

Abstract
High-frequency synaptic transmission is depressed by moderate rises in the extracellular potassium concentration ([K+]o). Previous reports have indicated that depression of action potential signaling may underlie the synaptic depression. Here, we investigated the specific contribution of K+-induced action potential changes to synaptic depression. We found that glutamatergic transmission in the hippocampal area CA1 was significantly depressed by 8-10 mm [K+]o, but that GABAergic transmission remained intact. Riluzole, a drug that slows recovery from inactivation of voltage-gated sodium channels (NaChs), interacts with subthreshold [K+]o to depress afferent volleys and EPSCs strongly. Thus, elevated [K+]o likely depresses synapses by slowing NaCh recovery from inactivation. It is unclear from previous studies whether [K+]o-induced action potential depression is caused by changes in initiation, reliability, or waveform. We investigated these possibilities explicitly. [K+]o-induced afferent volley depression was independent of stimulus strength, suggesting that changes in action potential initiation do not explain [K+]o-induced depression. Measurements of action potentials from single axons revealed that 8 mm [K+]o increased conduction failures in a subpopulation of fibers and depressed action potential amplitude in all fibers. Together, these changes quantitatively account for the afferent volley depression. We estimate that conduction failure explains more than half of the synaptic depression observed at 8 mm [K+]o, with the remaining depression likely explained by waveform changes. These mechanisms of selective sensitivity of glutamate release to [K+]o accumulation represent a unique neuromodulatory mechanism and a brake on runaway excitation.
Keywords: potassium, axon, action potential, propagation, conduction, hippocampus, synapse, CA1
Introduction
During strong stimulus trains, bulk [K+]o can rise by several millimolar in situ (Ransom and Goldring, 1973; Ballanyi et al., 1987). Sustained bulk [K+]o rises in the tens of millimolar can be associated with pathological states such as stroke/ischemia and seizures (Walz, 2000; Somjen, 2002). Despite the potential impact of [K+]o rises from physiological and pathophysiological activity on signaling, our understanding of the effects of [K+]o rises on synaptic function remains incomplete.
There is general agreement that in hippocampal area CA1, [K+]o rises cause depression of presynaptic afferent volley measurements (Sykova and Orkand, 1980; Hablitz and Lundervold, 1981; Poolos et al., 1987), a reflection of excitatory afferent activity. The effects of rises in [K+]o on action potentials in GABAergic axons and subsequent GABA release remain largely unknown, although effects on inhibition could be important for neuronal signaling in the face of [K+]o rises (Korn et al., 1987; McBain and Dingledine, 1992). In addition, whether [K+]o-induced presynaptic changes reflect alterations in action potential initiation, propagation, or waveform changes that depress Ca2+ influx are unknown.
Our previous work using solitary neurons in microcultures suggests that bulk [K+]o rises may selectively dampen evoked glutamatergic transmission, leaving evoked inhibitory transmission intact (He et al., 2002). Here, we explore whether these differences in susceptibility extend to intact circuits of hippocampal slices and whether changes in action potentials account for the susceptibility. If present in situ, selective regulation of evoked glutamate versus GABA signaling by [K+]o may be an important endogenous mechanism for maintaining relative levels of excitation and inhibition during high-frequency activity. Also, during pathological [K+]o accumulation, elevated [K+]o could paradoxically be an important brake on runaway excitation and could help limit excitotoxicity.
We found that [K+]o accumulation depressed EPSCs selectively relative to IPSCs in the hippocampal CA1 subfield, attributable to a differential sensitivity of glutamate-releasing versus GABA-releasing fibers to NaCh block and inactivation. Moderate increases in [K+]o that cause EPSC depression during high-frequency stimulus trains caused an increased probability of action potential conduction failure in single hippocampal axons and depressed action potential amplitudes. Conduction failure affected primarily a subpopulation of fibers, affected later action potentials in trains, and explained approximately half of the synaptic depression observed in elevated [K+]o during stimulus trains. Thus, despite recent evidence that action potential conduction is highly reliable under a variety of conditions, we conclude that action potential conduction failure participates in the synaptic depression observed during bulk [K+]o elevations.
Materials and Methods
Slice preparation. Sprague Dawley rats (12-16 d of age, unless otherwise noted) were anesthetized with halothane and decapitated. The brains were dissected into oxygenated ice-cold slicing buffer containing the following (in mm): 87 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3.0 MgCl2, 25 glucose, and 75 sucrose. Coronal slices (300 μm) were made through the hippocampus using a vibratome (World Precision Instruments, Sarasota, FL). Slices were transferred immediately to artificial CSF (aCSF) at 34°C for 30 min, and then incubated at room temperature (25°C) for at least 30 min before recording. The aCSF contained the following (in mm): 125 NaCl, 25 NaHCO3, 3 myoinositol, 3 Na-pyruvate, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, 0.4 ascorbic acid, and 10 glucose.
Electrophysiological recording. Slices were submerged in a perfusion chamber on an upright microscope (Nikon, Tokyo, Japan), and perfused with aCSF at room temperature (25°C) at a rate of 2-4 ml/min, unless otherwise stated. For experiments at 34°C, the temperature was set by a inline solution heater and heated chamber under the control of a dual-channel temperature control unit (Warner Instruments, Hamden, CT). CA1 pyramidal neurons were visualized in the slice using infrared differential interference contrast optics with a 40× water-immersion objective (Nikon). Bipolar stimulating electrodes (50 μm tip separation; Rhodes Medical Instruments, Woodland Hills, CA) were placed in the CA1 stratum radiatum within 150 μm of the recorded cell to stimulate afferent volley and synaptic responses.
Patch pipettes were pulled from filamented borosilicate glass capillaries to a tip diameter of 1-2 μm. Whole-cell recordings were made from neurons in CA1 stratum pyramidale using a patch-clamp amplifier (Axopatch 200B; Axon Instruments, Union City, CA), filtered at 5 kHz, digitized using a 16 bit analog/digital converter (Digidata 1322; Axon Instruments), and stored on a personal computer running acquisition software (pClamp 8.2-9.0; Axon Instruments). The whole-cell pipette solution contained the following (in mm): 130 cesium methanesulfonate, 4 NaCl, 0.5 CaCl2, 5 EGTA, 10 HEPES, 4 Mg-ATP, 0.3 Na2GTP, and 5 N-ethyl bromide quaternary salt (QX-314). Open-tip resistances were generally between 3 and 6 MΩ.
After establishing the whole-cell recording configuration, paired-pulses or stimulus trains were delivered at 20 or 50 Hz. Unless otherwise noted, stimulus intensity was varied to produce equivalent synaptic responses by altering stimulus amplitude (within the range of 80-160 μA) and/or duration (within the range of 60-160 μsec) until a reliable response to the conditioning pulse was achieved under control conditions. AMPA-receptor-mediated EPSCs were isolated with 50 μm d-APV and 100 μm picrotoxin in the superfusion solution. IPSCs were isolated with 50 μm d-APV and 1 μm 2,3-dihydroxy-6-nitro-7-sulfonyl-benzo[f] quinoxaline (NBQX). Holding potential was -70 mV for EPSCs and 0 mV for IPSCs. Access and input resistances were monitored throughout the recordings, but access resistance was not compensated. Afferent volley and field EPSP measurements were obtained with a patch pipette filled with extracellular saline and placed in the stratum radiatum within 150 μm of the stimulating electrode.
Paired-pulse modulation was always calculated as a ratio of the averaged waveforms of PSCs (Kim and Alger, 2001). Each averaged waveform was typically generated from 5 to 10 trials under each experimental condition. For PSCs whose decay spanned the paired-pulse interval, paired-pulse and frequency-dependent modulation was calculated by subtracting the decaying PSC immediately before the test PSC onset. An alternative method of fitting and subtracting an exponential decay from the preceding PSC was performed on a subset of 50 Hz IPSCs and EPSCs and, as expected, yielded slightly larger test response amplitudes in both control and elevated [K+]o conditions (16 ± 5% for IPSCs, n = 6; 7 ± 4% for EPSCs, n = 5). Thus, IPSC depression in particular may be slightly overestimated, but this did not affect the comparisons of different experimental conditions. All values are means ± SE. For statistical comparison of single responses, paired-pulse responses, or individual responses during a stimulus train, a paired, two-tailed Student's t test was performed, with the significance level set at p = 0.05. For comparison of overall synaptic or axonal responses to a stimulus train, a repeated-measures ANOVA was performed using SPSS (Chicago, IL) software, with the significance level set at p = 0.05.
Single-axon action potential recordings. Action potential recordings from single axons were made, similar to methods published recently (Raastad and Shepherd, 2003). Briefly, blunt patch pipettes (2-5 μm in diameter) filled with aCSF were lowered into the CA1 region 500-1500 μm from the stimulating electrode. Gentle suction was used to bring a small patch of tissue into the pipette and was maintained throughout recording. A slight increase in open tip resistance (less than threefold) was generally observed at this stage and was monitored throughout the recording. Single-axon action potentials were identified by their characteristic waveform (see Figs. 8 and 9 for examples), consistent latency from stimulus onset, and a step-like stimulus-response curve (Raastad and Shepherd, 2003). For recordings of single axons near the stimulating electrode, patch pipettes ∼1 μm in diameter (open tip resistance, ∼4-7 MΩ) were used, along with lower stimulus intensities (10-50 μA), to increase the likelihood of single axon detection. The bath for all single axon recordings included 100 μm picrotoxin, 25 μm d-APV, and 1 μm NBQX to eliminate postsynaptic currents.
Figure 8.

Effect of 8 mm [K+]o on action potentials from single CA1 axons. A, Responses of a representative low-reliability axon to a 10 pulse, 50 Hz stimulus train in 2.5 mm [K+]o (top) and 8 mm [K+]o (bottom). *Peak single-fiber action potential. Vertical spikes represent stimulus artifacts. B, Response of a representative high-reliability axon to a 10 pulse, 50 Hz stimulus train in 2.5 mm [K+]o (top) and 8 mm [K+]o (bottom). Note that although the fiber followed stimulation reliably in 8 mm [K+]o, there was more average depression of the waveform during the train than in the baseline condition. C, Stimulus input/output curve generated from the 2.5 mm [K+]o condition from the fiber in B. The multiple points reflect eight repetitions at each stimulus intensity, and variability reflects baseline noise, which did not appreciably change with successful action potential propagation. Note the all-or-none increase in response with increasing stimulus width, indicating the presence of a single activated axon. D, Failure analysis of all 17 axons analyzed (both high and low reliability) in 2.5 mm (filled circles represent initial trials, gray squares represent recovery) and 8 mm [K+]o (open circles). Note that the failure rate of the conditioning action potential was only slightly altered by 8 mm [K+]o. E, Summary waveform analysis of axons that continued to conduct in the 8 mm [K+]o condition (12 of the 17 axons shown in D and F). Action potential peak amplitude in these axons was depressed in later traces in the train (p < 0.05; repeated-measures ANOVA), but the amplitude of the conditioning action potential was not altered by 8 mm [K+]o. F, Summary of cumulative depression caused by both amplitude depression and increased failure rate (same 17 axons as in D). Note the quantitative similarity of these data to traces from afferent volleys shown in Figure 6B. Each response (PX) was normalized to the first peak of the stimulus train in 2.5 mm [K+]o (P1). Repeated-measures ANOVA revealed a significant interaction between [K+]o and stimulus number (p < 0.05).
Figure 9.

Failures can be attributed to a subset of axons with low reliability in elevated [K+]o. A, Failure analysis in 8 of 27 fibers that failed >40% in 8 mm [K+]o. Note that failures were still minimal (although sometimes detectable by the end of a train) in these fibers. B, In the remaining 19 fibers, conduction remained primarily faithful throughout the train even in elevated [K+]o. C, D, Correlation between baseline spike parameters and reliability in elevated [K+]o. The final responses of a 10 pulse, 50 Hz train were used to calculate baseline failures (C) and amplitude depression (D). C1, D1, Correlation between these baseline parameters and failure of transmission in 8 mm [K+]o. C2, D2, Cells divide into the low- and high-reliability groups, examining the probability of successful baseline action potential conduction at the 10th stimulus of the 50 Hz train (P10). Although eight fibers met the criterion for >40% failure in 8 mm K+ at some point during the train in 8 mm [K+]o (A), only seven lie above the 0.4 failure line in C2 and D2. This is because only the last response of a 10 pulse train is plotted in C2 and D2. One fiber met the criterion by failing >40% of the time at an earlier point in the train. Note that the correlation shows that failure during baseline transmission was weakly predictive of failure in elevated [K+]o (r2 = 0.27 for C1). This conclusion was supported by the averages shown in C2 (*p < 0.05). However, baseline amplitude alterations were not significantly correlated with reliability in elevated [K+]o (r2 = 0.10 for D1; p > 0.05 for D2).
For single axon recordings, full input/output (I/O) curves were generated in each experimental condition. Only trials at stimulus widths that were suprathreshold under all experimental conditions were analyzed. There was no consistent shift in the I/O relationship with elevated [K+]o, consistent with the macroscopic results shown in Figure 7. Responses of each axon to 16-32 stimuli under each experimental condition were analyzed. When no action potential (>2× SD of baseline noise) was present within a 4 msec window around the latency of action potentials evoked under control conditions, a failure was counted (Raastad and Shepherd, 2003). Action potential amplitude was measured as the difference between the peak and trough (Raastad and Shepherd, 2003) of the average action potential calculated from sweeps that contained no failures throughout the 10 pulse train. Amplitude-plus-failure measurements (see Fig. 8F) were made from averaged waveforms that included successes and failures.
Figure 7.

[K+]o-induced afferent volley depression is independent of stimulus intensity. A, Afferent volley stimulation input/output curve for conditioning pulses in a representative slice. Stimulus intensity was varied, whereas stimulus duration remained constant at 50 μsec. B-D, Modulation of afferent volley peak amplitudes during a 10 pulse, 50 Hz train at the indicated stimulus intensities (denoted by arrows in A). Responses during baseline and washout 2.5 mm [K+]o are indicated by filled circles and filled squares, respectively. Responses to 8 mm [K+]o are indicated by open circles. Frequency-dependent depression in 8 mm [K+]o remained quantitatively similar at all stimulus intensities. Similar effects were seen in three slices. NBQX (1 μm) was included in the bath to eliminate postsynaptic receptor activation.
Results
Selective modulation of glutamate release by elevated [K+]o
We examined the effect of exogenous bulk [K+]o elevations on evoked EPSCs and IPSCs in hippocampal slices. We found that 10 mm [K+]o significantly depressed EPSCs (Fig. 1A,C). In contrast to most presynaptic forms of depression, however, the EPSC depression was larger for the second of paired stimulus pulses than for the first, resulting in a decrease in paired-pulse facilitation (Fig. 1A,C). This effect contrasts with most presynaptic depressants, which increase paired-pulse facilitation (Thomson, 2000). For instance, we found in this preparation that decreasing [Ca2+]o from 2 to 1 mm produced 77 ± 6% depression of conditioning EPSCs and increased paired-pulse facilitation from 87 ± 42 to 245 ± 50% (n = 3). The stronger depression by elevated [K+]o of paired-pulse modulation is a hallmark of the effect of potassium (He et al., 2002) and low concentrations of NaCh blockers (Prakriya and Mennerick, 2000) at culture synapses.
Figure 1.

Effects of 10 mm [K+]o EPSCs and IPSCs. A, EPSCs in response to paired stimulation (50 msec paired-pulse interval). Thin traces are averaged EPSCs obtained in 2.5 mm [K+]o. Thick traces are average waveforms obtained in 10 mm [K+]o. Right, The 10 mm [K+]o traces have been scaled to the peak of the control conditioning EPSC to highlight the change in paired-pulse facilitation. B, A similar protocol to that depicted in A was applied to pharmacologically isolated IPSCs. The clamp potential was 0 mV. For A and B, sweeps are averages of 10-20 trials. C, D, Summary of the effect of 10 mm [K+]o on paired-pulse modulation of five EPSCs (C) and six IPSCs (D) compared with responses in 2.5 mm [K+]o. *p < 0.05, paired t test. Note that IPSCs tend to show paired-pulse depression in hippocampus, as described previously (Davies et al., 1990). For this figure and Figures 2, 3, 4, paired-pulse modulation is calculated by subtracting 1 from the ratio of second EPSC amplitude (P2) to the conditioning EPSC amplitude (P1) in the same condition. Thus, P2/P1 - 1 denotes the degree of paired-pulse modulation; depression is denoted by negative and facilitation by positive values.
Another hallmark of partial NaCh block or slowing of NaCh recovery from inactivation is the selective effect of these treatments on evoked EPSCs over IPSCs. In slices, 10 mm [K+]o, despite its strong depressant effect on EPSCs, had little effect on individual or paired GABAergic IPSCs (Fig. 1B,D). These results were similar to our previous work in culture, in which up to 12 mm [K+]o had no effect on conditioning or test IPSCs (He et al., 2002).
In the above experiments, equivalent stimuli were used to evoke EPSCs and IPSCs. However, it is possible that stimuli used to evoke moderate EPSCs evoked maximal IPSCs, resulting in a ceiling effect for IPSCs. If this were the case, an effect of elevated [K+]o on IPSCs might be unmasked by using submaximal IPSC stimuli. We performed stimulus I/O curves for evoked IPSCs (Fig. 2A) and repeated the elevated [K+]o experiment from Figure 1B using stimuli that elicited IPSCs ∼25% of their maximum amplitude (Fig. 2B). We found that 10 mm [K+]o did not enhance paired-pulse depression. In fact, 10 mm [K+]o under these conditions caused a significant reduction of IPSC paired-pulse depression (34 ± 6% control vs 15 ± 11% depression in 10 mm [K+]o; p < 0.05; n = 6) (Fig. 2B,D). This type of change typically results from a change in release probability, which presumably is not a major factor here. It is possible that elevated [K+]o results in a stimulus-dependent increase in excitability when fewer than the maximum number of fibers are stimulated by weak stimulus intensities. Similar interactions between modulators and stimulus intensity have been noted by others examining IPSC depression (Kim and Alger, 2001). Elevated [K+]o did not affect conditioning peak amplitude (Fig. 2C). These data suggest that the insensitivity of IPSCs to synaptic depression is not caused by supramaximal stimulation.
Figure 2.

Weak stimulation does not unmask IPSC depression by elevated [K+]o. A, Stimulus I/O curve for a representative IPSC. Stimulus intensity was varied from 70 to 160 μA, whereas the stimulus duration remained constant at 50 μsec. B, IPSCs from the same cell depicted in A. The IPSCs were obtained at a stimulus intensity indicated by the arrow in A. Top, Baseline IPSCs. Bottom, IPSCs in the presence of 10 mm [K+]o. Traces represent the average of eight sweeps in each condition. C, D, Summary results showing the effect on six IPSCs stimulated at less than half maximum stimulus intensity. C, Effect of 10 mm [K+]o on conditioning IPSC amplitude (Norm. 1st IPSC). Conditioning IPSCs in 10 mm [K+]o showed no significant change from control (p = 0.76; n = 6). D, Average effects on IPSC paired-pulse modulation. Note there was a slight decrease in paired-pulse depression of IPSCs, opposite of the effect of elevated [K+]o on EPSCs. *p < 0.05.
We considered the possibility that the selective effect of elevated [K+]o on glutamate release was caused by the indirect release of adenosine, which is known to be released in a depolarization-dependent manner (Mitchell et al., 1993) and which in hippocampus selectively diminishes glutamate release but not GABA release (Yoon and Rothman, 1991). We found that 1 μm of the A1 adenosine receptor antagonist 8-cyclopentyl-1,3-dipropylxanthine (DPCPX) did not affect the ability of 10 mm [K+]o to depress EPSCs. In six cells, 10 mm [K+]o depressed EPSCs by 61 ± 7%. When DPCPX was added to 10 mm [K+]o, the depression did not decrease significantly (67 ± 6%; n = 6; p = 0.46).
Involvement of NaCh inactivation in effect of [K+]o
Elevated [K+]o may depress excitatory transmission by slowing recovery from NaCh inactivation (He et al., 2002). Riluzole slows recovery from NaCh inactivation, and its actions are strongly dependent on membrane potential (Hebert et al., 1994). We hypothesized that if elevated [K+]o and riluzole share a mechanism of action, subthreshold concentrations of [K+]o and riluzole would show a synergistic interaction when applied in combination. In six cells, raising [K+]o from 2.5 to 8 mm caused no significant change in paired-pulse facilitation (Fig. 3A) or in conditioning EPSCs (31 ± 20% facilitation by 8 mm [K+]o). We found that 3 μm riluzole had no significant effect on conditioning EPSCs (25 ± 13% facilitation by riluzole) or on paired-pulse facilitation in 2.5 mm [K+]o (Fig. 3A). When 3 μm riluzole was coapplied with 8 mm [K+]o, paired-pulse facilitation was significantly attenuated (Fig. 3A). In some cases (two of six neurons), the combined treatment actually resulted in a small net paired-pulse depression rather than the usual facilitation. There was not a significant effect of the elevated [K+]o and riluzole combination on conditioning EPSCs (6 ± 19% increase relative to control). Together, these results suggest a synergistic interaction of these subthreshold [K+]o and riluzole concentrations. The data are consistent with the idea that slowing recovery from NaCh inactivation participates in the synaptic depression induced by elevated [K+]o. The combination of riluzole plus 8 mm [K+]o (n = 6), or even 10 mm [K+]o (n = 3), failed to affect IPSCs (Fig. 3B).
Figure 3.
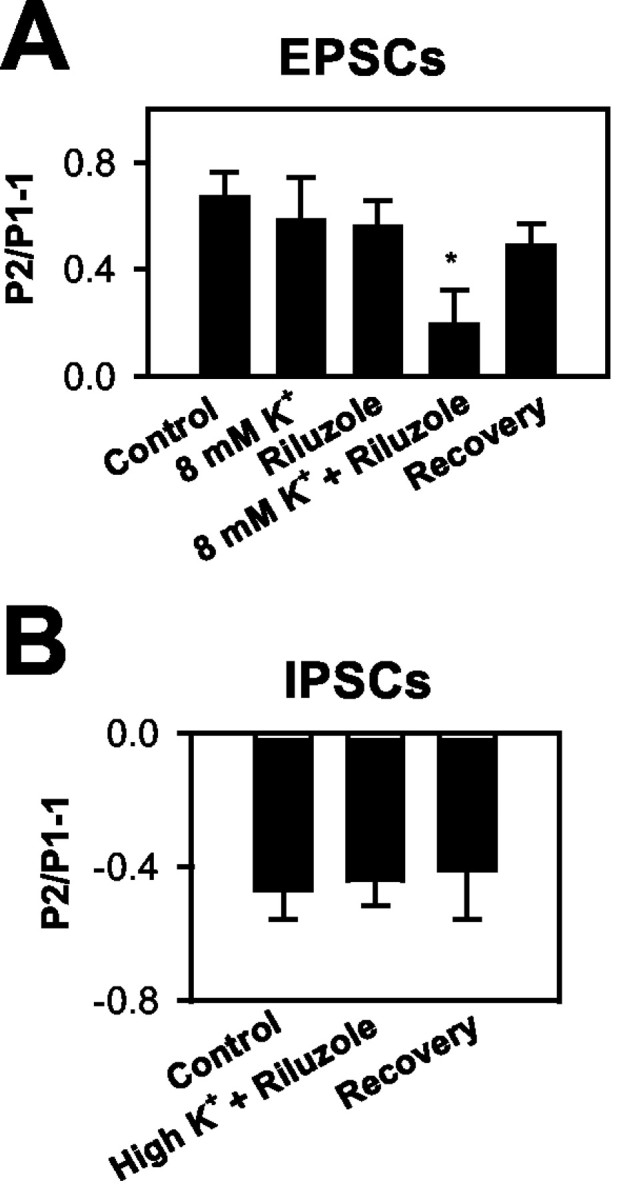
Elevated [K+]o interacts with a drug that slows NaCh inactivation. A, Effect on EPSC paired-pulse modulation. Neither 8 mm [K+]o nor 3 μm riluzole alone caused a significant change in paired-pulse facilitation. When the two conditions were combined, we recorded a significant reduction of paired-pulse facilitation. The paired-pulse interval was 50 msec, as in Figure 1. *Significantly different from control, 8 mm [K+]o alone, and riluzole alone (n = 6; p < 0.05). B, Effect on IPSCs. Neither 8 nor 10 mm [K+]o (high K+; n = 6 and 3, respectively; pooled data in the summary graph) combined with 3 μm riluzole affected IPSC paired-pulse modulation.
The experiments in Figure 3 suggest that action potential inhibition is a likely mechanism contributing to [K+]o-induced depression of EPSCs. To test this idea more directly, we examined extracellular presynaptic afferent volleys in response to stimulation with the same protocol used in Figure 3. Figure 4 shows that afferent volleys and field EPSPs were depressed in a frequency-dependent manner by the combination of 8 mm [K+]o with riluzole (Fig. 4B), similar to the effects on EPSCs shown in Figure 3. We also observed a change in the paired-pulse modulation of field EPSPs consistent with the EPSC effects (Fig. 4C). In both EPSC and field recordings, we found clear evidence of synergistic and stimulus-dependent effects of elevated [K+]o and riluzole. The changes in afferent volley amplitude induced by the combination of these conditions indicate that action potential initiation, propagation, or waveform (or a combination of these) is altered. However, afferent volley measurements, which sum the responses of many axons, cannot determine which of these effects on action potentials is responsible for the observed synaptic depression. We investigated these possibilities explicitly in later experiments.
Figure 4.
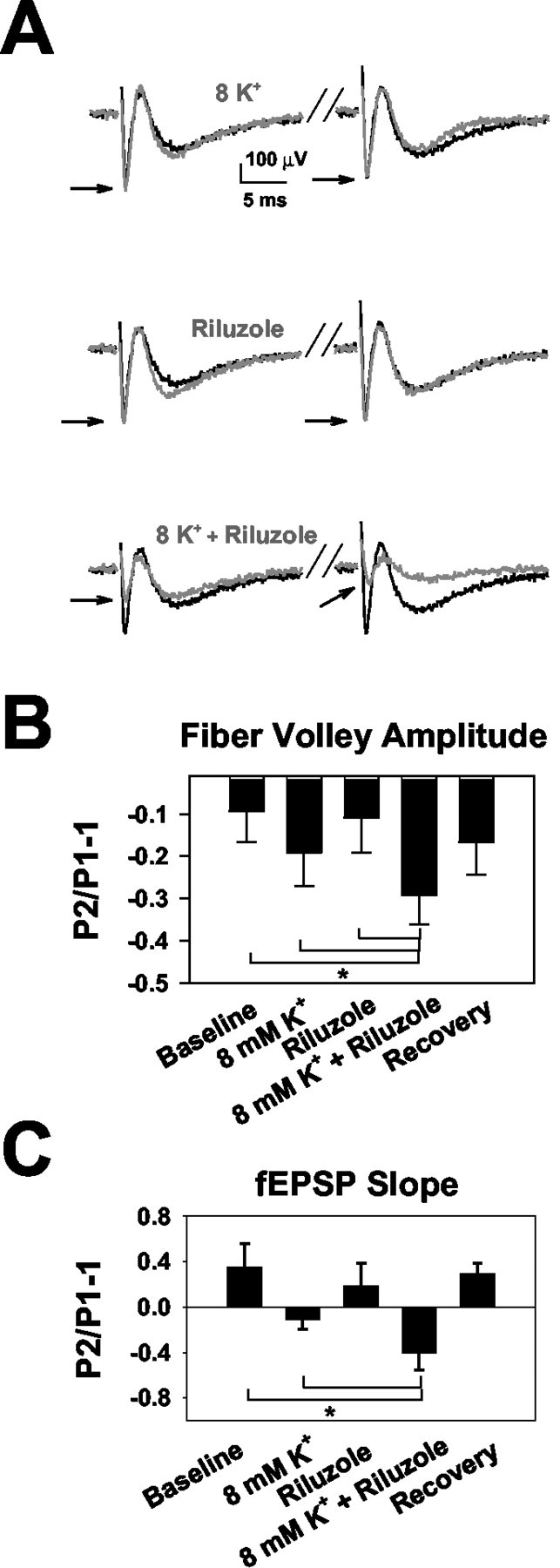
Effects of elevated [K+]o and riluzole on afferent volleys. A, Examples of afferent volley waveforms and the effect of the same conditions shown in Figure 3. Each trace represents the average of six trials under each condition. For each trace, the control (2.5 mm) [K+]o condition is plotted as the black trace. The gray trace represents the experimental conditions indicated by the gray text. Stimulus placement and intensity was optimized for isolation of the afferent volley, although synaptic field potentials are also visible. Arrows indicate afferent volley peaks in the test condition. In these experiments, neither 8 mm [K+]o nor riluzole alone affected the first (conditioning) afferent volley (8 mm [K+]o alone, 99 ± 6% of control; riluzole alone, 91 ± 6% of control; p = 0.98 and 0.21, respectively; n = 9). However, we did detect a significant depression of the conditioning volley in the presence of the 8 mm [K+]o and riluzole combination (31 ± 5% depression; p < 0.01). This was different from the EPSC experiments shown in Figure 3 and may reflect slightly different experimental conditions or differences in statistical power in the two types of experiments. Conditioning field potential slopes were also significantly depressed in the combined presence of riluzole and elevated [K+]o (p < 0.01; data not shown). B, Summary of effect of 8 mm [K+]o and riluzole on afferent volley amplitudes. The combination of 8 mm [K+]o and riluzole caused a significant depression of paired-pulse modulation. *p < 0.05 from control; 8 mm [K+]o only and riluzole only conditions (n = 9). C, Summary of effect of the experimental conditions on field potential slope. Eight millimolar [K+]o and riluzole produced significant depression of the field EPSP similar to effects seen on afferent volleys and EPSCs. *p < 0.05 from control and 8 mm [K+]o conditions. The combination was different from riluzole alone only at the trend level (p = 0.076; n = 8).
Effect of varied [K+]o on frequency trains of afferent volleys and EPSCs
In the paired-pulse experiments thus far, 8 mm [K+]o had little effect when administered alone. We reasoned that if NaCh inactivation underlies synaptic depression, 8 mm [K+]o or lower concentrations may have effects on EPSCs during longer, higher-frequency trains of stimuli, in which NaCh inactivation would likely accumulate more severely. Therefore, we examined the effect of 4-10 mm [K+]o on 50 Hz, 10 pulse stimulus trains. Consistent with our predictions, 8 mm [K+]o produced stimulus-dependent depression of EPSCs but not IPSCs during a 50 Hz train (Fig. 5A-D). In this data set, we also detected a small, but statistically significant depression of the conditioning EPSCs (Fig. 5B, top). At higher [K+]o (10 mm), depression was more severe (Fig. 5B,C), but at lower concentrations, conditioning EPSCs were not affected, as expected from the paired-pulse experiments. As also expected from Figures 1 and 2, IPSC trains at 50 Hz were not depressed by 8 mm [K+]o (Fig. 5D). As in Figure 2, we observed a slight reduction of depression by 8 mm [K+]o on the second pulse of the train (58 ± 3% depression in 2.5 mm [K+]o; 39 ± 9% depression in 8 mm [K+]o; n = 8; p < 0.05) (Fig. 5D), but subsequent responses showed no statistical difference from control. For EPSCs, the small effect of 4-8 mm [K+]o on conditioning responses is consistent with the idea that the accumulation of stimulus-associated inactivation, rather than an increase in steady-state inactivation, is most important in the synaptic effect of mild rises in [K+]o. The effect of higher concentrations of [K+]o on conditioning EPSCs is consistent with a sufficiently large increase in steady-state inactivation to diminish transmitter release.
Figure 5.

Concentration response for [K+]o on EPSC trains. A, Average waveforms depicting EPSC trains at 50 Hz in standard 2.5 mm [K+]o (thin traces) and in 8 mm [K+]o (thick traces, five sweeps averaged in each condition). Note that although there was little effect on the first EPSC of the train, the subsequent EPSCs were significantly depressed by 8 mm [K+]o. B, Summary of the effect of [K+]o at concentrations between 2.5 and 10 mm on the conditioning EPSC (1st Peak) and 10th EPSC peak. EPSCs of a 50 Hz train were evaluated. *p < 0.05; n = 14. C, EPSC amplitudes plotted over the course of the entire 50 Hz train at 2.5, 8, and 10 mm [K+]o (taken from the same data set as in B). To incorporate effects on conditioning pulses, responses to train stimulation (PX, where X denotes stimulus number in the train) in all conditions were normalized to the conditioning EPSC in control conditions (2.5 mm [K+]o, P1). The effects of [K+]o on conditioning responses can therefore be seen at the first point of subsequent conditions (P1experimental/P1control - 1). This type of analysis is also used in Figures 6, 7, 8, 9. D, Similar plot of IPSC amplitudes (n = 8).
Afferent volley amplitudes were depressed during 50 Hz stimulus trains (Fig. 6) in a manner similar to the depression of EPSCs observed in Figure 5. We found a weak effect of 8 mm [K+]o on initial conditioning afferent volleys (Fig. 6B) (9 ± 4% depression; n = 10), but subsequent volleys within the train were significantly depressed (p < 0.01; repeated-measures ANOVA). Thus, action potential signaling, which remains intact at 8 mm [K+]o over two-pulse stimulus trains at 20 Hz (Figs. 3, 4), is depressed over trains delivered at higher frequencies.
Figure 6.

Effect of 8 mm [K+]o on afferent volley frequency trains. A, Averaged traces showing the first conditioning (Stimulus 1) and last (Stimulus 10) response from a 10 pulse frequency train delivered at 50 Hz (traces represent six sweeps in each condition). Arrows highlight the afferent volley peak amplitude. Note the depression of the afferent volley by the 10th stimulus in the presence of 8 mm [K+]o. B, Summary of the effect of 8 mm [K+]o on afferent volley amplitudes over a 10 pulse, 50 Hz stimulus train (n = 10). Effects on conditioning peak during 8 mm [K+]o and washout conditions were different only at trend level (p = 0.05). Filled circles, Control amplitudes; open circles, responses in 8 mm [K+]o; filled squares (superimposed on control points), washout. There was a significant effect of elevated [K+]o and a significant interaction between elevated [K+]o and stimulus pulse (p < 0.05; repeated-measures ANOVA). C, Time course and reversibility of 8 mm [K+]o on stimulus-dependent depression. An example from the experiment in B shows the time course of wash-in and washout of the 8 mm [K+]o effect (denoted by solid horizontal bar) on frequency-dependent depression. The dotted line represents the degree of baseline depression before wash-in. D, Effect of elevated temperature on afferent volley trains. At 34°C elevation to 8 mm [K+]o produced statistically significant (p < 0.05; n = 7) frequency-dependent depression, but the effect was quantitatively similar to 25°C only when [K+]o was raised to 9 mm.
If recovery of NaCh inactivation is responsible for changes in the presynaptic volley and subsequent changes in glutamate release, then temperature increases might speed recovery and reduce the effect of elevated [K+]o on 50 Hz trains (Raastad and Shepherd, 2003). Consistent with this idea, we found that afferent volleys at 50 Hz and 34°C were less strongly (but still significantly) depressed by 8 mm [K+]o (Fig. 6D). Under these conditions, 9 mm [K+]o yielded quantitative depression similar to that elicited by 8 mm [K+]o at 25°C (Fig. 6D). Baseline depression in 2.5 mm [K+]o during the stimulus train was smaller at 34 than at 25°C (Fig. 6D) but increased with higher-frequency stimulation (75 and 100 Hz tested; data not shown). At 100 Hz, train depression in both 2.5 and 8 mm [K+]o was similar to 50 Hz, 25°C depression (data not shown). Thus, similar effects on afferent volleys were observed at both 34 and 25°C.
In other experiments, we examined whether increased [K+]o depressed afferent volleys in more mature tissue. Afferent volleys in 30-d-old hippocampus were also depressed in a stimulation-dependent manner at 50 Hz by [K+]o increased to 8 mm. For instance, the final afferent volley of a 10 pulse train, which showed minimal depression in 2.5 mm [K+]o (2 ± 1%), exhibited 43 ± 3% depression in 8 mm [K+]o (n = 4; p < 0.01). These results suggest that subsequent analyses pertain to both young and more mature tissue.
The above results help define the conditions and mechanisms by which synaptic depression is caused by moderately elevated [K+]o, but the results do not address the specific effect of [K+]o on the individual action potentials underlying the presynaptic afferent volley. To what extent does accumulating NaCh inactivation during a train increase action potential threshold, decrease propagation reliability, and/or depress the action potential waveform of individual fibers during the train? The ensuing experiments were designed to examine these three questions.
Elevated [K+]o causes frequency-dependent depression of action potential amplitude and conduction reliability
We first used afferent volley measurements to determine whether 8 mm [K+]o caused an increase in action potential threshold during the stimulus train, which would alter action potential initiation. If increases in initiation threshold explain the afferent volley amplitude depression, [K+]o-induced depression of afferent volley amplitude should be overcome by increasing stimulus intensity. In contrast with this expectation, we found that stimulus-dependent afferent volley depression was quantitatively similar at both low and high stimulus intensities (Fig. 7) (see also Hablitz and Lundervold, 1981). These results suggest that action potential changes are not caused by simple shifts in initiation threshold. We conclude that EPSC and afferent volley depression must either reflect depression of action potential conduction reliability or depression of individual action potential waveforms.
To determine the contribution of action potential conduction failure and waveform depression to the changes seen in afferent volleys and EPSCs at 8 mm [K+]o, we recorded action potentials from single axons in CA1 using methods described recently (Raastad and Shepherd, 2003). For interpreting these experiments we, like previous investigators (Raastad and Shepherd, 2003), assumed that the fibers recorded were Schaffer collateral axons and not interneuron fibers. This assumption appears valid because of the similar effect of elevated [K+]o on averaged single-fiber action potentials and afferent volleys (see below and Fig. 6), which are thought to reflect summed excitatory presynaptic axonal activity (Anderson et al., 1978). In addition, the distance between stimulating electrode and recording pipette averaged 1.0 mm for experiments in Figures 8 and 9, with the stimulating electrode placed in the stratum radiatum near the border of CA3 and CA1. There are few classes of interneuron with axonal arbors that encompass this distance (Freund and Buzsaki, 1996). Consideration of alternative interpretations of our results, based on the possibility that some GABAergic fibers were also recorded, is given in the Discussion. We found that at 2.5 mm [K+]o, single axons fired action potentials very reliably during suprathreshold stimulation, similar to previous reports (Raastad and Shepherd, 2003) (Fig. 8A,C). When 8 mm [K+]o was applied, reliability decreased significantly (Fig. 8A,D). Figure 8A shows an example of a fiber that was reliable in baseline conditions but became highly unreliable in 8 mm [K+]o after the conditioning stimulus. This example fiber fell into a group we arbitrarily classified as “low-reliability fibers,” which we defined as fibers failing to conduct action potentials ≥40% of the time (see below for additional discussion). As expected from afferent volley measurements, reliability changes were small for conditioning action potentials (Fig. 8A-D), consistent with afferent volley measurements and EPSC measurements that indicated little effect of 8 mm [K+]o on isolated action potentials. The small effect of 8 mm [K+]o on conditioning action potential reliability, even in unreliable fibers, supports the results of Figure 7 and further suggests that initiation failure cannot account for [K+]o-induced afferent volley depression.
To determine the effects of elevated [K+]o on action potential amplitude, we examined action potentials from single axons that responded reliably in 8 mm [K+]o. We found that peak amplitudes of action potentials later in stimulus trains were significantly depressed by 8 mm [K+]o (Fig. 8B,E). However, the peak amplitude of the conditioning action potential was unaffected by this [K+]o rise (90 ± 12% of control; p = 0.41; n = 12). Thus, axons that resisted [K+]o-induced conduction failure still showed depression of action potential amplitude. This indicates that amplitude depression (and resulting reduction of Ca2+ influx) could in principle also contribute to [K+]o-induced synaptic depression (see Discussion for estimates of the relative contributions).
Note that the overall effect of reliability changes plus waveform changes (Fig. 8F) (∼40%) was quantitatively similar to the depression in afferent volley measurements under these conditions (Fig. 6B). Thus, with our single axon measurements, we appear to have sampled from the same population of fibers that contribute to the macroscopic volleys (Fig. 6), which are likely to represent excitatory afferent activity (Anderson et al., 1978).
Our experiments in Figure 8 followed recently published methods (Raastad and Shepherd, 2003), in which large distances (>0.5 mm) were used between stimulus and recording electrodes to enhance the chances of recording a single activated fiber. We also explored whether propagation failures and waveform deficiencies occurred over shorter distances, comparable with those used for synaptic stimulation (within 150 μm). Using small recording pipettes, we found 10 axons that met the criteria for single activated axons within 150 μm of the stimulating electrode. Action potentials recorded from these axons failed similarly to the axons recorded further from the stimulus site. We also found that 3 of these 10 axons met the criteria for low-reliability fibers, a percentage similar to that observed at the greater recording distances described above. Overall, the failure rate by the 10th peak in the stimulus train was 26 ± 11% in 8 mm [K+]o, compared with <1% of the time in baseline conditions (n = 10; p < 0.05). Amplitudes of action potentials from reliable axons were depressed by 19 ± 7% by the 10th peak in 8 mm [K+]o but only 8 ± 4% in baseline (n = 7; p < 0.05). These data confirm that action potential conduction failure and amplitude depression likely participate in the synaptic depression observed in elevated [K+]o.
Over the 27 fibers examined in Figure 8 and in the experiment above, we noticed that the majority of axons continued to respond faithfully to stimulation even in elevated [K+]o. A minority of fibers seemed to account for the increased failures observed. As noted above, we formalized this observation by classifying fibers into two groups: high or low reliability. The classification was somewhat arbitrary, because the distribution of reliabilities was not clearly bimodal (Fig. 9C1). A low-reliability axon was defined as one that failed ≥40% of the time in any one experimental condition (invariably the 8 mm [K+]o condition). When fibers were sorted on this basis, 8 of 27 axons isolated were classified as low reliability (5 of 17 at recording distances >500 μm, 3 of 10 at distances <150 μm). Figure 9A shows the failure analysis from these eight fibers. Comparison of the failures in these axons with failures in the remaining 19 (Fig. 9B) shows that the increased failures observed clearly came from this minority of fibers. We asked whether either baseline failure or waveform changes might predict a change in reliability in increased [K+]o. Indeed, the baseline failure rate in low-reliability fibers appeared to be slightly higher by the end of the stimulus train than in the high-reliability fibers (Fig. 9A,B,C2). Linear regression between baseline failure rate and the 8 mm [K+]o failure rate showed a significant correlation (p < 0.05), but this relationship accounted for relatively little variance (Fig. 9C1) (r2 = 0.27). We also analyzed the baseline amplitude decreases over the course of the stimulus train; there was no significant ability of amplitude decrements in baseline conditions to predict failure in 8 mm [K+]o (Fig. 9D1,D2) (p > 0.05; r2 = 0.10). In summary, reliability and amplitude decreases predict subsequent failure in elevated [K+]o only weakly.
Discussion
CNS [K+]o is dynamic, and synaptic transmission may be both positively influenced (through increases in excitability and [Ca2+]i) and negatively influenced (through inactivation of channels necessary to sustain transmission) by [K+]o (Somjen, 2002). Estimates of resting [K+]o range from 2.5 to 4 mm (Somjen, 2002). Physiological, activity-dependent rises in bulk [K+]o depend on the region of the brain studied, and range from 0.1 to 1.5 mm. Much greater rises in bulk potassium occur during strong stimulation, during seizures, and during ischemia. Levels during seizures and during prolonged, strong stimulation reach a ceiling of ∼12 mm (Somjen, 2002). Therefore, the elevations explored in the present study are clearly relevant to pathophysiological [K+]o rises on nervous system function.
We focused on the influence of [K+]o on stimulus-evoked synaptic transmission in the CA1 region of rat hippocampus. Bulk [K+]o elevations up to 10 mm selectively depress stimulus-evoked EPSCs, leaving GABA transmission intact. The resistance of inhibitory transmission to elevated [K+]o was not caused by discrepancies in stimulation efficacy, because submaximal stimulation did not unmask depressant [K+]o effects. It should be noted, however, that our results do not preclude postsynaptic changes in GABA transmission that have been observed previously, related to the effects of elevated [K+]o on chloride equilibrium potential (ECl-) (Korn et al., 1987; DeFazio et al., 2000).
The mechanisms of selective EPSC depression remain to be fully elucidated, but appear to be related to the enhanced sensitivity of glutamate release to both pharmacological NaCh block (Martin et al., 1993; Prakriya and Mennerick, 2000) and depolarization-induced inactivation of NaChs (He et al., 2002). This selective sensitivity could be caused by differences in the pharmacological sensitivity, inactivation properties (Martina and Jonas, 1997), density, or localization of channels in principal cells versus interneurons. Studies are needed that compare action potential propagation in identified inhibitory axons with those in excitatory fibers to fully understand the differential effect of elevated [K+]o.
We explored possible mechanisms of [K+]o-induced depression of glutamate release and found evidence that action potential conduction failure participates in synaptic depression caused by elevated [K+]o at CA1 excitatory synapses. This result is perhaps somewhat surprising, because several recent results using imaging of activated boutons and electrophysiological techniques underscore a high reliability of action potential conduction under various conditions in the hippocampus and cortex (Allen and Stevens, 1994; Mackenzie et al., 1996; Mackenzie and Murphy, 1998; Cox et al., 2000; Koester and Sakmann, 2000), although reliability in the cortex may be altered under some conditions (Debanne et al., 1999). Several of the imaging studies also tested conditions expected to diminish action potential waveform and found reliability to remain intact (Mackenzie and Murphy, 1998; Cox et al., 2000). Our results agree that under baseline conditions, axonal reliability is very high. However, during repetitive stimulation, especially in a subset of fibers, rather modest increases in [K+]o significantly depress conduction reliability. Possible explanations for the apparent discrepancy between the present work and past work might include the selection bias inherent in imaging experiments, in which only boutons with sufficiently large baseline fluorescence signals can be used for study. Our own previous results in culture have suggested reliable invasion of synapses by action potentials under conditions in which NaChs are partially blocked (Prakriya and Mennerick, 2000). The difference between these results and present results could be attributable to anatomical differences between cultured neurons and neurons in situ.
Previous studies have not allowed the direct evaluation of the contributions of action potential initiation failures, conduction failures, or waveform changes to changes in afferent volleys. The quantitative similarity between our single-axon and afferent volley measurements suggests that our localized axonal measurements are microscopic reflections of the macroscopic afferent volley. This similarity also implies that single-fiber measurements are not biased any more than afferent volley measurements toward fibers of certain physiological or physical attributes. The response of individual axons to mild [K+]o elevations suggests that approximately half of the [K+]o-induced macroscopic afferent volley depression is caused by conduction failure, and furthermore that the majority of failures occurs in ∼30% of axons in the stratum radiatum.
Our single-axon recordings allow us to estimate the relative contributions of conduction failures and waveform changes to EPSC depression observed in elevated [K+]o. We found that synaptic depression by the end of a 10 pulse train at 50 Hz averaged ∼15% under baseline conditions of 2.5 mm [K+]o (Fig. 5C). Both action potential failure and waveform changes over the course of a baseline (i.e., 2.5 mm [K+]o) train were negligible (Fig. 8D-F). Therefore, as many studies have suggested previously, the synaptic depression observed in 2.5 mm [K+]o can be attributed primarily to synaptic mechanisms, such as the depletion of readily releasable transmitter stores (Zucker and Regehr, 2002), rather than to action potential changes. In the presence of 8 mm [K+]o, the EPSC depression increased to ∼55% total depression. Therefore, from a baseline of 15% depression (to a maximum of 100% depression), [K+]o depresses EPSCs by ∼47% [i.e., (55% - 15%) (100% - 15%)]. From our single-axon recordings, which reveal ∼30% overall failure rate, failing axons account for nearly two-thirds of the [K+]o-associated synaptic depression, assuming that the low- and high-reliability fibers contribute equally to baseline transmission. The remaining [K+]o-induced synaptic depression may therefore be attributed to action potential waveform changes, although we cannot exclude some effects on other channels essential for transmitter release. Interestingly, evidence from the calyx of Held (Borst and Sakmann, 1999) and from mossy fibers of hippocampal CA3 (Geiger and Jonas, 2000), in which the action potential invading synaptic terminals can be directly controlled and recorded, suggests that rather minor waveform changes can be associated with dramatic changes in transmitter release.
An alternative interpretation of our data could be that afferent volleys and single-axon measurements include a contribution from GABAergic axons. In fact, although afferent volleys correlate strongly with EPSP amplitude and are generally believed to reflect afferent excitatory activity (Anderson et al., 1978), we know of no data to exclude a contribution to presynaptic volleys from fibers of the bistratified cells and trilaminar classes of interneurons, whose axons course through the stratum radiatum (Freund and Buzsaki, 1996). One possibility is that some high-reliability fibers represent GABAergic fibers. In this case, waveform changes observed in high-reliability fibers appear to be ineffective in reducing GABA release, suggesting a safety factor for Ca2+ entry and transmitter release at GABA terminals. This interpretation would suggest that conduction failures are even more important in reducing EPSCs than the above calculations suggest.
Exogenous [K+]o rises to levels similar to those used in the present study have been used to generate seizure activity in hippocampal slices (Rutecki et al., 1985; Korn et al., 1987; Chamberlin and Dingledine, 1988; McBain and Dingledine, 1992). In general, we did not see evidence of seizure activity, although we sometimes observed an increased frequency of EPSCs and especially IPSCs. The absence of seizures likely reflects a combination of the young age of our tissue, the use of submerged slices (Alger et al., 1984), and the inclusion of QX-314 in whole-cell pipette solutions, which is expected to block the action currents that define ictal and interictal events in many studies (McBain and Dingledine, 1992). More generally, our observations of increased spontaneous transmission agree with the seizure literature in that we see signs of hyperexcitability when [K+]o is elevated. This raises an apparent paradox with the striking depression of evoked glutamate release observed in our experiments. We believe the resolution of the paradox lies in the well known overlap between the voltage range over which sodium channels inactivate and over which action potential threshold occurs. Individual fibers are likely to respond to [K+]o elevation with an increased probability of spontaneously spiking, caused by bringing the resting potential closer to threshold, but the mechanisms we explored in this study are likely to self-limit the effects of this hyperexcitability by decreasing conduction reliability and the amount of downstream synaptic excitation elicited.
In conclusion, we find that glutamate, but not GABA, transmission in CA1 of rat hippocampus is sensitive to bulk [K+]o increases. Furthermore, we provide evidence that action potential conduction failure underlies a substantial portion of [K+]o-dependent synaptic depression in the hippocampus. The action potential and synaptic changes resulting from [K+]o elevations above 8 mm are likely to disrupt normal transmission severely. However, because CA1 is particularly vulnerable to a variety of insults involving excess glutamate, including ischemia and seizures, the selective sensitivity of glutamate release may be an endogenous mechanism particularly important in this region for limiting runaway excitation and excitotoxicity.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS40488, the Klingenstein Fund (S.M.), and NIH Grant T32 GM08151-8 (J.P.M.). We thank Ann Benz and Amanda Shute for technical assistance and members of the laboratory for advice and criticism.
Correspondence should be addressed to Dr. Steven Mennerick, Washington University School of Medicine, Department of Psychiatry, 660 South Euclid Avenue, St. Louis, MO 63110. E-mail: menneris@psychiatry.wustl.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/240197-10$15.00/0
References
- Alger BE, Dhanjal SS, Dingledine R, Garthwaite J, Henderson G, King GL, Lipton P, North A, Schwartzkroin PA, Sears TA, Segal M, Whittingham TS, Williams J (1984) Brain slice methods. In: Brain slices (Dingledine R, ed), pp 381-435. New York: Plenum.
- Allen C, Stevens CF (1994) An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci USA 91: 10380-10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson P, Silfvenius H, Sunderberg SH, Sveen O, Wigstrom H (1978) Functional characteristics of unmyelinated fibres in the hippocampal cortex. Brain Res 144: 11-18. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P, ten Bruggencate G (1987) Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol (Lond) 382: 159-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JG, Sakmann B (1999) Effect of changes in action potential shape on calcium currents and transmitter release in a calyx-type synapse of the rat auditory brainstem. Philos Trans R Soc Lond B Biol Sci 354: 347-355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlin NL, Dingledine R (1988) GABAergic inhibition and the induction of spontaneous epileptiform activity by low chloride and high potassium in the hippocampal slice. Brain Res 445: 12-18. [DOI] [PubMed] [Google Scholar]
- Cox CL, Denk W, Tank DW, Svoboda K (2000) Action potentials reliably invade axonal arbors of rat neocortical neurons. Proc Natl Acad Sci USA 97: 9724-9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CH, Davies SN, Collingridge GL (1990) Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 424: 513-531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Kopysova IL, Bras H, Ferrand N (1999) Gating of action potential propagation by an axonal A-like potassium conductance in the hippocampus: a new type of non-synaptic plasticity. J Physiol (Paris) 93: 285-296. [DOI] [PubMed] [Google Scholar]
- DeFazio RA, Keros S, Quick MW, Hablitz JJ (2000) Potassium-coupled chloride cotransport controls intracellular chloride in rat neocortical pyramidal neurons. J Neurosci 20: 8069-8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freund TF, Buzsaki G (1996) Interneurons of the hippocampus. Hippocampus 6: 347-470. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Jonas P (2000) Dynamic control of presynaptic Ca(2+) inflow by fast-inactivating K(+) channels in hippocampal mossy fiber boutons. Neuron 28: 927-939. [DOI] [PubMed] [Google Scholar]
- Hablitz JJ, Lundervold A (1981) Hippocampal excitability and changes in extracellular potassium. Exp Neurol 71: 410-420. [DOI] [PubMed] [Google Scholar]
- He Y, Zorumski CF, Mennerick S (2002) Contribution of presynaptic Na(+) channel inactivation to paired-pulse synaptic depression in cultured hippocampal neurons. J Neurophysiol 87: 925-936. [DOI] [PubMed] [Google Scholar]
- Hebert T, Drapeau P, Pradier L, Dunn RJ (1994) Block of the rat brain IIA sodium channel alpha subunit by the neuroprotective drug riluzole. Mol Pharmacol 45: 1055-1060. [PubMed] [Google Scholar]
- Kim J, Alger BE (2001) Random response fluctuations lead to spurious paired-pulse facilitation. J Neurosci 21: 9608-9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B (2000) Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol (Lond) 529: 625-646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R (1987) Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol 57: 325-340. [DOI] [PubMed] [Google Scholar]
- Mackenzie PJ, Murphy TH (1998) High safety factor for action potential conduction along axons but not dendrites of cultured hippocampal and cortical neurons. J Neurophysiol 80: 2089-2101. [DOI] [PubMed] [Google Scholar]
- Mackenzie PJ, Umemiya M, Murphy TH (1996) Ca2+ imaging of CNS axons in culture indicates reliable coupling between single action potentials and distal functional release sites. Neuron 16: 783-795. [DOI] [PubMed] [Google Scholar]
- Martin D, Thompson MA, Nadler JV (1993) The neuroprotective agent riluzole inhibits release of glutamate and aspartate from slices of hippocampal area CA1. Eur J Pharmacol 250: 473-476. [DOI] [PubMed] [Google Scholar]
- Martina M, Jonas P (1997) Functional differences in Na+ channel gating between fast-spiking interneurones and principal neurones of rat hippocampus. J Physiol (Lond) 505: 593-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain C, Dingledine R (1992) Dual-component miniature excitatory synaptic currents in rat hippocampal CA3 pyramidal neurons. J Neurophysiol 68: 16-27. [DOI] [PubMed] [Google Scholar]
- Mitchell JB, Lupica CR, Dunwiddie TV (1993) Activity-dependent release of endogenous adenosine modulates synaptic responses in the rat hippocampus. J Neurosci 13: 3439-3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Mauk MD, Kocsis JD (1987) Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol 58: 404-416. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Mennerick S (2000) Selective depression of low release-probability excitatory synapses by sodium channel blockers. Neuron 26: 671-682. [DOI] [PubMed] [Google Scholar]
- Raastad M, Shepherd GM (2003) Single-axon action potentials in the rat hippocampal cortex. J Physiol (Lond) 548: 745-752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransom BR, Goldring S (1973) Slow depolarizations in cells presumed to be glia in the cerebral cortex of the cat. J Neurophysiol 36: 869-878. [DOI] [PubMed] [Google Scholar]
- Rutecki PA, Lebeda FJ, Johnston D (1985) Epileptiform activity induced by changes in extracellular potassium in hippocampus. J Neurophysiol 54: 1363-1374. [DOI] [PubMed] [Google Scholar]
- Somjen GG (2002) Ion regulation in the brain: implications for pathophysiology. Neuroscientist 8: 254-267. [DOI] [PubMed] [Google Scholar]
- Sykova E, Orkand RK (1980) Extracellular potassium accumulation and transmission in frog spinal cord. Neuroscience 5: 1421-1428. [DOI] [PubMed] [Google Scholar]
- Thomson AM (2000) Facilitation, augmentation and potentiation at central synapses. Trends Neurosci 23: 305-312. [DOI] [PubMed] [Google Scholar]
- Walz W (2000) Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36: 291-300. [DOI] [PubMed] [Google Scholar]
- Yoon KW, Rothman SM (1991) Adenosine inhibits excitatory but not inhibitory synaptic transmission in the hippocampus. J Neurosci 11: 1375-1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG (2002) Short-term synaptic plasticity. Annu Rev Physiol 64: 355-405. [DOI] [PubMed] [Google Scholar]