

Abstract
Protein kinase C (PKC) has been implicated in mediating ischemic and reperfusion damage in multiple organs. However, conflicting reports exist on the role of individual PKC isozymes in cerebral ischemic injury. Using a peptide inhibitor selective for δPKC, δV1-1, we found that δPKC inhibition reduced cellular injury in a rat hippocampal slice model of cerebral ischemia [oxygen-glucose deprivation (OGD)] when present both during OGD and for the first 3 hr of reperfusion. We next demonstrated peptide delivery to the brain parenchyma after in vivo delivery by detecting biotin-conjugatedδV1-1 and by measuring inhibition of intracellular δPKC translocation, an indicator of δPKC activity. Delivery of δV1-1 decreased infarct size in an in vivo rat stroke model of transient middle cerebral artery occlusion. Importantly, δV1-1 had no effect when delivered immediately before ischemia. However, delivery at the onset, at 1 hr, or at 6 hr of reperfusion reduced injury by 68, 47, and 58%, respectively. Previous work has implicated δPKC in mediating apoptotic processes. We therefore determined whether δPKC inhibition altered apoptotic cell death or cell survival pathways in our models. We found that δV1-1 reduced numbers of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive cells, indicating decreased apoptosis, increased levels of phospho-Akt, a kinase involved in cell survival pathways, and inhibited BAD (Bcl-2-associated death protein) protein translocation from the cell cytosol to the membrane, indicating inhibition of proapoptotic signaling. These data support a deleterious role for δPKC during reperfusion and suggest that δV1-1 delivery, even hours after commencement of reperfusion, may provide a therapeutic advantage after cerebral ischemia.
Keywords: ischemia, OGD, neuroprotection, hippocampus, peptide inhibitor, PKC
Introduction
Stroke is a leading cause of disability and death worldwide (National Institute of Neurological Disorders and Stroke, 1995). Both ischemia and the restoration of blood flow to ischemic tissue (reperfusion) cause cellular damage, possibly by different molecular mechanisms (Chan, 2001). Protein kinase C (PKC) has been implicated in mediating ischemic and reperfusion insults in multiple organs (Downey et al., 1994; Padanilam, 2001). However, much of this work has focused on the role of PKC in cardiac protection. Ischemic episodes to the brain also result in activation of several PKC isozymes (Wieloch et al., 1991; Sieber et al., 1998; Selvatici et al., 2003). However, there is no consensus on which isozymes are involved in mediating the cellular response or whether PKC activation is damaging or beneficial.
Recent reports have linked PKCs to modulation of several signaling pathways, including mediation of excitatory or inhibitory amino acid release (Nakane et al., 1998) and cytokine-induced superoxide production (Chao et al., 1995). In addition, a growing body of literature suggests that the δPKC isozyme is involved in apoptotic pathways, including mediating caspase activation and regulation of mitochondrial function (Brodie and Blumberg, 2003). This mode of cell death is observed during delayed cerebral reperfusion injury and contributes to the overall damage seen after stroke (for review, see Lipton, 1999). Importantly, δPKC mRNA and protein levels are increased in the cortex and hippocampus after global ischemia and in the compromised peri-infarct region after focal cerebral ischemia (Miettinen et al., 1996; Koponen et al., 2000). This penumbral area, where increased δPKC expression was noted, is prone to delayed damage (Phan et al., 2002), suggesting a role for this isozyme in reperfusion injury.
The aim of this study was to determine the role of δPKC in cerebral ischemic-reperfusion injury. To establish whether changes in δPKC activity contribute to the outcome from cerebral ischemia, we used the δPKC-selective inhibitor peptide δV1-1 (Chen et al., 2001). This rationally designed peptide was found to selectively inhibit δPKC translocation and induce cardioprotection from ischemia-reperfusion in isolated cells (Chen et al., 2001) and in intact hearts ex vivo and in vivo (Chen et al., 2001; Inagaki et al., 2003a,b). Intracellular delivery of the δV1-1 peptide was enabled by cross-linking it via a cysteine S-S (Cys S-S) bond to a membrane-permeable carrier peptide (Tat) derived from the Tat human immunodeficiency virus (HIV) protein (Schwarze et al., 1999).
Here, we report that treatment with δV1-1 protected against cerebral ischemic-reperfusion damage in both the in vitro and in vivo models. Delivery of δV1-1 during reperfusion reduced apoptotic cell death and enhanced prosurvival signaling, suggesting that inhibition of δPKC during the reperfusion period reduces injury in part by blocking intrinsic cell death pathways that contribute to secondary damage. Therefore, δPKC mediates, at least in part, reperfusion-induced injury after stroke.
Materials and Methods
Peptide preparation. Tat carrier peptide and δV1-1 (δPKC inhibitor peptide) were synthesized and conjugated via a Cys S-S bond as described previously (Chen et al., 2001). The δV1-1-Tat-conjugated peptide is referred to as δV1-1. δPKC isozyme-specific activity of the δV1-1 peptide has been demonstrated previously (Chen et al., 2001; Inagaki et al., 2003a,b). An important difference between Tat fusion proteins (Asoh et al., 2002; Cao et al., 2002; Kilic et al., 2002) and the Tat peptide mentioned here is the use of a cysteine S-S bond, which links the peptide to the Tat. This is to allow the peptide to be trapped inside the cells, because it is cleaved and released from the Tat carrier after intracellular delivery by reduction of the S-S bond.
Preparation of cultures. Organotypic hippocampal slice cultures were prepared as described by Bergold and Casaccia-Bonnefil (1997). Briefly, Sprague Dawley neonatal rats (9-11 d of age) were anesthetized by intraperitoneal injections of ketamine (1.0 mg for each pup). Pups were decapitated, and the hippocampi were dissected and transversely sliced (400 μm) on a McIlwain tissue chopper. Slices were placed in Gey's balanced salt solution (Invitrogen, San Diego, CA) supplemented with 6.5 mg/ml glucose (Sigma, St. Louis, MO) for 1 hr at 4°C. Slices were then transferred onto 30 mm diameter membrane inserts (Millicell-CM; Millipore, Bedford, MA) and placed into six-well culture trays with 1 ml of slice culture medium per well. The slice culture medium consisted of 50% Minimum Essential Medium, 25% HBSS, and 25% heat-inactivated horse serum (Invitrogen) supplemented with 6.5 mg/ml glucose and glutamine (1 mm). Cultures were maintained at 36°C in an incubator (100% humidity; 5% CO2) (CF autoflow; NuAire, Plymouth, MN) and kept in culture for 14-15 d before experiments.
Animal protocols were approved by the Stanford University Institutional Animal Care and Use Committee and the University of Miami Animal Care and Use Committee.
Oxygen and glucose deprivation. To model ischemia, cultures were exposed to oxygen and glucose deprivation (OGD) using an anaerobic chamber. The slices were washed three times with glucose-free HBSS, pH 7.4, containing (in mm): 1.26 CaCl2, 5.37 KCl, 0.44 KH2PO4, 0.49 MgCl2, 0.41 MgSO4, 136.9 NaCl, 4.17 NaHCO3, 0.34 Na2HPO4, 15 sucrose (all from Sigma). The slices were then placed into an airtight chamber, and 95% N2 5% CO2 gas (36°C) was circulated through the chamber for 5 min (4 L/min) to achieve anoxic conditions. After 5 min, the chamber was sealed and placed in the incubator (36°C) for 35 min (total of 40 min OGD). After OGD, the slices were placed back in the incubator in normal culture medium. Peptides Tat or δV1-1 were delivered in the culture medium at times stated. The organotypic slices were divided into groups (see Fig. 1): group 1, sham slices were incubated for 40 min with HBSS solution supplied with equimolar concentration of glucose instead of sucrose (sham OGD; n = 7); group 2, ischemia slices were exposed to 40 min of OGD (n = 15); group 3, Tat plus ischemia Tat peptide during OGD alone (n = 5); group 4, δV1-1 plus ischemia δV1-1 peptide during OGD alone (n = 6); group 5, Tat during ischemia and reperfusion Tat during OGD and the first 3 hr of reperfusion (n = 8); group 6, δV1-1 during ischemia and reperfusion δV1-1 during OGD and the first 3 hr of reperfusion (n = 12; all peptides 500 nm final concentration). For control, all six-well plates contained at least one well for sham and ischemia groups. Two hippocampal slices obtained from two different pups were placed on one insert. Therefore, the data obtained from each slice represents a different animal, and thus each slice is considered as n = 1.
Figure 1.

δPKC inhibition reduces cellular death after OGD and reperfusion in hippocampal slices. a, Experimental protocol. b, Bright-field images (top) and corresponding PI fluorescence images (bottom) from hippocampal slice cultures of sham, ischemic, and δV1-1-treated groups (left, middle, and right, respectively). Scale bar, 2.25 mm. c, Quantitative measurements of PI staining were performed 24 hr after ischemia (isch) in six experimental groups: sham (n = 7; group 1), ischemia (n = 15; group 2), Tat peptide treatment (n = 5; group 3), or δV1-1 peptide treatment (n = 6; group 4) during ischemia alone, Tat peptide treatment (n = 8; group 5) or δV1-1 treatment (n = 12; group 6) during ischemia and for the first 3 hr of reperfusion (3 hr rep). Significant values were compared with sham (*p < 0.001), ischemia (**p < 0.001), or Tat control (***p < 0.001). d, δPKC activity, as measured by translocation, increased after OGD followed by 3 hr of reperfusion. Delivery of δV1-1 during the ischemia and reperfusion period inhibits δPKC translocation in the in vitro slice model; n = 5, all groups; *p < 0.05 by Student's t test. e, TUNEL staining was used to assess apoptotic cell death in sham, control, and δV1-1-treated slices for groups 1, 5, and 6, respectively. TUNEL-positive cells are found in the CA1 region of hippocampus from organotypic slice cultures (magnification, 40×). Sham-treated slices contained few, if any, TUNEL-positive cells (a). A larger number of TUNEL-positive cells (arrow) was observed 24 hr after 40 min of OGD (b). δPKC inhibition reduced the TUNEL-positive cells 24 hr after 40 min of OGD (c).
Assessment of cell death by image analysis using propidium iodide. Propidium iodide (PI; Sigma), a cell impermeable DNA dye, was used to identify dead cells. Before OGD, slices were incubated in medium supplemented with 2 μg/ml PI for 1 hr and then replaced by regular media. Slices were studied using an inverted fluorescence microscope (Olympus IX 50; Olympus Optical, Tokyo, Japan), and pictures were taken using a SPOT CCD camera (Diagnostic Instruments, Sterling Heights, MI) and SPOT advanced software. Bright-field (0.254 sec exposure) and PI images (1.9 sec exposure; red filter) of slices were taken before the experiment. After imaging, the slices were exposed to OGD, and PI fluorescence images were taken 24 hr later. To quantitate total cell death in all slices, an overdose of NMDA (100 μm; Sigma) was applied to the culture medium. The last image was taken 24 hr after 1 hr NMDA treatment. Bright-field images were used to align the slice in the identical position during subsequent measurements. The intensity of PI fluorescence in the CA1 subfield of the hippocampal slices was used as an index of cell death.
For quantification purposes, fluorescence images were taken for each slice at 24 hr after NMDA treatment, 24 hr after OGD, and onset of experiment, and images were stacked using Scion Image software (Scion, Frederick, MD). The region of interest (ROI) was selected from the final image depicting total neuronal cell death, which remained constant for a particular slice. The ROI was subsequently superimposed on the fluorescence images taken 24 hr after OGD and at the onset of the experiment. Relative cell death was calculated from each ROI as follows: relative percentage of cell death = (Fexp - Fmin)/(Fmax - Fmin) × 100, where Fexp is the fluorescence of the test condition, Fmax is maximum fluorescence (NMDA treatment), and Fmin is background fluorescence. For all experimental analyses, the results are expressed as mean ± SEM. Statistical significance was determined with an ANOVA test followed by a Bonferroni's post hoc test.
Middle cerebral artery occlusion model. Ischemia was induced in male Sprague Dawley rats (290-320 gm) using an occluding intraluminal suture as described previously (Maier et al., 2001). Briefly, an uncoated 30 mm long segment of 3-0 nylon monofilament suture with the tip rounded by a flame was inserted into the stump of the external carotid artery and advanced into the internal carotid artery ∼19-20 mm from the bifurcation to occlude the ostium of the middle cerebral artery (MCA). At the end of the ischemic period (2 hr), the suture was removed and the animal was allowed to recover. Animals were maintained under isoflurane anesthesia during all surgical procedures. Tat or δV1-1 peptides were delivered as a bolus dose by intraluminal catheter directly into the internal carotid artery (pre-δV1-1 and post-δV1-1 groups; 0.06 mg/kg) or as an intraperitoneal dose (delayed-δV1-1 doses; 1 and 6 hr postreperfusion; 0.2 mg/kg). Physiological parameters including body temperature (35-38°C) and respiration rate were monitored and maintained using a heat blanket and anesthetic adjustment. For PKC translocation assays, rats were killed at the end of the ischemic period (without removal of the suture) after a 10 min or 24 hr reperfusion. The brain was sliced and striatum isolated and snap frozen.
Assessment of behavior after middle cerebral artery occlusion. Behavior was assessed at 24 hr of reperfusion using a scale modified from the study by Yang et al. (1994). After 24 hr of reperfusion, animals were graded on a scale of 1-4: grade 1, normal posture, spontaneous movement in any direction; grade 2, unilateral paw extension when lifted by the tail; grade 3, both paw extension and circling pattern when spontaneously walking; grade 4, abnormal posture, paw extension, circling pattern, cannot walk spontaneously. Animals from one of the four treatment time point groups (delivery of δV1-1 or control peptides at 6 hr reperfusion) were scored for behavior. Results were expressed as the mean ± SEM behavior score. Statistical analyses were performed using ANOVA followed by Fisher's post hoc test.
Assessment of brain infarct size. Animals were killed after 24 hr of reperfusion by an anesthetic overdose. Brains were quickly removed and sliced into 3 mm coronal sections, resulting in five slices of the brain. Slices were stained using 3% triphenyl tetrazolium chloride (TTC), and both faces of each slice were photographed for infarct assessment. Relative stroke area (ratio of the infarct size relative to the ipsilateral hemisphere, corrected for edema on the basis of measurement of the contralateral hemisphere) was measured to assess infarct size in the central three slices (two faces each; six faces total) of the five slices made from each brain. Results were expressed as the mean ± SEM infarct size of these six faces. All statistical analyses were performed using ANOVA followed by Bonferroni's post hoc test.
Measurement of apoptotic cell nuclei by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining. Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed both in the in vitro hippocampal slice model and the in vivo middle cerebral artery occlusion (MCAO) model. Briefly, hippocampal slices were subjected to OGD and treated with δV1-1 or control peptides as described above (groups 1, 5, and 6), followed by 24 hr of reperfusion. Slices were stained using TUNEL with tetramethyl-rhodamine red label (Roche, Hertforshire, UK). Slices were viewed on a Carl Zeiss Laser Scanning Microscope (LSM) 510 (Zeiss, Thornwood, NY). The images of the sections were analyzed using LSM 5 image browser. In the in vivo model, animals underwent a 2 hr MCAO occlusion, and Tat or δV1-1 peptides were delivered as an intra-arterial bolus dose at the onset of reperfusion as described above. Animals were killed after 72 hr of reperfusion and transcardially perfused with normal saline followed by 4% paraformaldehyde. Brains were isolated and fixed overnight in 4% paraformaldehyde, immersed in 30% sucrose for 2-3 d, and snap frozen in OCT cryoprotectant (Tissue-Tek, Miles, Elkhart, IN). Three 16 μm coronal slices were taken from equivalent midbrain regions in each brain, with each slice separated by at least 48 μm. Slides were stained after TUNEL kit protocol (Roche). Images were taken from four defined regions within the ipsilateral cortex of each slice. Cells were differentiated as being apoptotic both by positive staining and distinct morphology, including nuclear fragmentation suggestive of chromatin condensation; cells with diffuse, dim staining were not counted as apoptotic (Conti et al., 1998). Cell counts from the three slices taken from each brain were averaged to give a representative total number of cells within each image. An average cell count per brain was then calculated from each of the groups (n = 3-5 per group). Results were expressed as the percentage of change in TUNEL-positive cell number with respect to control animals.
Western blot analysis. Brains were quickly isolated, sliced, and striatal regions dissected from each central slice and frozen. Tissue fractionation, both from whole organotypic slices and striatal region of isolated brains, was performed to collect soluble (cytosolic) and particulate (membrane) fractions as described previously (Johnson and Mochly-Rosen, 1995). Protein concentration was assessed using Bradford reagent, and 10 μg of total lysate (40 μg from hippocampal slice lysate) from each fraction was subjected to gel electrophoresis (10% bis-acrylamide gel) and transferred to nitrocellulose membrane. Blots were blocked in 3% milk TBST, probed with an anti-phospho-serine 643 δPKC antibody (1:500; Santa Cruz Biotechnology, Santa Cruz, CA) in 2% BSA Tris-buffered saline Tween 20 (TBST) and probed with an anti-rabbit secondary (Amersham Biosciences, Arlington Heights, IL). Density of δPKC bands was measured to assess translocation of PKC from the cytosol to membrane fractions, a measure of activation (Kraft and Anderson, 1983). To assay Akt and BAD (Bcl-2-associated death protein) proteins, 20 μg of brain lysate was run on 12.5 and 15% gels, respectively, blotted to nitrocellulose and blocked as described above, and probed with anti-phospho-Akt antibody (1:500; Biosource International, Camarillo, CA) or anti-BAD antibody (1:250; Santa Cruz Biotechnology) followed by anti-rabbit secondary. Blots were then stripped and reprobed with anti-Akt antibody (1:500; Cell Signaling Technology, Beverly, MA) or β-actin (1:5000; Sigma) to confirm equal protein loading. Densities were measured from Western blot autoradiograms. All statistical analyses were performed using ANOVA followed by Fisher's post hoc tests.
Results
δPKC inhibition reduces reperfusion injury in hippocampal slice model of oxygen and glucose deprivation
We first determined whether delivery of δV1-1, a peptide inhibitor of δPKC, affected neuronal cell injury after OGD and reperfusion in an in vitro hippocampal slice model. This protocol leads to rapid death of CA1 pyramidal cells and granule cells, which manifests within 24 hr of reperfusion (Bergold and Casaccia-Bonnefil, 1997; Pringle et al., 1997; Raval et al., 2003). Propidium iodide fluorescence was measured to assess cell death (Fig. 1b). In all experiments, OGD-induced cell damage is expressed as a fraction of total cell death when an overdose of NMDA (100 μm) was applied to the culture medium. We found that 40 min of OGD followed by 24 hr of reperfusion (oxygen and glucose are reapplied) led to ∼70% cell death in CA1 pyramidal cells compared with 4% death in slices maintained in normoxic conditions (Fig. 1b,c).
When slices were treated with the δPKC-selective inhibitor δV1-1 (500 nm), during the ischemic period and during the first 3 hr of reperfusion, the percentage of neuronal death decreased by 43%, from 69 ± 3% in control to 39 ± 6% (Fig. 1c, group 6) (p < 0.05). However, treatment with δV1-1 during the ischemic period only did not confer any neuroprotection; 71 ± 2% neuronal death was observed (Fig. 1c, group 4). Tat control was not protective when added at either time point (Fig. 1c, groups 3, 5). These data demonstrate that the presence of the δPKC inhibitor during the ischemic period only conferred no protection. In contrast, inhibition of δPKC during the reperfusion period greatly reduced neuronal cell death in this in vitro model of ischemic-reperfusion injury.
δV1-1 peptide inhibits δPKC translocation in vitro
We next confirmed that δV1-1 peptide exerted its biological effect and inhibited δPKC activity. Redistribution of PKC from the soluble to the particulate fraction (PKC translocation) is a marker for PKC activation (Kraft and Anderson, 1983) and has been used in numerous studies to assess enzyme activity (Wieloch et al., 1991; Selvatici et al., 2002). We therefore assessed δPKC translocation in slices treated with the δV1-1 peptide for the first 3 hr of reperfusion. We found a 24% increase in δPKC translocation in slices subjected to OGD followed by 3 hr of reperfusion, indicating that δPKC was activated. In slices treated with δV1-1 peptide during ischemia and reperfusion, δPKC translocation was reduced by 45% (Fig. 1d) (p < 0.05).
δV1-1 peptide reduces number of apoptotic cells after OGD in vitro
To determine the mechanism by which δV1-1 confers neuroprotection, we assessed the number of cells undergoing apoptotic cell death after ischemic-reperfusion injury using TUNEL staining. In the hippocampal slice model, few, if any, TUNEL-positive cells were detected in sham-treated slices (Fig. 1ea). TUNEL-positive cells were abundantly detected in the CA1 region after 40 min OGD and 24 hr of reperfusion in the Tat control peptide-treated group (Fig. 1eb). The number of TUNEL-positive cells was markedly diminished in slices treated with δV1-1 peptide (Fig. 1ec).
δV1-1 peptide delivery to brain parenchyma
To determine whether δPKC inhibition protects the brain from reperfusion damage after prolonged ischemia in vivo, we first demonstrated that δV1-1 conjugated to Tat can be delivered across the blood-brain barrier. Peptides cannot be readily delivered across the blood-brain barrier into the brain parenchyma. However, previous work by Schwarze et al. (1999) demonstrated that a Tat peptide, derived from the HIV Tat protein, crosses biological membranes and can serve as a carrier to other cargo, including proteins. An intra-arterial bolus of biotinylated δV1-1 was injected into the internal carotid artery (2.5 μm final blood concentration), and the brains were harvested after 30 min. Using avidin-biotin complex, we detected the presence of the peptide in pyramidal cells in the cortex, suggesting effective delivery of the peptide into the brain parenchyma through the blood-brain barrier (Fig. 2a).
Figure 2.
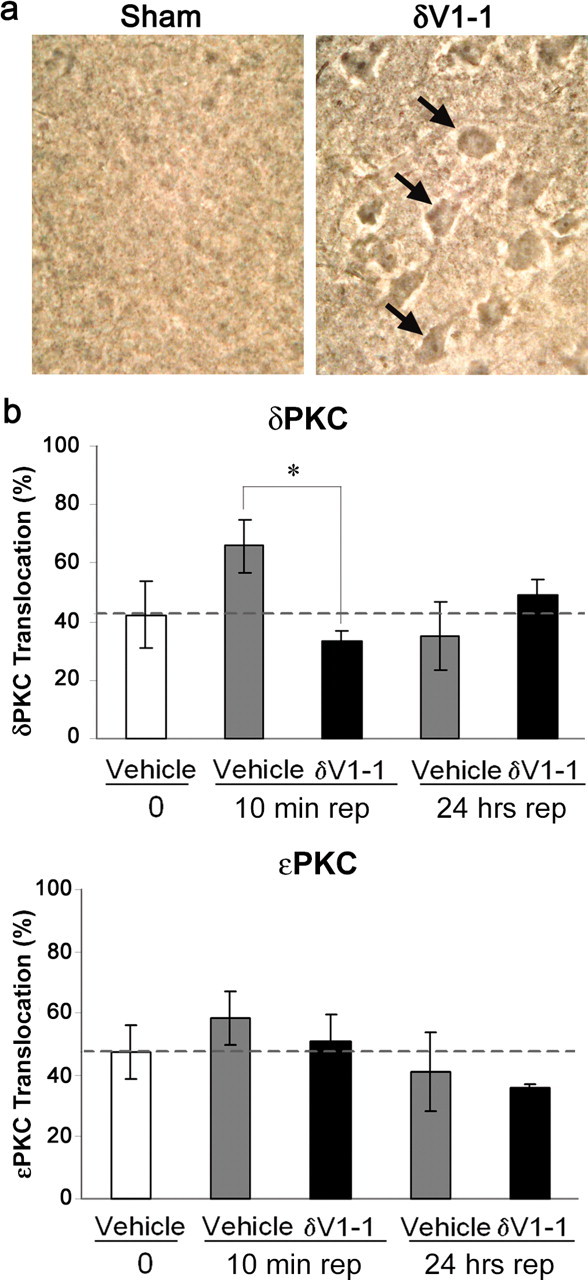
δV1-1 is delivered to the brain parenchyma in vivo after intra-arterial bolus. a, Background staining from sham brain (left) and corresponding staining of biotin-conjugated δV1-1 peptide in cortical neurons (right). b, Delivery of δV1-1 reduces δPKC translocation after 10 min of reperfusion (rep) (n = 3 per group; *p < 0.05). This effect was specific to δPKC; δV1-1 had no effect on ϵPKC translocation. Brain tissue was harvested from animals (δV1-1 or Tat treated) after ischemia and 10 min or 24 hr of reperfusion. Soluble and particulate fractions of brain tissue were subjected to Western blot analysis using anti-δPKC (phospho-ser643 δPKC) or anti-ϵPKC antibodies.
We next assessed the activity of δPKC in an in vivo transient focal ischemia model. Rats underwent 2 hr MCAO, followed by either 10 min or 24 hr of reperfusion. We found that δPKC was activated as measured by intracellular translocation after 10 min of reperfusion, and that this translocation was inhibited after in vivo delivery of δV1-1 at the onset of reperfusion (Fig. 2b) (50% inhibition; p < 0.05). However, at 24 hr of reperfusion, there was no difference in the δPKC translocation between the groups. Importantly, the effect of δV1-1 was selective (e.g., ϵPKC translocation was not altered by the δV1-1 treatment) (Fig. 2b).
δV1-1 reduced infarct size after in vivo delivery
We next determined whether inhibition of δPKC translocation affected cerebral ischemic and reperfusion injury in vivo using a 2 hr transient MCA occlusion model of stroke, followed by a 24 hr reperfusion period. Either δV1-1 or Tat (each at 250 nm final blood concentration) was delivered as a bolus dose via the internal carotid artery either immediately before the ischemic period (pre-δV1-1) or at the onset of reperfusion (post-δV1-1) (Fig. 3a). Delivery of the peptide did not alter standard physiological variables including blood pH, glucose, blood gases (pCO2, pO2), hematocrit, or mean arterial blood pressure. Brains were harvested after 24 hr of reperfusion, and stroke area was assessed after staining with TTC. Treatment with the δV1-1 peptide prior to the ischemic event (pre-δV1-1) did not confer protection from subsequent ischemia and reperfusion injury (30 ± 7%; n = 8). However, infarct areas were reduced by 68% in animals that were treated with δV1-1 at the onset of reperfusion (post-δV1-1; 8 ± 4%; n = 10) relative to Tat control-treated animals (vehicle; 26 ± 5%; n = 14; p < 0.05) (Fig. 3b,c).
Figure 3.

δV1-1 protects brain from reperfusion damage in vivo. a, Tat carrier peptide alone (n = 14) or δV1-1 were delivered intra-arterially through the internal carotid artery immediately before (δV1-1 preischemia; n = 8) or immediately after (δV1-1 postischemia; n = 10) a 2 hr occlusion of the MCA. Animals were killed after 24 hr of reperfusion, and brain slices were stained with TTC, which stains viable tissues red. Infarcted tissue appears white (marked with black border). b, Typical pictures from vehicle- and δV1-1-treated animals 24 hr after MCA occlusion. c, Delivery of δV1-1 after ischemia, but not before ischemia, reduced extent of brain infarction as measured after 24 hr of reperfusion. Significance was compared with control (*p < 0.05).
Delayed delivery of δV1-1-Tat during reperfusion confers protection
To determine whether δPKC activity was involved in nonacute events during reperfusion and to further define the therapeutic window for δV1-1 administration, we next determined whether δV1-1 provided a protective effect when delivered at delayed time points during reperfusion (Fig. 4a). We found that a single intraperitoneal dose of δV1-1 peptide (20 nmol bolus dose) at 1 hr after the onset of reperfusion also reduced infarct size compared with control (16 ± 3 vs 30 ± 5%, respectively; p < 0.05) (Fig. 4b). In addition, we found that δV1-1 delivery 6 hr after the onset of reperfusion (20 nmol) significantly reduced infarct size verses controls (14 vs 33%, respectively; p < 0.05) (Fig. 4c).
Figure 4.
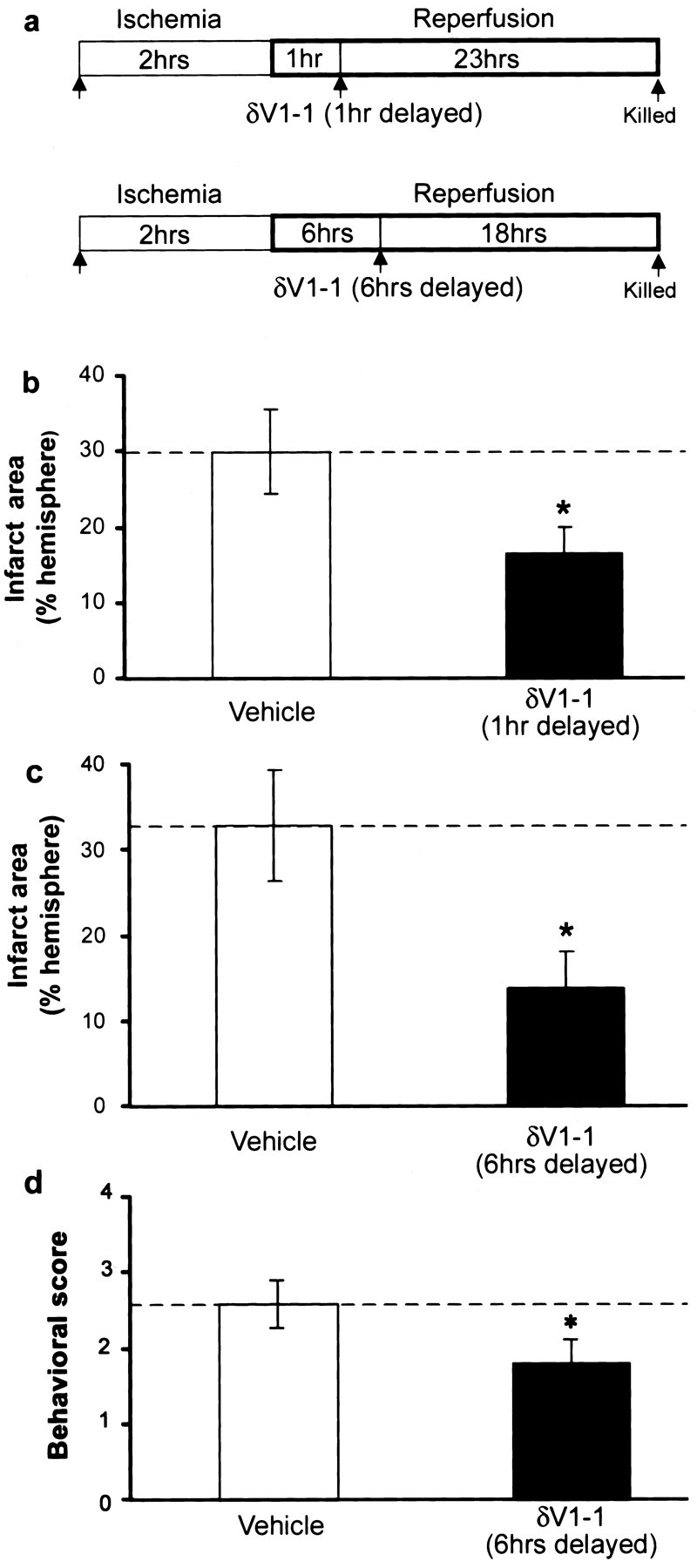
Delayed δV1-1 delivery during reperfusion reduces infarct size in MCAO model. a, Tat peptide alone or δV1-1 were delivered by intraperitoneal injection at 1 or 6 hr after the onset of reperfusion. Animals were assessed for behavior 23 hr later, killed, and brain infarct size was assessed using TTC. b, δV1-1 delivery 1 hr after the onset of reperfusion significantly reduced infarct size compared with Tat alone (vehicle, n = 11; δV1-11 hr, n = 13; *p < 0.05). c, δV1-1 delivery 6 hr after the onset of reperfusion significantly reduced infarct size compared with Tat peptide (vehicle, n = 9; δV1-1 6 hr, n = 9; *p < 0.05). d, δV1-1 peptide delivery 6 hr after the onset of reperfusion significantly decreases neurological deficit compared with animals treated with control peptide alone, as assessed at 24 hr of reperfusion (*p < 0.05).
As a component of our MCAO animal protocol, we used the inhalational anesthetic isoflurane for induction and maintenance of anesthesia during the ischemic period alone. We recognize that isoflurane, like other anesthetics, has been shown previously to confer protection from cerebral ischemia-reperfusion injury when delivered before ischemia (Kapinya et al., 2002; Sullivan et al., 2002). However, isoflurane was used for anesthesia in both control-treated and δV1-1-treated animals during the ischemic period. Moreover, we found that δV1-1 conferred neuroprotection without the use of isoflurane in the hippocampal slice model of ischemia-reperfusion and has been shown previously to confer cardioprotection in models of cardiac ischemia-reperfusion injury without the use of isoflurane (Chen et al., 2001). Finally, δV1-1 was administered during a period independent of isoflurane delivery (1 and 6 hr reperfusion) to confer protection and was administered during a period of isoflurane delivery (before ischemic onset) without conferring protection.
δV1-1 reduces neurological deficit in animals after MCAO injury
To determine whether delivery of δV1-1 lessens neurological deficit after MCAO injury, we selected one treatment group (δV1-1 or control peptide delivered at 6 hr postreperfusion) for behavioral assessment using a scale modified from the study by Yang et al. (1994). Animals were graded on a scale of 1-4, ranking increasing deficit as described above. Using this scoring system, we found that animals treated with δV1-1 peptide exhibited a significantly decreased behavioral score than control-treated animals, as assessed at 24 hr reperfusion (1.8 ± 0.3 vs 2.6 ± 0.3; p < 0.05) (Fig. 4d). Preserved neurological function in δV1-1-treated animals corresponded to a 57% decrease in weight loss compared with controls after this 24 hr period (p < 0.05), suggesting that δV1-1-treated animals may be feeding better, which is another indication of improved neurological function.
δV1-1 peptide reduces number of apoptotic cells after ischemic-reperfusion injury in vivo
We next assessed whether δV1-1 affected the number of apoptotic cells in the in vivo model of ischemia-reperfusion injury. The number of TUNEL-positive cells was markedly diminished in slices treated with δV1-1 peptide (Fig. 1ec). In the in vivo model, brains were isolated after 2 hr MCAO and 72 hr of reperfusion. TUNEL-positive apoptotic cells were found primarily in the peri-infarct region of the ipsilateral cortex (Fig. 5aa). However, in δV1-1-treated animals, apoptotic cell count was diminished (Fig. 5ab). TUNEL-positive cells in four defined regions of the ipsilateral cortex were counted in δV1-1- or control-treated animals. We found a reduction of 47% in δV1-1-treated animals compared with control-treated animals (Fig. 5b).
Figure 5.
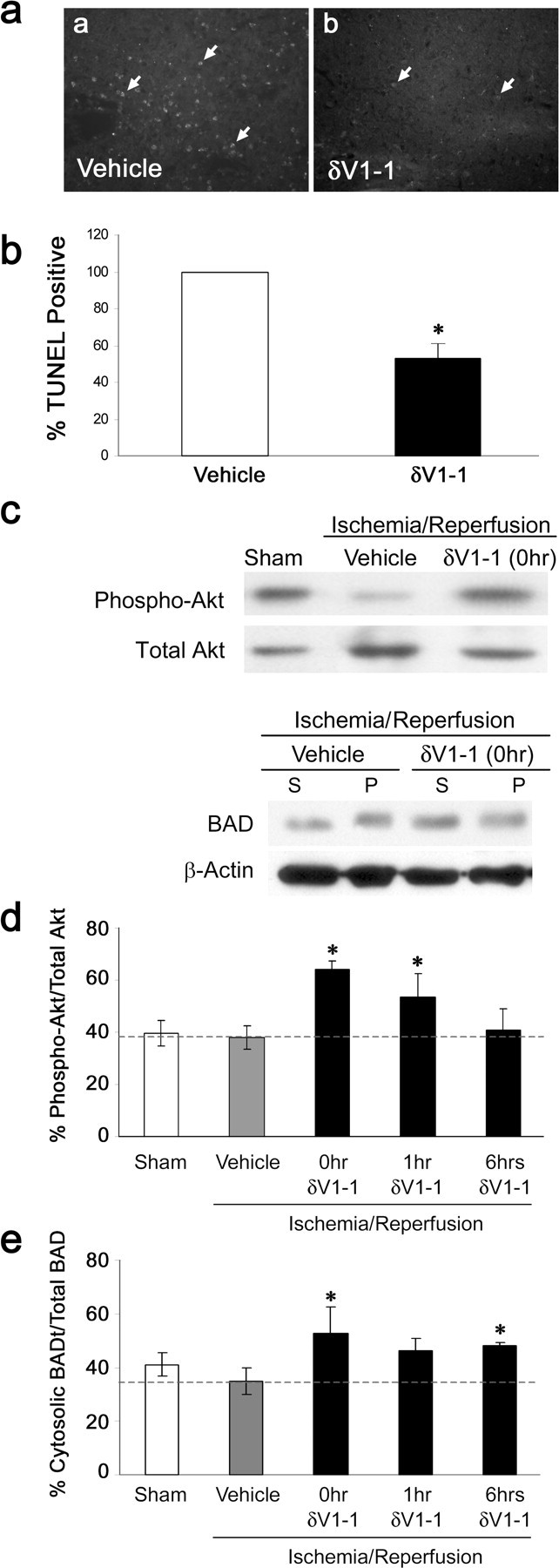
δV1-1 peptide treatment reduces apoptosis and alters levels of prosurvival and apoptotic markers after cerebral ischemia. After 72 hr of reperfusion, brains treated with either δV1-1 or Tat peptide at the onset of reperfusion were harvested and stained using TUNEL (Tat, n = 3; δV1-1, n = 4). a, b, Images were taken of ipsilateral cortex (100×) in control-treated (a) and δV1-1-treated (b) animals. *Arrows indicate TUNEL-positive cells. b, TUNEL-positive cells were counted in four defined regions of the ipsilateral cortex in each slice. A 47% decrease in TUNEL-positive cell count was seen in δV1-1-treated animals compared with control animals (*p < 0.05). c, After 24 hr of reperfusion, lysates from brains treated with Tat peptide alone (vehicle, n = 5) or δV1-1 at the onset (n = 5), at 1 hr (n = 4), or at 6 hr (n = 4) of reperfusion or brains from sham-treated animals (n = 4) were subject to Western blot analysis for phosphorylated Akt and BAD. β-actin was used to confirm protein levels. d, Levels of phospho-Akt were markedly increased with respect to total Akt in animals treated with δV1-1 peptide versus vehicle-treated animals at the onset of reperfusion and at 1 hr of reperfusion (*p < 0.05). Levels were not significantly different from controls in animals treated with δV1-1 at 6 hr into reperfusion. e, BAD protein translocation from the cytosol to the membrane bound fraction was inhibited in animals treated with δV1-1 at the onset of reperfusion (*p < 0.05) at 1 hr (did not reach statistical significance) or at 6 hr of reperfusion (*p < 0.05). Graph represents levels of cytosolic BAD protein with respect to total BAD protein.
δV1-1 treatment alters phospho-Akt levels and BAD protein levels in the in vivo ischemia model
To further determine the mechanism by which δV1-1 reduces cellular damage during reperfusion, we studied two markers for cell survival signaling and proapoptotic signaling, namely, activated Akt and the Bcl-2 family member BAD. Akt, the prosurvival enzyme, is active when phosphorylated on ser-473 and thr-308 (Alessi et al., 1996; Delcommenne et al., 1998). We found that there was a significant increase in phospho-Akt levels with respect to total Akt, when δV1-1 is delivered either at the onset of reperfusion (64 ± 3%; p < 0.05) or at 1 hr into reperfusion (54 ± 9%) verses control animals (38 ± 4%). When δV1-1 was delivered 6 hr after reperfusion, phospho-Akt was not significantly higher than control levels (Fig. 5c,d).
BAD protein becomes proapoptotic when it translocates from the cytosol to the mitochondria, where it associates with other members of the BCl-2 family of proteins (von Harsdorf et al., 1999). We found higher cytosolic levels of BAD with respect to total BAD protein in δV1-1-treated animals when treated at the onset of reperfusion (53 ± 10%; p < 0.05) and 6 hr into reperfusion (48 ± 1%; p < 0.05) compared with controls (35 ± 5%). When δV1-1 was delivered at 1 hr after ischemia, we observed increased cytosolic BAD; however, values did not reach statistical significance (46 ± 4%) (Fig. 5c,e).
Discussion
In this study, we demonstrated that inhibition of δPKC, using a δPKC-selective inhibitor peptide, significantly reduced cerebral tissue damage when administered at the onset of reperfusion. This neuroprotection was achieved using two models of ischemic and reperfusion injury: an in vitro hippocampal slice culture model and an in vivo transient focal ischemia model.
δV1-1 peptide delivery in vivo
δPKC exists in a dynamic state, rapidly cycling between an inactive and an active state, when it is conformationally able to bind to its isozyme-specific anchoring molecule δRACK (receptor for activated C kinase) and phosphorylate physiological substrates (Mochly-Rosen, 1995). The δV1-1 peptide was rationally designed to block binding of activated δPKC to δRACK and thereby inhibit its activity. For passage through the blood-brain barrier and intracellular delivery, the δV1-1 peptide was conjugated to the cell membrane-permeating Tat-carrier peptide, which was previously shown to deliver peptides (Aarts et al., 2002) and proteins (Asoh et al., 2002; Cao et al., 2002; Kilic et al., 2002) to the brain parenchyma in vivo.
We demonstrated that δPKC translocation was significantly reduced in the brain after both in vitro and in vivo delivery of δV1-1, consistent with access of the peptide to the brain parenchyma. In addition, the remarkably parallel protective effects of δV1-1 between the in vivo MCAO model and the in vitro hippocampal slice model, in which blood-brain barrier transport of the peptide is not a factor, suggest similar access and activity of the peptide to cells at risk in both models. Importantly, we also found that delivery of the peptide by an intra-arterial bolus or by intraperitoneal delivery greatly reduced infarct size, suggesting that peripheral routes of delivery are effective in administration of this peptide therapeutic to the CNS.
δV1-1 reduces damage from reperfusion injury
There is little consensus on whether individual PKC isozymes play a damaging or beneficial role in cellular injury after stroke. Here, we demonstrated that rapid activation of δPKC occurred in compromised tissue at the onset of reperfusion after MCA occlusion. Within 10 min of reperfusion after a 2 hr stroke, δPKC was activated, as shown by its translocation from the soluble to the membrane fraction. This rapid translocation suggests a role for δPKC in mediating reperfusion damage after ischemia and indicates an opportunity for intervention by selective inhibition of this enzyme.
The timing of δV1-1 delivery further supports our hypothesis that δPKC activity mediates reperfusion injury processes. We found that delivery of δV1-1 during the OGD period alone in the hippocampal slice model or before ischemia in the MCAO model provided no observable protection from cerebral injury. In contrast, delivery of δV1-1 during the reperfusion period significantly protected the brain. In the in vitro slice model, delivery during OGD and the first 3 hr of reperfusion decreased neuronal death. Similarly, in the in vivo model, delivery of δV1-1 at the onset of reperfusion, 1 hr, or 6 hr into reperfusion significantly reduced infarct size and corresponded with improved neurological outcome. We therefore suggest that δPKC-induced neuronal damage may occur for several hours during reperfusion. The protective effect of this peptide at 6 hr after the onset of reperfusion also suggests its potential clinical value for late reperfusion injury (Jonas et al., 2001).
δPKC in intracellular injury processes
Reperfusion injury is a major cause of cerebral damage after transient ischemia (Yang and Betz, 1994). The process of reperfusion is known to produce characteristic changes, including excitotoxic damage, inflammation, edema, and apoptosis (for review, see Lipton, 1999). Apoptosis involves a highly regulated system of cell death pathways and has been observed in multiple tissues in response to ischemic-reperfusion injury (Schumer et al., 1992; Gottlieb et al., 1994; Li et al., 1995). A growing body of literature suggests that δPKC plays a central role in mediating apoptosis (Gutcher et al., 2003). In a model of cardiac ischemia and reperfusion, we recently demonstrated a detrimental role for δPKC, promoting mitochondrial damage and induction of apoptosis (Inagaki et al., 2003b). Recent studies demonstrate that inhibition of ζPKC, but not δPKC, confers protection to neuron-astrocyte cultures exposed to a level of NMDA, which produced excitotoxic cell death (Koponen et al., 2003). The absence of protection conferred by the δPKC inhibitor may be attributable to the lack of apoptotic cell death in this model. These findings, together with our evidence that δPKC inhibitor peptide exhibited a CNS-protective effect when delivered during reperfusion, suggest that δV1-1 may confer protection, at least in part, by inhibiting proapoptotic processes. We therefore investigated whether δPKC alters markers for apoptotic cell change and cell survival.
We demonstrated that in both in vitro and in vivo models of cerebral ischemic-reperfusion injury, there was a significant reduction in the number of TUNEL-positive cells in δV1-1-treated slices (CA1 region) or animals (ipsilateral cortex), compared with control-peptide treatments. Interestingly, this reduction in apoptotic cell death was observed at 3 d postischemia in the MCAO model and is a first indication that protection conferred by δV1-1 is sustained for several hours (Valtysson et al., 1994).
Based on these findings, we sought to determine more precisely the role of δPKC in mediating apoptotic injury. We therefore assessed alterations in markers for cell survival and apoptotic pathways after ischemia-reperfusion. Akt (protein kinase B) mediates survival responses to a number of cell-death stimuli, including DNA damage, cell-cycle misregulation, and more recently, ischemia (Downward, 1998; Noshita et al., 2001). Thus, maintenance of Akt activity is important to cell survival. Using the 2 hr MCAO model, we found that cerebral ischemia-reperfusion injury caused a reduction in activated (phospho ser-473) Akt levels and that delivery of δV1-1 inhibited this reduction in active Akt. These data suggest that inhibition of δPKC in the first hour of reperfusion promoted cell-survival signals in the brain through preserving Akt activity. However, when δV1-1 was delivered after 6 hr of reperfusion, levels of active Akt were similar to the control group. This suggests that the neuroprotective activity of δV1-1 is not mediated by Akt at this later time window and that δPKC may be involved in alternative, or even multiple, signaling pathways to mediate cell death, according to the phase of reperfusion damage and cellular state (for review, see Lipton, 1999). Alternatively, this data may imply that the observed increase in active Akt is secondary to δV1-1 conferred protection.
Interestingly, Akt has also been linked to apoptotic cell-death pathways by phosphorylation of the Bcl-2 family member BAD at ser-136 (Datta et al., 1997). When phosphorylated, BAD is sequestered in the cytoplasm. After an apoptotic stimulus, BAD is dephosphorylated and subsequently translocates to the mitochondria, contributing to mitochondrial dysfunction (Yang et al., 1995; Zha et al., 1996). We were unable to determine the levels of phosphorylated BAD. However, consistent with the expected change in BAD distribution, we observed a decrease in cytosolic BAD and correlated increase in membrane-bound BAD in the brain of animals subject to ischemia followed by 24 hr of reperfusion. Importantly, in animals treated with δV1-1, the ratio of cytosolic- to membrane-bound BAD was increased. These data indicate that BAD translocation out of the cytoplasm, and thus its proapoptotic activity, was inhibited in animals treated with δV1-1. Together, these data suggest that inhibition of δPKC increased activity of the prosurvival enzyme Akt and inhibited the activity of the proapoptotic protein BAD. Although the signaling pathways between δPKC and downstream cell survival markers are only partly defined, the consistency of this observation in multiple models of ischemia and reperfusion support a role for δPKC upstream of cell survival (Murriel and Mochly-Rosen, 2004).
Reperfusion also promotes secondary damage through unregulated neurotransmitter release, decrease in cellular energy stores, and inhibition of electron transport, leading to changes in cell permeability and free radical generation (for review, see Lipton, 1999). Previous work has outlined a role for PKC isozymes in these responses and suggests a correlation between sustained glutamate release, PKC activity, and downstream neuronal damage (Miettinen et al., 1996; Nakane et al., 1998; Skeberdis et al., 2001). These studies raise the possibility that other pathways may also be involved in δPKC-mediated reperfusion damage and suggests additional avenues by which inhibition of δPKC may confer neuroprotection. Alternatively, the protective effect of the δV1-1 peptide may occur in part through altering cerebral blood flow (CBF) after ischemia. However, as the peptide was protective in an in vitro model, where vascular function does not mediate outcome, we suggest the neuroprotective effect cannot be caused by its effect on CBF alone.
Our data indicate that δPKC mediates cerebral reperfusion damage and is the first report that inhibition of this enzyme greatly improves histopathologic and behavioral outcome after stroke in vivo. We suggest that multiple PKC isozymes play a role in cerebral ischemic-reperfusion injury with unique windows of activity during the ischemic and reperfusion periods. Here, we show a selective role for δPKC as a mediator of reperfusion damage but not ischemic injury. These data underscore the importance of using isozyme-selective tools to assess the role of individual PKC isozymes in mediating ischemic and reperfusion damage of the CNS and the therapeutic potential of a selective δPKC inhibitor on injury progression after cerebral ischemia and reperfusion.
Footnotes
This work was supported by National Institutes of Health (NIH) Research Grants NS40516 (M.A.Y.), P01 NS37520 and R01 NS27292 (G.K.S.), NS44350 (D.M.-R.), and NS34773 (M.A.P.-P.) and by State of California Funds for medical research on alcohol and substances of abuse (D.M.-R.), American Heart Association Grant 0225227B, a Florida-Puerto Rico affiliates postdoctoral fellowship (A.P.R.), and NIH-National Institute of Neurological Disorders and Stroke Predoctoral Fellowship NS45413-01 (R.B.). We gratefully acknowledge the assistance of Beth Hoyte for figure preparation. D.M.-R. is a founder of KAI Pharmaceuticals, whose goal is to bring peptide regulators of protein kinase C to the clinic. However, the research described in this study was performed in her laboratory at the university and with sole support from the NIH and State of California Funds to her university activities.
Correspondence should be addressed to Daria Mochly-Rosen, Department of Molecular Pharmacology, Stanford University School of Medicine, Stanford, CA 94305-5174. E-mail: mochly@stanford.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/246880-09$15.00/0
References
- Aarts M, Liu Y, Liu L, Besshoh S, Arundine M, Gurd JW, Wang YT, Salter MW, Tymianski M (2002) Treatment of ischemic brain damage by perturbing NMDA receptor-PSD-95 protein interactions. Science 298: 846-850. [DOI] [PubMed] [Google Scholar]
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA (1996) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15: 6541-6551. [PMC free article] [PubMed] [Google Scholar]
- Asoh S, Ohsawa I, Mori T, Katsura K, Hiraide T, Katayama Y, Kimura M, Ozaki D, Yamagata K, Ohta S (2002) Protection against ischemic brain injury by protein therapeutics. Proc Natl Acad Sci USA 24: 17107-17112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergold PJ, Casaccia-Bonnefil P (1997) Preparation of organotypic hippocampal slice cultures using the membrane filter method. Methods Mol Biol 72: 15-22. [DOI] [PubMed] [Google Scholar]
- Brodie C, Blumberg PM (2003) Regulation of cell apoptosis by protein kinase C delta. Apoptosis 8: 19-27. [DOI] [PubMed] [Google Scholar]
- Cao G, Pei W, Ge H, Liang Q, Luo Y, Sharp FR, Lu A, Ran R, Graham SH, Chen J (2002) In vivo delivery of Bcl-xL fusion protein containing TAT protein transduction domain protects against ischemic brain injury and neuronal apoptosis. J Neurosci 22: 5423-5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Peterson PK (1995) Modulation of human microglial cell superoxide production by cytokines. J Leukoc Biol 58: 65-70. [DOI] [PubMed] [Google Scholar]
- Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21: 2-14. [DOI] [PubMed] [Google Scholar]
- Chen L, Hahn H, Wu G, Chen CH, Liron T, Schechtman D, Cavallaro G, Banci L, Guo Y, Bolli R, Dorn GW II, Mochly-Rosen D (2001) Opposing cardioprotective actions and parallel hypertrophic effects of delta PKC and epsilon PKC. Proc Natl Acad Sci USA 98: 11114-11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti AC, Raghupathi R, Trojanowski JQ, McIntosh TK (1998) Experimental brain injury induces regionally distinct apoptosis during the acute and delayed post-traumatic period. J Neurosci 18: 5663-5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to cell-intrinsic death machinery. Cell 91: 231-241. [DOI] [PubMed] [Google Scholar]
- Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S (1998) Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/Akt by the integrin-linked kinase. Proc Natl Acad Sci USA 15: 11211-11216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey JM, Cohen MV, Ytrehus K, Liu Y (1994) Cellular mechanisms in ischemic preconditioning: the role of adenosine and protein kinase C. Ann NY Acad Sci 17: 82-98. [PubMed] [Google Scholar]
- Downward J (1998) Mechanisms and consequences of activation of Protein kinase B/Akt. Curr Opin Cell Biol 10: 262-267. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA, Burleson KO, Kloner RA, Babior BM, Engler RL (1994) Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest 94: 1621-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutcher I, Webb PR, Anderson NG (2003) The isoform-specific regulation of apoptosis by protein kinase C. Cell Mol Life Sci 60: 1061-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki K, Chen L, Ikeno F, Lee FH, Imahashi K, Bouley DM, Rezaee M, Yock PG, Murphy E, Mochly-Rosen D (2003a) Inhibition of delta protein kinase C against reperfusion injury of the ischemic heart, in vivo Circulation 108: 2304-2307. [DOI] [PubMed] [Google Scholar]
- Inagaki K, Hahn HS, Dorn GW II, Mochly-Rosen D (2003b) Additive protection of the ischemic heart ex vivo by combined treatment with delta-protein kinase C inhibitor and epsilon-protein kinase C activator. Circulation 19: 869-875. [DOI] [PubMed] [Google Scholar]
- Johnson JA, Mochly-Rosen D (1995) Inhibition of the spontaneous rate of contraction of neonatal cardiac myocytes by protein kinase C isozymes. A putative role for the epsilon isozyme. Circ Res 76: 654-663. [DOI] [PubMed] [Google Scholar]
- Jonas S, Aiyagari V, Vieira D, Figueroa M (2001) The failure of neuronal protective agents versus the success of thrombolysis in the treatment of ischemic stroke Ann NY Acad Sci 939: 257-267. [DOI] [PubMed] [Google Scholar]
- Kapinya KJ, Lowl D, Futterer C, Maurer M, Waschke KF, Isaev NK, Dirnagl U (2002) Tolerance against ischemic neuronal injury can be induced by volatile anesthetics and is inducible NO synthase dependent. Stroke 33: 1889-1898. [DOI] [PubMed] [Google Scholar]
- Kilic E, Dietz GP, Hermann DM, Bahr M (2002) Intravenous TAT-Bcl-Xl is protective after middle cerebral artery occlusion in mice. Ann Neurol 52: 617-622. [DOI] [PubMed] [Google Scholar]
- Koponen S, Goldsteins G, Keinanen R, Koistinaho J (2000) Induction of protein kinase C delta subspecies in neurons and microglia after transient global brain ischemia. J Cereb Blood Flow Metab 20: 93-102. [DOI] [PubMed] [Google Scholar]
- Koponen S, Kurkinen K, Akerman KEO, Mochly-Rosen D, Chan PH, Koistinaho J (2003) Prevention of NMDA-induced death of cortical neurons by inhibition of protein kinase C zeta. J Neurochem 86: 442-450. [DOI] [PubMed] [Google Scholar]
- Kraft AS, Anderson WB (1983) Phorbol esters increase the amount of Ca2+, phospholipid-dependent protein kinase associated with plasma membrane. Nature 301: 621-623. [DOI] [PubMed] [Google Scholar]
- Li Y, Sharov VG, Jiang N, Zaloga C, Sabbah HN, Chopp M (1995) Ultra-structural and light microscopic evidence of apoptosis after middle cerebral artery occlusion in the rat. Am J Pathol 146: 1045-1051. [PMC free article] [PubMed] [Google Scholar]
- Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. [DOI] [PubMed] [Google Scholar]
- Maier CM, Sun GH, Kunis D, Yenari MA, Steinberg GK (2001) Delayed induction and long-term effects of mild hypothermia in a focal model of transient cerebral ischemia: neurological outcome and infarct size. J Neurosurg 94: 90-96. [DOI] [PubMed] [Google Scholar]
- Miettinen S, Roivainen R, Keinanen R, Hokfelt T, Koistinaho J (1996) Specific induction of protein kinase C delta subspecies after transient middle cerebral artery occlusion in the rat brain: inhibition by MK-801. J Neurosci 16: 6236-6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochly-Rosen D (1995) Localization of protein kinases by anchoring proteins: a theme in signal transduction. Science 268: 247-251. [DOI] [PubMed] [Google Scholar]
- Murriel CL, Mochly-Rosen D (2004) Opposing roles of delta and epsilon PKC in cardiac ischemia and reperfusion; targeting the apoptotic machinery. Arch Biochem Biophys 420: 246-254. [DOI] [PubMed] [Google Scholar]
- Nakane H, Yao H, Ibayashi S, Kitazono T, Ooboshi H, Uchimura H, Fujishima M (1998) Protein kinase C modulates ischemia-induced amino acids release in the striatum of hypertensive rats. Brain Res 26: 290-296. [DOI] [PubMed] [Google Scholar]
- National Institute of Neurological Disorders and Stroke (1995) Tissue plasminogen activator for acute ischemic stroke. The National Institute of Neurological Disorders and Stroke rt-PA stroke study group. N Engl J Med 333: 1581-1587. [DOI] [PubMed] [Google Scholar]
- Noshita N, Lewen A, Sugawara T, Chan PH (2001) Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab 21: 1442-1450. [DOI] [PubMed] [Google Scholar]
- Padanilam BJ (2001) Induction and subcellular localization of protein kinase C isozymes following renal ischemia. Kidney Int 59: 1789-1797. [DOI] [PubMed] [Google Scholar]
- Phan TG, Wright PM, Markus R, Howells DW, Davis SM, Donnan GA (2002) Salvaging the ischaemic penumbra: more than just reperfusion? Clin Exp Pharmacol Physiol 29: 1-10. [DOI] [PubMed] [Google Scholar]
- Pringle AK, Iannotti F, Wilde GJ, Chad JE, Seeley PJ, Sundstrom LE (1997) Neuroprotection by both NMDA and non-NMDA receptor antagonists in in vitro ischemia. Brain Res 25: 36-46. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Mochly-Rosen D, Sick TJ, Perez-Pinzon MA (2003) Epsilon PKC is required for the induction of tolerance by ischemic and NMDA-mediated preconditioning in the organotypic hippocampal slice. J Neurosci 23: 384-391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumer M, Colombel MC, Sawczuk IS, Gobe G, Connor J, O'Toole KM, Olsson CA, Wise GJ, Buttyan R (1992) Morphologic, biochemical, and molecular evidence of apoptosis during the reperfusion phase after brief periods of renal ischemia. Am J Pathol 140: 831-838. [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF (1999) In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285: 1569-1572. [DOI] [PubMed] [Google Scholar]
- Selvatici R, Marino S, Piubello C, Rodi D, Beani L, Gandini E, Siniscalchi A (2003) Protein kinase C activity, translocation, and selective isoform subcellular redistribution in the rat cerebral cortex after in vitro ischemia. J Neurosci Res 71: 64-71. [DOI] [PubMed] [Google Scholar]
- Sieber FE, Traystman RJ, Brown PR, Martin LJ (1998) Protein kinase C expression and activity after global incomplete cerebral ischemia in dogs. Stroke 29: 1445-1452. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Lan J, Opitz T, Zheng X, Bennett MV, Zukin RS (2001) mGluR1-mediated potentiation of NMDA receptors involves a rise in intracellular calcium and activation of protein kinase C. Neuropharmacology 40: 856-865. [DOI] [PubMed] [Google Scholar]
- Sullivan BL, Leu D, Taylor DM, Fahlman CS, Bickler PE (2002) Isoflurane prevents delayed cell death in an organotypic slice culture model of cerebral ischemia. Anesthesiology 96: 189-195. [DOI] [PubMed] [Google Scholar]
- Valtysson J, Hillered L, Andine P, Hagberg H, Persson L (1994) Neuro-pathological endpoints in experimental stroke pharmacotherapy: the importance of both early and late evaluation. Acta Neurochir 129: 58-63. [DOI] [PubMed] [Google Scholar]
- von Harsdorf R, Li PF, Dietz R (1999) Signaling pathways in reactive oxygen species-induced cardiomyocyte apoptosis. Circulation 99: 2934-2941. [DOI] [PubMed] [Google Scholar]
- Wieloch T, Cardell M, Bingren H, Zivin J, Saitoh T (1991) Changes in the activity of protein kinase C and the differential subcellular redistribution of its isozymes in the rat striatum during and following transient forebrain ischemia. J Neurochem 56: 1227-1235. [DOI] [PubMed] [Google Scholar]
- Yang G-Y, Betz AL (1994) Reperfusion-induced injury to the blood-brain barrier after middle cerebral artery occlusion in rats. Stroke 25: 1658-1664. [DOI] [PubMed] [Google Scholar]
- Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H (1994) Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 25: 165-170. [DOI] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, Korsmeyer SJ (1995) Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80: 285-291. [DOI] [PubMed] [Google Scholar]
- Zha J, Harada H, Yang E, Jockel J, Korsmeyer SJ (1996) Serine phosphorylation of death agonist BAD in response to survival factor results in binding to 14-3-3 not BCL-X(L). Cell 87: 619-628. [DOI] [PubMed] [Google Scholar]