

Abstract
Here, we show that postsynaptic p21-activated kinase (Pak) signaling diverges into two genetically separable pathways at the Drosophila neuromuscular junction. One pathway controls glutamate receptor abundance. Pak signaling within this pathway is specified by a required interaction with the adaptor protein Dreadlocks (Dock). We demonstrate that Dock is localized to the synapse via an Src homology 2-mediated protein interaction. Dock is not necessary for Pak localization but is necessary to restrict Pak signaling to control glutamate receptor abundance. A second genetically separable function of Pak kinase signaling controls muscle membrane specialization through the regulation of synaptic Discs-large. In this pathway, Dock is dispensable. We present a model in which divergent Pak signaling is able to coordinate two different features of postsynaptic maturation, receptor abundance, and muscle membrane specialization.
Keywords: GTPase, synaptic homeostasis, postsynaptic density, neurotransmitter receptor, synapse, Drosophila
Introduction
Synapse development involves coordinated changes in synapse function and morphology. For example, at the vertebrate neuromuscular junction (NMJ), synapse maturation involves expansion of the acetylcholine receptor (AChR) field to keep pace with a growing muscle and nerve terminal. At the same time, the postsynaptic muscle membrane becomes highly specialized, including the formation of postsynaptic muscle membrane folds (Sanes and Lichtman, 1999). These muscle folds are subdivided into specialized zones. AChRs localize to the top of the folds adjacent to the presynaptic terminal, whereas other cytoskeletal proteins and signaling molecules localize to the base of the muscle folds (Sanes and Lichtman, 1999). A large number of resident synaptic proteins have been identified and their functions assessed genetically or biochemically. However, relatively little is known about how synaptic signaling pathways are organized to coordinately control different features of the synapse (Sheng and Pak, 1999; Allison et al., 2000; Husi et al., 2000; Walikonis et al., 2000).
Synapse maturation at the Drosophila NMJ shares many features with the maturation of the vertebrate NMJ. The nascent embryonic synapse grows tremendously over the course of 4 d of larval development. During this time, the postsynaptic receptor field increases in size to keep pace with muscle growth and the elaboration of the presynaptic nerve terminal. At the same time, the postsynaptic muscle membrane develops into a highly convoluted series of folds termed the subsynaptic reticulum (SSR). Mechanistically, the development of the SSR requires the presence of Discs-large (Dlg), the Drosophila homolog of PSD-95 (postsynaptic density-95) (Lahey et al., 1994). However, it remains unknown how developmental changes in glutamate receptor (GluR) abundance are coordinated with the formation of the SSR.
Previous work at the Drosophila NMJ has supported a model in which p-21 activated kinase (Pak) interacting exchange factor (Pix), a guanine nucleotide exchange factor, and Pak, a serine threonine kinase, are generally required for postsynaptic maturation. In the absence of Pix, many synaptic proteins are absent from the synapse, including Pak (localized by Pix) and Dlg (Parnas et al., 2001). In the absence of these proteins, glutamate receptor levels are also decreased, and the SSR does not form. Mutations that delete Pak cause a similar disruption of postsynaptic development. Based on these data, we proposed a model that Pix recruits Pak to the synapse, and that Pak signaling is subsequently required for postsynaptic development. However, Pak signaling was not previously investigated, because only the Pak null mutation was analyzed in detail.
Here, we demonstrate that Pak signaling diverges into two independent, genetically separable signaling pathways that are capable of coordinating glutamate receptor abundance with the formation of SSR at the Drosophila NMJ. One pathway regulates glutamate receptor abundance. Within this pathway, Pak activity is specified by a required interaction with the adaptor protein Dreadlocks (Dock; Nck homolog), which we demonstrate is a synaptic protein at the Drosophila NMJ. A second Pak-signaling pathway controls the synaptic abundance of Dlg and the elaboration of SSR. Pak activity within this signaling pathway is independent of Dock function and requires an intact kinase domain.
Materials and Methods
Fly stocks. Flies were maintained at 25°C on normal food. The Pak mutants (Pak3, Pak4, Pak6, Pak11) and upstream activation sequence (UAS) myristilated-Pak were a gift from Larry Zipursky (University of California, Los Angeles, Los Angeles, CA) and Huey Hing (University of Illinois at Urbana-Champaign, Urbana, IL). dock4 and trioP3 lines were gifts from Barry Dickson (Institute for Molecular Pathology, Vienna, Austria). Df(3R)Win11, Df(2L)ast2, dockP1, and the Rac and Cdc42 dominant-negative lines UAS-RacN17 and UAS-Cdc42N17 were obtained from the Bloomington Stock Center (Bloomington, IN). Wild-type flies were w1118 for first-instar experiments and yw for third-instar experiments.
UAS-PakSH was constructed by performing site-directed mutagenesis (M504A and T566A) of a Pak cDNA (generous gift from H. Hing), ligating into pUAST and then transforming into the fly using standard germ-line transformation techniques. Mutations made were consistent with the creation of an ATP analog-sensitive Pak allele without perturbation of wild-type function (Weiss et al., 2000). As such, expression of UAS-PakSH can be used to rescue Pak expression in the Pak mutant background and restore Pak activity. In addition, this Pak mutation may also enable future experiments to specifically inhibit Pak kinase activity through the application of membrane permeable, inhibitory ATP analogs as done previously for cla4 in yeast (Weiss et al., 2000).
Immunohistochemistry and imaging. Wandering third-instar larva were dissected in hemolymph-like 3 (HL3) saline and fixed in Bouin's fixative (Sigma, St. Louis, MO) for 2 min. For comparison of fluorescence intensities, mutant larval fillets were always stained in the same reaction tube with wild-type controls, and fluorescence intensities were normalized to these wild-type controls. Genotypes being directly compared were imaged identically. All images presented for comparison in this study are calibrated identically.
The rabbit anti-Dock antibody (1:500) was a generous gift from Jack Dixon (University of California, San Diego, La Jolla, CA). The rabbit anti-Pak antibody (1:500) was a gift from L. Zipursky. The rabbit anti-Dlg antibody was a gift from Vivian Budnik (University of Massachusetts, Worcester, MA). Monoclonal antibodies (mAbs) anti-GluRIIA (8B4D2; 1:10) and mAb-Dlg (1:50) were from the Developmental Studies Hybridoma Bank (University of Iowa, Iowa City, IA). Tetramethylrhodamine isothiocyanate-conjugated anti-HRP (1:500) and secondary antibodies (1:200), FITC-labeled anti-mouse and cyanine 5-labeled anti-rabbit, were provided by Jackson ImmunoResearch (West Grove, PA).
Images were digitally captured using a cooled CCD camera (Quantix camera with Koda 1401E chip) mounted on a Zeiss (Thornwood, NY) Axiovert 200 equipped with Nomarski and epifluorescent illumination. Images were acquired and analyzed using Slidebook software (Intelligent Imaging Innovations, Denver, CO). Individual synapses were optically sectioned at 0.2 μm (18-25 sections per synapse) using a piezo-electric driven z-drive controlling the position of a Zeiss 100× oil immersion objective. The intensity of immunostaining was quantified as follows: a two-dimensional projection of the maximum fluorescence at the NMJ (muscle 4 in third instar) was created from a series of 0.2 μm synaptic sections. The average fluorescence was calculated over the entire synaptic area. For GluRIIA levels, we defined the synaptic area as delimited by HRP immunoreactivity and then averaging the GluRIIA staining intensity within this synaptic area. Anti-HRP staining recognizes presynaptic epitopes that are virtually unaffected in the Pak mutant background (see Fig. 1 B). Axonal staining was eliminated from the analysis. The synaptic area defined by anti-HRP encompasses the vast majority of GluRIIA staining (see Fig. 1, merged images). Rare GluRIIA clusters that lay outside anti-HRP immunostaining were included manually. This technique was also used to quantify Pak immunoreactivity in each mutant background. For analysis of Dlg levels, we quantified the average maximum fluorescence from the synapse delimited by Dlg. Dlg staining is present throughout the muscle membrane folds.
Figure 1.

Decreased GluRIIA abundance at the third-instar NMJ in Pak mutant animals. A, Representative NMJ from wild-type (wt) and Pak mutant animals are shown that are costained with anti-GluRIIA and anti-HRP. At the Pak mutant NMJ, synaptic morphology is grossly normal, but GluRIIA abundance is substantially decreased. B, Quantification of the fluorescence intensity of anti-GluRIIA, anti-HRP, and anti-PAK staining at the NMJ of wild-type, control, and Pak mutant animals. Data are expressed as percentage of wild-type fluorescence intensity. The genotypes corresponding to each bar are shown below the graph. There is a statistically significant decrease in GluRIIA fluorescence intensity in the Pak mutant combinations compared with wild-type and control NMJ. Pak3/Df(3R)Win11, 57 ± 2.3%, n = 17; Pak4/Df(3R)Win11, 72 ± 4.4%, n = 17; Pak6/Df(3R)Win11, 56 ± 3.8%, n = 18; and Pak3/Pak6, 72 ± 2.2%, n = 6. The average anti-HRP fluorescence varies <10% across all genotypes. Anti-Pak fluorescence is significantly decreased only in the Pak6/Df(3R)Win11 combination compared with wild type (23.7 ± 4.7%). In all graphs, statistical significance is indicated as *p < 0.05; **p < 0.00005.
Electrophysiology. Wandering third-instar larvae were selected after leaving the food. Larvae were dissected in HL3 saline in 0.5 mm Ca2+. Whole-muscle recordings were made from muscle 6, abdominal segment A3, of female larvae as described previously (Davis et al., 1996). Only recordings with a resting potential of at least -60 mV and input resistances of at least 7 MΩ were included in our analysis. Quantal content was calculated by dividing the maximal EPSP amplitude by the average amplitude of the spontaneous miniature release events (mEPSP). Measurements of maximal EPSP and input resistance were done by hand using the cursor option in Clampfit (Axon Instruments, Union City, CA). Measurements of spontaneous miniature release events were semiautomated using MiniAnalysis software (Synaptosoft, Decatur, GA). For each recording, 100-300 mEPSP events were averaged to determine the average mEPSP amplitude.
RNA extraction and cDNA preparation. Total RNA was extracted from 10 wandering third-instar larvae with CNS removed per genotype using Trizol (Invitrogen, San Diego, CA) according to the instructions of the manufacturer. For preparation of cDNA for real-time PCR analysis, 1 μl of total RNA was transcribed using an iScript cDNA synthesis kit (Bio-Rad, Hercules, CA).
Primer design. Primers were designed using PrimerQuest (Integrated DNA Technologies, Coralville, IA). Each primer was designed to produce a ∼100 bp amplicon. Primer sequences are as follows: GluRIIA forward (GACCATTTCCGAGGATGATGTGGA), GluRIIA reverse (CATCATTGGTTCGTTCACCGTTGG), RpL32 forward (CCACCAGTCGGATCGATATGCTAA), and RpL32 reverse (TTGGGCATCAGATACTGTCCCTTG).
Real-time reverse transcription-PCR. Real-time reverse transcription (RT)-PCR assays were performed using an iCycler (Bio-Rad) with SYBR Green fluorescence. Real-time PCR amplification was performed after an initial denaturation of 8 min at 95°C, followed by 50 cycles of 20 sec denaturation at 95°, 30 sec annealing at 60°C, and 30 sec extension at 72°C. Fluorescent detection was performed at the annealing stage. The reaction was done in 50 μl using iQ SYBR Green Supermix (Bio-Rad) with 500 nm primer concentration and 1 ng to 1 μg of cDNA. The threshold cycle was determined by the user and placed above baseline activity within the exponential increase phase. To look for changes in transcript levels in samples derived from control and experimental larvae, we compared the threshold cycles (Ct) for our genes of interest (GluRIIA) with a control housekeeping gene (RpL32) to determine ΔCt. The difference between ΔCt values for a wild-type and mutant genotype (ΔΔCt) represents the degree of induction or inhibition of GluRIIA transcript. The relative value of this fold difference of induction can be determined using the equation: fold induction = 2-ΔΔCt. Data reported were analyzed as fold induction between mutant and wild-type animals. Gene expression was measured in triplicate or duplicate for each genotype and repeated at multiple template concentrations.
Results
Pak kinases are a family of serine threonine kinases that are defined by their binding to, and activation by, the Rho family small GTPases, Rac, and Cdc42 (Daniels and Bokoch, 1999). Pak(s) can affect the actin cytoskeleton through the phosphorylation of proteins such as myosin light chain kinase (MLCK) and LIM (the three gene products Lin-11, Isl-1, and Mec-3) kinase (Manser et al., 1997; Li et al., 2001). Paks can also function as a Map4K within the MAP kinase cascade in vertebrates, Drosophila, and yeast (Dan et al., 2001). Pak and related family members, including the yeast Ste20 gene, have a conserved domain structure. The N-terminal half of Pak includes a proline-rich domain that, in Drosophila, binds the second SH3 domain of Dock (Hing et al., 1999). The N-terminal portion of Pak also includes an autoinhibitory domain, a Cdc42/Rac interaction domain, a proline-rich domain demonstrated to bind the Rho-type guanine nucleotide exchange factor (GEF) Pix, and a domain recently implicated in the dimerization of two inactive Pak monomers (Parrini et al., 2002). The C-terminal region of Pak encompasses the kinase domain.
In our genetic analysis of Pak signaling, we have taken advantage of previously characterized point mutations that disrupt the Pak gene (Table 1). These mutations have been used to investigate Pak-mediated signaling at the growth cone (Hing et al., 1999). The Pak6 mutation induces a premature stop codon that truncates the Pak protein within the Cdc42/Rac interaction domain and eliminates the Pix interaction domain as well as the kinase domain (Hing et al., 1999). Pak6 is reported to be a severe loss-of-function mutation or a genetic null (Hing et al., 1999; Parnas et al., 2001). The Pak3 and Pak5 alleles are point mutations that specifically disrupt the kinase domain of Pak (Hing et al., 1999). The Pak4 allele is a mutation that disrupts an N-terminal proline-rich domain necessary for Pak binding to the second SH3 domain of the adaptor protein Dock (Hing et al., 1999). Importantly, the Pak4 mutation has been biochemically shown to block the Pak-Dock interaction (Hing et al., 1999).
Table 1.
Mutant alleles of dock and Pak
|
Alleles |
Molecular lesion |
References |
|---|---|---|
| dockP1 | P element insertion in first intron | Garrity et al., 1996 |
| dock4 | C320Y, disruption of SH2 domain | Newsome et al., 2000 |
| Pak3 | G569D, disruption of kinase domain | Hing et al., 1999 |
| Pak4 | P9L, disruption of dock interaction domain | Hing et al., 1999 |
| Pak5 | D553N, disruption of kinase domain | Hing et al., 1999 |
|
Pak6
|
R113Stop, truncation in CRIB domain |
Hing et al., 1999 |
We first analyzed GluRIIA abundance in Pak mutations at the mature third-instar synapse. In this experiment, we analyzed each Pak mutation over a deficiency that uncovers the Pak locus. We also examined the heteroallelic combination Pak3/Pak6. The gross morphology of the Pak mutant synapses is normal, although there appears to be an increase in the variability of bouton size and number. It was previously reported that certain Pak allelic combinations caused the muscles to become thin and degenerate (Parnas et al., 2001). We observed a similar effect in the Pak11/Pak6 allelic combination. However, when these Pak mutations are placed over a deficiency chromosome that uncovers the Pak locus, the muscles appear grossly normal, suggesting that the muscle defects are attributable to second site mutations on the Pak chromosomes. Thus, in all of our experiments, we include an analysis of Pak (or Dock) mutations over deficiency chromosomes.
The kinase and Dock interaction domains of Pak are necessary postsynaptically for normal GluRIIA abundance
We assessed the abundance of GluRIIA at the third-instar NMJ using previously characterized antibodies (Petersen et al., 1997; Parnas et al., 2001). Here, we demonstrate a significant decrease in GluRIIA abundance in all Pak mutant combinations compared with wild-type and heterozygous controls (Fig. 1A,B). Interestingly, GluRIIA abundance is decreased similarly in the Pak null and Pak kinase domain mutations. Quantitatively similar changes in GluRIIA abundance were also observed at the first-instar NMJ (supplemental Fig. 1, available at www.jneurosci.org/cgi/content/full/24/31/6871/DC1). The change in GluRIIA fluorescence intensity could reflect a change in GluRIIA abundance per receptor cluster or could reflect a selective elimination of clusters within the synapse. Therefore, we quantified GluRIIA fluorescence intensity per cluster within wild-type and Pak mutant synapses. We show a significant decrease in the fluorescence intensity within individual GluR clusters (supplemental Fig. 1D, available at www.jneurosci.org/cgi/content/full/24/31/6871/DC1). In addition, we quantified the density of GluRIIA puncta in wild-type and Pak mutant synapses and demonstrate that there is no change in this parameter (supplemental Fig. 1E, available at www.jneurosci.org/cgi/content/full/24/31/6871/DC1). These data indicate that signaling via the Pak kinase domain is necessary to specify GluRIIA abundance within the normal complement of GluRIIA clusters at the Drosophila NMJ.
Analysis of Pak kinase mutations suggests that Pak kinase activity is necessary for normal GluRIIA abundance. However, an alternate possibility is that Pak localization to the synapse requires an intact kinase domain. Therefore, we also determined the Pak protein levels at the synapse in each of the Pak mutant combinations (Fig. 1B). Synaptic Pak staining levels are severely decreased only in the null mutant background [Pak6/Df(3R)Win11]. Pak staining at the synapse is wild type in heterozygous controls [Df(3R)Win11/+] and in every other mutant combination tested, including two independent mutant combinations that specifically disrupt the Pak kinase domain [Pak3/Pak6 and Pak3/Df(3R)Win11]. Because kinase domain mutations impair GluRIIA levels without altering Pak levels at the synapse, this supports the conclusion that Pak kinase signaling is necessary to specify GluRIIA levels. As an additional control for these experiments, we demonstrate that there is less than a 10% change in anti-HRP immunoreactivity in any genetic background (Fig. 1B).
In our analysis, we also examined a point mutation that disrupts the proline-rich Dock interaction domain of Pak [Pak4/Df(3R)Win11]. We show that GluRIIA levels are decreased in Pak4/Df(3R)Win11, even though Pak protein is present at the synapse at wild-type levels (Fig. 1B). In combination with our analysis of mutations that affect the Pak kinase domain, these data suggest that both Pak kinase activity and a protein-protein interaction mediated by the proline-rich Dock interaction domain are necessary for normal GluRIIA abundance. In experiments described below, we provide evidence that a Pak-Dock interaction is indeed required to achieve normal GluRIIA abundance.
The colocalization of Pak with the postsynaptic glutamate receptors and the effects of the Pak mutations on GluRIIA abundance argue that Pak functions postsynaptically in muscle. However, Pak and Pix proteins are found in the nerve as well as the muscle (Parnas et al., 2001). Therefore, we attempted to rescue Pak activity selectively in muscle or nerve by expressing a Pak cDNA under UAS control. Antibody staining using anti-Pak demonstrated high levels of muscle overexpression and synaptic targeting of the transgenic Pak protein, whereas neuronally expressed Pak did not localize synaptically (data not shown). Expression of UAS-PakSH in either nerve or muscle in a wild-type background did not have any morphologic effects (data not shown). We then overexpressed UAS-PakSH in muscle using a muscle-specific GAL4 driver (G14-GAL4). Muscle overexpression of UAS-PakSH in the Pak mutant background (Pak3/Pak6) rescues GluRIIA levels toward wild-type abundance (rescue of 43.82 ± 7.92%; p < 0.002). Neuronal overexpression of UAS-PakSH using a neuronal-specific GAL4 driver (1407-GAL4) did not significantly rescue GluRIIA levels (rescue of 10.2 ± 6.41%; p = 0.35). Although we cannot rule out a function of Pak in additional tissues, these data demonstrate an essential function of Pak postsynaptically. These data also demonstrate that although Pak is necessary for normal GluRIIA abundance, Pak overexpression is not sufficient to increase GluRIIA levels. Consistent with this conclusion, we overexpressed a myristolated Pak transgene, which functions as an activated kinase in growth cones (Hing et al., 1999). Overexpression of this transgene in muscle does not alter GluRIIA abundance (data not shown). Together, these data argue that Pak is necessary postsynaptically for normal GluRIIA abundance.
An additional set of experiments provides evidence that Pak may function postsynaptically to control GluRIIA abundance. In most systems, Pak is activated by Rac and Cdc42 (Daniels and Bokoch, 1999). We found that muscle overexpression of either dominant-negative Cdc42 (UAS-Cdc42N17) or dominant-negative Rac (UAS-RacN17) had no effect on synapse development or GluRIIA abundance as observed previously (Parnas et al., 2001). However, when UAS-Cdc42N17 and UAS-RacN17 are coexpressed in muscle, we observed a significant decrease in GluRIIA abundance without alteration of gross synapse development (Fig. 2A,B). It is unclear why coexpression of the dominant-negative transgenes is necessary for the decrease in GluRIIA levels. However, because Pak can bind to both Rac and Cdc42, there may be some redundancy that is overcome by coexpression of these transgenes. Pak is also known to interact with Trio (a Rac GEF) in photoreceptor guidance (Newsome et al., 2000). We found no change in GluRIIA levels in Trio mutations (trioP3; data not shown). Thus, although we do not directly demonstrate that Cdc42/Rac activate Pak, these data, in combination with our structure function analysis and biochemical evidence from other systems, support a model in which Pak is activated postsynaptically to control GluR levels at the NMJ.
Figure 2.
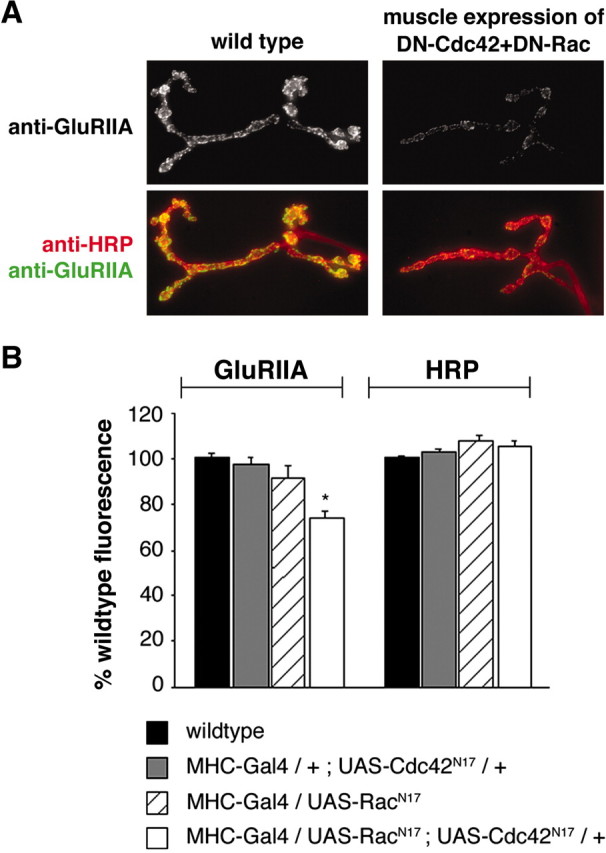
Simultaneous disruption of Cdc42 and Rac in muscle leads to decreased GluRIIA levels. A, Wild-type (left) and transgenic larvae (right) that simultaneously overexpress dominant-negative Rac and dominant-negative Cdc42 [myosin heavy chain (MHC)-Gal4/UAS-Racn17; UAS-Cdc42N17/+] are shown stained with anti-HRP and anti-GluRIIA. B, GluRIIA levels are significantly decreased only in MHC-Gal4/UAS-Racn17; UAS-Cdc42N17/+, when both dominant-negative constructs are postsynaptically expressed (73.6 ± 3.3%; n = 17). Significance is denoted as *p < 0.0001.
Finally, we performed experiments to address whether Pak controls GluRIIA abundance transcriptionally. In other systems, it has been shown that Pak can function as a Map4K and could therefore modulate GluRIIA transcription (Dan et al., 2001). However, it is well established that Pak can also function as a cytoskeletal regulator through the phosphorylation of Lim kinase and myosin light chain kinase (Manser et al., 1997; Li et al., 2001). In this capacity, Pak could function locally at the synapse to control GluRIIA levels. To distinguish between these alternatives, we performed real-time RT-PCR on cDNA samples derived from wild-type, Df(3R)Win11/+, and Pak6/Df(3R)Win11animals and calculated the fold induction of GluRIIA transcript levels for the mutant genotypes compared with wild type. We found no evidence of altered GluRIIA levels on the basis of this analysis [Df(3R)Win11/+ has fold induction of 0.98 ± 0.2; Pak6/Df(3R)Win11 has fold induction of 1.2 ± 0.2]. Thus, we favor the hypothesis that Pak acts locally at the synapse to control GluRIIA stabilization or turnover.
Impaired GluRIIA abundance alters postsynaptic quantal size but does not impair synaptic homeostasis
To confirm that changes in GluRIIA antibody staining intensity corresponded to a functional absence of GluRIIA, we next assayed synaptic function in the Pak mutant background. Quantal size was determined as the average amplitude of the spontaneous miniature EPSPs (mEPSPs). Quantal size is similar in the wild type and two heterozygous genetic controls. However, quantal size was significantly reduced in Pak6/Df(3R)Win11 animals, demonstrating that reduced GluRIIA staining correlates with a functional deficit that is consistent with less GluRIIA at the synapse (Fig. 3A). There was no change in the average muscle input resistance or resting membrane potential in any genetic background. These data support the conclusion that Pak is necessary for the regulation of quantal size by controlling GluRIIA abundance at the synapse. Furthermore, the positive correlation between quantal size and the anti-GluRIIA staining levels supports the use of anti-GluRIIA as a reliable reporter of GluRIIA abundance.
Figure 3.

Decreased quantal size and normal homeostatic compensation at Pak mutant NMJ. A, Quantification of quantal size in control (filled bar) and experimental genotypes (open bars). Wild-type and heterozygous controls [Df(3R)Win11/+ and Pak6/+; GluRIIASP16/+] have quantal sizes equal to 1.1 ± 0.1 mV (n = 6), 0.98 ± 0.7 mV (n = 6), and 0.96 ± 0.83 mV (n = 5), respectively. Experimental genotypes all showed significant decreases in quantal size: Pak6/Df(3R)Win11, 0.73 ± 0.07 mV, n = 6; GluRIIASP16, 0.51 ± 0.01 mV, n = 18; GluRIIASP16; Pak6/+, 0.46 ± 0.02 mV, n = 5; GluRIIASP16; Pak3/Pak6, 0.4 ± 0.01, n = 7. There is also a small, yet statistically significant, difference between GluRIIASP16 and GluRIIASP16; Pak3/Pak6. B, Quantification of quantal content in control (filled bars) and experimental genotypes (open bars). There is no difference in quantal content comparing the experimental genotype Pak6/Df(3R) Win11 with wildtype or experimental controls. Values are as follows: wildtype, 34.5 ± 1.5, n = 6; Df(3R)Win11/+, 32.1 ± 2.8, n = 6; Pak6/+; GluRIIASP16/+, 39.3 ± 5.2, n = 5; Pak6/Df(3R)Win11, 40.7 ± 3.7, n = 6. The experimental genotypes GluRIIASP16, GluRIIASP16; Pak6/+, and GluRIIASP16; Pak3/Pak6 all showed significant increases in quantal content compared with wild type and genetic controls, indicating that homeostatic compensation has occurred. Values are as follows: GluRIIASP16, 58 ± 3.7 mV, n = 15; GluRIIASP16; Pak6/+, 48.8 ± 5.2 mV, n = 5; and GluRIIASP16; Pak3/Pak6, 63.6 ± 8.7, n = 6. C, Representative traces of evoked potentials (left; each trace represents the average of 10 individual traces) and spontaneous miniature potentials (right) from control [Df(3R)Win11/+] and Pak mutant NMJ [Pak6/Df(3R)Win11]. The traces show the reduction in quantal size in Pak mutant animals and the wild-type EPSP amplitude indicative of effective synaptic homeostasis. Calibration: 10 mV, 50 msec (for evoked release); 1 mV, 250 msec (for spontaneous traces). Significance is denoted as *p < 0.05; **p < 0.002; ***p < 0.00002.
We then tested whether Pak specifically controls GluRIIA levels versus those of other glutamate receptor subunits at the synapse. There are two additional known GluR subunits expressed at the Drosophila NMJ, although antibodies are readily available only for GluRIIA. In a previous study of the Drosophila Pix mutation, it was shown that although GluRIIA levels are decreased, overexpressed GluRIIB was normally localized and its levels were unchanged (Parnas et al., 2001). However, the abundance of an overexpressed protein can be misleading, particularly because it was not previously determined whether the changes in GluR levels were the consequence of altered receptor transcription.
In our analysis, we first compared quantal size in Pak6/Df(3R)Win11 and GluRIIA knock-out animals. The synapse in the GluRIIASP16 null mutant is morphologically wild type, and two remaining glutamate receptor subunits are responsible for synaptic conductances that achieve a quantal size that is ∼50% of that observed in wild type (Petersen et al., 1997; DiAntonio et al., 1999). Consistent with some GluRIIA staining remaining in the Pak6/Df(3R)Win11 animal, the GluRIIASP16 null mutation reveals a greater reduction in quantal size compared with Pak6/Df(3R)Win11 (Fig. 3A). We then assayed quantal size in a GluRIIASP16;Pak3/Pak6 double mutant. If Pak specifically controls GluRIIA, we would expect that quantal size in the double mutant would be identical to that observed in the GluRIIA knock-out alone. In the GluRIIASP16;Pak3/Pak6 double mutation, we observed a small but statistically significant further reduction in quantal size compared with the GluRIIA null mutation alone (Fig. 3A). Thus, Pak may affect the abundance of other GluR subunits. However, because the additional reduction in quantal size in the double mutation is so small, it argues that the primary function of Pak signaling is to control the abundance of GluRIIA at the synapse. This argument is further supported by the observation that EPSP and mEPSP half-widths are decreased in the Pak mutant animals (Fig. 3C), similar to what is observed in mutations that specifically delete GluRIIA (Petersen et al., 1997).
We next assayed the physiological consequence of altered GluRIIA abundance during evoked stimulation at the NMJ. Robust homeostatic signaling mechanisms have been demonstrated to increase presynaptic release in response to a decrease in postsynaptic quantal size at the Drosophila NMJ (Petersen et al., 1997; Davis et al., 1998; Paradis et al., 2001). In a previous study of Pix mutations, it was suggested that homeostatic signaling might be impaired by lack of Pix and Pak signaling (Parnas et al., 2001). To directly test whether synaptic homeostasis occurs in a Pak mutant background, we quantified presynaptic release (quantal content) calculated by dividing the average EPSP amplitude by the average quantal size (Davis et al., 1998; Paradis et al., 2001). Quantal content is not statistically increased in the Pak6Df(3R)Win11 animal, although there is a trend in this direction (Fig. 3B). This result might suggest that Pak is required for synaptic homeostasis. However, we observed a robust increase in presynaptic quantal content in the GluRIIASP16;Pak3/Pak6 double mutant (Fig. 3B). This result clearly demonstrates that Pak does not disrupt the putative retrograde signaling system that is thought to be required for synaptic homeostasis at the Drosophila NMJ.
At this point, our data support a model in which Pak is localized to the synapse via an interaction with Pix. Subsequent Pak activation by Rac/Cdc42 is necessary to control GluRIIA abundance at the NMJ. However, as shown previously, a structure function analysis reveals that a point mutation in the proline-rich Dock-interaction domain of Pak is also necessary for normal GluRIIA abundance. We therefore turned our attention to an analysis of Dock at the Drosophila NMJ.
Dock localizes to the NMJ and controls GluRIIA abundance
Previous experiments suggest that Dock may be present at the Drosophila NMJ (Desai et al., 1999). However, the function of Dock during synapse formation is not understood, nor has the function of Dock during subsequent synapse development been assessed. We therefore tested whether the Dock protein is present at the larval NMJ and whether Dock is required for regulation of GluRIIA abundance at the NMJ.
Antibody staining with a previously characterized Dock antibody demonstrates that Dock localizes to the NMJ throughout larval development (Fig. 4) (data not shown). A previously characterized P-element insertion in dock is viable to the late larval stages and eliminates Dock immunoreactivity at the NMJ when placed over a deficiency chromosome that uncovers the dock locus (Fig. 4). This is consistent with previous studies demonstrating that dockP1 is a strong loss-of-function or null allele (Garrity et al., 1996).
Figure 4.

Synaptic localization of Dock. A wild-type (left) and a dock mutant NMJ [right; dockP1/Df(2L)ast2] are shown stained with anti-HRP and anti-Dock. Anti-Dock immunoreactivity is localized to the NMJ in wild type. In dock null animals, anti-Dock staining is completely absent from the NMJ (top right). The images shown are calibrated identically.
We next assayed GluRIIA abundance in two independent Dock mutations: dockP1 and dock4. The dock4 mutation specifically disrupts the SH2 domain of Dock. We found that GluRIIA levels are significantly reduced in the dockP1 and dock4 mutant backgrounds compared with wild type and heterozygous controls (Fig. 5A,B). As with Pak mutations, the gross morphology of the synapse is unaffected. Thus, Dock is necessary for normal GluRIIA abundance at the synapse. Interestingly, we found that GluRIIA staining is decreased to levels that are quantitatively similar to those observed in the Pak mutant background (compare Figs. 5B and 1B). Together with the observation that GluRIIA abundance is decreased in the Pak4 mutation (a point mutation in the Pak-Dock interaction domain), these data strongly suggest that a Pak-Dock interaction is necessary to control GluRIIA levels.
Figure 5.
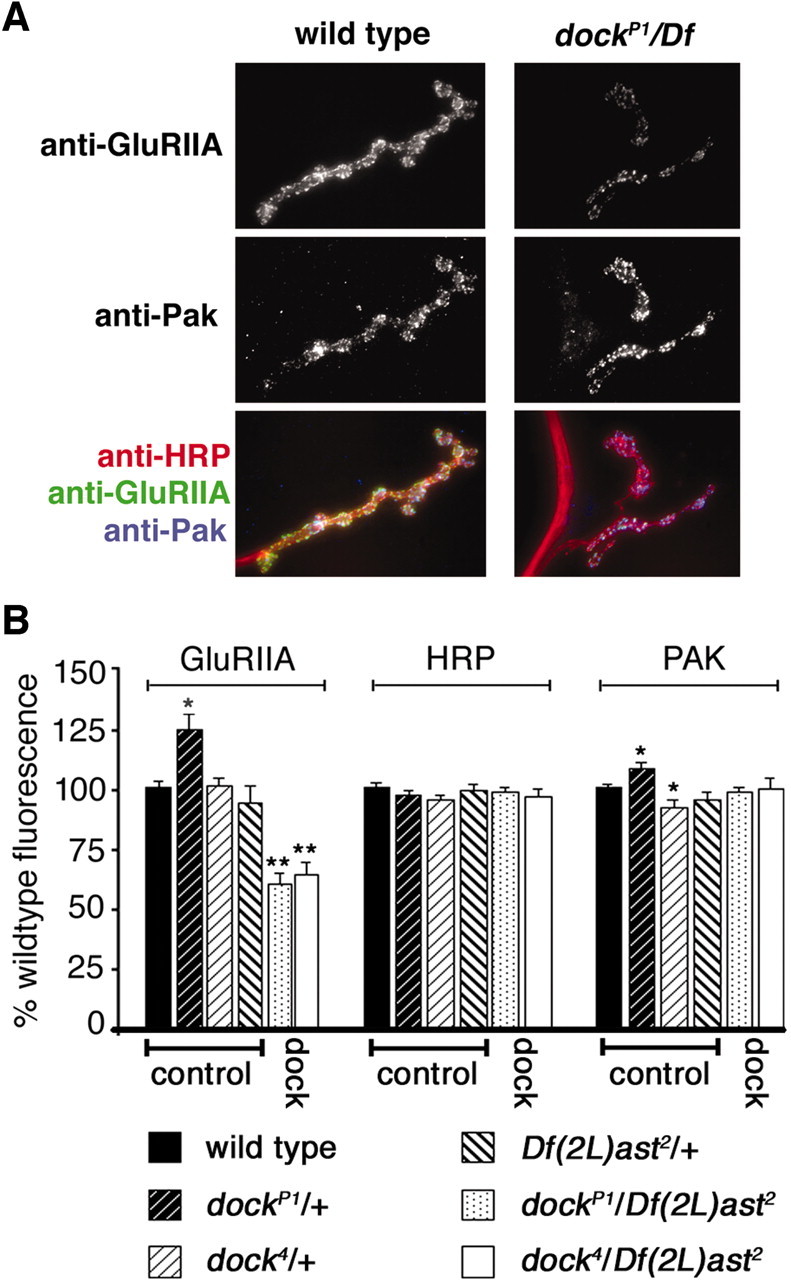
Synaptic localization of GluRIIA is decreased in dock mutants. A, Representative images of a wild-type (left) and a dock null NMJ [dockP1/Df(2L)ast2] stained with anti-GluRIIA, anti-Pak, and anti-HRP. GluRIIA staining is reduced in the dock mutant animals without a corresponding decrease in Pak levels at the synapse. B, Quantification of the fluorescence intensity of anti-GluRIIA, anti-HRP, and anti-PAK staining. There is a significant decrease in the intensity of GluRIIA staining in dockP1/Df(2L)ast2 null animals (61.2 ± 3.6%; n = 8) and dock4/Df(2L)ast2 (64.3 ± 4.8%; n = 6) compared with wild-type and the heterozygous controls dockP1/+, dock4/+, and Df(2L)ast2/+. Additionally, we observed a significant increase in GluRIIA staining and Pak staining in the dockP1/+ animals (*p < 0.05; **p < 0.0002).
Dock localization requires an intact SH2-domain and is not required for Pak localization to the NMJ
It has been suggested that Dock localizes Pak to the membrane of the Drosophila growth cone. This is based on, in part, the demonstration that myristolated (activated) Pak can bypass the requirement for Dock during axon pathfinding in the Drosophila visual system (Hing et al., 1999). At the Drosophila NMJ, however, data suggest that membrane-associated Pix is necessary for the localization of Pak to the synapse (Parnas et al., 2001). We therefore assayed Pak localization in the dock mutant background and Dock localization in the Pak mutant background. We found that Pak is normally localized in synaptic puncta in the Dock mutant background (Fig. 6B). We also found that Dock is normally localized in a Pak mutant background (Fig. 6C). These data demonstrate that Pak and Dock are independently localized to the synapse. These data also support the previous conclusion that Pak is localized to the synapse via an interaction with membrane-associated Pix (Parnas et al., 2001).
Figure 6.

Dock and Pak are synaptically localized independently of each other. A, Representative images of a wild-type (left) and a dock4/Df(2L)ast2 NMJ stained with anti-dock and anti-HRP. Dock protein, with a point mutation in the SH2 domain, is no longer highly localized at the synapse. B, Representative images of wild-type (left) and a dockP1/Df(2L)ast2 NMJ stained with anti-Pak. Pak is localized properly in dock null mutants. C, Representative images of wild-type (left) and Pak4/Df(3R)Win11 NMJ stained with anti-dock. Dock is localized properly in Pak mutants.
The question remains as to how Dock is localized to the synapse. Dock is an adaptor protein that is known to bind transmembrane receptors via a conserved SH2 domain (Schmucker et al., 2000; Song et al., 2003). Here, we demonstrate that a mutation in the SH2 domain of Dock severely impairs the synaptic localization of Dock (Fig. 6A). These data suggest that Dock may be localized to the synapse via a persistent interaction with a synaptic transmembrane protein.
Evidence that signaling via dock and Pak converge to control GluRIIA levels
Despite the observation that Pak and Dock are localized to the synapse by independent mechanisms, several lines of evidence indicate that Pak and Dock function together to control GluRIIA abundance. We have shown that mutations that disrupt the ability of Pak to bind Dock (Pak4) reduce GluRIIA abundance to levels that are quantitatively similar to that observed in Dock mutations alone (Pak4) (Fig. 1). To further test whether Dock and Pak function together to control GluRIIA levels, we quantified GluRIIA levels in double mutant combinations of Pak and dock (Fig. 7). We observed that GluRIIA levels are significantly decreased in a transheterozygous combination of dockP1 and Pak6 compared with wild-type animals and animals that are heterozygous for either mutation alone (Figs. 1B, 5D). Furthermore, one mutant copy of dock in a Pak mutant background does not enhance the reduction in GluRIIA levels seen in the Pak null mutant alone, and one mutant copy of Pak in the dock null mutant background creates only a small additional change in GluRIIA levels (Fig. 7). Although the absence of a strong dominant interaction supports the idea that dock and Pak interact genetically, a better test would be to examine GluRIIA abundance in the double-mutant background. Unfortunately, we were unable to attain viable homozygous double mutations. Because dock and Pak have functions in other tissues during embryonic and larval development, we suspect that the lethality of the double mutant is attributable to pleiotropic effects of the double mutants. Given that Pak and Dock are localized independently at the synapse and are known to physically and genetically interact, we propose that Dock functions to localize activated Pak signaling to control GluRIIA abundance at the synapse.
Figure 7.

Dock and Pak interact genetically to regulate synaptic GluRIIA levels. Quantification of anti-GluRIIA and anti-Pak fluorescent intensities in Pak and dock double-mutant combinations is shown. Compared with wild type, the dockP1/+; Pak6/+ transheterozygous animals show a decrease in GluRIIA staining at the synapse (72 ± 2.2%; n = 6). A further reduction in GluRIIA staining is seen in a dock null animal with one copy of a Pak mutant gene, dockP1/Df(2L)ast2; Pak6/+ (49 ± 3.0%; n = 6), and in the Pak mutant animal with one mutant copy of dock, dockP1/+; Pak6/Df(3L)Win11 (57 ± 2.4%; n = 6). For comparison, bars representing the single mutants, dockP1/Df(2L)ast2 and Pak6/Df(3L)Win11, are included from Figures 2 and 5. As an additional control, Pak6/+ animals are shown, which have a reduction in GluRIIA levels (89 ± 2.2%; n = 7). Levels of significance are *p < 0.05; **p < 0.0005; ***p < 0.000005.
Pak signaling controls synaptic Dlg and postsynaptic muscle development independently of dock
It was shown previously that Pix and Pak null mutations cause a decrease in Dlg abundance at the NMJ (Parnas et al., 2001). In every case where Dlg levels were decreased, there was a parallel reduction in the elaboration of the postsynaptic muscle membrane folds, the SSR. Because Dlg mutations impair SSR development (Lahey et al., 1994), it was hypothesized that the decrease in Dlg observed in Pix and Pak mutations causes impaired development of the postsynaptic SSR.
However, previously published data demonstrate that not all Pak mutations cause the same decrease in synaptic Dlg levels (Parnas et al., 2001). Specifically, Dlg levels were shown to be normal in the Pak4 mutation, and there was no change in the elaboration of the SSR. The reason for this discrepancy comparing the Pak null mutation and the Pak4 mutation was not explored previously (Parnas et al., 2001). Here, we demonstrate that Pak signaling and an intact Pak kinase domain are necessary for normal Dlg levels, but that Dock (and the Pak-Dock interaction) is not necessary for normal Dlg levels.
We first demonstrated that Dlg levels are severely decreased in the Pak kinase domain mutations (Fig. 8 A, B). Thus, the kinase domain is necessary for normal Dlg levels. We then confirmed previously published data demonstrating that the Pak4 mutation, which disrupts binding to Dock, causes only a slight change in Dlg levels (no change in Dlg was previously reported by Parnas et al., 2001). We further demonstrated that Dlg levels are unchanged compared with heterozygous controls in two independent Dock mutations (Fig. 8 B), including the presumed null allele [dockP1/Df(2L)ast2]. Note that Dlg levels are slightly decreased when comparing the heterozygous control [Df(2L)ast2/+] to wild type. However, the appropriate comparisons are between dock4/Df(2L)ast2 and Df(2L)ast2/+, which controls for the genetic background of the Df chromosome. Also, note that Dlg levels in the dock null, dockP1/Df(2L)ast2, are not statistically different from wild type. Thus, it appears that Pak signaling diverges downstream of Pix. One branch of the Pak signaling pathway interacts with Dock and is restricted to control GluRIIA levels, whereas a genetically separable Pak signaling pathway specifies synaptic Dlg levels and governs postsynaptic muscle development. Finally, the minor change in Dlg levels seen in the Pak4 could indicate that a second, unknown, SH3 domain-containing protein is involved in Pak signaling to control Dlg levels.
Figure 8.

Synaptic localization of Dlg is decreased in dock and Pak mutants. A, Representative images of wild-type (left), Pak4/Df(3R)Win11 (center), and Pak3/Df(3R)Win11 (right) NMJs stained with anti-Dlg. Dlg staining is severely reduced only in the Pak kinase domain point mutation (Pak3) and not the dock interaction domain point mutation (Pak4). B, Quantification of the fluorescence intensity of anti-Dlg staining in Pak and dock mutants. There is a strong decrease in the intensity of Dlg staining in Pak3/Df(3R)Win11 (50.5 ± 2.0%; n = 16) and Pak5/Df(3R)Win11 (55.5 ± 2.9%; n = 6) animals compared with wild type. There is a smaller decrease in intensity of Dlg staining in Pak4/Df(3R)Win11 animals (79.1 ± 2.4%; n = 11), which is significantly different from the decrease in Pak5/Df(3R)Win11 animals as indicated in the graph. Dlg intensity among dock heterozygous controls exhibits a small yet significant decrease compared with wild type (dock4/+, 89.43 ± 2.5%, n = 9; Df(2L)ast2/+, 86 ± 2.7%, n = 8). dock4/Df(2L)ast2 NMJs also exhibit a small yet significant decrease in Dlg staining compared with wild-type (80.8 ± 3.0%; n = 10) NMJs. Compared with heterozygous controls, as indicated on the graph, there is no significant decrease in Dlg staining in dock mutant synapses. Levels of significance are *p < 0.005; **p < 0.0000001.
Discussion
Here, we have defined a postsynaptic signaling network that can coordinate the regulation of glutamate receptor abundance at the active zone with the developmental elaboration of the postsynaptic muscle membrane specialization termed the subsynaptic reticulum (Fig. 9). This signaling network is centered on the Pak kinase. In our model, Pak is localized to the postsynaptic membrane via an interaction with Pix as suggested previously (Parnas et al., 2001). Pak is likely activated by Rac and/or Cdc42 in muscle (Fig. 2). During activation, Pak signaling appears to diverge into two genetically separable pathways. One branch of Pak signaling converges with Dock-mediated signaling to specify the abundance of GluRIIA (Figs. 1, 4, 5, 7). Dock itself is necessary for normal GluRIIA abundance and is recruited to the postsynaptic membrane via an independent SH2-mediated interaction with an as yet unidentified synaptic protein, perhaps a receptor tyrosine kinase (Figs. 5, 6). The second branch of Pak signaling controls the synaptic levels of Dlg, which subsequently specify the development of the postsynaptic muscle membrane folds (Fig. 8). This pathway requires the Pak kinase activity but is independent of synaptic Dock. Because Pak-dependent regulation of GluRIIA abundance alters synaptic function (Fig. 3), this signaling system can independently specify and possibly coordinate structural and functional synapse development at the Drosophila NMJ.
Figure 9.

The postsynaptic Pix-Pak-Dock signaling system. Pix localizes Pak to the synaptic membrane. After activation, Pak signaling diverges. In one branch, Pak binds to Dock. Dock itself is recruited to the synapse via an essential SH2-mediated interaction with an unknown synaptic protein. Dock-Pak binding is required for normal GluR abundance. Pak signaling also diverges to control Dlg levels and thereby regulates the formation of the postsynaptic muscle membrane folds.
This represents a simple, linear model for the coordinate regulation of structural and functional synapse development. However, there are indications that this signaling system includes additional complexity. There is a small reduction in Dlg levels in the Pak4 mutation, although the change in GluRIIA levels are more severe. This could indicate that Pak-dependent regulation of Dlg includes an additional, as yet unidentified, SH3 domain-containing protein. Dock and its vertebrate homolog Nck are know to interact with a variety of signaling molecules, and Dock could be localized to the synapse via interactions other than a receptor tyrosine kinase or transmembrane protein. Finally, the changes in GluRIIA and Dlg have been quantified for the Pix mutation and are observed to be consistently more severe than the changes documented in our study (Parnas et al., 2001). This could represent differences in the methods of visualization and quantification. Alternatively, Pix may have additional outputs, independent of Pak, that function in parallel to control Dlg and GluRIIA levels.
Mechanisms of neurotransmitter receptor regulation
There are interesting parallels and obvious differences between our model of synapse development at the Drosophila NMJ compared with synapse formation at the vertebrate NMJ. At the vertebrate NMJ, the signaling system that controls the initial clustering of AChRs, and the subsequent expansion of these clusters during synapse development, requires the activation of a receptor tyrosine kinase (MuSK) and the effector protein Rapsyn. In addition to controlling receptor clustering and abundance, MuSK and Rapsyn are also required for general postsynaptic differentiation (Sanes and Lichtman, 1999). Our data demonstrating that the Dock SH2 domain is necessary for GluR abundance implicates an as yet unidentified receptor tyrosine kinase at the Drosophila NMJ. However, there is no clear MuSK homolog in Drosophila. Furthermore, although synapse development in Drosophila is compromised, it proceeds in the absence of Pak or Dock, whereas synapse development at the vertebrate NMJ is blocked in MuSK and Rapsyn knock-out animals. Indeed our electrophysiological data demonstrate that glutamate receptor clusters must persist in Pak mutant animals, because quantal events and evoked synaptic transmission persist despite a severe decrease in the levels of GluRIIA. Thus, in Drosophila, it appears that Pak-Dock signaling is necessary for the developmental maturation of the postsynaptic receptor field as opposed to initial synapse assembly.
At central synapses in C. elegans, glutamate receptor abundance is regulated by ubiquitin-mediated signaling (Burbea et al., 2002). At these synapses, GluR-1 is ubiquinated in vivo, and receptor abundance can be bidirectionally modulated by manipulation of the ubiqutin signaling system (Burbea et al., 2002). In Drosophila, however, pharmacological manipulation of the ubiquitin-proteosome system does not alter GluR abundance at the synapse (Speese et al., 2003). Furthermore, a C-terminal sequence of GluR-1 that is ubiquinated in C. elegans is conserved from worm to vertebrate AMPA receptors but is not conserved in Drosophila GluRIIA. Thus, signaling via Pak and Dock may represent an alternative or additional mechanism to control glutamate receptor abundance at the synapse.
The means by which Pak signaling affects GluRIIA abundance is not clear. We demonstrate that receptor transcription is not altered in the Pak mutant background. GluRIIA abundance requires the convergence of Dock and Pak signaling as well as the presence of Pix, and all of these proteins are localized to the postsynaptic membrane. Thus, it seems likely that Pak functions locally at the synapse to control receptor abundance.
One means by which Pak could influence the abundance of receptors at postsynaptic membrane is through modulation of the synaptic cytoskeleton. Consistent with this hypothesis, Pak is known to signal to the actin cytoskeleton through the phosphorylation of MLCK and LIM kinase (Manser et al., 1997; Li et al., 2001). There is increasing evidence for a role of actin in the stabilization and maintenance of neurotransmitter receptors in other systems (Lisman and Zhabotinsky, 2001). In this context, Dock may be required to restrict Pak-mediated regulation of the actin cytoskeleton to the postsynaptic density and glutamate receptor clusters.
Regulation of receptor abundance during development and plasticity
Regulated changes in the stoichiometry of transmitter receptor subunits is a well-established phenomenon in both the CNS and peripheral nervous system (Wu et al., 1996; Sanes and Lichtman, 2001). Although developmental changes in receptor subunit composition have not been documented at the Drosophila NMJ, we hypothesize that convergent signaling, acting through Dock and Pak, could define a signaling system important for such phenomena. In support of such a possibility, our genetic analysis indicates that the regulation of GluRIIA is quite specific because changes in GluRIIA abundance can account for the majority of the decrease in postsynaptic quantal size observed in the Pak mutant background. In GluRIIA null mutations, quantal size is reduced by ∼50% (DiAntonio et al., 1999). In the GluRIIASP16; Pak6/Df(3R)Win11 double mutants, there is only a slight further reduction in quantal size beyond that observed in the GluRIIA null mutation alone (Fig. 3A). This suggests that the primary effect of Pak signaling is to regulate GluRIIA with only a slight additional effect on other GluRII subunits. This possibility is further supported by the previous demonstration that overexpressed GluRIIB is normally localized in the Pix mutation, which lacks synaptic Pak (Parnas et al., 2001).
The regulation of GluR subunit composition is also an essential mechanism underlying homeostatic quantal scaling at vertebrate central synapses. The molecular mechanisms underlying quantal scaling of AMPA-type receptors are essentially unknown. However, the regulated trafficking of NMDA receptors has been proposed as a mechanism to account for the scaling of the NMDA receptor current (Mu et al., 2003). In Drosophila, homeostatic changes to postsynaptic quantal size have been observed in response to altered muscle innervation (Davis et al., 1998). It is therefore interesting to speculate that Pak and Dock signaling may be involved in the mechanisms of quantal scaling.
Footnotes
This work was supported by a grant from the National Institutes of Health to G.W.D. S.D.A. is supported by a National Science Foundation predoctoral fellowship. We thank Larry Zipursky, Jack Dixon, Huey Hing, Barry Dickson, and Vivian Budnik for reagents; Ellie Heckscher, Kurt Marek, Catherine Pawson, Kira Poskanzer, and Sean Sweeney for comments on this manuscript; Nathan Okerlund and Sean Sweeney for making the UAS-PakSH transgenic line; Andy Chang for initial characterization of the Pak phenotype; and Patrick Haddick and Kimberly Raab-Graham for assistance with real-time PCR analysis.
Correspondence should be addressed to Graeme W. Davis, Department of Biochemistry and Biophysics, Program in Neuroscience, University of California, San Francisco, 1550 4th Street, San Francisco, CA 94143. E-mail: gdavis@biochem.ucsf.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246871-09$15.00/0
References
- Allison DW, Chervin AS, Gelfand VI, Craig AM (2000) Postsynaptic scaffolds of excitatory and inhibitory synapses in hippocampal neurons: maintenance of core components independent of actin filaments and microtubules. J Neurosci 20: 4545-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbea M, Dreier L, Dittman JS, Grunwald ME, Kaplan JM (2002) Ubiquitin and AP180 regulate the abundance of GLR-1 glutamate receptors at postsynaptic elements in C. elegans Neuron 35: 107-120. [DOI] [PubMed] [Google Scholar]
- Dan I, Watanabe NM, Kusumi A (2001) The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol 11: 220-230. [DOI] [PubMed] [Google Scholar]
- Daniels RH, Bokoch GM (1999) p21-activated protein kinase: a crucial component of morphological signaling? Trends Biochem Sci 24: 350-355. [DOI] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS (1996) Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron 17: 669-679. [DOI] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS (1998) Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila Neuron 20: 305-315. [DOI] [PubMed] [Google Scholar]
- Desai CJ, Garrity PA, Keshishian H, Zipursky SL, Zinn K (1999) The Drosophila SH2-SH3 adapter protein Dock is expressed in embryonic axons and facilitates synapse formation by the RP3 motoneuron. Development 126: 1527-1535. [DOI] [PubMed] [Google Scholar]
- DiAntonio A, Petersen SA, Heckmann M, Goodman CS (1999) Glutamate receptor expression regulates quantal size and quantal content at the Drosophila neuromuscular junction. J Neurosci 19: 3023-3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrity PA, Rao Y, Salecker I, McGlade J, Pawson T, Zipursky SL (1996) Drosophila photoreceptor axon guidance and targeting requires the dreadlocks SH2/SH3 adapter protein. Cell 85: 639-650. [DOI] [PubMed] [Google Scholar]
- Hing H, Xiao J, Harden N, Lim L, Zipursky SL (1999) Pak functions downstream of Dock to regulate photoreceptor axon guidance in Drosophila Cell 97: 853-863. [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG (2000) Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci 3: 661-669. [DOI] [PubMed] [Google Scholar]
- Lahey T, Gorczyca M, Jia XX, Budnik V (1994) The Drosophila tumor suppressor gene dlg is required for normal synaptic bouton structure. Neuron 13: 823-835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Fan J, Woodley DT (2001) Nck/Dock: an adapter between cell surface receptors and the actin cytoskeleton. Oncogene 20: 6403-6417. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM (2001) A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron 31: 191-201. [DOI] [PubMed] [Google Scholar]
- Manser E, Huang HY, Loo TH, Chen XQ, Dong JM, Leung T, Lim L (1997) Expression of constitutively active alpha-PAK reveals effects of the kinase on actin and focal complexes. Mol Cell Biol 17: 1129-1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Otsuka T, Horton AC, Scott DB, Ehlers MD (2003) Activity-dependent mRNA splicing controls ER export and synaptic delivery of NMDA receptors. Neuron 40: 581-594. [DOI] [PubMed] [Google Scholar]
- Newsome TP, Schmidt S, Dietzl G, Keleman K, Asling B, Debant A, Dickson BJ (2000) Trio combines with dock to regulate Pak activity during photoreceptor axon pathfinding in Drosophila Cell 101: 283-294. [DOI] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW (2001) Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron 30: 737-749. [DOI] [PubMed] [Google Scholar]
- Parnas D, Haghighi AP, Fetter RD, Kim SW, Goodman CS (2001) Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron 32: 415-424. [DOI] [PubMed] [Google Scholar]
- Parrini MC, Lei M, Harrison SC, Mayer BJ (2002) Pak1 kinase homodimers are autoinhibited in trans and dissociated upon activation by Cdc42 and Rac1. Mol Cell 9: 73-83. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A (1997) Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron 19: 1237-1248. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (1999) Development of the vertebrate neuromuscular junction. Annu Rev Neurosci 22: 389-442. [DOI] [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW (2001) Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci 2: 791-805. [DOI] [PubMed] [Google Scholar]
- Schmucker D, Clemens JC, Shu H, Worby CA, Xiao J, Muda M, Dixon JE, Zipursky SL (2000) Drosophila Dscam is an axon guidance receptor exhibiting extraordinary molecular diversity. Cell 101: 671-684. [DOI] [PubMed] [Google Scholar]
- Sheng M, Pak DT (1999) Glutamate receptor anchoring proteins and the molecular organization of excitatory synapses. Ann NY Acad Sci 868: 483-493. [DOI] [PubMed] [Google Scholar]
- Song J, Wu L, Chen Z, Kohanski RA, Pick L (2003) Axons guided by insulin receptor in Drosophila visual system. Science 300: 502-505. [DOI] [PubMed] [Google Scholar]
- Speese SD, Trotta N, Rodesch CK, Aravamudan B, Broadie K (2003) The ubiquitin proteasome system acutely regulates presynaptic protein turnover and synaptic efficacy. Curr Biol 13: 899-910. [DOI] [PubMed] [Google Scholar]
- Walikonis RS, Jensen ON, Mann M, Provance Jr DW, Mercer JA, Kennedy MB (2000) Identification of proteins in the postsynaptic density fraction by mass spectrometry. J Neurosci 20: 4069-4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss EL, Bishop AC, Shokat KM, Drubin DG (2000) Chemical genetic analysis of the budding-yeast p21-activated kinase Cla4p. Nat Cell Biol 2: 677-685. [DOI] [PubMed] [Google Scholar]
- Wu G, Malinow R, Cline HT (1996) Maturation of a central glutamatergic synapse. Science 274: 972-976. [DOI] [PubMed] [Google Scholar]