

Abstract
The cyclooxygenases COX-1 and COX-2 catalyze the first committed step of prostaglandin synthesis from arachidonic acid. Previous studies in rodent stroke models have shown that the inducible COX-2 isoform promotes neuronal injury, and the administration of COX-2 inhibitors reduces infarct volume. We investigated the function of PGE2, a principal prostaglandin product of COX-2 enzymatic activity, in neuronal survival in cerebral ischemia. PGE2 exerts its downstream effects by signaling through a class of four distinct G-protein-coupled EP receptors (for E-prostanoid: EP1, EP2, EP3, and EP4) that have divergent effects on cAMP and phosphoinositol turnover and different anatomical distributions in brain. The EP2 receptor subtype is abundantly expressed in cerebral cortex, striatum, and hippocampus, and is positively coupled to cAMP production. In vitro studies of dispersed neurons and organotypic hippocampal cultures demonstrated that activation of the EP2 receptor was neuroprotective in paradigms of NMDA toxicity and oxygen glucose deprivation. Pharmacologic blockade of EP2 signaling by inhibition of protein kinase A activation reversed this protective effect, suggesting that EP2-mediated neuroprotection is dependent on cAMP signaling. In the middle cerebral artery occlusion-reperfusion model of transient forebrain ischemia, genetic deletion of the EP2 receptor significantly increased cerebral infarction in cerebral cortex and subcortical structures. These studies indicate that activation of the PGE2 EP2 receptor can protect against excitotoxic and anoxic injury in a cAMP-dependent manner. Taken together, these data suggest a novel mechanism of neuroprotection mediated by a dominant PGE2 receptor subtype in brain that may provide a target for therapeutic intervention.
Keywords: hippocampus, cerebral cortex, neuroprotection, prostaglandin, PGE2, EP2 receptor, cAMP, stroke
Introduction
The inducible isoform of cyclooxygenase, COX-2, is rapidly upregulated in neurons after NMDA receptor-dependent synaptic activity (Yamagata, 1993), consistent with a physiologic role in modulating synaptic plasticity (Amateau, 2002; Chen et al., 2002). COX-2 expression and activity are also induced in neurons in vivo in acute paradigms of excitotoxicity (Yamagata, 1993; Adams et al., 1996; Miettinen et al., 1997), and can promote injury in cerebral ischemia (Nogawa et al., 1997; Nakayama et al., 1998; Dore, 2003) and possibly in neurodegenerative disorders such as ALS (Drachman et al., 2002; Pompl, 2003), Parkinson's disease (Feng, 2002; Teismann, 2003), and Alzheimer's disease (Pasinetti, 1998). Thus, COX-2 activity and prostaglandin production function physiologically in neuroplasticity and pathologically in promoting neuronal injury, depending on the magnitude of COX-2 induction.
Prostaglandins (PGs) are derived from the metabolism of arachidonic acid by cyclooxygenase to the intermediate PGH2, which then serves as the substrate for generation of five prostanoids: PGE2, PGF2α, PGD2, PGI2 (prostacyclin), and TxA2 (thromboxane A2). These prostanoids bind to specific G-protein-coupled receptors designated EP (for E-prostanoid), FP, DP, IP, and TP receptors, respectively (for review, see Breyer et al., 2001). PG receptor subtypes are distinguished by which PG they bind and by the signal transduction pathway that is activated after ligand binding. Activation of PG receptors leads to changes in the production of cAMP and/or phosphoinositol turnover and Ca2+ mobilization. Further complexity occurs in the case of PGE2, which can bind four receptor subtypes (EP1, EP2, EP3, and EP4) with distinct and potentially antagonistic signaling cascades. EP2 and EP4 receptors couple positively to Gs to increase cAMP formation, whereas EP3 couples negatively to cAMP through Gi; EP1 couples to Gq, and activation results in increased intracellular calcium concentrations.
From ample precedent supporting a neurotoxic role of COX-2, we hypothesized that PGE2, which is dominantly induced and functionally coupled to COX-2 activity (Huang et al., 1998; Brock et al., 1999; Yamagata et al., 2001; Vidensky, 2003) might mediate the toxic effects of COX-2 in brain. Previous in vitro studies of PGE2 have been controversial, showing both toxic and protective effects on neuronal survival (Akaike, 1994; Cazevielle, 1994; Prasad, 1998a,b; Takadera, 2002; Carlson, 2003). In this study, to investigate the downstream effects of PGE2 on neuronal survival in cerebral ischemia, we focused on the function of the EP2 receptor, which is abundantly expressed in cerebral cortex, hippocampus, and striatum. We found that activation of the EP2 receptor paradoxically promoted a marked neuroprotective effect in paradigms of excitotoxicity in organotypic hippocampal slices. Moreover, this protective effect could be abolished by blocking the activation of protein kinase A, suggesting that EP2-mediated neuroprotection is dependent on cAMP signaling. We confirmed this protective effect in vivo in a model of transient focal ischemia and found significant increases in cerebral infarction in mice that lacked the EP2 receptor as compared with wild-type (WT) mice. Taken together, these data demonstrate a novel neuroprotective pathway mediated by a dominantly expressed PGE2 receptor.
Materials and Methods
Animals. This study was conducted in accordance with the National Institutes of Health guidelines for the use of experimental animals. Protocols were approved by the Institutional Animal Care and Use Committee at Johns Hopkins University. Sprague Dawley rats were obtained from Charles River Laboratories (Wilmington, MA) for organotypic and dispersed neuronal cultures. EP2 receptor -/+ C57B6 mice (Sheller, 2000; Breyer, 2002) backcrossed for >10 generations on a C57B6 background were used to avoid artifactual differences caused by genetic background and bred to generate homozygous -/- mice.
Prostaglandin receptor agonists and other reagents. Prostaglandin agonists were prepared as a stock of 10 mm in 100% ethanol and frozen at -70°C until use. The COX-2 inhibitors NS398 (Biomol Research Labs, Plymouth Meeting, PA) and SC58236 (a generous gift from Pfizer, St. Louis, MO) were diluted in DMSO. PGE2 or the EP2 receptor agonist butaprost (Cayman Chemicals, Ann Arbor, MI) was added at concentrations of 1 nm to 1 μm.
Neuronal cultures and assessment of neuronal survival. Pure hippocampal cultures were prepared from E17 rat embryos and plated at a density of 400,000 cells per well in 24-well plates coated with poly-d-lysine (BD Biosciences, Bedford, MA) in Neurobasal medium, 1× B27, penicillin-streptomycin (Invitrogen, Carlsbad, CA), and media was refreshed twice a week by replacing half the media with fresh media. The percentage of neuronal and astrocytic cells was determined as follows: neuronal cultures were fixed for 30 min at 4°C with 4% paraformaldehyde, 4% sucrose, and 0.1 m PBS, pH 7.4; cells were washed, permeabilized with 0.3% Triton X-100, blocked with 10% serum, and incubated overnight at 4°C in anti-NeuN monoclonal antibody (1:1000; Chemicon, Temecula, CA) and anti-GFAP polyclonal antibody (1:4000; Dako, Carpinteria, CA) with 3% serum; cells were then washed, and incubated with anti-rabbit Alexa Fluor 488 and anti-mouse Alexa Fluor 546 secondary fluorescent antibodies (Molecular Probes, Eugene, OR). The number of NeuN- and GFAP-positive cells was counted in eight separate fields over two wells, and the percentage of NeuN-positive neurons and GFAP-positive astrocytes was determined. Purity of neuronal cultures was 94.6% neurons and 5.2% astrocytes. Excitotoxicity assays were begun at 14 d in vitro (DIV), when neurons are expressing all glutamate receptors (Rao, 1998). All cultures were maintained at 37°C in a humidified incubator with 5% CO2. Cell death was quantified 24 hr after excitotoxic stimulation by cell counting or lactose dehydrogenase assay (LDH) of culture medium. For cell counting, quantification of cell death was performed in cultures stained with the fluorescent DNA binding dye Hoescht 33342 (1 μm). Nuclei were visualized and photographed under epifluorescence illumination (340 nm excitation and 510 nm barrier filter) using a 40× oil immersion objective. Cells in which nuclear staining was diffuse were considered viable, and cells in which nuclear staining was condensed or fragmented were considered apoptotic. Two hundred cells from three fields in three separate cultures per experimental condition were counted without knowledge of experimental conditions. For LDH measurements, culture media was diluted 1/3 and assayed for LDH (Roche, Indianapolis, IN) per the manufacturer's instructions. Averages of three or more experiments were combined. In studies examining the contribution of protein kinase A (PKA), cultures were pretreated for 30 min with H89 (Sigma, St. Louis, MO) or KT 5720 (Tocris, Ellisville, MO) before glutamate stimulation.
Reverse transcription-PCR of EP receptors in culture. Total RNA was extracted from cultured hippocampal neurons with Trizol (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. RNA was then treated with DNase (Invitrogen) to eliminate any contaminating genomic DNA. The reverse transcription (RT) reaction was performed using Superscript one-step RT-PCR followed by PCR amplification using Platnium TaqDNA Polymerase (Invitrogen) according to the manufacturer's instructions. Primers were designed from divergent regions of prostaglandin receptor sequences, as determined by alignment of PG receptor cDNA sequences (Gene Works; Intelligenetics, Campbell, CA). Nested outer and inner 5′ and 3′ primers were as follows: EP2 outer 5′GGAGAACTGCGAGAGTCGTC, outer 3′CCTTTACGTTCCTCCAACGA, inner 5′ GTGCTGGTAACGGAACTGGT, and inner 3′AGCGGCAGAGAACAGAAGAG. RT was performed at 50°C for 30 min. PCR using outer primers was performed in a volume of 25 μl (1× reaction buffer, 0.2 mm deoxynucleotide triphosphates, 2.0 mm MgCl2, forward and reverse primers (100 nm), 1 μl of RT product, 0.3 μl of RT/Platnium Taq) and consisted of 30 cycles (94°C for 40 sec, 54°C for 45 sec, and 72°C for 60 sec), followed by a 10 min extension cycle at 72°C. The PCR mix was then diluted 1:1000 and amplified using the inner nested primers. Amplification using the inner primers was also performed in a volume of 25 μl [1× reaction buffer, 0.2 mm deoxynucleotide triphosphates, 2.5 mm MgCl2, forward and reverse primers (100 nm), 10 μl of diluted outer PCR mix, and 0.3 μl of RT/Platnium Taq] for 30 cycles (94°C for 40 sec, 55°C for 45 sec, and 72°C for 60 sec) followed by a 10 min extension at 72°C. The expected size of the EP2 fragment was 311 bp, and the identity of this 312 bp fragment was confirmed by sequencing. Control reactions for outer and inner primers were performed without RT to control for genomic DNA contamination, and no bands were observed for these reactions (see Fig. 4 A).
Figure 4.

Neuroprotective effect of EP2 receptor activation. A, RT-PCR of the EP2 receptor in hippocampal neurons at 14 d in vitro. Left lane (-) indicates the negative control in which no reverse transcriptase was added, and the right lane (+ RT) demonstrates EP2 receptor band (arrow). B, A 200× fluorescent microscopy confirms expression of EP2 receptor in hippocampal neurons at DIV 14. Double immunofluorescent staining with anti-EP2 and anti-NeuN antibodies confirms the neuronal expression of the EP2 receptor in these cultures. Control cultures (no primary antibody) did not show staining (data not shown). C, EP2 receptor activation by addition of the selective EP2 agonist butaprost (10 nm) protects hippocampal neurons from glutamate toxicity. Glutamate (50 μm) ± butaprost or vehicle was added overnight, and nuclei were stained with Hoechst and counted. The percentage of apoptotic nuclei was quantified as a percentage of the total number of nuclei counted (#p < 0.001; *p < 0.001; representative experiment of three experiments, n = 4-6 wells per condition). D, In organotypic hippocampal cultures, EP2 receptor activation protects CA1 neurons treated with NMDA (10 μm for 1 hr). Percentage of maximal PI fluorescence was calculated (see Materials and Methods) in slices treated with NMDA ± butaprost (0 nm to 1 μm); mean ± SE; **p < 0.01; *p < 0.05. Unlike the PGE2 experiments (Fig. 2), addition of butaprost and activation of the EP2 receptor result in neuroprotection in both dispersed and organotypic cultures. E, EP2 receptor activation also protects CA1 neurons in hippocampal slices subjected to OGD for 1 hr. Slices were subjected to 1 hr of OGD and then incubated with vehicle or butaprost (1 nm to 1 μm) for 24 hr. Butaprost at concentrations >10 nm significantly protected neurons, as determined by percentage of maximal PI fluorescence, *p < 0.05; **p < 0.01. F, EP2 receptor activation by butaprost significantly increases cytosolic levels of cAMP in 5-10 min; n = 3; mean ± SD; **p < 0.01. DIV 14 hippocampal neurons were stimulated with 10 nm butaprost, and cells were collected at 5 min intervals. G, The protective effects of EP2 receptor activation by butaprost are abolished with treatment with the PKA inhibitor H89 (1 μm) in hippocampal neurons treated with glutamate (50 μm for 18 hr). #p < 0.001 control versus glutamate; **p < 0.001 glutamate versus glutamate plus butaprost; p < 0.001 glutamate-butaprost versus glutamate-butaprost-H89. H, Administration of a second PKA inhibitor, KT 5720, also reverses EP2 neuroprotection in hippocampal neurons treated with glutamate (50 μm for 18 hr); **p < 0.01 control versus glutamate; #p < 0.02 glutamate versus glutamate-butaprost; *p < 0.05 comparing glutamate-butaprost to glutamate-butaprost-KT 5720 (0.6 and 1.2 μm).
Organotypic hippocampal slice model. Hippocampal organotypic cultures were prepared according to the methods of Stoppini et al. (1991). Coronal hemispheric slices of 350 μm were derived from postnatal day 7 rat pups using a tissue chopper (McILwain tissue chopper; Vibratome Company, St. Louis, MO). Hippocampal slices were transferred onto 30 mm Millicell membrane inserts (0.4 μm; Millipore, Bedford, MA), permeable membranes in six well plates, with each well containing 1 ml of medium (50% MEM, 25% HBSS, 25% heat-inactivated horse serum, 6.5 mg/ml d-glucose, 5 U/ml penicillin G, and 5 μg/ml streptomycin sulfate). Cultures were maintained in a humidified incubator under 5% CO2 at 37°C for 13 d, and medium was changed twice a week. At 13 d in culture, medium was replaced with fresh medium containing propidium iodide (PI; 5 μg/ml; Sigma), a fluorescent vital dye for 24 hr, and basal PI fluorescence was imaged the next day to confirm that slices were viable and healthy (this time point referred to as “basal” or time = 0 hr). The time point of DIV 14 for stimulation has been used in a number of studies of glutamate receptor toxicity (Vornov, 1991, 1995; Abdel-Hamid and Tymianski, 1997; Jakobsen, 2001; Vincent, 2002a), synaptic plasticity (Lu, 2001), neuroinflammation (Schmidt, 2001), and effects of Aβ peptide (Vincent, 2002b). The microglial activation that occurs after the preparation of the slices has subsided by DIV 14, and microglia return to their resting ramified state between days 6 and 10 in vitro (Hailer, 1996; Czapiga, 1999). This organotypic culture system allows for the examination of neuronal, astrocytic, and microglial effects, but as an ex vivo preparation, does not address effects of blood flow or recruitment of peripheral inflammatory cells. The incubation of slices with PI does not exert any harmful effects because it does not enter intact cells, but with sufficient membrane injury, PI will enter dying cells, bind to nucleic acids, and cause the injured cells to fluoresce (Tasker, 1992). Slices were then stimulated with 10 μm NMDA for 1 hr or subjected to oxygen glucose deprivation (see below) for 1 hr in the presence of PG receptor agonists or vehicle. After stimulation, media was replaced with fresh media containing either PG receptor agonist or vehicle (ethanol ≤0.1%) and PI (5 μg/ml), and slices were incubated another 24 hr, at which time they were imaged again for PI fluorescence (referred to as the 24 hr time point). Medium was then removed and replaced with fresh medium containing a lethal dose of 20 μm NMDA and PI and incubated overnight (time point referred to as t = max). This NMDA treatment has been shown to induce selective neuronal death and represents maximal neuronal death for each slice (Abdel-Hamid and Tymianski, 1997). Control and NMDA-treated slices were included in each experiment with triplicate wells or 15 slices per condition for each experimental group.
Oxygen-glucose deprivation. Induction of anoxia was performed at 13 d in vitro. Slice cultures were rinsed twice with warm BSS (in mm: 125 NaCl, 5 KCl, 1.2 NaH2PO4, 26 NaHCO3, 1.8 CaCl2, 0.9 MgCl2, 10 HEPES, and 10 glucose), and returned to the incubator to equilibrate for 30 min. Slice cultures were rinsed twice with warm deoxygenated glucose-free BSS, transferred to an air-tight chamber, and flushed with anoxic gas (5% CO2, 85% N2, and 10% H2) for 1 hr at 37°C. Sixty minutes of anoxia has been determined to cause increased PI fluorescence and neuronal death in the CA1 to CA3 regions and dentate gyrus (Abdel-Hamid and Tymianski, 1997). Controls were rinsed with BSS and transferred to a normal aerated incubator for 1 hr. The anoxia was terminated by returning the cultures to fresh medium (50% MEM, 25% HBSS, 25% HIHS, 6.5 mg/ml d-glucose, 5 U/ml penicillin G, and 5 μg/ml streptomycin sulfate) containing PI. Cultures were imaged for PI fluorescence 24 hr after oxygen-glucose deprivation (OGD), and then maximally stimulated with 20 μm NMDA overnight to obtain maximal PI fluorescence and imaged as described above.
Measurement of neuronal death in organotypic hippocampal slices. Neuronal death was assayed by quantification of mean PI fluorescence in the CA1 subregion of each hippocampal slice. Although PI fluorescence is a marker of overall cell death, changes in PI fluorescence in the CA1 region will mainly reflect changes in neuronal viability because of the high density of neurons that makes up this hippocampal subregion, the observation that PI staining is restricted to this neuronal cell layer, and correlation of PI staining with subsequent staining with Nissl (Vornov, 1991, 1995; Abdel-Hamid and Tymianski, 1997; Jakobsen, 2001; Schmidt, 2001; Vincent, 2002a,b). Astrocytic and microglial cell death may also occur, however, because these cell types are more resistant to excitotoxic insults (David, 1996), and undergo a reactive and proliferative response after tissue injury (Wu, 1998; Streit, 2000), so they are unlikely to contribute significantly to the CA1 PI fluorescence.
Mean PI fluorescence intensity represents the mean of the fluorescence intensity values of each pixel in the image of the subregion measured (for example CA1). This value is proportional to the number of injured cells (Newell et al., 1995). Sequential fluorescence is measured at three time points: (1) at t = 0, before NMDA or OGD; stimulation to measure basal levels of spontaneous neuronal death; (2) at t = 24 hr, or 24 hr after stimulation with NMDA or OGD; and (3) at t = max, after a final treatment of slices with a 24 hr overnight lethal treatment with 20 μm NMDA, which produces the maximal amount of neuronal death. This maximal PI value represents total neuronal loss from fluorescence remaining from the 24 hr time point and new neuronal death from the lethal NMDA challenge, and does not reflect neuronal death that has been cleared from the culture. Slices were imaged using an inverted Nikon Diaphot microscope connected to a Cool Snap CCD camera (Roper). The same exposure time and gain were used in all experiments; these were set such that individual PI fluorescent nuclei were clearly visualized, and maximal cell death did not saturate the camera. The images were collected and analyzed using the Open Lab imaging software (Improvision) on a Macintosh G4. Images were digitized at 8 bits/pixel, and mean fluorescence values of the region were measured for t = 0 (basal), t = 24 hr (after a 1 hr treatment with NMDA or OGD), and t = max (after 24 hr treatment with NMDA 20 μm). The t = 0 background fluorescence was subtracted from the t = 24 hr and the t = max values, and the percentage of neuronal death was then calculated by normalizing the 24 hr fluorescence value to the t = max value for each individual slice per the formula: (t = 24 hr) - (t = 0)/(t = max) - (t = 0). To ensure that the same subregion (consisting of CA1) is selected in all three images from time points t = 0, t = 24 hr, and t = max, the CA1 region was outlined first using the t = max image of the slice, where PI fluorescence is maximal and the boundaries of CA1 are readily apparent. With this outline, a region of interest (ROI) is created. Using the OpenLab software, the ROI outline can be “grabbed” and applied to the remaining two time point images (t = 0 and t = 24 hr), ensuring that the same CA1 area is being quantified in all three images for each slice. Each experiment was done in triplicate, with n = 10-15 sections per condition per experiment. Control slices were processed in parallel with slices subjected to NMDA or OGD and subjected to all washes and changes in medium. Percentage of maximal PI fluorescence levels for control slices reflects basal cell death that occurs in vehicle-treated hippocampal slices in response to media changes and PI imaging during the 3 d experiment.
Immunostaining of EP2 receptor subtype. Adult C57B6 mice and Sprague Dawley rats were anesthetized with a lethal dose of pentobarbital and perfused with 0.1 m PBS followed by 4% paraformaldehyde—PBS, pH 7.4. Brains were postfixed in the same fixative overnight, and 40 μm coronal free-floating sections were generated. Sections were incubated in 0.3% Triton X-100 and 10% blocking serum overnight, then incubated with primary polyclonal antibody to EP2 (Cayman Chemicals) at 1:4000 in 3% serum block overnight at 4°C, washed in PBS, and incubated with biotinylated secondary antibody, washed in PBS, incubated with avidin-biotin complex (Vector Laboratories, Burlingame, CA) and developed with nickel-cobalt DAB (Pierce, Rockford, IL). Specificity of the EP2 polyclonal antibody was confirmed with control experiments in which the primary antibody was omitted and in separate experiments comparing EP2 receptor -/- and wild-type mice that demonstrated absent staining in EP2-/- brain. For immunostaining of EP2 receptor in cultured neurons, hippocampal neurons cultured on glass coverslips were fixed for 30 min in 4% paraformaldehyde and 4% sucrose in PBS, rinsed in PBS, and incubated with 0.3% Triton X-100 and 10% blocking serum overnight at 4°C, then incubated in primary antibody with 3% blocking serum overnight at 4°C; cells were then washed in PBS, and incubated with anti-rabbit Alexa Fluor 488 secondary antibody (Molecular Probes), rinsed, and mounted on slides for visualization by microscopy.
Prostaglandin measurement. Prostaglandin measurements were performed on culture media 24 hr after stimulation with glutamate +/- COX-2 inhibitor. PGE2 levels were measured by ELISA (Cayman Chemicals; sensitivity 30 pg/ml) as previously described (Andreasson et al., 2001; Vidensky, 2003).
Determinations of cAMP. Hippocampal neuronal cultures were plated in 24 well tissue culture dishes. Direct cAMP measurements were performed using the nonacetylated version of a commercial assay kit (Assay Designs, Inc., Ann Arbor, MI) according to the manufacturer's protocol. cAMP concentrations were standardized to protein content using the Pierce BCA kit. Phosphodiesterase was inhibited by the addition of 3 isobutyl-1-methylxanthine (50 μm) to cultures for 30 min. The EP2 receptor agonist butaprost (100 nm) was added to cultures for 5, 10, 15, 20, and 25 min before lysis of cells. Forskolin (100 nm), a direct and potent activator of adenylyl cyclase, was used as a positive control.
Transient focal ischemia. Transient focal ischemia was induced by middle cerebral artery occlusion (MCAO) for 90 min (Sampei, 2000; McCullough, 2003) followed by 22.5 hr of reperfusion (total 24 hr survival) in male EP2-/- and wild-type mice weighing 21-25 gm at 10-12 weeks of age. Under halothane anesthesia (5.0% for induction, 1.0% for maintenance), mice were spontaneously ventilated with oxygen-enriched (100%) air via a nose cone. Rectal muscle temperatures were monitored with a MONO-THERM system and maintained at 37°C during surgery by a thermostatically controlled heating pad. A small incision was made in the skin over the right side of the skull, a laser Doppler probe (model MBF3D; Moor Instruments, Wilmington, DE) was placed directly on the right parietal skull surface 2 mm posterior and 3 mm lateral to the bregma, and a relative laser Doppler flow (LDF) was measured. A midline ventral neck incision was then made, and unilateral MCAO was performed by inserting a 6.0 nylon monofilament (with a heat-blunted tip coated with cyanoacrylate) into the internal carotid artery 6 mm from the internal carotid/pterygopalatine artery bifurcation via an external carotid artery stump. The LDF signal was monitored before and during the onset of occlusion to ensure an adequate blood flow reduction with passage of the suture. Once the vessel was occluded and the LDF was recorded, the probe was removed, skin incisions were sutured, the animal was allowed to awaken from anesthesia (usually within 5 min), and the animal was scored for neurological deficits 1 hr after ischemic onset. Intra-ischemic neurological deficit was confirmed and scored as follows: 0, no deficit; 1, forelimb weakness and torso turning to the ipsilateral side when held by tail; 2, circling to affected side; 3, unable to bear weight on affected side; and 4, no spontaneous locomotor activity or barrel rolling (Longa, 1989). If no deficit was observed, the animal was removed from further study. Mice were then reanesthetized for removal of the suture at the end of a 90 min occlusion. Rectal temperature was maintained at 37° during surgery, stroke, and reperfusion. At 22.5 hr of reperfusion, unanaesthetized animals were rapidly decapitated, and the brain was harvested for pathological examination.
In a separate nonsurvival cohort of animals, the femoral artery was cannulated for measurement of arterial blood gases and mean arterial pressure (MAP). Arterial blood gases, glucose, and hemoglobin were measured at baseline and 60 min into MCAO. Intra-ischemic occlusion was monitored with a LDF probe (Moor Instruments, Wilmington, DE). The skull was exposed, and the laser Doppler probe was positioned at 2 mm posterior and 3 mm lateral to the bregma over the parietal skull surface. After placement of the probe, the MCA was occluded, and the LDF was monitored both during ischemia and for 45 min after reperfusion. These animals were not awakened during ischemia and were killed after reperfusion. Therefore, all invasive blood pressure measurements were obtained in anesthetized animals. To assess large vessel anatomy in wild-type and EP2-/- mice, three unoperated mice of each genotype were deeply anesthetized and perfused via the left ventricle with 5 ml of ice-cold saline followed by 3 ml of FeSO3 solution (elemental iron: 2 gm/20 ml saline). Mice were decapitated, and brains were removed from the skull with the circle of Willis intact and placed in 10% formalin for 24 hr for examination of the large cerebral vessel anatomy.
Quantification of infarct volume. After 22 hr of reperfusion, mice were killed, and brain tissue was harvested. Brains were sectioned coronally into five 2 mm sections and incubated with 1% 2,3,5-triphenyl-tetrazolium chloride (TTC) in saline for 30 min at 37°C. The area of infarct, identified by the lack of TTC staining, was measured on the rostral and caudal surfaces of each slice and numerically integrated across the thickness of the slice to obtain an estimate of infarct volume in each slice (OpenLab software; Improvision). Volumes from all slices were summed to calculate total infarct volume over the entire infarcted hemisphere. Infarct volume was measured separately in the cerebral cortex, striatum, and hemisphere. Infarct volume was corrected for swelling by comparing the volume of neocortex in the infarcted hemisphere and the noninfarcted hemisphere (Lin et al., 1993).
Regional cerebral blood flow assessment. Regional cerebral blood flow (CBF) was measured at end-ischemia in a separate cohort of animals using [14C] iodoantipyrine ([14C]-IAP) autoradiography (Jay, 1988), as previously described in rat (Alkayed, 1998; McCullough, 2001) and modified for mouse (Sawada, 2000). MCAO was performed as above with additional placement of both arterial (PE 10; Clay-Adams, Sparks MD; 0.28 mm inner diameter, 0.61 mm outer diameter, 15 cm long) and venous (PE 10, 10-cm-long) femoral catheters. At 75 min of MCAO, arterial blood pressure, pH, PCO2, and PO2 were measured, then 15 min later, intravenous administration of C14 IAP was begun (n = 5 EP2-/-; four WT male mice). A total of 4 μCi of [C14] IAP in 81 μl of isotonic saline (1 mCi/10 ml stock diluted in equal volume of saline to obtain 5 μCi/100 μl) was infused intravenously over 45 sec at a constant infusion rate of 6.48 ml/hr (Harvard 22 pump; Harvard Apparatus, Holliston, MA; 2 ml glass Popper syringe with 8.92 diameter). Simultaneously, free-flowing blood was obtained from the distal end of the arterial catheter, and blood was collected into heparinized saline drops of known volume placed in paraffin wells. Nine blood samples were collected at 5 sec intervals and promptly covered to avoid evaporation of the sample. Sample volumes were measured using a pipette and calculated by subtracting the volume of a saline drop from the total volume of blood sample plus saline. Parallel time control saline drops were used to account for change in volume caused by evaporation. The mice were rapidly decapitated at 45 sec, and the brains were removed (within 60 sec) and flash frozen in 2-methylbutane chilled in dry ice (-50°C) and stored at -80°C. Each brain was sectioned on a cryostat (20-μm- thick coronal sections at -18°C) and thaw-mounted onto glass coverslips. Sections were apposed to film for 10 d (SB-5; Kodak, Rochester, NY) with 14C standards. Concentration of [14C] IAP was determined by liquid scintillation spectroscopy (model 3801; Beckman, Somerset, NJ) after decolorization with 0.2 ml of tissue solubilizer (Soluene-350; Packard Instruments, Downers Grove, IL). Autoradiographic images representing six coronal sections (+3, +2, +1, 0, -1, and -2 mm from the bregma, three images at each level) were digitized, and absolute regional CBF was determined by image analysis software (Inquiry; Loates Associates, Westminster, MD). Rates of regional CBF were calculated by the Kety-Schmidt modification of the Fick principle, as previously described (Jay et al., 1988). Two methods of IAP data analysis were used. First, CBF was sampled at discrete 0.08 mm2 regions within the cortical and striatal regions that are vulnerable in the MCAO model. The areas sampled included penumbral (frontal cortex and medial striatum) and core ischemic (parietal cortex and lateral striatum) areas. The penumbral regions of frontal cortex and medial striatum lie at the junction of the anterior communicating artery (ACA) and MCA, and at the junction of the MCA and posterior cerebral artery (PCA), respectively. The parietal cortex and lateral striatum constitute core areas that reside fully in the MCA vascular territory. Measurements from 6 coronal areas (+3, +2, +1, 0, -1, -2 mm from the bregma) with three images at each level yielded 18 data points for each of the four brain areas examined. These measurements were then averaged over three to seven consecutive coronal slices, as described in Alkayed et al. (1998), to yield values of absolute CBF in each of the four brain areas. In the second mode of analysis of IAP autoradiographic data, the distribution of brain volume in the ischemic hemisphere (in cubic millimeters) over incremental levels of absolute CBF was determined for both genotypes. This complementary method of analysis examines whether there are differences in volumes of brain (per cubic millimeter of tissue) that experience a particular range of CBF. The images of all coronal slices were scanned, and pixels were stratified according to the corresponding CBF rates (divided in increments of 0-20, 21-50, 51-90, and 91-130 ml/min/100 gm). Pixels with flow rates falling within a range of blood flow were summed, converted to volume units, and stratified according to the corresponding cerebral blood flow rate, as described in Rusa et al. (1999), to yield absolute values of brain tissue volume (in cubic millimeters) in the ischemic hemisphere for both genotypes for each range of CBF.
Statistical analysis. We used p < 0.05 as a significance level. Statistical analysis was performed by one-way ANOVA, followed by Tukey post hoc analysis. All data are reported as mean ± SEM. Regional CBF was assessed by two-way ANOVA with a post hoc Newman-Keuls test to evaluate differences between groups in both the striatum and cortex at end-ischemic time points.
Results
COX-2 is induced after glutamate stimulation and mediates neuronal death in dispersed hippocampal neurons
We first confirmed in our dispersed culture system the recent findings of Hewett et al. (2000), who demonstrated that COX-2 inhibition could rescue neurons after in vitro NMDA toxicity. In our cultures, PGE2 levels were significantly induced after administration of 100 μm glutamate for 18 hr, and this induction was completely abolished if glutamate-stimulated cultures were treated with the COX-2 inhibitor NS398 (10 μm) (Fig. 1A), indicating that the increase in PGE2 occurred as a result of induced COX-2 activity. The COX-2 inhibitor NS398 rescued neurons in a dose-dependent manner (Fig. 1B) in hippocampal and cerebral cortical cultures, and protection was also seen in a dose-dependent manner with a second COX-2 inhibitor SC58236 (Fig. 1C), indicating that COX-2 enzymatic activity promotes cell death in dispersed pure neuronal cultures.
Figure 1.

COX-2 inhibition protects cultured neurons and hippocampal slices from glutamate-mediated toxicity and OGD. A, PGE2 levels in control (c), glutamate (gl; 100 μm), and glutamate + the COX-2 inhibitor NS 398 (gl/ns; NS 398 10 μm)-treated hippocampal cultures 24 hr after stimulation (c vs gl: *p < 0.001; gl/ns vs gl: #p < 0.001). B, Cell death was assayed by LDH release in hippocampal and cortical cultures 24 hr after glutamate stimulation (100 μm) ± NS 398 (10 and 50 μm; *p < 0.01). C, LDH assay demonstrates neuroprotection mediated by a second COX-2 inhibitor SC 58236 (0.01-10 μm) in hippocampal neurons stimulated with glutamate (50 μm; *p < 0.01). After subtraction of basal LDH release, cell death was calculated as the percentage of the LDH value from cultures treated with glutamate alone. D, The COX-2 inhibitor SC58236 protects neurons in hippocampal slices. Representative panels of sequential fluorescent images from control slice (a-c, NMDA + vehicle), and slice treated with 10 nm SC58236 (d-f, NMDA + SC 58236). Basal fluorescence images (a, d) do not show spontaneous neuronal degeneration in CA1 (outlined in white); 24 after 1 hr treatment of 20 μm NMDA, there is a significant increase in neuronal death in the control (b) slice, but an attenuated amount in slice cotreated with SC58236 (e). Maximal fluorescence images are obtained after complete killing of neurons with a further 24 hr treatment with NMDA (c, f). E, Neuroprotection in the CA1 region of organotypic hippocampal slices at 1 nm SC 58236, with increasing protection with higher doses, up to 1 μm. Slices were challenged at 14 DIV with NMDA 20 μm for 1 hr (n = 10-15 slices per condition). ANOVA followed by Newman-Keuls post hoc test shows differences between control and all NMDA groups (*p < 0.001), differences between treatment groups and NMDA group (#p < 0.05; ## p < 0.005). F, COX-2 inhibition protects against 1 hr OGD in hippocampal slices. Neuronal death (PI fluorescence) was quantified 24 hr after OGD. Slices were administered 2 mm 2-deoxy-d-glucose (DG) +/- 100 nm SC58236 (difference between OGD +/- inhibitor, *p < 0.01; difference between control and OGD, #p < 0.01).
To model this neuroprotection in a system that is closer to the in vivo state, we tested whether COX-2 inhibition would also be protective in hippocampal organotypic slice cultures (Fig. 1D). Hippocampal slices were stimulated at day 14 in vitro with either NMDA (Fig. 1E) or OGD (Fig. 1F) in the presence or absence of the COX-2 inhibitor SC58236 (1 nm to 1 μm), and the percentage of maximal PI fluorescence was measured for the CA1 region of hippocampus. NMDA receptor activation triggers the release of arachidonic acid from membrane phospholipids (Dumuis, 1988) and induces the expression of COX-2 in neurons in vivo (Yamagata, 1993) and in vitro (Hewett, 2000). In organotypic hippocampal slices, COX-2 induction also induced selective death of neurons, and inhibition of COX-2 enzymatic activity rescued neurons, as measured by CA1 PI fluorescence after either NMDA stimulation or OGD. The neuroprotective effects of COX-2 inhibitors in this model recapitulate those seen in vivo in models of stroke and excitotoxin injection (Miettinen et al., 1997; Nogawa et al., 1997; Nakayama et al., 1998) and confirm that hippocampal organotypic slices are a suitable model in which to explore mechanisms of COX-2-mediated neurotoxicity.
Effects of PGE2 administration on neuronal survival in glutamate toxicity
We investigated whether PGE2, the dominantly induced prostaglandin metabolite of COX-2 enzymatic activity, would promote neuronal injury in the setting of excitotoxicity. Elevated PGE2 levels follow acute in vivo insults such as cerebral ischemia, and pharmacologic inhibition of COX-2 activity leads to reduction in levels of PGE2 levels and neuroprotection (Nogawa et al., 1997; Nakayama et al., 1998; Dore, 2003), leading to the hypothesis that PGE2 may mediate the toxic effects of COX-2 activity on neurons. In hippocampal neuronal cultures, the effects of PGE2 at physiologic low nanomolar concentrations were paradoxically protective after administration of 100 μm glutamate (Fig. 2A). This finding suggests that in this culture assay, COX-2 neurotoxicity is not mediated by PGE2, but by other product or products of COX-2 enzymatic activity. We then tested whether the protective effect of PGE2 would be preserved in organotypic hippocampal cultures, where there is a relative preservation of neuronal and astrocytic architecture and presumably a preservation of the interactions between astrocytes and neurons (Fig. 2B). Interestingly, the protective effect of PGE2 was abolished in organotypic cultures, and increasing concentrations of PGE2 had no effect on neuronal survival after NMDA administration. This suggests that activation of PGE2 receptors on astrocytes may counteract the protective effects of PGE2 on neurons. The Ki of PGE2 for its receptors is 22, 6.8, 0.9, and 1.1 nm for EP1, EP2, EP3, and EP4, respectively (Boie, 1997), and the stimulation paradigms in this series of experiments were well within this range. Notably, the effects of PGE2 were tested at physiologic low concentrations to avoid the cross-activation of other PG receptors, notably FP (Ki, 100 nm) that can occur at submicromolar concentrations (Kiriyama, 1997). We conclude that the effects of PGE2 on neuronal viability differ significantly in dispersed neuronal cultures and organotypic cultures, likely because of the preservation of physiologic astrocytic/neuronal interactions in organotypic cultures (Stoppini et al., 1991; Vornov, 1991). Although in dispersed pure neurons, PGE2 elicits a neuroprotective effect, in organotypic slices PGE2 interacts with multiple EP receptors on neurons as well as astrocytes, resulting in a net neutral effect on neuronal survival. These findings are relevant to studies that have found that PGE2 can stimulate astrocytic glutamate release (Nishihara, 1995; Bezzi, 1998; Sanzgiri, 1999).
Figure 2.

PGE2 is neuroprotective in cultured hippocampal neurons but not in organotypic hippocampal slices. A, PGE2 administration at submicromolar concentrations promotes protection in hippocampal cultures stimulated with glutamate (100 μm) for 24 hr (n = 4 wells per condition; *p < 0.02; **p < 0.01; representative of 3 experiments). B, Increasing concentrations of PGE2 had no effect on neuronal viability when assayed in organotypic hippocampal slices treated with 1 hr NMDA (10 μm), suggesting a negative effect of PGE2 at the level of astrocytes in neutralizing the protective effects of PGE2 on pure hippocampal cultures. Neuronal death in CA1 is measured by percentage of maximal PI fluorescence (n = 10-15 slices per condition; n = 6 experiments). Concentrations of PGE2 were used at submicromolar concentrations to avoid the cross-activation of other PG receptors at higher concentrations (Breyer et al., 2001).
Localization of the EP2 receptor in forebrain
In vivo, the specificity of the downstream effects of PGE2 is determined by the anatomic distribution of its EP receptors. Previous studies of mRNA localization of EP receptors have shown that EP2 and EP3 receptor expression occur in cerebral cortex, hippocampus, thalamus, and hypothalamus, whereas EP1 and EP4 receptors are mostly concentrated in hypothalamus (Sugimoto et al., 1994; Batshake et al., 1995; Zhang and Rivest, 1999; Ek et al., 2000). In this study, we focused on the function of the EP2 receptor on neuronal survival in cerebral ischemia and accordingly investigated its protein expression in relevant cortical and subcortical structures. Immunocytochemical staining of mouse brain for EP2 demonstrated widespread expression in cerebral cortex, hippocampus, and striatum with cellular expression in both neurons and astrocytes (Fig. 3). No staining was seen in white matter tracts. The subcellular localization of EP2 was perinuclear in cortical, hippocampal, and striatal neurons, and localized to dendritic processes of pyramidal neurons in hippocampus and cerebral cortex (Fig. 3B-D). Although we cannot rule out terminal staining along the dendrites from afferent inputs, double immunofluorescent colocalization studies do not show punctate colocalization of EP2 receptor with synaptophysin in vitro in cultured neurons (data not shown). These studies indicate that the EP2 receptor is robustly expressed in relevant cortical and subcortical structures in both neurons and astrocytes.
Figure 3.

Immunocytochemical distribution of the EP2 receptor. A, Macroscopic distribution of EP2 receptor immunoreactivity at 100× magnification in a sagittal section showing hippocampus and overlying cerebral cortex immunostained with anti-EP2 antibody (brown) and counterstained with cresyl violet (stains nuclei blue). Note parenchymal distribution of EP2 receptors, with sparing of white matter tracts (VI and V: layers VI and V of cerebral cortex). B, A 400× magnification of the CA3 region of hippocampus shows abundant perinuclear (thin arrow) and dendritic (large arrow) staining in CA3 pyramidal neurons, and diffuse astrocytic staining. C, A 400× magnification of layer V pyramidal neurons in cerebral frontal cortex, demonstrating staining of neuronal perikarya and apical dendrites (thick arrow). D, A 400× magnification of EP2 staining in striatal neurons shows abundant staining in perinuclear distribution (thin arrows).
Activation of the EP2 receptor in vitro confers neuroprotection in the setting of excitotoxicity and OGD
Using the EP2 receptor selective agonist butaprost (Gardiner, 1986), we tested the effect of EP2 receptor activation on neuronal survival. In dispersed hippocampal cultures, RT-PCR confirmed the presence of EP2 receptor mRNA (Fig. 4A), and protein expression was confirmed with immunostaining for EP2, which colocalized with the neuronal marker NeuN (Fig. 4B). EP2 receptor was expressed in neuronal processes, consistent with the in vivo immunocytochemical findings. In dispersed hippocampal cultures, activation of the EP2 receptor (10 nm butaprost) significantly protected neurons from glutamate-mediated toxicity (Fig. 4C) (EC50 of butaprost is 23 nm) (Tani, 2002). These results were confirmed in hippocampal organotypic cultures, in which EP2 receptor activation by butaprost in nanomolar concentrations also significantly protected hippocampal pyramidal neurons after NMDA and OGD (Fig. 4D,E). Thus, activation of the EP2 receptor was directly protective in pure neuronal cultures, but also in organotypic slices, where receptor was present on both neurons and astrocytes. This effect differed from that of PGE2, which in binding to all four receptor subtypes elicited a net neutral effect on survival.
The neuroprotection of EP2 receptor activation is mediated by cAMP-PKA pathway
Ample precedent exists in neurons that activation of cAMP signaling is protective (Rydel, 1988; D'Mello, 1993; Hanson, 1998; Walton, 2000). We confirmed the positive coupling of EP2 receptor activation and cAMP production in pure hippocampal neurons stimulated with 10 nm butaprost (Fig. 4F). cAMP levels peaked at 5 min after stimulation and returned to baseline at 15 min. We then tested whether the protective effect of EP2 receptor activation depended on an intact cAMP signaling pathway in dispersed hippocampal neurons. Administration of the PKA antagonist H89 (Chijiwa, 1990) disrupted the neuroprotective effect of EP2 receptor activation in neurons stimulated with toxic doses of glutamate (Fig. 4G), indicating that the neuroprotective effects of EP2 receptor activation are mediated by cAMP and activation of PKA. We also tested the effects of a second PKA inhibitor, KT 5720 (Kase, 1987), and found that it too could reverse the neuroprotection of EP2 receptor activation (Fig. 4H). The reversal of protection using two distinct PKA inhibitors suggests that EP2 mediates this effect via cAMP and PKA activation.
Genetic deletion of EP2 results in a greater infarct volume in a model of transient focal ischemia
From the preceding in vitro studies that demonstrated a neuroprotective effect of pharmacological EP2 receptor activation in OGD and NMDA toxicity, we hypothesized that genetic deletion of the EP2 receptor in vivo would lead to increased infarct volumes after stroke. A model of transient focal ischemia with MCAO followed by reperfusion was selected to test this hypothesis using male EP2-/- and EP2+/+ WT mice (Fig. 5). EP2-/- male mice do not show differences in mean arterial pressures (MAP) at baseline compared with WT littermates (Kennedy, 1999), and this finding was confirmed in this study (MAP, 77.2 ± 4.1 for WT, 77.8 ± 4.4 for EP2-/- mice). Physiological measurements as well as continuous LDF were monitored at baseline, during ischemia, and after reperfusion (n = 5 per genotype). There were no significant differences between EP2-/- and WT mice in MAP, pH, pCO2, pO2 or blood glucose levels before, during, or after MCAO (see Table 1 for intraischemic values). LDF measurements demonstrated that MCAO resulted in similar decreases in cortical perfusion throughout the ischemic period and similar increases in blood flow after reperfusion in both genotypes, suggesting that deletion of the EP2 receptor does not alter the severity of the ischemic insult (Fig. 5B) (11.2% of baseline for EP2-/-, 11.4% of baseline for WT). Examination of the large cerebral vessels did not show anatomical differences in the circle of Willis or in the origins of the cerebral arteries (Fig. 5A).
Figure 5.
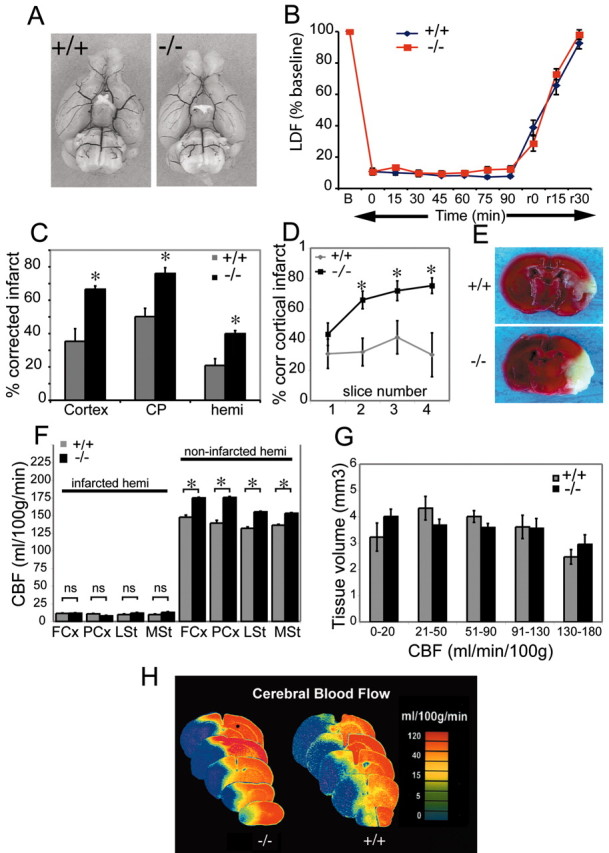
Deletion of the EP2 receptor results in marked increase in infarct volume. A, Macroscopic analysis of cerebral arterial vasculature does not show differences between genotypes. There are no differences in the circle of Willis and major cerebral arteries. B, Cortical perfusion during ischemia as estimated by laser Doppler flowmetry (%LDF) shows that the percentage of reduction in perfusion during ischemia and recovery of blood flow during reperfusion do not differ between genotypes. LDF measured over ischemic cortex indicates a rapid decrease in flow to 11.2% of baseline for wild-type and 11.4% for -/- mice after insertion of the filament. Flow is stable during ischemia and recovers to baseline after the filaments are withdrawn. C, The volume of infarcted brain tissue after 90 min of ischemia and 22.5 hr of reperfusion was measured and expressed as the percentage of corrected infarct in WT+/+ and EP2-/- male mice. Deletion of the EP2 receptor results in significantly larger stroke volumes in cerebral cortex, caudate-putamen (CP), and hemisphere (hemi). There is a 0.88-, 0.51-, and 0.91-fold increase in stroke volume in EP2-/- mice over wild-type for cerebral cortex, caudate-putamen, and hemisphere, respectively (*p < 0.001, 0.01, and 0.05, respectively). D, Percentage of corrected cortical infarct volume was measured over four coronal levels in wild-type and EP2-/- mice at 22.5 hr after MCAO and shows a significant increase in percentage of corrected cortical volume (p < 0.0005, 0.005, and 0.005 for slice number 2, 3, and 4, respectively). E, Representative TTC-stained coronal sections at the level of the striatum shows large infarct in EP2-/- mouse. F, End-ischemic CBF in WT (+/+) and EP2-/- (-/-) male mice. Mice were subjected to 90 min of MCAO, and CBF was measured at end-ischemia with [14C] iodoantipyrine autoradiography. rCBF measurements within the infarcted and noninfarcted hemispheres (infarcted hemi and noninfarcted hemi) are regionally quantified in cerebral cortex and striatum (FCx and PCx: frontal and parietal cortex, respectively; LSt and MSt: lateral and medial striatum, respectively) in WT and EP2-/- mice. No significant differences were seen in the level of rCBF reduction between genotypes (n = 4 WT; 5 EP2-/-) in the ischemic infarcted hemisphere, indicating an equivalence of vascular tone during MCAO. In the noninfarcted hemisphere, there is an increase in CBF in the EP2-/- as compared with WT; *p < 0.01. G, Brain volumes at incremental levels of absolute regional cerebral blood flow in EP2-/- versus wild-type male mice. Mice were subjected to 90 min of MCAO, and CBF was measured at end-ischemia with [14C] iodoantipyrine autoradiography. There were no differences between groups at any flow increment, suggesting that the loss of the EP2 receptor does not alter the distribution of tissue volume with low to near-zero perfusion during middle cerebral artery occlusion. H, Colorized autoradiograms from a pair of representative WT and EP2-/- brains are shown with sections taken at -2, -1, 0, +1, and +2 mm from the bregma showing changes in CBF at 90 min of MCAO.
Table 1.
Physiological variables in WT and EP2-/-mice intraischemically at 60 min of MCAO
|
Group |
pH |
PaCO2 |
PaO2 |
Hg |
Glucose |
MAP |
|---|---|---|---|---|---|---|
| WT | 7.39 ± 0.02 | 41 ± 1.1 | 158 ± 10 | 12.6 ± 1.2 | 143 ± 18 | 74 ± 4 |
| EP2-/- |
7.39 ± 0.03 |
45 ± 2 |
145 ± 6 |
12.3 ± 2.1 |
129 ± 14 |
76 ± 3 |
There were no differences in pH, arterial CO2 or O2 tension, blood pressure, or glucose levels between genotypes either at baseline or 60 min into occlusion.
The volume of infarcted brain tissue after 90 min of ischemia and 22.5 hr of reperfusion was measured in a second cohort of mice (n = 10 EP2-/- and n = 9 WT mice). Quantification of infarct with TTC demonstrated a significantly larger infarct size in EP2-/- mice compared with wild-type control mice (Fig. 5C-E) for corrected cortical, caudate-putamen, and hemispheric infarct volumes (cortex: p < 0.001; caudate-putamen: p < 0.01; hemisphere: p < 0.05). Figure 5D shows the percentage of cerebral cortical infarction in rostral to caudal slices that are in the territory of the MCA, including the frontal and parietal cortex in slices 1 through 4. The cerebral occipital cortex in the most posterior slice (slice 5; data not shown) is not supplied by the MCA, but by the PCA, and this accordingly results in an abrupt drop off in infarct size. In WT mice, cortical infarction volume peaks in slice 3 and comes down in a bell-shaped curve in slice 4. In EP2-/- mice, the increase in infarct size continues into slice 4, a result of the marked exacerbation of infarction in these EP2-/- mice. LDF measurements for the relative reduction in CBF at ischemic onset were similar in both WT and EP2-/- mice, (EP2-/- 13.8% of baseline, WT 14.1% of baseline). No differences were observed for rectal and temporalis temperature between genotypes. Mean neurological deficits were significantly different between genotypes (NS = 3.1 ± 0.01 and 2.2 ± 0.02 for EP2-/- and WT, respectively), reflective of the increased ischemic damage seen in the EP2-/- mice.
Although LDF measures the advent of occlusion and reperfusion and is an adequate marker of relative changes in CBF, it may not fully capture regional differences in CBF in the penumbra that can have important physiological consequences in stroke. To ascertain that the EP2-/- mice sustained a similar intraischemic insult as compared with WT mice, and to rule out enhanced vasoconstriction as a mechanism leading to an increased infarct size in EP2-/- mice, absolute CBF was calculated using quantitative [14C]-IAP autoradiography at 90 min of MCAO. Potential differences in vascular responsiveness between EP2-/- and WT mice were examined by quantifying absolute regional CBF in the frontal and parietal cortex and the lateral and medial striatum (Fig. 5F-H, FCx and PCx; LSt and MSt;) in the ischemic and in the contralateral nonischemic hemisphere (n = 5 EP2-/- and 4 WT per genotype). There were no differences in absolute end-ischemic CBF in the ipsilateral ischemic hemispheres (Fig. 5F) (p = ns) between WT and EP2-/- mice in any of the brain regions examined, suggesting equivalence of vascular tone during ischemia in both genotypes. CBF in the nonischemic contralateral hemisphere was higher in EP2-/- mice compared with WT (p < 0.01 for parietal and frontal cortex, as well as medial and lateral striatum). The significance of this increase in the noninfarcted hemisphere is not clear, given the equivalent reductions in ischemic blood flow in the infarcted hemispheres of both genotypes. In the nonischemic hemisphere, a difference in flow redistribution after MCAO is a possible explanation for the differences in CBF between genotypes, however this is not addressed in this study. To further examine the differences in end-ischemic CBF distribution in these same animals, we quantified brain tissue volume that experienced near-zero CBF as well as tissue volumes experiencing less severely reduced CBF. Figure 5G shows the results of this partitioning of brain volumes into incremental levels of absolute regional CBF in EP2-/- and WT animals in the ischemic hemisphere. There were no differences in brain volumes at incremental levels of absolute regional CBF between genotypes, suggesting that the loss of the EP2 receptor does not alter the distribution of tissue area with low to near-zero perfusion during MCAO. These data suggest that the increased stroke injury in EP2-/- mice involve mechanisms that do not involve exacerbation of intraischemic CBF.
Discussion
In this study, we examined the effects of PGE2 on neuronal survival, and whether PGE2 might mediate the toxic effects of COX-2 enzymatic activity. Of the four EP receptors that bind PGE2, EP2 and EP3 are expressed in telencephalic structures and are likely to mediate downstream effects of COX-2 enzymatic activity in cerebral ischemia. In this study, we focused on the function of the EP2 receptor in neuronal survival in vitro and in vivo in a model of transient focal ischemia. Using dispersed neuronal cultures and confirmatory studies in organotypic hippocampal cultures, we demonstrate that activation of the PGE2 EP2 receptor subtype paradoxically protects against excitotoxic and anoxic injury in vitro. Subsequent studies suggest that this protective action is dependent on cAMP-mediated signaling. We further confirm the protective effect of the EP2 receptor in vivo in wild-type and EP2-/- mice using the MCAO/reperfusion model of transient focal ischemia, in which deletion of the EP2 receptor results in a significant increase in infarct size. EP2-/- and WT mice had similar reductions in absolute CBF during ischemia, as measured by [14C]-IAP, suggesting that differences in vascular tone do not contribute to the increase in stroke volume in EP2-/- mice. These data demonstrate a novel neuroprotective pathway mediated by a highly abundant PGE2 receptor subtype, in which activation of the EP2 receptor can prevent injury in paradigms of acute excitotoxicity and ischemia.
Immunocytochemical studies of EP2 receptor distribution confirm at the protein level a robust and widespread expression of EP2 receptor in forebrain, with high expression in neurons and astrocytes in cerebral cortex, striatum, and hippocampus (Caggiano, 1999; Zhang and Rivest, 1999). Using the selective EP2 receptor agonist butaprost, we observed a marked protective effect in pure neuronal cultures and organotypic hippocampal cultures subjected to glutamate or OGD toxicity. Activation of the EP2 receptor resulted in an increase in cAMP levels, consistent with previous studies of this receptor in non-neuronal cells. Ample evidence has accumulated, suggesting a neuroprotective effect of cAMP in a variety of neurotoxic paradigms, so we tested whether inhibition of the downstream effects of increased cAMP could reverse the neuroprotective effect of EP2 activation. The PKA inhibitors H89 and KT 5720 abolished the neuroprotective effect of the EP2 receptor, suggesting that cAMP signaling is required for the EP2-mediated protective effect. Given that other prostaglandin receptors, such as DP, IP, and EP4 are also coupled to cAMP, it is possible that these receptors as well might promote neuroprotective effects when selectively activated.
The protective effect of the EP2 receptor was confirmed in vivo using the MCAO/reperfusion model of transient cerebral ischemia in EP2-/- and WT mice. Physiological parameters were similar at baseline and during ischemia in both genotypes. Regional CBF assessed by LDF did not differ between genotypes, suggesting that the increased stroke volume in EP2-/- mice was not caused by differences in the severity of the ischemic insult. To quantify regional differences in absolute CBF, particularly in the penumbra, where enhanced vasoconstriction could lead to increased infarct size, [14C]-IAP autoradiography was performed and demonstrated that EP2-/- and WT mice had equivalent reductions in absolute CBF in the ischemic hemisphere. These data suggest that increased vasoconstrictor tone during ischemia does not account for the increased damage seen in the EP2-/-mice, but indicates rather that the EP2 receptor mediates a survival effect.
The EP2 receptor can participate in arteriolar vasodilation (Kennedy, 1999), although other prostaglandins also function in regulation of vascular tone (for review, see Breyer et al., 2001). The effects on absolute and relative CBF in mice lacking PG receptor subtypes have not been previously characterized. Pharmacologic approaches have demonstrated rapid and transient reductions in MAP in response to infusions of PGE2 or the EP2 receptor agonist butaprost in WT mice, a response that is absent in EP2-/- mice (Kennedy, 1999), suggesting that the EP2 receptor participates in regulation of vascular tone. The lack of differences in ischemic CBF may in part be attributable to preserved vasodilatory and vasoconstrictor responses mediated by prostacyclin and EP1/EP3 receptors in EP2-/- mice, and suggest that overall vascular responsiveness remains intact in these mice (Kennedy, 1999). Interestingly, there is an increase in the contralateral noninfarcted hemisphere CBF in EP2-/- mice, and although this marginal elevation is significant, it is unclear what relevance this has to the increased infarct volume in the ischemic hemisphere. These experiments do not address the possibility that resting as well as reperfusion CBF may differ between genotypes, and the possibility of subtle changes in cortical or striatal blood flow cannot be completely excluded at time points during reperfusion because these were not evaluated in this study. In addition, our reperfusion paradigm lasted 24 hr, and in these experiments, we cannot address the function of EP2 receptor at later times, specifically its role in the recruitment and activation of immune cells in the postischemic inflammatory phase of stroke. This is of relevance, given that PGE2 has been shown to inhibit lymphocytic and microglial activation and the expression of neurotoxic cytokines and inflammatory mediators (Fedyk, 1996; Caggiano, 1999; Montine, 2002).
How then is the toxicity of COX-2 enzymatic activity mediated? PG receptor subtypes that are not positively coupled to cAMP could mediate toxic effects, for example the EP3 or the EP1 receptor, or other prostanoids such as the FP receptor, which promotes neuronal death in spinal cord injury (Kitanaka et al., 1993; Liu et al., 2001) and is coupled to increased PI turnover and elevation of intracellular Ca2+. PGF2α levels increase in models of excitotoxicity (Seregi et al., 1985; Ogawa et al., 1987; Nakagomi et al., 1990) and after catecholamine-mediated neurotransmission in hippocampal mossy fibers (Separovic and Dorman, 1993). Lerea (1997) has further demonstrated that PGF2α couples NMDA receptor activation to induction of the immediate early gene c-fos in neurons, underscoring the importance of prostaglandin signaling in NMDA receptor-mediated synaptic plasticity as well as injury. The mechanism by which PGD2 could promote toxicity is less clear. Although PGD2 has been immunolocalized to all regions of the brain (Watanabe et al., 1985), the DP receptor localizes predominantly to the leptomeninges (Oida et al., 1997; Mizoguchi et al., 2001) and is positively coupled to cAMP. A second receptor for PGD2 has been identified (Abe et al., 1999) that is coupled to Gαi and induces intracellular Ca2+ mobilization (Hirai et al., 2001), however its expression pattern and function in brain have not been defined. The PGI2 IP receptor is positively coupled to cAMP and is present in brain at highest concentration in sensory integration areas in spinal cord and brainstem (Matsumura, 1995) and in blood vessels where it functions as a vasodilator. More recently, specific binding of a PGI2 analog suggests the existence of a second IP receptor that is highly concentrated in telencephalic structures (Watanabe, 1999). Finally, the TXA2 TP receptor, which is coupled to Gq and phospholipase activation, is present in endothelium and functions in vascular tone and platelet aggregation. TP receptors are also expressed in hippocampal neurons and can promote glutamate (Hsu et al., 1996) and noradrenaline release (Nishihara, 1999) and inhibit voltage-sensitive Ca2+ channels (Hsu and Kan, 1996). In addition, TP receptors are present on astrocytes (Nakahata, 1992) and oligodendrocyte and myelin tracts (Blackman, 1998). The complex effects of TXA2 will depend on the cross-activation of TP receptors on multiple cell types. Taken together, the complexity of the COX-2 effect will arise out of the integration of multiple PG receptor pathways, each with a distinct signaling and downstream genetic signature. Recent studies focusing on downstream effects of COX-2 activity point to a potential effect on cell cycle gene transcription (Trifan, 1999; Mirjany, 2002; Xiang, 2002), a finding of relevance given the neurotoxic potential of COX-2 activity.
COX-2 activity may exert effects on neuronal survival alternatively in a PG-independent manner through the elaboration of free radical species. The COX enzyme possesses two active sites, a cyclooxygenase site, which catalyzes the conversion of arachidonic acid to the PGG2 intermediate, and a peroxidase site, which reduces the PGG2 intermediate to PGH2 and can increase NF-κB signaling independently of the cyclooxygenase activity (Munroe, 1995). The peroxidase component of the COX enzyme requires a reducing cosubstrate to donate a single electron to the peroxidase intermediate, and a cosubstrate free radical is then produced for each reaction (Eling, 1990; Smith, 1991). Thus, increased arachidonic acid metabolism by COX-2 can produce reactive oxygen species as a byproduct of the peroxidase activity of the enzyme, a potential mechanism that is being explored in models of neurodegenerative disease (Hastings, 1995; Teismann, 2003).
In conclusion, in vitro and in vivo studies of the PGE2 EP2 receptor subtype demonstrate that the EP2 signaling cascade mediates a significant neuroprotective effect in the setting of glutamate toxicity and ischemia. The dependence on cAMP-mediated signaling for this neuroprotective effect suggests that other PG receptors that are similarly coupled to cAMP may also protect neurons. These novel findings have broad implications for potential new therapeutic strategies in stroke that target specific PG receptor pathways.
Footnotes
This work was supported in part by the American Federation for Aging Research and the American Heart Association and by National Institutes of Health Grants GM15431 and DK46205. We thank J. Dyhr, D. Eve, and R. Chivukula for technical assistance and K. Conant, P. Beachy, and V. Dawson for helpful comments.
Correspondence should be addressed to K. Andreasson, 600 North Wolfe Street, Meyer 5-119B, Baltimore, MD 21205. E-mail: kandreas@jhmi.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240257-12$15.00/0
References
- Abdel-Hamid KM, Tymianski M (1997) Mechanisms and effects of intracellular calcium buffering on neuronal survival in organotypic hippocampal cultures exposed to anoxia/aglycemia or to excitotoxins. J Neurosci 17: 3538-3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe H, Takeshita T, Nagata K, Arita T, Endo Y, Fujita T, Takayama H, Kubo M, Sugamura K (1999) Molecular cloning, chromosome mapping and characterization of the mouse CRTH2 gene, a putative member of the leukocyte chemoattractant receptor family. Gene 227: 71-77. [DOI] [PubMed] [Google Scholar]
- Adams J, Collaco-Moraes Y, de Belleroche J (1996) Cyclooxygenase-2 induction in cerebral cortex: an intracellular response to synaptic excitation. J Neurochem 66: 6-13. [DOI] [PubMed] [Google Scholar]
- Akaike A, Kaneko S, Tamura Y, Nakata N, Shiomi H, Ushikubi F, Narumiya S (1994) Prostaglandin E2 protects cultured cortical neurons against N-methyl-d-aspartate receptor-mediated glutamate cytotoxcity. Brain Res 663: 237-244. [DOI] [PubMed] [Google Scholar]
- Alkayed NJ, Harakumi I, Kimes AS, London ED, Traystman RJ, Hurn PD (1998) Gender-linked injury in experimental stroke. Stroke 29: 159-165. [DOI] [PubMed] [Google Scholar]
- Amateau SK, McCarthy MM (2002) A novel mechanism of dendritic spine plasticity involving estradiol induction of prostaglandin-E2. J Neurosci 22: 8586-8596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasson KI, Savonenko A, Vidensky S, Goellner JJ, Zhang Y, Shaffer A, Kaufmann WE, Worley PF, Isakson P, Markowska AL (2001) Age-dependent cognitive deficits and neuronal apoptosis in cyclooxygenase-2 transgenic mice. J Neurosci 21: 8198-8209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batshake B, Nilsson C, Sundelin J (1995) Molecular characterization of the mouse prostanoid EP1 receptor gene. Eur J Biochem 231: 809-814. [DOI] [PubMed] [Google Scholar]
- Bezzi P, Carmignoto G, Pasti L, Vesce S, Rossi D, Rizzini BL, Pozzan T, Volterra A (1998) Prostaglandins stimulate calcium dependent glutamate release in astrocytes. Nature 391: 281-285. [DOI] [PubMed] [Google Scholar]
- Blackman S, Dawson G, Antonakis K, Le Breton G (1998) The identification and characterization of oligodendrocyte thromboxane A2 receptors. J Biol Chem 273: 475-483. [DOI] [PubMed] [Google Scholar]
- Boie YSR, Sawyer N, Slipetz DM, Ungrin MD, Neuschafer-Rube F, Puschel GP, Metters KM, Abramovitz M (1997) Molecular cloning and characterization of the four rat prostaglandin E2 prostanoid receptor subtypes. Eur J Pharmacol 340: 227-241. [DOI] [PubMed] [Google Scholar]
- Breyer RM, Bagdassarian CK, Myers SA, Breyer MD (2001) Prostanoid receptors: subtypes and signaling. Annu Rev Pharmacol Toxicol 41: 661-690. [DOI] [PubMed] [Google Scholar]
- Breyer RM, Kennedy CR, Zhang Y, Guan Y, Breyer MD (2002) Targeted gene disruption of the prostaglandin E2 EP2 receptor. Adv Exp Med Biol 507: 321-326. [DOI] [PubMed] [Google Scholar]
- Brock TG, McNish RW, Peters-Golden M (1999) Arachidonic acid is preferentially metabolized by cyclooxygenase-2 to prostacyclin and prostaglandin E2. J Biol Chem 274: 11660-11666. [DOI] [PubMed] [Google Scholar]
- Caggiano AO, Kraig RP (1999) Prostaglandin E receptor subtypes in cultured rat microglia and their role in reducing lipopolysaccharide-induced interleukin-1β production. J Neurochem 72: 565-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson NG (2003) Neuroprotection of cultured cortical neurons mediated by the cyclooxygenase-2 inhibitor APHS can be reversed by a prostanoid. J Neurosci Res 71: 71-88. [DOI] [PubMed] [Google Scholar]
- Cazevielle C, Muller A, Meynier F, Dutrait N, Bonne C (1994) Protection by prostaglandins from glutamate toxicity in cortical neurons. Neurochem Int 24: 156-159. [DOI] [PubMed] [Google Scholar]
- Chen C, Magee JC, Bazan NG (2002) Cyclooxygenase-2 regulates prostaglandin E2 signaling in hippocampal long-term synaptic plasticity. J Neurophysiol 87: 2851-2857. [DOI] [PubMed] [Google Scholar]
- Chijiwa T, Mishima A, Hagiwara N, Sano M, Hayashi K, Inoue Y, Naito K, Toshioka T, Hidaka H (1990) Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase (H89) of PC12D pheochromocytoma cells. J Biol Chem 265: 5267-5272. [PubMed] [Google Scholar]
- Czapiga M, Colton CA (1999) Function of microglia in organotypic slice cultures. J Neurosci Res 56: 644-651. [DOI] [PubMed] [Google Scholar]
- D'Mello S, Galli C, Ciotti T, Calissano P (1993) Induction of apoptosis in cerebellar granule cells by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA 23: 10989-10993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David JC, Yamada KA, Bagwe MR, Goldberg MP (1996) AMPA receptor activation is rapidly toxic to cortical astrocytes when desensitization is blocked. J Neurosci 16: 200-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore S, Otsuka T, Mito T, Sugo N, Hand T, Wu L, Hurn P, Traystman R, Andreasson K (2003) Neuronal overexpression of cyclooxygenase-2 increases cerebral infarction. Ann Neurol 54: 155-162. [DOI] [PubMed] [Google Scholar]
- Drachman DB, Frank K, Dykes-Hoberg M, Teismann P, Almer G, Przedborski S, Rothstein JD (2002) Cyclooxygenase 2 inhibition protects motor neurons and prolongs survival in a transgenic mouse model of ALS. Ann Neurol 52: 771-778. [DOI] [PubMed] [Google Scholar]
- Dumuis A, Sebben M, Haynes L, Pin JP, Bockaert J (1988) NMDA receptors activate the arachidonic acid cascade system in striatal neurons. Nature 336: 68-70. [DOI] [PubMed] [Google Scholar]
- Ek M, Arias C, Sawchenko P, Ericsson-Dahlstrand A (2000) Distribution of the EP3 prostaglandin E(2) receptor subtype in the rat brain: relationship to sites of interleukin-1-induced cellular responsiveness. J Comp Neurol 428: 5-20. [DOI] [PubMed] [Google Scholar]
- Eling TE, Thompson DC, Foureman GL, Curtis JF, Hughes MF (1990) Prostaglandin H synthase and xenobiotic oxidation. Annu Rev Pharmacol Toxicol 330: 1-45. [DOI] [PubMed] [Google Scholar]
- Fedyk ER, Ripper JM, Brown DM, Phipps RP (1996) A molecular analysis of PGE receptor (EP) expression on normal and transformed B lymphocytes: coexpression of EP1, EP2, EP3beta and EP4. Mol Immunol 33: 33-45. [DOI] [PubMed] [Google Scholar]
- Feng ZH, Wang, TG, Li DD, Fung P, Wilson BC, Liu B, Ali SF, Langenbach R, Hong JS (2002) Cyclooxygenase-2-deficient mice are resistant to 1-methyl-4-phenyl1, 2, 3, 6-tetrahydropyridine-induced damage of dopaminergic neurons in the substantia nigra. Neurosci Lett 329: 354-358. [DOI] [PubMed] [Google Scholar]
- Gardiner PJ (1986) Characterization of prostanoid relaxant/inhibitory receptors using a highly selective agonist, TR4979. Br J Pharmacol 87: 45-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailer NP, Jarhult JD, Nitsch R (1996) Resting microglial cells in vitro: analysis of morphology and adhesion molecule expression in organotypic hippocampal slice cultures. Glia 18: 319-331. [DOI] [PubMed] [Google Scholar]
- Hanson M, Shen S, Wiemelt A, McMorris F, Barres B (1998) Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro J Neurosci 18: 7361-7371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings TG (1995) Enzymatic oxidation of dopamine: the role of prostaglandin H synthase. J Neurochem 64: 919-924. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Uliasz T, Vidwans A, Hewett JA (2000) Cyclooxygenase-2 contributes to N-methyl-d-aspartate mediated neuronal cell death in primary cortical cultures. J Pharmacol Exp Ther 293: 417-425. [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, Nagata K (2001) Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med 193: 255-261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu K, Kan W (1996) Thromboxane A2 agonist modulation of excitatory synaptic transmission in the rat hippocampal slice. Br J Pharmacol 118: 2220-2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu K, Huang C, Kan W, Gean P (1996) TXA2 agonists inhibit high-voltage-activated calcium channels in rat hippocampal CA1 neurons. Am J Physiol 271: C1269-C1277. [DOI] [PubMed] [Google Scholar]
- Huang M, Stolina M, Sharma S, Mao JT, Zhu L, Miller PW, Wollman J, Herschman H, Dubinett SM (1998) Non-small cell lung cancer cyclooxygenase-2-dependent regulation of cytokine balance in lymphocytes and macrophages: up-regulation of interleukin 10 and down-regulation of interleukin 12 production. Cancer Res 58: 1208-1216. [PubMed] [Google Scholar]
- Jakobsen B, Zimmer J (2001) Chronic exposure of kainate and NBQX changes AMPA toxicity in hippocampal slice cultures. NeuroReport 12: 3593-3597. [DOI] [PubMed] [Google Scholar]
- Jay TM, Lucignani G, Crane AM, Jehle J, Sokoloff L (1988) Measurement of local cerebral blood flow with [14C]iodoantipyrine in the mouse. J Cereb Blood Flow Metab 8: 121-129. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M (1987) K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochem Biophys Res Commun 142: 436-440. [DOI] [PubMed] [Google Scholar]
- Kennedy CR, Zhang Y, Brandon S, Guan Y, Coffee K, Funk CD, Magnuson MA, Oates JA, Breyer MD, Breyer RM (1999) Salt-sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med 5: 217-220. [DOI] [PubMed] [Google Scholar]
- Kiriyama M, Ushikubi F, Kobayashi T, Hirata M, Sugimoto Y, Narumiya S (1997) Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br J Pharmacol 122: 217-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitanaka J, Ishibashi T, Baba A (1993) Phloretin as an antagonist of prostaglandin F2 alpha receptor in cultured rat astrocytes. J Neurochem 60: 704-708. [DOI] [PubMed] [Google Scholar]
- Lerea LS, Carlson NG, Simonato M, Morrow J, Roberts JL, McNamara JO (1997) Prostaglandin F 2alpha is required for NMDA receptor-mediated induction of c-fos mRNA in dentate gyrus neurons. J Neurosci 17: 117-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin TN, He YY, Wu G, Khan M, Hsu CY (1993) Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 24: 117-121. [DOI] [PubMed] [Google Scholar]
- Liu D, Li L, Augustus L (2001) Prostaglandin release by spinal cord injury mediates production of hydroxyl radical, malondialdehyde and cell death: a site of the neuroprotective action of methylprednisolone. J Neurochem 77: 1036-1047. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R (1989) Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1: 84-91. [DOI] [PubMed] [Google Scholar]
- Lu X, Wyszynski M, Sheng M, Baudry M (2001) Proteolysis of glutamate receptor-interacting protein by calpain in rat brain: implications for synaptic plasticity. J Neurochem 77: 1553-1560. [DOI] [PubMed] [Google Scholar]
- Matsumura K, Watanabe Y, Onoe H, Watanabe Y (1995) Prostacyclin receptor in the brain and central terminals of the primary sensory neurons: an autoradiographic study using a stable prostacyclin analogue [3H]iloprost. Neuroscience 65: 493-503. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Alkayed N, Williams M, Traystman RJ, Hurn PD (2001) Post-ischemic estrogen reduces hypoperfusion and secondary ischemia after experimental stroke. Stroke 32: 796-802. [DOI] [PubMed] [Google Scholar]
- McCullough LD, Blizzard K, Simpson ER, Oz OK, Hurn PD (2003) Aromatase cytochrome P450 and extragonadal estrogen play a role in ischemic neuroprotection. J Neurosci 23: 8701-8705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen S, Fusco FR, Yrjanheikki J, Keinanen R, Hirvonen T, Roivainen R, Narhi M, Hokfelt T, Koistinaho J (1997) Spreading depression and focal brain ischemia induce cyclooxygenase-2 in cortical neurons through N-methyl-d-aspartic acid-receptors and phospholipase A2. Proc Natl Acad Sci USA 94: 6500-6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirjany M, Ho L, Pasinetti GM (2002) Role of cyclooxygenase-2 in neuronal cell cycle activity and glutamate-mediated excitotoxicity. J Pharmacol Exp Ther 301: 494-500. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A, Eguchi N, Kimura K, Kiyohara Y, Qu WM, Huang ZL, Mochizuki T, Lazarus M, Kobayashi T, Kaneko T, Narumiya S, Urade Y, Hayaishi O (2001) Dominant localization of prostaglandin D receptors on arachnoid trabecular cells in mouse basal forebrain and their involvement in the regulation of non-rapid eye movement sleep. Proc Natl Acad Sci USA 98: 11674-11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine TJ, Milatovic D, Gupta RC, Valyi-Nagy T, Morrow JD, Breyer RM (2002) Neuronal oxidative damage from activated innate immunity is EP2 receptor-dependent. J Neurochem 83: 463-470. [DOI] [PubMed] [Google Scholar]
- Munroe DG, Wang EY, MacIntyre JP, Tam SS, Lee DH, Taylor GR, Zhou L, Plante RK, Kazmi SM, Bauerle PA, et al. (1995) Novel intracellular signaling function of prostaglandin H synthase-1 in NF-kappa B activation. J Inflamm 45: 260-268. [PubMed] [Google Scholar]
- Nakagomi T, Sasaki T, Ogawa H, Noguchi M, Saito I, Takakura K (1990) Immunohistochemical localization of prostaglandin F2 alpha in reperfused gerbil brain. Neurol Med Chir (Tokyo) 30: 223-228. [DOI] [PubMed] [Google Scholar]
- Nakahata N, Ishimoto H, Kurita M, Ohmori K, Takahashi A, Nakanishi H (1992) The presence of thromboxane A2 receptors in cultured astrocytes from rabbit brain. Brain Res 583: 100-104. [DOI] [PubMed] [Google Scholar]
- Nakayama M, Uchimura K, Zhu RL, Nagayama T, Rose ME, Stetler RA, Isakson PC, Chen J, Graham SH (1998) Cyclooxygenase-2 inhibition prevents delayed death of CA1 hippocampal neurons following global ischemia. Proc Natl Acad Sci USA 95: 10954-10959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell DW, Barth A, Papermaster V, Malouf AT (1995) Glutamate and non-glutamate receptor mediated toxicity caused by oxygen and glucose deprivation in organotypic hippocampal cultures. J Neurosci 15: 7702-7711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishihara I, Minami T, Watanabe Y, Ito S, Hayaishi O (1995) Prostaglandin E2 stimulates glutamate release from synaptosomes of rat spinal cord. Neurosci Lett 18: 57-60. [DOI] [PubMed] [Google Scholar]
- Nishihara M, Yokotani K, Inoue S, Osumi Y (1999) U-46619, a selective Thromboxane A2 mimetic, inhibits the release of endogenous noradrenaline from the rat hippocampus in vitro. Jpn J Pharmacol 82: 226-231. [DOI] [PubMed] [Google Scholar]
- Nogawa S, Zhang F, Ross ME, Iadecola C (1997) Cyclo-oxygenase-2 gene expression in neurons contributes to ischemic brain damage. J Neurosci 17: 2746-2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H, Sasaki T, Kassell NF, Nakagomi T, Lehman RM, Hongo K (1987) Immunohistochemical demonstration of increase in prostaglandin F2-alpha after recirculation in global ischemic rat brains. Acta Neuropathol 75: 62-68. [DOI] [PubMed] [Google Scholar]
- Oida H, Hirata M, Sugimoto Y, Ushikubi F, Ohishi H, Mizuno N, Ichikawa A, Narumiya S (1997) Expression of messenger RNA for the prostaglandin D receptor in the leptomeninges of the mouse brain. FEBS Lett 417: 53-56. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM, Aisen PS (1998) Cyclooxygenase-2 expression is increased in frontal cortex of Alzheimer's disease brain. Neuroscience 87: 319-324. [DOI] [PubMed] [Google Scholar]
- Pompl PN, Ho L, Bianchi M, McManus T, Qin W, Pasinetti GM (2003) A therapeutic role for cyclooxygenase-2 inhibitors in a transgenic mouse model of amyotrophic lateral sclerosis. FASEB J 10: 1096. [DOI] [PubMed] [Google Scholar]
- Prasad KN, Hovland AR, La Rosa FG, Hovland PG (1998a) Prostaglandins as putative neurotoxins in Alzheimer's disease. Proc Soc Exp Biol Med 219: 120-125. [DOI] [PubMed] [Google Scholar]
- Prasad KN, La Rosa FG, Prasad JE (1998b) Prostaglandins act as neurotoxin for differentiated neuroblastoma cells in culture and increase levels of ubiquitin and beta-amyloid. In Vitro Cell Dev Biol Anim 34: 265-274. [DOI] [PubMed] [Google Scholar]
- Rao A, Kim E, Sheng M, Craig AM (1998) Heterogeneity in the molecular composition of excitatory postsynaptic sites during development of hippocampal neurons in culture. J Neurosci 18: 1217-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusa R, Alkayed NJ, Crain B, Traystman RJ, Kimes AS, London ED, Klaus JA, Hurn PD (1999) 17β-Estradiol reduces stroke injury in estrogen-deficient female animals. Stroke 30: 1665-1670. [DOI] [PubMed] [Google Scholar]
- Rydel R, Greene L (1988) cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA 85: 1257-1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampei KG, S., Alkayed N, Crain B, Korach K, Traystman R, Demas G, Nelson R, Hurn P (2000) Stroke in estrogen receptor-alpha-deficient mice. Stroke 31: 738-743. [DOI] [PubMed] [Google Scholar]
- Sanzgiri R, Araque A, Haydon P (1999) Prostaglandin E2 stimulates glutamate receptor dependent astrocyte neuromodulation in cultured hippocampal cells. J Neurobiol 41: 221-229. [PubMed] [Google Scholar]
- Sawada M, Alkayed NJ, Goto S, Crain BJ, Traystman RJ, Shaivitz A, Nelson RJ, Hurn PD (2000) Estrogen receptor antagonist ICI182, 780 exacerbates ischemic injury in female mouse. J Cereb Blood Flow Metab 20: 112-118. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Tlustochowska A, Stuertz K, Djukic M, Gerber J, Schutz E, Kuhnt U, Nau R (2001) Organotypic hippocampal cultures. A model of brain tissue damage in Streptococcus pneumoniae meningitis. J Neuroimmunol 113: 30-39. [DOI] [PubMed] [Google Scholar]
- Separovic D, Dorman RV (1993) Prostaglandin F2 alpha synthesis in the hippocampal mossy fiber synaptosomal preparation: I. Dependence in arachidonic acid, phospholipase A2, calcium availability and membrane depolarization. Prostaglandins Leukot Essent Fatty Acids 48: 127-137. [DOI] [PubMed] [Google Scholar]
- Seregi A, Forstermann U, Heldt R, Hertting G (1985) The formation and regional distribution of prostaglandins D2 and F2 alpha in the brain of spontaneously convulsing gerbils. Brain Res 337: 171-174. [DOI] [PubMed] [Google Scholar]
- Sheller JR, Mitchell D, Meyrick B, Oates J, Breyer R (2000) EP(2) receptor mediates bronchodilation by PGE(2) in mice. J Appl Physiol 88: 2214-2218. [DOI] [PubMed] [Google Scholar]
- Smith WL, Marnett LJ, DeWitt DL (1991) Prostaglandin and thromboxane biosynthesis. Pharmacol Ther 48: 153-179. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37: 173-182. [DOI] [PubMed] [Google Scholar]
- Streit WJ (2000) Microglial response to brain injury. Toxicol Pathol 28: 28-30. [DOI] [PubMed] [Google Scholar]
- Sugimoto Y, Shigemoto R, Namba T, Negishi M, Mizuno N, Narumiya S, Ichikawa A (1994) Distribution of the messenger RNA for the prostaglandin E receptor subtype EP3 in the mouse nervous system. Neuroscience 62: 919-928. [DOI] [PubMed] [Google Scholar]
- Takadera T, Yumoto H, Tozuka Y, Ohyashiki T (2002) Prostaglandin E2 induces caspase-dependent apoptosis in rat cortical cells. Neurosci Lett 317: 61-64. [DOI] [PubMed] [Google Scholar]
- Tani KNA, Ishida A, Egashira H, Sagawa K, Harada H, Ogawa M, Maruyama T, Ohuchida S, Nakai H, Kondo K, Toda M (2002) Development of a highly selective EP2-receptor agonist. Part 2: identification of16-hydroxy-17, 17-trimethylene 9beta-chloro PGF derivatives. Bioorg Med Chem 10: 1107-1114. [DOI] [PubMed] [Google Scholar]
- Tasker RC, Coyle JT, Vornov JJ (1992) The regional vulnerability to hypoglycemia-induced neurotoxicity in organotypic hippocampal culture: protection by early tetrodotoxin or delayed MK-801. J Neurosci 12: 4298-4308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann P, Tieu K, Choi DK, Wu DC, Naini A, Hunot S, Vila M, Jackson-Lewis V, Przedborski S (2003) Cyclooxygenase-2 is instrumental in Parkinson's disease neurodegeneration. Proc Natl Acad Sci USA 100: 5473-5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifan OC, Smith RM, Thompson BD, Hla T (1999) Overexpression of cyclooxygenase-2 induces cell cycle arrest. Evidence for a prostaglandin-independent mechanism. J Biol Chem 274: 34141-34147. [DOI] [PubMed] [Google Scholar]
- Vidensky S, Zhang Y, Hand T, Goellner J, Isakson P, Andreasson K (2003) Neuronal overexpression of COX-2 results in dominant production of PGE2 and altered fever response. Neuromol Med 3: 15-27. [DOI] [PubMed] [Google Scholar]
- Vincent VA, Robinson CC, Simsek D, Murphy GM (2002a) Macrophage colony stimulating factor prevents NMDA-induced neuronal death in hippocampal organotypic cultures. J Neurochem 82: 1388-1397. [DOI] [PubMed] [Google Scholar]
- Vincent VA, Selwood SP, Murphy Jr GM (2002b) Proinflammatory effects of M-CSF and A beta in hippocampal organotypic cultures. Neurobiol Aging 23: 349-362. [DOI] [PubMed] [Google Scholar]
- Vornov JJ, Tasker RC, Coyle JT (1991) Direct observation of the agonist-specific regional vulnerability to glutamate, NMDA, and kainate neurotoxicity in organotypic hippocampal cultures. Exp Neurol 114: 11-22. [DOI] [PubMed] [Google Scholar]
- Vornov JJ, Tasker RC, Park J (1995) Neurotoxicity of acute glutamate transport blockade depends on coactivation of both NMDA and AMPA/kainate receptors in organotypic hippocampal cultures. Exp Neurol 133: 7-17. [DOI] [PubMed] [Google Scholar]
- Walton MR, Dragunow I (2000) Is CREB a key to neuronal survival? Trends Neurosci 23: 48-53. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Tokumoto H, Yamashita A, Narumiya S, Mizuno N, Hayaishi O (1985) Specific bindings of prostaglandin D2, E2 and F2 alpha in postmortem human brain. Brain Res 342: 110-116. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Matsumura K, Takechi H, Kato K, Morii H, Bjorkman M, Langstrom B, Noyori R, Suzuki M, Watanabe Y (1999) A novel subtype of prostacyclin receptor in the central nervous system. J Neurochem 72: 2583-2592. [DOI] [PubMed] [Google Scholar]
- Wu VW, Schwartz JP (1998) Cell culture models for reactive gliosis: new perspectives. J Neurosci Res 51: 675-681. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Ho L, Valdellon J, Borchelt D, Kelley K, Spielman L, Aisen PS, Pasinetti GM (2002) Cyclooxygenase (COX)-2 and cell cycle activity in a transgenic mouse model of Alzheimer's disease neuropathology. Neurobiol Aging 23: 327-334. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Andreasson K, Kaufmann WE, Barnes CA, Worley PF (1993) Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron 11: 371-386. [DOI] [PubMed] [Google Scholar]
- Yamagata K, Matsumura K, Inoue W, Shiraki T, Suzuki K, Yasuda S, Sugiura H, Cao C, Watanabe Y, Kobayashi S (2001) Coexpression of microsomal-type prostaglandin E synthase with cyclooxygenase-2 in brain endothelial cells of rats during endotoxin- induced fever. J Neurosci 21: 2669-2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Rivest S (1999) Distribution, regulation and colocalization of the genes encoding the EP2- and EP4-PGE2 receptors in the rat brain and neuronal responses to systemic inflammation. Eur J Neurosci 11: 2651-2668. [DOI] [PubMed] [Google Scholar]