Ca2+ spikes (∼24% of the cells) or a single long-lasting (>500 msec) Ca2+ plateau (∼1% of the cells).
Figure 1.

Characterization of HVA Ca2+ spike in layers V-VI PFC pyramidal neurons using incremental intracellular depolarizing pulse amplitude and duration in the presence of TTX (1 μm) and TEA (200 μm). Three types of Ca2+ spikes were observed. A-D, Representative traces from neurons that fired single, double, or multiple Ca2+ spikes only, regardless of depolarizing pulse duration (33.7, 28.6, and 26.5% of the population, respectively). Note the sudden jump of spike amplitude (♦) when the all-or-nothing Ca2+ spike was elicited by 150 pA current pulse for this neuron (A, B), but a linear increase in the overall integrated area (•) of the Ca2+ spike and the ensuing post-spike plateau potential (C, D). E, In a small number of neurons (11.2% of the population), a small depolarizing pulse (50 msec) was sufficient to trigger a prolonged Ca2+ plateau that typically lasted for >1.5 sec (outlasting the short depolarizing pulse) followed by an abrupt repolarization. Incremental duration of suprathreshold pulse injected into these neurons did not elicit a longer Ca2+ plateau. These small number of neurons (n = 11) were present mostly in deep layer VI PFC pyramidal neurons.
On the other hand, a “subthreshold” current was determined by manually clamping the membrane potential (with DC injection) to -40 mV after TTX and TEA application and then determining the minimum current to evoke a Ca2+ spike. Once this Ca2+ spike threshold was determined, the subthreshold current was chosen as the current amplitude that was two to three decremental steps (0.01 pA steps) below that which caused the first Ca2+ spike but capable of evoking a hump potential (Seamans et al., 1997). The established Ca2+ spike or hump potentials were then evoked at a rate of 0.033 Hz throughout the experiment.
For both suprathreshold and subthreshold Ca2+ potential recordings, the input resistance of the neuron was also monitored continuously throughout these experiments by a hyperpolarizing (-50 pA, 100 msec) prepulse 100-250 msec before a subthreshold or suprathreshold depolarizing pulse was injected into the neuron.
Drugs. Most drugs were bath-applied by gravity. Complete exchange of the bathing solution took ∼1.5 min. TEA (20 mm) and TTX (1 μm) (Sigma, St. Louis, MO) were bath-applied to block K+ and Na+ channels, respectively. Before being diluted to the appropriate final concentration in the perfusate, the stock solutions of the D1/D5 agonist dihydrexidine (DHX) [Tocris (Mottola et al., 1992)] and R-(+)SKF81291 (Sigma) were made up fresh in ascorbic acid (10 mm) (final concentration of ascorbic acid, 10 μm); the L-type Ca2+ channel blocker nimodipine (Tocris) was dissolved in 100% ethanol, and the D1 antagonist SCH23390 (Tocris) was dissolved in distilled water.
The selective N-type Ca2+ channels blocker ω-conotoxin GVIA (Calbiochem, La Jolla, CA) and the selective P-type Ca2+ channel blocker ω-agatoxin IVA (Calbiochem) were made up in concentrated aliquots and stored at -20°C before use. Direct application of the selective Ca2+ channels blockers onto the slice was made via a bolus injection into the perfusion in-line: (1) 100 μl from a 10 μm solution of ω-conotoxin GVIA, (2) a 1 mm solution of ω-agatoxin, or (3) a 10 μm solution of nimodipine. The estimated final, local bath concentration of (1) ω-conotoxin was 1 μm, (2) ω-agatoxin was 100 nm, and (3) nimodipine was 1 μm. Twenty seconds after the bolus infusion, inflow of ACSF was stopped for 3 min to allow the toxin to equilibrate in the bath and to take effect, after which the ACSF inflow resumed.
BAPTA (Sigma), PKC inhibitory peptide (PKCi[19-36]) (Calbiochem), and PKA inhibitory peptide (PKAi[5-24]) (Calbiochem) were included in the patch pipette. Dantrolene, caffeine (Sigma), N-[2-((o-bromocinamyl)amino)ethyl]-5-isoquinolinesulfonamide (H-89), and bisindoylmaleimide (BiM1) (Calbiochem) were made up fresh in ACSF and diluted to appropriate concentrations for the experiments.
Biocytin staining. After electrophysiological recording, PFC slices were immediately fixed for 1 hr with 4% paraformaldehyde (<2 week old; stored at 4°C) and then permeabilized overnight in phosphate buffer containing 0.1% Triton X-100. The slices were incubated in 0.3% (v/v) hydrogen peroxide for 30 min followed by a 60 min incubation in peroxidase-linked streptavidin diluted at 1:100. After extensive washing, the slices were visualized following the directions on a DAB kit (Vector Laboratories, Burlingame, CA). The slices were then wet-mounted on slides, allowed to air dry, and then coverslipped in DMSO before images were captured with a Zeiss microscope-CCD color Spot Insight camera (Diagnostic Instruments, Sterling Heights, MI) with a water-immersion 100× lens.
Data analyses. The peak amplitude and integrated area of Ca2+ potentials were measured using pClamp 8.0 software (Axon Instruments). Mean peak amplitude from each cell was averaged from 5-10 traces. For each neuron, Cd2+ was applied at the end of the experiment to block all Ca2+ potentials and reveal the membrane capacitance changes caused by the depolarizing pulse. For experiments using suprathreshold evoked Ca2+ spikes, the post-Cd2+ peak and integrated area were subtracted from the data. Group data measurements at 18-23 min were made after DHX application when the post-DHX response reached steady state. For experiments using subthreshold evoked Ca2+ hump potential, subtraction of the post-Cd2+ peak and area was not used. One-way ANOVA and post hoc Dunnett's or Tukey's tests were used to compare multiple group data with each other or with control group, and paired Student's t test was used to compare differences within-subject design group data. All group data were expressed as means ± SEM. Differences between control and experimental responses with p < 0.05 were deemed significant.
Results
Stable somatic whole-cell patch-clamp recordings (lasting 0.5-1 hr) were obtained from 141 neurons located in layers V-VI of the prelimbic and dorsal infralimbic regions of the PFC. Only PFC neurons with action potentials >70 mV, overshot 0 mV (before subsequent TTX applications), and resting membrane potentials more negative than -60 mV were included in the analysis. In this voltage range, there was a general lack of spontaneous firing in most PFC neurons recorded.
Electrophysiological characterization of high-threshold Ca2+ spikes
Suprathreshold depolarizing current pulses evoked either a fast single Ca2+ spike (lasting ∼100 msec) with occasional regenerative spike bursts (Fig. 2A,C) or a long-lasting Ca2+ spike (lasting >1 sec) (Fig. 2B,D). After application of TTX and TEA, the membrane potential was current clamped (via DC injection) continuously at -40 mV near the firing threshold of Ca2+ spikes. To determine the soma-dendritic site of electrogenesis of the evoked Ca2+ spikes, brief intrasomatic hyperpolarizing pulses (ISHPs) (-0.4 nA, 10 msec) were superimposed at different time points within the depolarizing pulse that was used to evoke the Ca2+ spikes (Fig. 2). Neurons were classified as having a soma-proximal-basal dendritic site of Ca2+ spike electrogenesis if the short ISHPs (-0.4 nA, 10 msec) could effectively terminate the evoked Ca2+ spike. In contrast, for neurons in which longer duration ISHPs (-0.4 nA, >20 msec) were needed to terminate the Ca2+ potential, the Ca2+ potential was likely generated at a more distal site from the somatic recording pipette. This latter type of neuron was classified as having a distal dendritic site of Ca2+ electrogenesis (Fig. 2). Of the 141 neurons tested, three-fourths (74%) had a proximal site of Ca2+ spike electrogenesis, whereas only approximately one-fourth of the neurons studied (26%) had a distal site of Ca2+ spike electrogenesis. These include both short-duration Ca2+ spikes and longer-duration post-Ca2+ spike plateau potentials (Fig. 2).
Figure 2.

Site of evoked Ca2+ spike electrogenesis determined by fast ISHPs. A, A short ISHP (-0.4nA, 10 msec) delivered in the midst of a suprathreshold depolarizing current pulse that previously evoked a robust Ca2+ spike, now effectively terminated the Ca2+ spike. This suggests that the Ca2+ spike was generated at a site relatively close to the somatic recording pipette. Such neurons were classified as having a “soma-proximal-basal dendritic site” of Ca2+ spike electrogenesis. B, In other neurons, longer-duration ISHPs (15-30 msec, -0.4 nA) delivered in the midst of a strong depolarizing current pulse were needed to terminate the Ca2+ spikes evoked. This suggests that the Ca2+ potential was generated relatively far from the somatic recording pipette. These neurons are classified as having a “distal site” of Ca2+ spike electrogenesis. C, D, In PFC neurons that elicit a prolonged Ca2+ plateau potential, in some (C), ISHPs (5-10 msec, -0.4 nA) abruptly terminate the Ca2+ plateau potentials (suggesting a “proximal site” of Ca2+ spike electrogenesis). On the other hand, there are also PFC neurons that can generate a prolonged Ca2+ plateau potential that failed to be terminated by strong and longer-duration ISHPs (D) (suggesting a distal site of Ca2+ spike electrogenesis).
To study the specific Ca2+ channel subtypes that mediate high-threshold Ca2+ spikes, the L-type-specific channel blocker nimodipine (1 μm) was bath-applied in TTX and TEA. Addition of nimodipine (eight of nine neurons) completely blocked the evoked Ca2+ spike, resulting in a voltage response resulting mainly from passive membrane responses to depolarizing current pulses. In the majority of the neurons studied, complete blockade of the high-threshold Ca2+ spikes was achieved after 10 min of nimodipine application, suggesting that the Ca2+ spikes recorded were mediated almost entirely by L-type Ca2+ channels.
Because blockade of L-type Ca2+ channels may also indirectly block other voltage-dependent Ca2+ channels, we performed additional experiments using the selective N-type Ca2+ blocker ω-conotoxin GVIA and the specific P-type Ca2+ channel blocker ω-agatoxin IVA. In five of five cells studied, addition of ω-conotoxin reduced the evoked Ca2+ spike to 80.7% of the control peak (an insignificant reduction of 19.3%; p > 0.05). Subsequent application of ω-agatoxin reduced the Ca2+ spike to 71.0% of the control peak (a reduction of 9.7% from the ω-conotoxin response; p > 0.05). Further addition of nimodipine reduced the attenuated spike to 33.9% of the original spike (a significant reduction of 37.1% from conotoxin plus agatoxin, or from control; p < 0.001), suggesting that the L-type Ca2+ channel comprises the largest component of the Ca2+ spike, followed by the N-type and P-type Ca2+ channels (Fig. 3).
Figure 3.

Pharmacological dissection to show that L-type Ca2+ channels comprise the major component of the evoked Ca2+ spike. A, B, Representative traces showing a successive reduction of the suprathreshold evoked Ca2+ spike after the sequential application of a selective N-type Ca2+ channel blocker ω-conotoxin GVIA (Cono; 1 μm) (A) and a selective P-type Ca2+ channel blocker ω-agatoxin IVA (Aga; 100 nm) (B). D, E, Subsequent application of a specific L-type Ca2 blocker nimodipine (Nimo; 1 μm) greatly suppressed the Ca2 spike, whereas further exposure to cadmium (200 μm), a selective blocker of Ca2 channels, completely blocked any residual component of the Ca2+ potential so that only the membrane capacitative response remained. F, Histograms of group data showing that L-type Ca2+ channels account for a majority of the Ca2+ spike, and this is followed by N- and then P-type Ca2+ channels. *p < 0.05; **p < 0.001.
We then tested whether the evoked suprathreshold Ca2+ spikes could “run down” over time. Control traces of the Ca2+ spike (evoked every 30 sec) were recorded over the course of 30-60 min from five neurons (Fig. 4B). The peak spike amplitude was unchanged, suggesting that the Ca2+ spikes did not run down over time in our slice preparation for at least 1 hr.
Figure 4.

D1/D5R activation attenuates suprathreshold intrasomatic current evoked Ca2+ spike. A, Representative voltage traces showing a reduction of the suprathreshold evoked Ca2+ spike after D1/D5R activation by dihydrexidine (1 μm). Note that after a large suppression of the evoked Ca2+ spike by DHX, further addition of Cd2+ (200 μm) blocked a residual component of the Ca2+ potential so that only the membrane capacitative response remained. B, Open circles show no change in the amplitude of the Ca2+ spikes (evoked every 30 sec by a 50 msec intracellular depolarizing current pulse) over a typical recording period of >30 min, suggesting that under our recording condition, there was no run down of the evoked Ca2+ spike potentials. Filled triangles show that D1/D5R activation by DHX (10 μm) induced a prolonged (>30 min) reduction of the evoked Ca2+ spike. C, Graphic plots show that DHX dose dependently suppressed the peak but had little effect on the integrated area of the evoked Ca2+ spikes. Cn, Control.
D1R activation suppresses suprathreshold Ca2+ spikes
Previous studies have already established that D1R, but not D2R, functionally regulates dendritic Ca2+ spikes (Yang and Seamans, 1996). We therefore focus on investigating the nature of this modulation. Brief exposure (5 min) of different bath concentrations of DHX (0.01, 0.1, 1, and 10 μm for 5 min) resulted in both a dose- and time-dependent change in the Ca2+ spikes (Fig. 4C). Group data showed that DHX at 1 and 10 μm significantly suppressed the peak amplitude of Ca2+ spikes from control (F(4,20) = 4.55; p < 0.009), with maximum reduction of Ca2+ amplitude occurring at ∼20 min post-DHX (Fig. 4B). We also found that evoked Ca2+ spike amplitude was suppressed by another D1/D5 receptor agonist (R-(+)SKF81297, 10 μm; n = 3; data not shown). These findings using patch-clamp recordings also replicated the findings from a previous sharp intracellular electrode study that used another D1 agonist, SKF38393 (Yang and Seamans, 1996).
To test that the DHX effect is mediated by D1/D5 receptors, the selective D1/D5 receptor antagonist SCH23390 (1 μm) was bath-applied during evoked Ca2+ spike recordings. SCH23390 alone did not change the suprathreshold evoked Ca2+ spike (Fig. 5A) but significantly blocked the suppressive action of DHX on the Ca2+ spike (n = 2) (Fig. 5B,D), suggesting that the suppressive action of DHX on suprathreshold evoked Ca2+ spikes is mediated by D1/D5 receptor activation.
Figure 5.
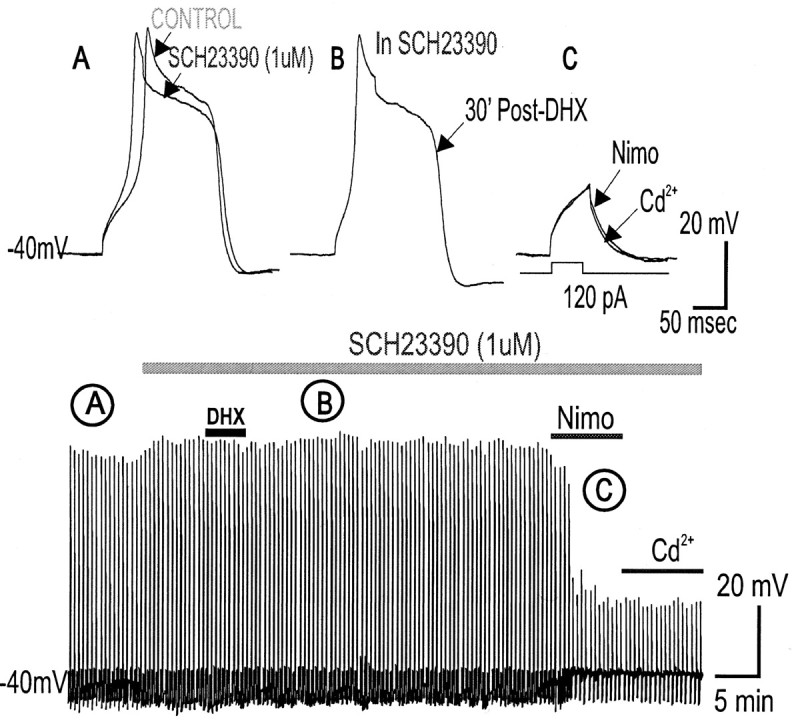
Blocking D1/D5 receptors with SCH23390 prevents DHX-mediated suppression of high-threshold Ca2+ spikes. A, Application of the selective D1/D5 receptor blocker SCH23390 (1 μm) alone did not change the suprathreshold evoked Ca2+ spiking properties (peak and area) in PFC neurons. B, D1/D5 receptor activation by DHX (10 μm) in SCH23390 failed to attenuate the Ca2+ spike, suggesting that DHX-mediated attenuation of suprathreshold evoked Ca2 spikes requires D1/D5R activation. C, Subsequent application of a specific L-type Ca2 blocker nimodipine (10 μm) suppressed the Ca2 spike, whereas further exposure to cadmium (200 μm), a selective blocker of Ca2 channels, blocked a residual component of the Ca2+ potential so that only the membrane capacitative response remained. The bottom time-compressed traces indicate the time course of the response. D, Histograms of group data showing that D1/D5R activation (DHX alone) suppresses the Ca2+ spike but DHX application in the presence of SCH23390 prevents the suppression, suggesting that DHX-mediated suppression of suprathreshold evoked Ca2+ spikes depends on D1/D5R activation. Nimo, Nimodipine.
In eight neurons studied, the L-type Ca2+ channel blocker nimodipine immediately reduced the suprathreshold current pulse evoked Ca2+ spike so that only the passive membrane response to the injected depolarizing pulse remained. This suggests that the high-threshold Ca2+ spike was generated primarily through L-type channels. In the continuous presence of nimodipine, when longer duration depolarizing pulses were injected, the Ca2+ spikes could be reinstated partially in only one of eight cells, suggesting that non-L-type channels are responsible for this reinstated component. Addition of DHX resulted in a suppression of this reestablished component, suggesting that D1/D5R activation also directly affects non-L-type Ca2+ channels (data not shown).
D1/D5R suppression of evoked Ca2+ spike is dependent on [Ca2+]i, but not on release of intracellular Ca2+ via IP3R of internal Ca2+ stores
Each evoked suprathreshold Ca2+ spike leads to a significant influx of Ca2+. One possible mechanism for D1 suppression of the suprathreshold Ca2+ spikes is that D1/D5R activation may augment [Ca2+]i, which in turn activates intracellular Ca2+-dependent mechanisms to cause functional attenuation of L-type Ca2+ channel activity. To test this hypothesis, the Ca2+ chelater BAPTA (5 mm) was included in the recording pipette to buffer [Ca2+]i levels. We found that 15 min after patching with a BAPTA-filled pipette, once a suprathreshold current pulse that could reliably evoke a Ca2+ spike was achieved, the ability of this depolarizing pulse to elicit a Ca2+ spike declined over time (Fig. 6A,C). The Ca2+ spike could be reinstated after injection of a higher amplitude current pulse, however, suggesting that BAPTA prevented a [Ca2+]i facilitation of suprathreshold Ca2+ spike maintenance over time. After several short rounds of this reestablishment of an evoked Ca2+ spike, a steady-state evoked Ca2+ spike could finally be achieved over time in the presence of internal BAPTA. Under this condition, the addition of DHX (10 μm) did not result in the D1/D5R suppression of the suprathreshold evoked Ca2+ spike for ≥20 min (n = 6) (Fig. 6B,D,E), suggesting that D1/D5R activation requires an intracellular Ca2+-dependent mechanism(s) to suppress the evoked high-threshold Ca2+ spike. Although the Ca2+ spike can be reestablished using stronger depolarizing pulse in the [BAPTA]i-alone condition (suggesting that BAPTA prevents a [Ca2+]i facilitation of L-type Ca2+ channels), Ca2+ spikes (in the absence of [BAPTA]i) cannot be reestablished after D1/D5 agonist application (suggesting that DHX may be acting through a different mechanism).
Figure 6.

Intracellular chelation by BAPTA prevented D1/D5R suppression of high-threshold Ca2+ spikes. A, Ca2+ spikes evoked by suprathreshold depolarizing pulses a using BAPTA-filled pipette exhibit attenuated responses over time. C, Vertical lines are Ca2+ spikes (displayed in slower chart speed) that show that an increase in current pulse intensity is necessary to reestablish the same Ca2+ spike. This suggests that there may be a constitutively active intracellular Ca2+-dependent facilitation of evoked Ca2+ spikes. B, D, After achieving a steady-state evoked Ca2+ spike over time, D1/D5R activation by DHX (10 μm) failed to suppress the evoked Ca2+ spike recorded by the BAPTA-filled electrode. E, Histograms summarizing group data that illustrate that the suppression of the evoked Ca2+ spike was blocked with steady-state intracellular Ca2+ chelation by BAPTA. This suggests that D1/D5R suppression of suprathreshold-evoked Ca2+ spikes is Ca2+ dependent.
Brain D1/D5R stimulation has been linked to activation of Gq-protein and subsequent increase in IP3 turnover (Wang et al., 1995; Jin et al., 2001). It is conceivable that D1R-Gq activation can suppress L-type Ca2+ channels through a phospholipase C (PLC)β-IP3-calcineurin signaling pathway, as shown in 5-HT2-Gq receptors in PFC neurons (Day et al., 2002). To test the possibility that D1/D5R stimulation leads to a mobilization of [Ca2+]i, which then activates intracellular mechanisms that inhibit L-type Ca2+ channels, we included heparin in the recording pipette to specifically block the release of Ca2+ via IP3R of internal Ca2+ stores. The addition of heparin did not prevent the D1/D5R-induced suppression of the Ca2+ spike (n = 3) (Fig. 7C), suggesting that the mechanism for D1/D5R suppression of the suprathreshold evoked Ca2+ spike is independent of [Ca2+]i release via IP3R of internal Ca2+ stores. Because heparin may have nonspecific effects other than blocking the release of Ca2+ from IP3R internal stores, we also used Xestospongin C, a selective IP3R blocker (De Smet et al., 1999). The addition of Xestospongin C also failed to prevent the D1/D5R-induced suppression of the Ca2+ spike (n = 3) (Fig. 7A-D), suggesting that the mechanism for D1/D5R suppression of the suprathreshold evoked Ca2+ spike is independent of release of Ca2+ from IP3R of internal Ca2+ stores and thus does not involve a PLCβ-IP3-calcineurin signaling pathway.
Figure 7.

Blocking intracellular Ca2+ release from IP3R store by Heparin or Xestospongic C does not change the D1/D5R-mediated suppression of suprathreshold Ca2+ spikes. A, Presence of the selective IP3R inhibitor Xestospongin C in the recording pipette did not change the suprathreshold evoked Ca2+ spiking properties in PFC neurons. B, D1/D5R activation by DHX (10 μm) in the presence of Xestospongin C did not prevent D1/D5R-mediated suppression of the suprathreshold evoked Ca2+ spike, suggesting that D1/D5R-mediated suppression of suprathreshold evoked Ca2+ spikes does not depend on intracellular Ca2+ release from IP3R stores. C, Time course of the evoked Ca2+ spike response to DHX in the presence of Xestospongin C. A and B represent the time point at which representative traces were taken above. D, Histograms of group data showing that D1/D5R activation still suppresses the Ca2+ spike in the presence of a nonselective IP3R inhibitor (heparin) or a specific IP3R inhibitor (Xestospongin C), thus suggesting that D1/D5R-mediated suppression of suprathreshold evoked Ca2+ spikes does not depend on intracellular Ca2+ release from IP3R stores.
D1/D5R suppression of evoked Ca2+ spike is dependent on PKC activation
To test whether D1/D5R activation suppresses L-type Ca2+ spikes through activation of Ca2+-dependent intracellular enzymes, we first looked at PKC, because PKA activation is known to potentiate (rather than suppress) neuronal L-type channels (Sculptoreanu et al., 1995; Surmeier et al., 1995). D1/D5R stimulation may activate PKC, which then inactivates the L-type Ca2+ channel by phosphorylating the channel (Obejero-Paz et al., 1998; McHugh et al., 2000). Bath application of the specific PKC inhibitor BiM1 (25 nm) alone did not change the suprathreshold evoked Ca2+ spike (Fig. 8A-C) but significantly blocked the suppressive action of DHX on the Ca2+ spike (n = 5) (Fig. 8G), suggesting that D1/D5R suppression of suprathreshold evoked Ca2+ spike depends on PKC activation.
Figure 8.

Inhibiting PKC activation by BiM1 or PKCi[19-36] prevented D1/D5R-mediated suppression of evoked suprathreshold Ca2+ spikes. A, Bath application of the selective PKC inhibitor BiM1 (25 nm) alone did not change the suprathreshold evoked Ca2+ spiking properties (peak and area) in PFC neurons. B, D1/D5R activation by DHX (10 μm) in the presence of BiM1 failed to attenuate the Ca2+ spike, suggesting that D1/D5R activation attenuates suprathreshold evoked Ca2 spikes through PKC activation. C, Subsequent application of a specific L-type Ca2 blocker nimodipine (10 μm) suppressed the Ca2 spike, whereas further exposure to cadmium (200 μm), a selective blocker of Ca2 channels, blocked a residual component of the Ca2+ potential so that only the membrane capacitative response remained. Bottom time-compressed traces indicate the time course of the response. D, Using PKCi [19-36] (10 μm) in the patch pipette did not change the suprathreshold evoked Ca2+ spiking properties in PFC neurons. E, D1/D5R activation by DHX (10 μm) with PKCi[19-36] (10 μm) in the patch pipette failed to attenuate the Ca2+ spike, suggesting that D1/D5R activation attenuates suprathreshold evoked Ca2 spikes through PKC activation. F, Subsequent application of a specific L-type Ca2 blocker nimodipine (10 μm) suppressed the Ca2 spike, whereas further exposure to Cd2+ (200 μm), a nonselective blocker of all Ca2 channels, blocked a residual component of the Ca2+ potential so that only the membrane capacitative response remained. Bottom time-compressed traces indicate the time course of the response. G, Histograms of group data showing that D1/D5R activation (DHX alone) suppresses the Ca2+ spike, but D1/D5R activation in the presence of a PKC inhibitor (BiM1) or PKCi[19-36] (10 μm) in the patch pipette prevents the suppression, suggesting that D1/D5R-mediated suppression of suprathreshold evoked Ca2+ spikes depends on PKC activation. *p < 0.05.
To ensure complete PKC inactivation, additional experiments included the PKC inhibitory peptide[19-36] (PKCi[19-36]; 10 μm) in the patch pipette that, by itself, did not change the suprathreshold evoked Ca2+ spike (Fig. 8D-F), but significantly blocked the suppressive action of DHX on the Ca2+ spike (n = 3) (Fig. 8G). Addition of the L-type Ca2+ blocker nimodipine still abolished the majority of the evoked Ca2+ spike (Fig. 8C,F). Additional application of Cd2+ (200 μm) showed a slight decrease in the remaining small Ca2+ spike peak, suggesting again that the spikes were recorded almost entirely from L-type Ca2+ channels (Fig. 8C,F). These findings suggest the D1 agonist may stimulate a D1-Gq-coupled receptor to activate PKC to suppress the suprathreshold Ca2+ spike.
D1R activation transiently potentiates subthreshold Ca2+ hump potentials
During the above experiments when we were testing the effects of D1 agonist on suprathreshold Ca2+ spikes, we observed that a subthreshold hump potential secondary to the primary evoked spike was transiently potentiated to the point of triggering full Ca2+ spikes in some neurons during the first few minutes of DHX application (Fig. 9). Nevertheless, although this potentiation of the subthreshold hump potential to a Ca2+ spike eventually subsided within ∼7 min, full suppression of the primary first suprathreshold Ca2+ spike occurred much later (Fig. 9A,B). This finding suggests that the D1/D5R activation led to a transient emergence and subsequent potentiation of a Ca2+ hump potential that occurs within a time window when primary full Ca2+ spikes are not yet fully suppressed.
Figure 9.

During suprathreshold depolarizations, D1/D5R activation transiently potentiates a secondary Ca2+ hump potential that led to spike firing. A, Representative voltage traces showing a single suprathreshold evoked Ca2+ spike (arrow). After D1/D5R activation, a Ca2+ hump potential emerges that trails the first Ca2+ spike. This Ca2+ hump potential eventually leads to spiking after 9′ post-D1/D5R activation (open arrow). Eventually, this potentiation subsides and completely disappears before the first Ca2+ spike is completely suppressed. B, Graphic representation of the peak amplitude of the first Ca2+ spike (filled circle) and the potentiated second Ca2+ spike (open circle) after brief D1/D5R activation by DHX for 5 min.
Seamans et al. (1997) first showed that during somatic or proximal dendrite stem recordings, a Ca2+ hump potential is evoked by intracellular current pulses subthreshold to triggering a full Ca2+ spike. An increase in sustained membrane depolarization, however, or a stepwise increase in the amplitude of the depolarizing current can eventually trigger a Ca2+ spike (Seamans et al., 1997). The hump potential was not mediated by a Na+ current (TTX insensitive), a T-type Ca2+ current (Ni2+ insensitive), or NMDA receptors (APV insensitive), but was generated by Cd2+-sensitive HVA Ca2+ channels electrotonically close to the soma (Seamans et al., 1997). This Ca2+ hump potential may have a functional role in amplifying incoming EPSPs en route from the apical dendrites to reach the soma. In this study, we found that nimodipine (1 μm) completely suppressed the Ca2+ hump potential (Fig. 10A), suggesting that L-type Ca2+ channel may also mediate the hump potential.
Figure 10.

D1/D5R activation transiently potentiates a Ca2+ hump potential triggered by subthreshold depolarizing pulses. A, Ca2+ hump potential evoked by subthreshold depolarizing pulses was blocked by the L-type Ca2+ channel antagonist nimodipine (1 μm), suggesting that the Ca2+ hump potential is mediated by a low-threshold L-type Ca2+ channel subtype. B-D, Time course of the effects of D1/D5R stimulation to subthreshold Ca2+ hump potential. Note that occasional Ca2+ spikes are observed in the control. Representative traces from the control are shown (B). After DHX application, subthreshold hump potential now elicited a full Ca2+ spike (C), and the frequency of occurrence of full Ca2+ spikes is increased transiently (for ∼15 min). This enhancement was gradually reduced over time. After the potentiation had subsided, only the membrane capacitative response was evoked by the subthreshold depolarizing pulse. Further addition of nimodipine (1 μm) and cadmium (200 μm) (D) did not change the profile of the evoked response, suggesting that the spikes were recorded entirely from L-type Ca2+ channels. E, Time-compressed traces indicate the time course of the response. F, There was no change in the membrane resistance that was continuously being monitored by a hyperpolarizing (-50 to -150 pA, 100 msec) prepulse before each weak depolarizing pulse delivered to evoke a subthreshold Ca2+ hump potential. G, Histograms of group data showing that D1/D5R activation potentiated the subthreshold evoked Ca2+ hump potential to Ca2+ spike firing, resulting in an increase in Ca2+ spike amplitude after 5 min of DHX application. *p < 0.05.
We then examined the effects of D1 agonist on the hump potential. Subthreshold depolarizing current pulse injection triggered, ∼10% of the time, full high-threshold Ca2+ spikes. After 5 min of DHX application, there was an increased probability of evoking a full Ca2+ spike by the same subthreshold pulses (Fig. 10B). Overall, the subthreshold Ca2+ hump potential was transiently increased by 37.9 ± 4.2% (peak) and 44.9 ± 9.5% (area), leading to the firing of full suprathreshold Ca2+ spikes (Fig. 10E). The duration of this full Ca2+ spike was also increased. Unlike the prolonged suppressive effects of the D1 agonist on suprathreshold Ca2+ spikes, the D1 potentiation of the subthreshold Ca2+ hump potential lasted for 7-15 min after DHX application (n = 8 of 9) and eventually returned back to passive membrane responses (Fig. 10B,E). One neuron had a long-lasting potentiation after DHX (>30 min). There was no overall change in the input resistance during and after the DHX (10 μm) application period (Fig. 10F), and thus it is unlikely that any increase of input resistance augmented the membrane voltage deflection to a sufficiently depolarized range to trigger the high-threshold post-DHX Ca2+ spikes. Furthermore, application of nimodipine resulted in an immediate block of the Ca2+ spike, suggesting that D1/D5R activation potentiated an L-type Ca2+ channel-mediated Ca2+ spike evoked by previously subthreshold depolarizing pulses (Fig. 10D,E).
Transient D1 potentiation of subthreshold Ca2+ hump is independent of a release of internal Ca2+ from intracellular Ca2+ stores or D1 blockade of K+ currents
D1/D5R activation may lead to a Ca2+-dependent facilitation (CDF) of L-type Ca2+ channels through an increased release of Ca2+ from internal stores. Bath-applied caffeine (20 μm) alone to release Ca2+ from internal [Ca2+]i stores failed to potentiate subthreshold evoked Ca2+ spike (n = 3; data not shown), suggesting that a release of Ca2+ from [Ca2+]i stores is ineffective in potentiating a Ca2+ hump potential to a Ca2+ spike.
Dantrolene is known to specifically block release of Ca2+ via ryanodine receptors of internal Ca2+ stores (Zhao et al., 2001). We tested whether dantrolene could block DHX potentiation of subthreshold Ca2+ hump potentials. After a 5 min application of DHX in the subthreshold evoked Ca2+ hump condition, when frequency and duration of the Ca2+ spike began to increase, dantrolene was introduced in two different ways: (1) direct fast application onto the slice via a bolus injection into the perfusion in-line (100 μl from a 0.5 mm solution of dantrolene, with estimated diluted, final, local bath concentrations of 20-60 μm) (Fig. 11A) or (2) included in the recording pipette (10 μm) (Fig. 11B). In both cases, the addition of dantrolene did not prevent the D1/D5R-induced potentiation of the Ca2+ spike, suggesting that D1/D5R potentiation of the subthreshold evoked Ca2+ spike is independent of release of Ca2+ via ryanodine receptor from internal Ca2+ stores (n = 6) (Fig. 11C).
Figure 11.

Blocking intracellular Ca2+ release from ryanodine receptor stores by dantrolene does not change the transient D1/D5R-mediated potentiation of the subthreshold evoked Ca2+ spike. A-D, Time course of subthreshold evoked Ca2+ potential in response to DHX with internal Ca2+ release is blocked by either focal (A) or bath application (C) of dantrolene. D1/D5R activation led to a transient increase in peak amplitude and integrated area of the resulting evoked Ca2+ spike, suggesting that release of Ca2+ from internal stores is not required for the potentiation of the subthreshold spike (nor for the inevitable termination of the potentiation). Representative traces are shown for focal (B) and bath (D)-applied dantrolene. Note: After DHX application, the apparent increase in the downward deflection of vertical lines (A, C) below -40 mV represents an increase in the afterhyperpolarizing potential immediately after the Ca2+ spikes. E, Histogram showing that both DHX alone and DHX with dantrolene resulted in increased Ca2+ peak amplitude after 5 min post-DHX application, suggesting that Ca2+ release from intracellular stores is not required for this D1/D5R mediated potentiation. *p < 0.05. NIMO, Nimodipine.
The activation of PKA or PKC can downregulate K+ channels by shifting the activation of the channels to more positive potentials, thus making the cell easily excitable (Hoffman and Johnston, 1998). Because D1/D5R activation has been shown to activate both PKA and PKC mechanisms (Surmeier et al., 1995; Chergui and Lacey, 1999; Wang et al., 2002), we wanted to explore whether the D1/D5R-mediated potentiation of subthreshold Ca2+ spikes occurred independent from any TEA-independent K+ channel modulation. We blocked most K+ channels by bath applying Cs+ (2 mm) and included Cs+ (10 mm) in the recording pipette. PFC neurons were voltage-clamped at -40 mV (in TTX and TEA), and low-intensity depolarizing voltage pulses (50 msec) were used to evoke small inward Ca2+ currents. After the 5 min DHX application in continuous Cs+, the small Ca2+ current transiently potentiated (n = 3; data not shown). The time course of occurrence of this transient Ca2+ current potentiation was similar to the potentiation of Ca2+ hump potential to Ca2+ spikes under current clamp. Because internal and external Cs+ (in TEA) would have blocked most K+ currents, these data illustrate that D1/D5R-mediated potentiation of Ca2+ spikes was independent of a D1/D5R blockade of TEA-insensitive K+ channels. Moreover, voltage-clamp control would have been adequate because electrophysiological and immunohistochemical evidence suggest that the somatic L-type Ca2+ channel is mediating this spike generation (Fig. 2) (Hell et al., 1993).
Transient D1/D5R potentiation of subthreshold Ca2+ potentials to spikes depends on PKA activation
D1/D5R activation alone has been shown in chromaffin cells and striatal neurons to potentiate L-type Ca2+ channels through cAMP and PKA pathways (Artalejo et al., 1990, 1992; Surmeier et al., 1995). To test whether this intracellular pathway also mediates D1/D5R action on L-type Ca2+ channels in PFC pyramidal neurons, we bath-applied a selective PKA inhibitor H-89 (10 μm). After application of H-89 alone, the subthreshold Ca2+ hump potential and the probability of firing of the occasional Ca2+ spikes were unchanged (n = 6). After DHX application in the continuous presence of H-89, however, the previous transient potentiation of the subthreshold Ca2+ potential responses and the increased probability of occurrence of full Ca2+ spikes to DHX no longer occurred (n = 4) (Fig. 12A-C). Because H-89 may have nonspecific effects other than blocking PKA activation, we included the PKA inhibitory peptide[5-24] (PKAi[5-24]; 10 μm)in the patch pipette. In this case, DHX application resulted in a much attenuated potentiation of the subthreshold evoked Ca2+ potentials (n = 2) (Fig. 12D-G). These data suggest that D1/D5R potentiation of the subthreshold evoked Ca2+ potentials to full Ca2+ spikes depends on PKA activation.
Figure 12.
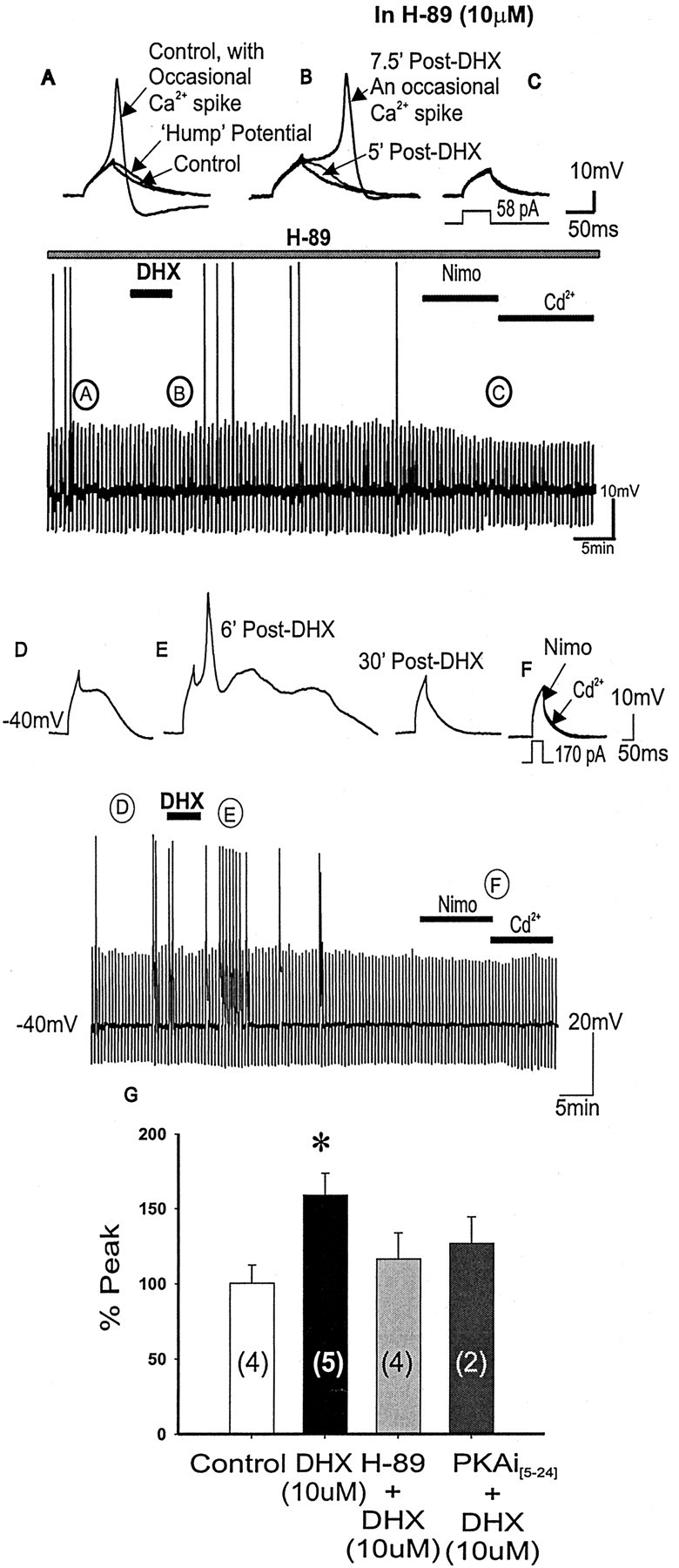
Blocking PKA activation by H-89 or PKAi[5-24] prevented D1/D5R-mediated enhancement of the evoked subthreshold Ca2+ hump potential. A-C, Representative traces corresponding to the continuous time-compressed traces of evoked subthreshold Ca2+ potentials or occasional evoked Ca2+ spikes with application of the specific PKA inhibitor H-89 alone. Subthreshold Ca2+ potentials or occasional Ca2+ spikes are observed in the control state (A), suggesting that spike firing properties are unchanged with H-89 (10 μm) application. After DHX application (B), the frequency of Ca2+ spikes is unchanged and no potentiation is observed, suggesting that D1/D5R potentiation of subthreshold Ca2+ hump potentials depends on PKA activation. Further addition of nimodipine and cadmium (C) resulted in membrane capacitive responses. Bottom time-compressed traces indicate the time course of the response. D-F, When using PKAi[5-24] (10 μm) in the patch pipette, application of DHX resulted in blocking most of the expected potentiation by the D1 agonist (E); however, a few hump potentials could still be potentiated to Ca2+ spikes (E). Further addition of nimodipine and cadmium (F) resulted in membrane capacitive responses. G, Histograms summarizing group data showing that although D1/D5R activation (with DHX alone) potentiated subthreshold Ca2+ plateaus, D1/D5R activation in the presence of PKA inhibitor H-89 or PKAi[5-24] (10 μm) in the patch pipette did not potentiate the Ca2+ potentials, suggesting that D1/D5R-mediated potentiation depends on PKA activation. The bottom time-compressed traces indicate the time course of the response.
Suppression of D1/D5R-PKA potentiated subthreshold evoked Ca2+ hump potential depends on PKC activation
As shown above, D1/D5R activation leads to a PKA-dependent potentiation of previously subthreshold Ca2+ potentials, resulting in a transient period of full Ca2+ spike firing triggered by a subthreshold depolarizing current pulse. On the basis of what we have shown above, that D1/D5R activation could also lead to a Ca2+-dependent PKC-mediated suppression of full Ca2+ spikes (evoked by suprathreshold depolarizing current pulses), we hypothesize that once enough Ca2+ influx is achieved during the PKA-mediated potentiation, a Ca2+-dependent PKC mechanism may terminate the D1/D5R-PKA potentiated Ca2+ spike firing.
To test this hypothesis, PKCi[19-36] (10 μm) was included in the patch pipette, and Ca2+ hump potentials were evoked by subthreshold depolarizing pulses (Fig. 13). PKCi[19-36] alone did not alter the subthreshold Ca2+ potential; however, after bath application of DHX, full Ca2+ spikes were evoked by the same subthreshold current pulses. Unlike the transient (∼7 min) Ca2+ spike firing responses in DHX without PKCi[19-36] (Fig. 10), the full Ca2+ spike firing persisted for >20 min (n = 4) (Fig. 13A-E), suggesting that termination of the transient D1/D5R-PKA potentiated subthreshold evoked Ca2+ potentials to full Ca2+ spike relies on a Ca2+-dependent PKC activation in the manner that we showed above (Fig. 8). The firing of full Ca2+ spikes evoked by previously subthreshold current pulses continued for as long as the recording until the application of nimodipine (1 μm), which immediately blocked the full Ca2+ spike (Fig. 13C), suggesting that D1/D5R potentiated an L-type Ca2+ channel-mediated Ca2+ spike. Hence, D1/D5R stimulation activates PKA to potentiate a subthreshold Ca2+ hump potential to full Ca2+ spike firing, which can then be switched off by a Ca2+-dependent PKC activation over time.
Figure 13.

When blocking PKC activation by PKC inhibitory peptide[19-36], D1/D5R stimulation on subthreshold Ca2+ hump potential resulted in a long-lasting enhancement of the hump potential to evoked sustained Ca2+ spike firing. A-D, Representative traces corresponding to the continuous time-compressed traces of evoked subthreshold Ca2+ potentials or occasional evoked Ca2+ spikes with the specific PKC inhibitory peptide[19-36] in the patch pipette (D). After DHX application (A), the frequency of occurrence of evoked Ca2+ spikes is increased through an increase in the area of the hump potential, creating two Ca2+ spikes. Multiple Ca2+ spikes ride on the potentiated hump potential (B), resulting in four Ca2+ spikes. The potentiation is long lasting (>20 min), and the spike is blocked by addition of nimodipine (C), suggesting that the spikes were recorded from L-type Ca2+ channels. Further addition of cadmium blocked a small residual component of the Ca2+ potential so that only the membrane capacitative response remained, suggesting that the spikes were recorded mainly from L-type Ca2+ channels. E, Histograms summarizing group data showing that in the presence of PKCi[19-36], the D1/D5R-mediated potentiation of the subthreshold Ca2+ hump potentials begins ∼5 min post-DHX and led to a long-lasting enhancement and spike firing past 30 min post-DHX.
Discussion
The results from this study showed the following. (1) High-threshold evoked Ca2+ spikes generated from soma-basal-proximal dendrites (readily terminated by brief intrasomatic hyperpolarizing pulses) are mediated by nimodipine-sensitive L-type Ca2+ channels; (2) Suprathreshold Ca2+ spike firing is dependent on an intracellular Ca2+-dependent facilitation of Ca2+ channel activity because intracellular BAPTA suppresses these suprathreshold Ca2+ spike firings; (3) D1/D5R stimulation failed to suppress steady-state suprathreshold Ca2+ spike firing when intracellular Ca2+ was buffered by BAPTA or when PKC activity was inhibited by BiM1 or PKC inhibitory peptide[19-36]. (4) Subthreshold Ca2+ hump potential was transiently potentiated by a D1/D5R-PKA mechanism because both the PKA inhibitor H-89 and PKA inhibitory peptide[5-24] block the D1/D5R-mediated potentiation of the hump potential. This potentiation does not rely on intracellular Ca2+ release because dantrolene failed to block the D1/D5R-mediated potentiation. (5) D1/D5R-mediated potentiation was not dependent on a D1/D5R modulation of K+ channels because intracellular Cs+ failed to block the D1/D5R-mediated potentiation of subthreshold Ca2+ hump potentials. (6) When PKC was inhibited by bath-applied BiM1 or internally by PKC inhibitory peptide[19-36], the D1/D5R-PKA mechanism potentiated the subthreshold Ca2+ hump potentials to sustained Ca2+ spike firing.
Dendritic Ca2+ spike electrogenesis in rat PFC pyramidal neurons
The Ca2+ spikes and the post-spike Ca2+ plateau are activated in topographically different sites of electrogenesis in multiple dendrites and their branches in single pyramidal PFC neurons. On the basis of the vulnerability of the HVA Ca2+ spike to injection of brief hyperpolarizing current pulses, two major areas of Ca2+ electrogenesis, with one located near the soma and another at a considerable electrotonic distance (e.g., up to 600 μm) from the soma, have been implicated (Reuveni et al., 1993; Yuste et al., 1994; Schiller et al., 1997). Our data are consistent with the Ca2+ spike being generated at, or very near, the soma where the recording was made (78%), with a minority group (22%) of neurons having a “distal” site of Ca2+ electrogenesis (i.e., cannot be terminated by brief large-amplitude intrasomatic hyperpolarizing pulses). This finding is also consistent with localization data showing a somatic location of L-type Ca2+ channels (Westenbroek et al., 1992; Hell et al., 1993).
L-type subunit expression in developing and mature rat brain
For the development of the key subunits of the L-type Ca2+ channels (the α1 subunits that associate with multiple β subunits), it is notable that by P14 onward, the expression of α1 [specifically, the α1C and α1D that form the pore of L-type Ca2+ channels (Chin et al., 1992; Tanaka et al., 1995; Ludwig et al., 1997)] and β subunits [such as the β2a subunit that plays a role in the formation and targeting of functional L-type Ca2+ channels to the membrane surface (Chien et al., 1995; Gao et al., 1999)] may have achieved adult levels for functional L-type Ca2+ channels. These findings suggest that the brains of the juvenile rats (P20-24) that we used have a complete complement of the adult L-type Ca2+ channels.
D1/D5R suppression of a suprathreshold evoked L-type Ca2+ spike
The suprathreshold Ca2+ spikes shown in this study are primarily L-type Ca2+ channel mediated because the dihydropyridine channel blocker nimodipine abolished them, leaving a small (<10%) residual component that can be completely abolished by Cd2+. Although our protocol of holding the membrane at a depolarized potential (VHold = -40 mV) will inactivate most of the N- and T-type Ca2+ channels and allow the study of the L-type Ca2+ channel (Fox et al., 1987), the addition of the specific N-type Ca2+ blocker ω-conotoxin still reduced the suprathreshold evoked Ca2+ spike by 19.3%. Further reductions were seen after subsequent application of the specific P-type Ca2+ blocker ω-agatoxin (by 9.7%) and nimodipine (by 37.1%), suggesting that although most of the suprathreshold evoked Ca2+ spike is attributable to L-type Ca2+ channels, a smaller but evident portion of the spike is composed of N-type Ca2+ channels with marginal contribution by P-type Ca2+ channels.
D1 receptor stimulation induced different changes to current-evoked dendritic Ca2+ potentials in neocortical layers V-VI neurons from rat PFC. In Ca2+ spikes evoked by suprathreshold currents, D1/D5R activation resulted in a dose-dependent reduction in the peak amplitude of the nimodipine-sensitive Ca2+ spike, suggesting that D1/D5R activation suppresses activation of high voltage-activated L-type Ca2+ channels. This suppressive action by D1/D5R agonist on the suprathreshold Ca2+ spikes in this study is consistent with a DA-mediated suppression of high-threshold Ca2+ currents in other vertebrate and invertebrate neurons (Paupardin-Tritsch et al., 1985; Marchetti et al., 1986; Williams et al., 1990; Nussinovitch and Kleinhaus, 1992; Surmeier et al., 1995; Yang and Seamans, 1996).
Influx of Ca2+ ions via L-type channels is known to exert profound feedback effects on the subsequent opening and closing of the channel via Ca2+ interaction at the pore-forming α1C subunit of the L-type Ca2+ channel (McDonald et al., 1994; Dolphin, 1996). When using BAPTA to buffer internal Ca2+, we found that the evoked Ca2+ spike firing was reduced by BAPTA, suggesting that a constitutively active CDF of the evoked Ca2+ spikes occurs. After a gradual increment of the depolarizing current pulse amplitude, steady-state evoked Ca2+ spikes can eventually be reestablished and maintained. Under this condition, D1/D5R activation now failed to suppress the dendritic Ca2+ potential, suggesting that the action of D1/D5R is [Ca2+]I dependent and may be caused by a D1/D5R suppression of the CDF that constitutively facilitates Ca2+ spike firing. Consistent with this finding is the report by Perrier et al. (2000) who found in turtle motoneurons that L-type Ca2+ plateau potentials are selectively inhibited when [Ca2+]i is chelated by intracellular BAPTA. Because the D1/D5R-mediated suppression of L-type channels is [Ca2+]i dependent, studies that use barium (to substitute for Ca2+) and BAPTA (to buffer internal Ca2+) to study dopamine actions on L-type channels and neuronal excitability are problematic because such D1/D5R-mediated effects would be masked by such approaches.
To test whether D1/D5R activation suppresses L-type Ca2+ spikes through activation of Ca2+-dependent intracellular enzymes, we first looked at PKC, because PKA activation is known to potentiate (rather than suppress) neuronal L-type channels (Sculptoreanu et al., 1995; Surmeier et al., 1995). Inhibition of the L-type Ca2+ spikes may be mediated through a PKC pathway because we found that the D1/D5R-mediated suppression of the Ca2+ spike was blocked by a PKC inhibitor (BiM1) or PKC inhibitory peptide[19-36]. PKC activation decreases cardiac and neuronal L-type Ca2+ potentials and currents (Doerner et al., 1988; Zhang et al., 1997) or results in a biphasic change (increase followed by a decrease) in Ca2+ current amplitude (Lacerda et al., 1988; Boixel et al., 2000). Two N-terminal isoforms (a long- and a short-NT isoform) of α1c subunit of the L-type Ca2+ channel are known (Snutch et al., 1991). Through patch-clamp recording techniques and site-directed mutagenesis, McHugh et al. (2000) has found that Thr27 and Thr31 of the α1c long-NT domain of the Ca2+ channel are two sites required for PKC-mediated inhibition of the L-type Ca2+ channel current. The α1c protein containing the long-NT isoform is found in both heart and brain of rats (Shistik et al., 1999). Whether these two specific sites on the L-type Ca2+ channel are phosphorylated by PKC in PFC neurons to cause changes in Ca2+ current is not known.
Although D1R is known to be coupled physically and functionally to a Gs-protein (Kimura et al., 1995; Sidhu and Kimura, 1997), coimmunoprecipitation and receptor-binding studies indicate that D1/D5R receptors are associated with both Gs- and Gq-proteins in the frontal cortex (Jin et al., 2001), opening the possibility that PFC D1/D5R sites may also activate phospholipase C through Gq-proteins. Recently, an intermediate molecule, calcyon, was discovered to functionally couple D1R with Gq and may aid to couple D1R to other functionally important intracellular proteins such as PKC (Lezcano et al., 2000). Functional coupling of calcyon-D1R-Gq requires “priming” through an increase of intracellular [Ca2+]i. Our suprathreshold Ca2+ spike activation may ensure an influx of Ca2+ to activate the actions of calcyon. Under this condition, D1/D5R activation could lead to a calcyon-D1R-Gq coupling to activate PLC and subsequently PKC stimulation, which resulted in L-type channel inactivation in PFC neurons.
D1/D5R potentiation of subthreshold evoked L-type Ca2+ current
The subthreshold Ca2+ potential shown in this study is primarily L-type Ca2+ channel mediated because the dihydropyridine channel blocker nimodipine abolished it (see Results) (Fig. 9A). Weak dendritic depolarization frequently evokes Ca2+ hump potentials but is subthreshold to evoking a suprathreshold Ca2+ spike (Seamans et al., 1997). We found that D1/D5R activation leads to a transient augmentation of the Ca2+ hump potentials to a full Ca2+ spike that typically lasts for 7-15 min. Dopamine in retinal horizontal cells has also been shown to transiently (lasting ∼7 min) facilitate P- and L-type Ca2+ currents, and this is mediated by a cAMP-second messenger system (Pfeiffer-Linn and Lasater, 1996). D1/D5R activation may recruit L-type Ca2+ channels as described in bovine chromaffin cells. These channels are normally quiescent but can be activated by a large depolarizing prepulse or by repetitive depolarization in the physiological range (Artalejo et al., 1992).
PKA activation is known to be necessary for potentiation of neuronal L-type channels (Sculptoreanu et al., 1995; Surmeier et al., 1995). D1/D5 receptor activation alone can potentiate L-type Ca2+ channels in the absence of prepulses or repetitive activity, and this activation by D1/D5R agonists is mediated by cAMP-dependent protein kinase A as shown in chromaffin cells (Artalejo et al., 1990, 1992). Recent findings using single-cell PCR, immunocytochemistry, and D1R knock-out mice show that D5, and not D1, receptors potentiate burst firing in subthalamic neurons through a PKA-mediated modulation of L-type Ca2+ conductance (Baufreton et al., 2003). In the present study, we found that the PKA inhibitor H-89 and the PKA inhibitory peptide[5-24] blocked the D1/D5R agonist potentiation of the L-type Ca2+ hump potentials to Ca2+ spike firing. During intracellular delivery of subthreshold depolarizing pulses, when PKC is blocked, D1/D5 agonist application led to sustained evoked Ca2+ spiking that arose from potentiated Ca2+ hump potentials. On the other hand, when PKC is blocked, during suprathreshold depolarizing pulses, repetitive evoked Ca2+ spiking masks any possible D1/D5 receptor-mediated PKA potentiation of subthreshold hump potentials. These data suggests that PKA activation by D1/D5R stimulation critically mediates D1/D5R potentiation of subthreshold L-type Ca2+ potentials. Site-directed mutagenesis, combined with electrophysiological recordings, showed that Ser1901 in the α1c subunit of the Ca2+ channel is the site required for PKA-mediated enhancement of the L-type Ca2+ channel current (Naguro et al., 2001). Hence, it is tempting to suggest that D1/D5R activation led to PKA activation and subsequent phosphorylation of Ser1901 in an α1c subunit of L-type Ca2+ channel to cause augmentation of channel conductance that led to full Ca2+ spike firing in PFC neurons.
A bi-directional dopamine D1/D5R modulation Of state-dependent switching of soma-dendritic Ca2+ potentials
A major finding of this paper is that D1/D5 receptor stimulation activates PKA to potentiate a subthreshold L-type Ca2+ hump potential (Fig. 14A,B), thus leading to L-type Ca2+ spike firing, which then can be terminated by a D1/D5R-mediated Ca2+-dependent PKC suppression of L-type Ca2+ channels over time (Fig. 14A,C). Functionally, soma-dendritic populations of L-type Ca2+ channels can be modulated spatially and temporally by a depolarizing state-dependent D1/D5R modulation of PKA-PKC pathways to integrate incoming synaptic signals.
Figure 14.

Schematic model that illustrates the state-dependent bi-directional D1/D5R modulation of L-type Ca2+ potentials through PKA potentiation and PKC suppression. Functional incoming synaptic signals to the distal dendrites and reaching the soma represent numerous temporally and spatially contiguous synaptic signals from diverse inputs. Such synaptic signals can be categorized as either strong or weak inputs, evoking suprathreshold Ca2+ spikes and plateaus or subthreshold hump potentials, respectively. A critical frequency of back-propagating Na+ spikes can evoke large, regenerative, voltage-gated Ca2+ spikes in the distal dendritic initiation zone (Schiller et al., 1997; Larkum et al., 1999a), whereas a single back-propagating Na+ spike generated in the axon facilitates the initiation of voltage-gated Ca2+ spikes when it is coincidental with synaptic input to the same distal dendritic site (Larkum et al., 1999b). For synaptic integration, the timing and location of evoked voltage-gated Ca2+ responses in dendrites by strong or weak inputs is important (Oakley et al., 2001). Nevertheless, an active participation of dendritic Ca2+ in amplifying distal inputs may occur mainly when powerful repolarizing dendritic K+ channels are suppressed (Gonzalez-Burgos and Barrionuevo, 2001). Dopamine D1/D5 receptor activation is known to suppress several subtypes of soma-dendritic K+ currents (Kitai and Surmeier, 1993; Nisenbaum et al., 1998; Dong and White, 2003), thus enabling the actions of dendritic Ca2+ potential to contribute actively in synaptic signal amplification and integration. A, Weak synaptic inputs lead to weak dendritic depolarizations that frequently evoke Ca2+ hump potentials that are subthreshold to evoking a suprathreshold Ca2+ spike (Seamans et al., 1997). B, D1/D5R activation results in a transient amplification or “boost” of the weak synaptic signal through a D1R-Gs-PKA pathway that potentiates L-type Ca2+ channels over time. C, The D1/D5R-PKA potentiation of the Ca2+ hump led to full Ca2+ spike firing via L-type Ca2+ channel activation in soma-proximal-basal dendrites. This Ca2+ spike firing is temporary, however, because it is eventually suppressed as greater Ca2+ influx activates a Ca2+-dependent D1-Gq-calcyon-PKC suppression of the L-type Ca2+ channel, thereby reducing Ca2+ influx (A). D, On the other hand, incoming strong synaptic inputs or back-propagating Na+ spikes can evoke full suprathreshold Ca2+ spikes. Optimal stimulation of D1/D5R via a [Ca2+]i-dependent D1-Gq-calcyon-PKC mechanism may lead to a differential suppression of the L-type Ca2+ channel-mediated Ca2+ spikes in various regions of the dendrites. This may serve functionally to “sharpen” incoming synaptic signals before they are integrated in the soma. Strong stimulation of D1/D5 receptor leads to severe suppression of dendritic Ca2+ spikes and may serve to prevent any synaptic signals from being integrated or amplified.
Strong synaptic input or back-propagated Na+ spikes can evoke L-type Ca2+ spikes in soma-proximal-basal dendritic branches (Stuart et al., 1997; Svoboda et al., 1997; Helmchen et al., 1999; Larkum et al., 1999b). Moderate D1/D5R stimulation may attenuate the signal-amplifying actions of dendritic suprathreshold Ca2+ spikes, and the synaptic signals arriving at multiple dendrites may be differentially suppressed spatially. This can result in the soma receiving an optimal level of synaptic signals, whereas other signals will fail to be integrated by the soma (Fig. 14, SIGNAL SHARPENING). A very strong D1/D5R attenuation of the dendritic Ca2+ mechanism may also contribute in part to the inverted “U” dose-response curve of the effects of D1 agonists on PFC-mediated cognitive performance when a large portion of dendritic Ca2+ channels are functionally suppressed by D1/D5 receptor stimulation for a prolonged period of time (Yang and Seamans, 1996; Zahrt et al., 1997; Goldman-Rakic et al., 2000). Furthermore, D1/D5R activation may act to modulate the ability of the neuron to generate voltage-gated Ca2+-dependent regenerative potentials in a use-dependent manner and thereby also determine optimal intracellular Ca2+ mechanisms in the induction mechanism for long-term depression (LTD) (180-500 nm [Ca2+]i) and long-term potentiation (LTP) (>500 nm [Ca2+]i) (Cormier et al., 2001). Conceivably, D1/D5R-PKC activation can lead to a suppressed Ca2+ entry, resulting in LTD, whereas D1/D5R-PKA activation can increase the dendritic Ca2+ entry and lead to LTP induction (for long-term memory consolidation).
Differential voltage-dependent DA modulation of neuronal excitability exists in striatal and PFC neurons. When held at either approximately -87 mV (mimicking a “down” state) or approximately -57 mV (mimicking an “up” state of striatal or PFC neurons), D1/D5R activation (by SKF81297) or intracellular elevation of cAMP also enhanced the depolarizing EPSPs or neuronal excitability (Hernandez-Lopez et al., 1997; Lewis and O'Donnell, 2000; Lavin and Grace, 2001). In the context of the findings in our present study, we found that the depolarizing states by which dendritic Ca2+ potentials are activated (i.e., Ca2+ hump or Ca2+ spike, both triggered at -40 mV) determined the direction that they are going to be switched by dopamine. D1/D5 receptor stimulation can boost subthreshold EPSPs via potentiating a subthreshold Ca2+ hump potential (Fig. 14A-C), especially when K+ currents are suppressed, e.g., via D1/D5 receptor stimulation (Kitai and Surmeier, 1993; Yang and Seamans, 1996; Hoffman and Johnston, 1998; Nisenbaum et al., 1998; Gonzalez-Burgos and Barrionuevo, 2001; Dong et al., 2002; Dong and White, 2003).
The potentiation of Ca2+ hump potentials to Ca2+ spike firing by a D1-PKA mechanism may boost incoming weak synaptic inputs to threshold for firing Na+ and Ca2+ dendritic spikes. The modulation of subthreshold Ca2+ potentials by DA may coincidentally increase the responsiveness of these neurons to NMDA-mediated synaptic responses (Galarraga et al., 1997; Hernandez-Lopez et al., 1997; Cepeda et al., 1998; Seamans et al., 2001; Wang and O'Donnell, 2001). Because L-type Ca2+ channels and NMDA receptors enable a large influx of cationic currents at depolarized potentials, the modulation of these currents by DA may allow PFC neurons to be more responsive to excitatory inputs when they are in a depolarized state and also provide sufficient [Ca2+]i for LTP induction.
After D1 stimulation, as repetitive Na+ and Ca2+ spike firing are induced, the slow D1-PKC mechanism will attenuate the Ca2+ spikes effectively to limit the duration of persistent Ca2+ spike bursts (Fig. 14D). Because calcyon functionally couples Gq to D1R in dendrites, an upregulation of calcyon (Koh et al., 2003) in schizophrenic PFC neurons can lead to D1-PKC hyperfunction, resulting in a hypothesized severe suppression of dendritic Ca2+ potentials and disruptions of normal synaptic signal amplification mechanisms. Hence, the bi-directional nature of the D1/D5 receptor-mediated dual PKC-PKA modulation of somadendritic L-type Ca2+ spike potentials may mean that state-dependent D1/D5R activation could shift the PKC-PKA equilibrium and thereby influence overall PFC functions, impacting both normal behavior and neuropsychiatric states.
Note added in proof. In a recent study published after our galley proof submission, Gulledge and Stuart (2003) showed that in the apical dendrites and tufts (up to 480 μm from the soma) of layer V pyramidal neurons of juvenile Wistar rats, no Cd2+-sensitive Ca2+ spikes could be evoked by strong distal synaptic stimulation or by back-propagating Na+ spikes. They found that DA (10-100 μm) failed to suppress imaged distal dendritic Ca2+ signal triggered by single or multiple back-propagated spikes. It is notable that the investigators did not use any blockers of dendritic K+ channels to remove strong inactivation of the small (but present) dendritic Ca2+ spikes by K+ currents (Seamans et al., 1997; Bekkers, 2000; Johnston et al., 2003). Moreover, a BAPTA-based Ca2+ sensor (Oregon Green BAPTA) was used to capture imageable dendritic Ca2+ signals for their DA modulation study. As shown in our data from the present study, an intracellular Ca2+-dependent PKC activation is crucial for mediating D1 receptor suppression of soma-dendritic Ca2+ spike. If this soma-dendritic D1 mechanism is applicable to distal dendrites and tufts, our data may suggest that the lack of K+ channel blockade (resulting in no evoked Ca2+ spike) and the use of Oregon Green BAPTA that captures most functional intracellular Ca2+ (resulting in no PKC activation) in the study by Gulledge and Stuart (2003) may account for the reasons why they failed to evoke distal dendritic Ca2+ spikes and DA modulation of distal dendritic Ca2+ signaling.
Footnotes
C.E.Y. is the recipient of an Eli Lilly postdoctoral fellowship. We appreciate the feedback and suggestions from Drs. Jeremy Seamans and Emmanuel Sher on an earlier draft of this manuscript.
Correspondence should be addressed to Charles R. Yang, Neuroscience Discovery, Eli Lilly & Company, Lilly Corporate Center, Indianapolis, IN 46285-0510. E-Mail: cyang@lilly.com.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240008-16$15.00/0
References
- Artalejo CR, Ariano MA, Perlman RL, Fox AP (1990) Activation of facilitation calcium channels in chromaffin cells by D1 dopamine receptors through a cAMP/protein kinase A-dependent mechanism. Nature 348: 239-242. [DOI] [PubMed] [Google Scholar]
- Artalejo CR, Rossie S, Perlman RL, Fox AP (1992) Voltage-dependent phosphorylation may recruit Ca2+ current facilitation in chromaffin cells. Nature 358: 63-66. [DOI] [PubMed] [Google Scholar]
- Baufreton J, Garret M, Rivera A, de la Calle A, Gonon F, Dufy B, Bioulac B, Taupignon A (2003) D5 (not D1) dopamine receptors potentiate burst-firing in neurons of the subthalamic nucleus by modulating an L-type calcium conductance. J Neurosci 23: 816-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM (2000) Distribution and activation of voltage-gated potassium channels in cell-attached and outside-our patches from large layer 5 cortical pyramidal neurons of the rat. J Physiol (Lond) 525: 611-620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger B, Gaspar P, Verney C (1991) Dopaminergic innervation of the cerebral cortex: unexpected differences between rodents and primates. Trends Neurosci 14: 21-27. [DOI] [PubMed] [Google Scholar]
- Boixel C, Tessier S, Pansard Y, Lang-Lazdunski L, Mercadier JJ, Hatem SN (2000) Tyrosine kinase and protein kinase C regulate L-type Ca(2+) current cooperatively in human atrial myocytes. Am J Physiol 278: H670-676. [DOI] [PubMed] [Google Scholar]
- Brown AM, Schwindt PC, Crill WE (1993) Voltage dependence and activation kinetics of pharmacologically defined components of the high-threshold calcium current in rat neocortical neurons. J Neurophysiol 70: 1530-1543. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Colwell CS, Itri JN, Chandler SH, Levine MS (1998) Dopaminergic modulation of NMDA-induced whole cell currents in neostriatal neurons in slices: contribution of calcium conductances. J Neurophysiol 79: 82-94. [DOI] [PubMed] [Google Scholar]
- Chergui K, Lacey MG (1999) Modulation by dopamine D1-like receptors of synaptic transmission and NMDA receptors in rat nucleus accumbens is attenuated by the protein kinase C inhibitor Ro 32-0432. Neuropharmacology 38: 223-231. [DOI] [PubMed] [Google Scholar]
- Chien AJ, Zhao X, Shirokov RE, Puri TS, Chang CF, Sun D, Rios E, Hosey MM (1995) Roles of a membrane-localized beta subunit in the formation and targeting of functional L-type Ca2+ channels. J Biol Chem 270: 30036-30044. [DOI] [PubMed] [Google Scholar]
- Chin H, Smith MA, Kim HL, Kim H (1992) Expression of dihydropyridine-sensitive brain calcium channels in the rat central nervous system. FEBS Lett 299: 69-74. [DOI] [PubMed] [Google Scholar]
- Cormier RJ, Greenwood AC, Connor JA (2001) Bidirectional synaptic plasticity correlated with the magnitude of dendritic calcium transients above a threshold. J Neurophysiol 85: 399-406. [DOI] [PubMed] [Google Scholar]
- Day M, Olson PA, Platzer J, Striessnig J, Surmeier DJ (2002) Stimulation of 5-HT(2) receptors in prefrontal pyramidal neurons inhibits Ca(v)1.2 L type Ca(2+) currents via a PLCbeta/IP3/calcineurin signaling cascade. J Neurophysiol 87: 2490-2504. [DOI] [PubMed] [Google Scholar]
- De Smet P, Parys JB, Callewaert G, Weidema AF, Hill E, De Smedt H, Erneux C, Sorrentino V, Missiaen L (1999) Xestospongin C is an equally potent inhibitor of the inositol 1,4,5-trisphosphate receptor and the endoplasmic-reticulum Ca(2+) pumps. Cell Calcium 26: 9-13. [DOI] [PubMed] [Google Scholar]
- Doerner D, Pitler TA, Alger BE (1988) Protein kinase C activators block specific calcium and potassium current components in isolated hippocampal neurons. J Neurosci 8: 4069-4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (1996) Facilitation of Ca2+ current in excitable cells. Trends Neurosci 19: 35-43. [DOI] [PubMed] [Google Scholar]
- Dong Y, White FJ (2003) Dopamine d1-class receptors selectively modulate a slowly inactivating potassium current in rat medial prefrontal cortex pyramidal neurons. J Neurosci 23: 2686-2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Nasif F, Cooper DC, Hu X-T, White FJ (2002) Repeated cocaine administration alters dopamine modulation of K+ current in rat prefrontal cortex pyramidal neurons. Soc Neurosci Abstr 28: 898.9. [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW (1987) Single-channel recordings of three types of calcium channels in chick sensory neurones. J Physiol (Lond) 394: 173-200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galarraga E, Hernandez-Lopez S, Reyes A, Barral J, Bargas J (1997) Dopamine facilitates striatal EPSPs through an L-type Ca2+ conductance. NeuroReport 8: 2183-2186. [DOI] [PubMed] [Google Scholar]
- Gao T, Chien AJ, Hosey MM (1999) Complexes of the alpha1C and beta subunits generate the necessary signal for membrane targeting of class C L-type calcium channels. J Biol Chem 274: 2137-2144. [DOI] [PubMed] [Google Scholar]
- Garey LJ, Ong WY, Patel TS, Kanani M, Davis A, Mortimer AM, Barnes TR, Hirsch SR (1998) Reduced dendritic spine density on cerebral cortical pyramidal neurons in schizophrenia. J Neurol Neurosurg Psychiatry 65: 446-453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA (2000) Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57: 65-73. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS (1998) The cortical dopamine system: role in memory and cognition. Adv Pharmacol 42: 707-711. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Muly III EC, Williams GV (2000) D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev 31: 295-301. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Barrionuevo G (2001) Voltage-gated sodium channels shape subthreshold EPSPs in layer 5 pyramidal neurons from rat prefrontal cortex. J Neurophysiol 86: 1671-1684. [DOI] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ (2003) Action potential initiation and propagation in layer 5 pyramidal neurons of the rat prefrontal cortex: absence of dopamine modulation. J Neurosci 23: 11363-11372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser M, Spruston N, Stuart GJ (2000) Diversity and dynamics of dendritic signaling. Science 290: 739-744. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA (1993) Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 123: 949-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmchen F, Svoboda K, Denk W, Tank DW (1999) In vivo dendritic calcium dynamics in deep-layer cortical pyramidal neurons. Nat Neurosci 2: 989-996. [DOI] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Bargas J, Surmeier DJ, Reyes A, Galarraga E (1997) D1 receptor activation enhances evoked discharge in neostriatal medium spiny neurons by modulating an L-type Ca2+ conductance. J Neurosci 17: 3334-3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman D, Chen S, Aung TT, Cherksey B, Sugimori M, Llinas RR (1991) Localization of P-type calcium channels in the central nervous system. Proc Natl Acad Sci USA 88: 7076-7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Johnston D (1998) Downregulation of transient K+ channels in dendrites of hippocampal CA1 pyramidal neurons by activation of PKA and PKC. J Neurosci 18: 3521-3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LQ, Wang HY, Friedman E (2001) Stimulated D(1) dopamine receptors couple to multiple Galpha proteins in different brain regions. J Neurochem 78: 981-990. [DOI] [PubMed] [Google Scholar]
- Johnston D, Christie BR, Frick A, Gary R, Hoffman DA, Schexnayder LK, Watanabe S, Yuan LL (2003) Active dendrites, potassium channels and synaptic plasticity. Philos Trans R Soc Lond B Biol Sci 358: 667-674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura K, White BH, Sidhu A (1995) Coupling of human D-1 dopamine receptors to different guanine nucleotide binding proteins. Evidence that D-1 dopamine receptors can couple to both Gs and G(o). J Biol Chem 270: 14672-14678. [DOI] [PubMed] [Google Scholar]
- Kitai ST, Surmeier DJ (1993) Cholinergic and dopaminergic modulation of potassium conductances in neostriatal neurons. Adv Neurol 60: 40-52. [PubMed] [Google Scholar]
- Koh PO, Undie AS, Kabbani N, Levenson R, Goldman-Rakic PS, Lidow MS (2003) Up-regulation of neuronal calcium sensor-1 (NCS-1) in the prefrontal cortex of schizophrenic and bipolar patients. Proc Natl Acad Sci USA 100: 313-317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krimer LS, Jakab RL, Goldman-Rakic PS (1997) Quantitative three-dimensional analysis of the catecholaminergic innervation of identified neurons in the macaque prefrontal cortex. J Neurosci 17: 7450-7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulisevsky J (2000) Role of dopamine in learning and memory: implications for the treatment of cognitive dysfunction in patients with Parkinson's disease. Drugs Aging 16: 365-379. [DOI] [PubMed] [Google Scholar]
- Lacerda AE, Rampe D, Brown AM (1988) Effects of protein kinase C activators on cardiac Ca2+ channels. Nature 335: 249-251. [DOI] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ (2002) Signaling of layer 1 and whisker-evoked Ca2+ and Na+ action potentials in distal and terminal dendrites of rat neocortical pyramidal neurons in vitro and in vivo. J Neurosci 22: 6991-7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Kaiser KM, Sakmann B (1999a) Calcium electrogenesis in distal apical dendrites of layer 5 pyramidal cells at a critical frequency of back-propagating action potentials. Proc Natl Acad Sci USA 96: 14600-14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B (1999b) A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature 398: 338-341. [DOI] [PubMed] [Google Scholar]
- Lavin A, Grace AA (2001) Stimulation of D1-type dopamine receptors enhances excitability in prefrontal cortical pyramidal neurons in a state-dependent manner. Neuroscience 104: 335-346. [DOI] [PubMed] [Google Scholar]
- Levy F, Farrow M (2001) Working memory in ADHD: prefrontal/parietal connections. Curr Drug Targets 2: 347-352. [DOI] [PubMed] [Google Scholar]
- Lewis BL, O'Donnell P (2000) Ventral tegmental area afferents to the prefrontal cortex maintain membrane potential “up” states in pyramidal neurons via D(1) dopamine receptors. Cereb Cortex 10: 1168-1175. [DOI] [PubMed] [Google Scholar]
- Lezcano N, Mrzljak L, Eubanks S, Levenson R, Goldman-Rakic P, Bergson C (2000) Dual signaling regulated by calcyon, a D1 dopamine receptor interacting protein. Science 287: 1660-1664. [DOI] [PubMed] [Google Scholar]
- Lorenzon NM, Foehring RC (1995) Characterization of pharmacologically identified voltage-gated calcium channel currents in acutely isolated rat neocortical neurons. I. Adult neurons. J Neurophysiol 73: 1430-1442. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Flockerzi V, Hofmann F (1997) Regional expression and cellular localization of the α1 and β subunit of high voltage-activated calcium channels in rat brain. J Neurosci 17: 1339-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee J, Hoffman D, Colbert C, Johnston D (1998) Electrical and calcium signaling in dendrites of hippocampal pyramidal neurons. Annu Rev Physiol 60: 327-346. [DOI] [PubMed] [Google Scholar]
- Marchetti C, Carbone E, Lux HD (1986) Effects of dopamine and noradrenaline on Ca channels of cultured sensory and sympathetic neurons of chick. Pflügers Arch 406: 104-111. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ (1994) Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev 74: 365-507. [DOI] [PubMed] [Google Scholar]
- McHugh D, Sharp EM, Scheuer T, Catterall WA (2000) Inhibition of cardiac L-type calcium channels by protein kinase C phosphorylation of two sites in the N-terminal domain. Proc Natl Acad Sci USA 97: 12334-12338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK, Cohen JD (2001) An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24: 167-202. [DOI] [PubMed] [Google Scholar]
- Mills LR, Niesen CE, So AP, Carlen PL, Spigelman I, Jones OT (1994) N-type Ca2+ channels are located on somata, dendrites, and a subpopulation of dendritic spines on live hippocampal pyramidal neurons. J Neurosci 14: 6815-6824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottola DM, Brewster WK, Cook LL, Nichols DE, Mailman RB (1992) Dihydrexidine, a novel full efficacy D1 dopamine receptor agonist. J Pharmacol Exp Ther 262: 383-393. [PubMed] [Google Scholar]
- Naguro I, Nagao T, Adachi-Akahane S (2001) Ser(1901)of alpha(1C) subunit is required for the PKA-mediated enhancement of L-type Ca(2+) channel currents but not for the negative shift of activation. FEBS Lett 489: 87-91. [DOI] [PubMed] [Google Scholar]
- Nisenbaum ES, Mermelstein PG, Wilson CJ, Surmeier DJ (1998) Selective blockade of a slowly inactivating potassium current in striatal neurons by (+/-) 6-chloro-APB hydrobromide (SKF82958). Synapse 29: 213-224. [DOI] [PubMed] [Google Scholar]
- Nussinovitch I, Kleinhaus AL (1992) Dopamine inhibits voltage-activated calcium channel currents in rat pars intermedia pituitary cells. Brain Res 574: 49-55. [DOI] [PubMed] [Google Scholar]
- Oakley JC, Schwindt PC, Crill WE (2001) Initiation and propagation of regenerative Ca(2+)-dependent potentials in dendrites of layer 5 pyramidal neurons. J Neurophysiol 86: 503-513. [DOI] [PubMed] [Google Scholar]
- Obejero-Paz CA, Auslender M, Scarpa A (1998) PKC activity modulates availability and long openings of L-type Ca2+ channels in A7r5 cells. Am J Physiol 275: C535-543. [DOI] [PubMed] [Google Scholar]
- Paupardin-Tritsch D, Colombaioni L, Deterre P, Gerschenfeld HM (1985) Two different mechanisms of calcium spike modulation by dopamine. J Neurosci 5: 2522-2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) The rat brain in stereotaxic coordinates, Ed 4. New York: Academic.
- Perrier JF, Mejia-Gervacio S, Hounsgaard J (2000) Facilitation of plateau potentials in turtle motoneurones by a pathway dependent on calcium and calmodulin. J Physiol (Lond) 528: 107-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer-Linn CL, Lasater EM (1996) Dopamine modulates unitary conductance of single PL-type calcium channels in Roccus chrysops retinal horizontal cells. J Physiol (Lond) 496: 607-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuveni I, Friedman A, Amitai Y, Gutnick MJ (1993) Stepwise repolarization from Ca2+ plateaus in neocortical pyramidal cells: evidence for nonhomogeneous distribution of HVA Ca2+ channels in dendrites. J Neurosci 13: 4609-4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Maravall M, Svoboda K (2001) Ca(2+) signaling in dendritic spines. Curr Opin Neurobiol 11: 349-356. [DOI] [PubMed] [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B (1997) Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. J Physiol (Lond) 505: 605-616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sculptoreanu A, Figourov A, De Groat WC (1995) Voltage-dependent potentiation of neuronal L-type calcium channels due to state-dependent phosphorylation. Am J Physiol 269: C725-732. [DOI] [PubMed] [Google Scholar]
- Seamans JK, Gorelova NA, Yang CR (1997) Contributions of voltage-gated Ca2+ channels in the proximal versus distal dendrites to synaptic integration in prefrontal cortical neurons. J Neurosci 17: 5936-5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seamans JK, Durstewitz D, Christie BR, Stevens CF, Sejnowski TJ (2001) Dopamine D1/D5 receptor modulation of excitatory synaptic inputs to layers V prefrontal cortex neurons. Proc Natl Acad Sci USA 98: 301-306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selemon LD, Goldman-Rakic PS (1999) The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry 45: 17-25. [DOI] [PubMed] [Google Scholar]
- Shistik E, Keren-Raifman T, Idelson GH, Blumenstein Y, Dascal N, Ivanina T (1999) The N terminus of the cardiac L-type Ca(2+) channel alpha(1C) subunit. The initial segment is ubiquitous and crucial for protein kinase C modulation, but is not directly phosphorylated. J Biol Chem 274: 31145-31149. [DOI] [PubMed] [Google Scholar]
- Sidhu A, Kimura K (1997) A novel regulation of expression of the alpha-subunit of the G stimulatory protein by dopamine via D1 dopamine receptors. J Neurochem 68: 187-194. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Goldman-Rakic PS (1993) Heterogeneous targets of dopamine synapses in monkey prefrontal cortex demonstrated by serial section electron microscopy: a laminar analysis using the silver-enhanced diaminobenzidine sulfide (SEDS) immunolabeling technique. Cereb Cortex 3: 223-238. [DOI] [PubMed] [Google Scholar]
- Smiley JF, Levey AI, Ciliax BJ, Goldman-Rakic PS (1994) D1 dopamine receptor immunoreactivity in human and monkey cerebral cortex: predominant and extrasynaptic localization in dendritic spines. Proc Natl Acad Sci USA 91: 5720-5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snutch TP, Tomlinson WJ, Leonard JP, Gilbert MM (1991) Distinct calcium channels are generated by alternative splicing and are differentially expressed in the mammalian CNS. Neuron 7: 45-57. [DOI] [PubMed] [Google Scholar]
- Stuart G, Schiller J, Sakmann B (1997) Action potential initiation and propagation in rat neocortical pyramidal neurons. J Physiol (Lond) 505: 617-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC Jr, Nairn AC, Greengard P (1995) Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 14: 385-397. [DOI] [PubMed] [Google Scholar]
- Svoboda K, Denk W, Kleinfeld D, Tank DW (1997) In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385: 161-165. [DOI] [PubMed] [Google Scholar]
- Tanaka O, Sakagami H, Kondo H (1995) Localization of mRNAs of voltage-dependent Ca(2+)-channels: four subtypes of alpha 1- and beta-subunits in developing and mature rat brain. Brain Res Mol Brain Res 30: 1-16. [DOI] [PubMed] [Google Scholar]
- Tzschentke TM (2001) Pharmacology and behavioral pharmacology of the mesocortical dopamine system. Prog Neurobiol 63: 241-320. [DOI] [PubMed] [Google Scholar]
- Usowicz MM, Sugimori M, Cherksey B, Llinas R (1992) P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron 9: 1185-1199. [DOI] [PubMed] [Google Scholar]
- Wang HY, Undie AS, Friedman E (1995) Evidence for the coupling of Gq protein to D1-like dopamine sites in rat striatum: possible role in dopamine-mediated inositol phosphate formation. Mol Pharmacol 48: 988-994. [PubMed] [Google Scholar]
- Wang J, O'Donnell P (2001) D(1) dopamine receptors potentiate NMDA-mediated excitability increase in layers V prefrontal cortical pyramidal neurons. Cereb Cortex 11: 452-462. [DOI] [PubMed] [Google Scholar]
- Wang Z, Feng XQ, Zheng P (2002) Activation of presynaptic D1 dopamine receptors by dopamine increases the frequency of spontaneous excitatory postsynaptic currents through protein kinase A and protein kinase C in pyramidal cells of rat prelimbic cortex. Neuroscience 112: 499-508. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, Egan MF, Bertolino A, Callicott JH, Mattay VS, Lipska BK, Berman KF, Goldberg TE (2001) Prefrontal neurons and the genetics of schizophrenia. Biol Psychiatry 50: 825-844. [DOI] [PubMed] [Google Scholar]
- Westenbroek RE, Hell JW, Warner C, Dubel SJ, Snutch TP, Catterall WA (1992) Biochemical properties and subcellular distribution of an N-type calcium channel alpha 1 subunit. Neuron 9: 1099-1115. [DOI] [PubMed] [Google Scholar]
- Williams PJ, MacVicar BA, Pittman QJ (1990) Synaptic modulation by dopamine of calcium currents in rat pars intermedia. J Neurosci 10: 757-763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seamans JK (1996) Dopamine D1 receptor actions in layers V-VI rat prefrontal cortex neurons in vitro: modulation of dendritic-somatic signal integration. J Neurosci 16: 1922-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seaman JK, Gorelova N (1996) Electrophysiological and morphological properties of layer V-VI principal pyramidal cells in rat prefrontal cortex in vitro. J Neurosci 16: 1904-1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Seamans JK, Gorelova N (1999) Developing a neuronal model for the pathophysiology of schizophrenia based on the nature of electrophysiological actions of dopamine in the prefrontal cortex. Neuropsychopharmacology 21: 161-194. [DOI] [PubMed] [Google Scholar]
- Ye JH, Akaike N (1993) Calcium currents in pyramidal neurons acutely dissociated from the rat frontal cortex: a study by the nystatin perforated patch technique. Brain Res 606: 111-117. [DOI] [PubMed] [Google Scholar]
- Young CE, Yang CR (2002) Differential dopamine D1 receptor (D1R) modulation of soma-dendritic, nimodipine-sensitive Ca2+ potentials in rat prefrontal cortical (PFC) neurons in vitro. Soc Neurosci Abstr 28: 437.21. [Google Scholar]
- Yuste R, Tank DW (1996) Dendritic integration in mammalian neurons, a century after Cajal. Neuron 16: 701-716. [DOI] [PubMed] [Google Scholar]
- Yuste R, Gutnick MJ, Saar D, Delaney KR, Tank DW (1994) Ca2+ accumulations in dendrites of neocortical pyramidal neurons: an apical band and evidence for two functional compartments. Neuron 13: 23-43. [DOI] [PubMed] [Google Scholar]
- Zahrt J, Taylor JR, Mathew RG, Arnsten AF (1997) Supranormal stimulation of D1 dopamine receptors in the rodent prefrontal cortex impairs spatial working memory performance. J Neurosci 17: 8528-8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Johnson JA, Chen L, El-Sherif N, Mochly-Rosen D, Boutjdir M (1997) C2 region-derived peptides of beta-protein kinase C regulate cardiac Ca2+ channels. Circ Res 80: 720-729. [DOI] [PubMed] [Google Scholar]
- Zhao F, Li P, Chen SR, Louis CF, Fruen BR (2001) Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J Biol Chem 276: 13810-13816. [DOI] [PubMed] [Google Scholar]