

Abstract
Alzheimer's disease (AD) may be caused by the abnormal processing of the amyloid precursor protein (APP) and the accumulation of β-amyloid (Aβ). The amyloid precursor protein can be proteolytically cleaved into multiple fragments, many of which have distinct biological actions. Although a high level of Aβ can be toxic, the α-secretase cleaved APP (sAPPα) is neuroprotective. However, the mechanism of sAPPα protection is unknown. Here, we show that sAPPα increases the expression levels of several neuroprotective genes and protects organotypic hippocampal cultures from Aβ-induced tau phosphorylation and neuronal death. Antibody interference and small interfering RNA knock-down demonstrate that the sAPPα-driven expression of transthyretin and insulin-like growth factor 2 is necessary for protection against Aβ-induced neuronal death. Mice overexpressing mutant APP possess high levels of sAPPα and transthyretin and do not develop the tau phosphorylation or neuronal loss characteristic of human AD. Chronic infusion of an antibody against transthyretin into the hippocampus of mice overexpressing APP with the Swedish mutation (APPSw) leads to increased Aβ, tau phosphorylation, and neuronal loss and apoptosis within the CA1 neuronal field. Therefore, the elevated expression of transthyretin is mediated by sAPPα and protects APPSw mice from developing many of the neuropathologies observed in AD.
Keywords: Alzheimer's disease, α-secretase, neuropathology, neurotrophic, neurodegeneration, neuroprotection, organotypic slice culture, β-amyloid
Introduction
Alzheimer's disease (AD) is characterized by β-amyloid (Aβ) accumulation and plaque formation, abnormal phosphorylation and aggregation of the microtubule-associated protein tau, and massive neuronal loss. Mutations identified in hereditary forms of AD as well as abundant animal models and in vitro data strongly implicate Aβ and the protein from which it is derived, the amyloid precursor protein (APP), as the principal factor driving the development of AD (Hardy and Selkoe, 2002). However, Aβ induction of the major AD neuropathologies, including phosphorylation of endogenous tau and in vivo neuronal loss, has yet to be convincingly demonstrated.
In fact, although mice overexpressing mutant forms of APP accumulate high levels of Aβ, they do not develop the tau phosphorylation or severe neuronal loss observed in AD (Irizarry et al., 1997a,b; Takeuchi et al., 2000). We have shown previously that mice overexpressing APP with the Swedish mutation (APPSw) markedly upregulate several neuroprotective genes and proteins (Stein and Johnson, 2002). These include transthyretin (TTR), a protein that can bind Aβ and prevent Aβ fibril formation in vitro (Schwarzman et al., 1994), and insulin-like growth factor-2 (IGF-2), a protein demonstrated to protect against Aβ toxicity in vitro (Dore et al., 1997). It remains to be demonstrated whether upregulation of TTR and/or IGF-2 in fact protects APPSw mice from Aβ-induced tau phosphorylation and neuronal degeneration. In addition, the factor driving the expression of these neuroprotective genes is unknown.
Cleavage of APP by α-secretase occurs within the Aβ sequence and thus precludes Aβ formation while generating an NH2 terminal, secreted protein termed sAPPα (α-secretase cleaved APP). Mice that overexpress a mutant APP as well as a protein with α-secretase activity [a disintegrin and metalloprotease 10 (ADAM10)] display reduced plaque deposition, whereas a dominant-negative ADAM10 enhanced plaque formation (Postina et al., 2004). In addition, several studies have demonstrated that sAPPα can protect cells in vitro (Mattson et al., 1993; Goodman and Mattson, 1994; Furukawa et al., 1996). This protection suggests a complex level of control located within the APP molecule that, when gone awry, may result in the pathologies associated with AD (Stein and Johnson, 2003). Here, we provide evidence to support the amyloid hypothesis in ex vivo hippocampal slice cultures as well as in vivo. In addition, we show that sAPPα can induce the expression of many neuroprotective genes and proteins, including TTR and IGF-2, and protect against Aβ toxicity.
Materials and Methods
Animals. Tg2576 mice were created as described previously (Hsiao et al., 1996). Briefly, they contain the human amyloid precursor protein 695 with the double mutation K670N and M671L (Swedish mutation) and driven by the prion protein promoter. In this study, transgenic and nontransgenic control mice were generated from C57B6/SJL N2 generation Tg2576 mice back-crossed to C57B6/SJL breeders. Mice were killed at 12 and 18 months of age. All animal protocols were approved by the Institutional Animal Use and Care Committee at the University of Wisconsin-Madison.
Hippocampal organotypic cultures. Pups from C57B6/SJL mice were anesthetized and decapitated at postnatal day 15. The brain was removed and placed in ice-cold dissecting medium containing 50% Minimum Essential Medium (Invitrogen, Carlsbad, CA), 50% HBSS (Invitrogen), 25 mm HEPES, and 36 mm glucose. Both hippocampi were dissected under a dissecting microscope and cut at 400 μm on a McIlwain tissue chopper. Slices were separated and placed on filter inserts with a 30 mm diameter and a 0.4 μm filter pore size (Millipore, Billerica, MA) held in six-well culture plates. For the first 3 d, slices were kept in Neurobasal media with B-27 supplement, 1 mm glutamine, and 1% penicillin/streptomycin (Invitrogen). Subsequently, the media were changed every 3 d using media without antibiotics. Organotypic cultures were maintained in a humidified incubator at 37°Cin5%O2 and 5% CO2 for 14-21 d.
Cell death and viability were determined on live slices using the fluorescent probes from a Live/Dead Kit (Molecular Probes, Eugene, OR). After a 40 min incubation in calcein AM (1:400) and ethidium homodimer (EthD-1; 1:1000) in PBS, slices were visualized at 200× magnification with an inverted Nikon (Tokyo, Japan) Diaphot 200 microscope using the Bio-Rad (Hercules, CA) MRC-1024 laser scanning confocal system. Random images of the neuronal fields were captured at the emission spectrum of each probe, and Scion Image software (Frederick, MD) was used to quantify the number of live and dead cells. At least three areas each, including a neuronal field, were imaged and used to determine the percentage of death for each slice. The percentage of death was calculated as the number of EthD-1-positive cells divided by the total number of EthD-1 and calcein AM-positive cells.
Detection of cell death by terminal deoxynucleotidyl transferase-mediated deoxyUTP nick end labeling and Nissl staining. Hippocampal slice cryosections from three animals per treatment were stained with terminal deoxynucleotidyl transferase-mediated deoxyUTP (dUTP) nick end labeling (TUNEL). Cells with DNA fragmentation were determined by the terminal deoxynucleotidyl transferase incorporation of FITC-12-dUTP into DNA (In Situ Cell Death Detection kit; Roche Products, Indianapolis, IN). Other sections were stained with cresyl violet acetate, and the neurons with healthy nuclei as well as the neurons with condensed and clumped chromatin were counted within the neuronal fields.
Immunohistochemistry. Mice were killed with CO2 and immediately perfused through the heart with PBS. The right hemispheres were fixed in 4% paraformaldehyde (PFA) overnight, sunk in 30% sucrose, and frozen in optimal cutting temperature (OCT) embedding medium (Tissue-Tek, Torrance, CA). Hippocampal slices were fixed in 4% PFA for 20 min, incubated in 30% sucrose overnight, and frozen in OCT embedding medium. Frozen sections with a width of 10 μm were taken through the hippocampus. Postmortem human tissue was formalin fixed, cut at 10 μm, and boiled for 10 min in a 10 mm Tris buffer, pH 1, for antigen retrieval. IGF-2 and TTR were detected with a 1:200 dilution of the polyclonal antibody against IGF-2 (F-20) or TTR (C-20; Santa Cruz Biotechnology, Santa Cruz, CA). Phospho-BAD [B-cell leukemia/lymphoma 2 (Bcl-2)-associated death protein] (Ser112; Cell Signaling Technology, Beverly, MA) and BAD (Stressgen, Victoria, British Columbia, Canada) were detected with a 1:250 dilution of the respective polyclonal antibody. Phosphorylated tau was detected with the monoclonal AT8 antibody (1:200; Research Diagnostics, Flanders, NJ) and anti-phosphotau[threonine 231 (Thr231)] (1:500; Calbiochem, La Jolla, CA). As a control, preimmune rabbit or mouse IgG (Vector Laboratories, Burlingame, CA) or goat IgG (Santa Cruz Biotechnology) was used in place of the primary antibody. A biotinylated anti-goat IgG, anti-rabbit IgG, or anti-mouse IgG was used as the secondary antibody. Finally, the Vectastain Elite ABC kit (Vector Laboratories) and tyramide conjugated to either Alexa Fluor 488 or 568 (Molecular Probes) were used to visualize the antibody staining. Anti-neuronal-specific nuclear protein (NeuN) (1:250; Chemicon, Temecula, CA) and 4G8 (1:250; Signet, Dedham, MA) together with an anti-mouse IgG secondary antibody conjugated to Alexa Fluor 488 were used to detect NeuN and Aβ. The DNA binding dyes ToPro3 (Molecular Probes) or Hoechst 33258 (Sigma, St. Louis, MO) were used to visualize nuclei. Sections were imaged using either the Bio-Rad Laser Scanning Confocal system or epifluorescence (Zeiss, Thornwood, NY). The figures are representative of the results obtained from three APPSw mice and three nontransgenic controls, three animals per treatment for hippocampal slices, or from four goat IgG and four anti-TTR antibody-infused mice.
Treatments. β-amyloid1-42 and reverse Aβ (Aβ42-1) were obtained from BACHEM (Torrance, CA). These peptides were dissolved in 0.1% NH3OH at 222 μm. For treatments, Aβ was aggregated at 50 μm in neurobasal media with B-27 supplement and 1 mm glutamine at 37°C for 24 hr. The slice culture media was then removed, and the slices were treated with 1 ml of 25 or 50 μm Aβ, reverse Aβ, or vehicle for 24 hr. Human TTR (Calbiochem) or IGF-2 (Peprotech, Rocky Hill, NJ) was added to slices together with the Aβ treatment at final concentrations of 3 μm (TTR) or 500 nm (IGF-2). sAPPα was prepared by S.L.C. and M.P.M. as described previously (Mattson et al., 1993). Unless otherwise stated, all sAPPα treatments were performed at 1 nm 48 hr before the addition of Aβ. Antibodies against TTR (C-20), IGF-2 (F-20; 4 μg/ml; Santa Cruz Biotechnology), or sAPPα (6E10; 10 μg/ml; Chemicon) were added to slices together with the sAPPα treatment. Goat IgG (4 μg/ml; Santa Cruz Biotechnology) or mouse IgG1 (10 μg/ml; Chemicon) was added as a control. The 10 aa fragment of the C-terminal region of sAPPα (592-601 of APP695; SEVKMDAEFR; Sigma) was added according to the same protocol for sAPPα.
Small interfering RNAs (siRNAs) were created based on the mouse mRNA sequences using the Ambion Silencer siRNA Construction kit (Ambion, Austin, TX). Several siRNAs were created by in vitro transcription for each silenced gene. Transfection was performed for 4 hr at 37°C before vehicle or sAPPα treatment using 25 nm siRNA combined with the polyamines in the Ambion siPORT amine transfection agent. Transfections were performed in neurobasal media with 1 mm glutamine (Invitrogen). The successful mRNA target sequences are as follows: TTR, 5′-AATCCAAATGTCCTCTGATGG-3′ and 5′-AACTGGACACCAAATCGTACT-3′; IGF-2, 5′-AAGGGGATAGAGATGTGAGAG-3′ and 5′-AAATTATGTGGTAATTCTGCA-3′; IGF-1 receptor (IGF-1R), 5′-AACGACTATCAGCAGCTGAAG-3′ and 5′-AACAGCTGGAACATGGTGGAT-3′
Microarray analysis. Total RNA was extracted from hippocampal slices of male nontransgenic mice treated with either vehicle or 1 nm sAPPα for 24 hr. Total RNA was isolated with Trizol (Invitrogen) and used to synthesize cRNA as described previously (Stein and Johnson, 2002). Fifteen micrograms of fragmented cRNA were hybridized for 16 hr at 45°C to a MG-U74Av2 array (Affymetrix, Santa Clara, CA). Affymetrix Microarray Suite 5.0 was used to scan and analyze the relative abundance of each gene from the intensity signal value. Significantly changed genes were determined using the Wilcoxon signed rank test for each comparison. Probe sets with p < 0.01 were called “increased/decreased,” probe sets with p values in the range of 0.01 < p < 0.05 were called “marginally increased/decreased,” and the remaining probe sets were called “no change.” An additional level of ranking was used to incorporate multiple comparisons such that no change = 0, marginal increase/decrease = 1/-1, and increase/decrease = 2/-2 (Li and Johnson, 2002). The final rank equaled the sum of the ranks from the nine comparisons, and the value varied from -18 to 18 for the 3 × 3 comparison in hippocampal slices. The cutoff values for the final determination of increased or decreased gene expression were set as rank ≥9 and fold change (FC) ≥1.2 for increased genes and rank less than or equal to -9 and FC less than or equal to -1.2 for decreased genes. Intensity values for gene expression were normalized to mean = 0 and variance = 1 and clustered using the self-organized map (SOM) algorithm from the Affymetrix Data Mining Tool. Data from this study are available from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO; available at http://www.ncbi.nlm.nih.gov/geo) and are listed under the following accession numbers: GSE1555 (for the hippocampal slice data series), GSM26700-5 (for the expression data from the individual arrays), and GPL81 (for the array platform).
Infusions. Eighteen-month-old APPSw mice were deeply anesthetized with an isoflurane gas anesthesia system and placed in a stereotaxic apparatus (Stoelting, Wood Dale, IL). An incision was made to expose the cranium, and a Dremel drill was used to drill through the skull. Cannulas from the Alzet Brain Infusion kit (Alzet, Cupertino, CA) were stereotaxically implanted within the right hippocampi of APPSw mice (coordinates relative to bregma: antero-posterior, -2.7 mm; mediolateral, -3.0 mm; dorsoventral, -3.0 mm) and cemented in place with cyanoacrylate. Osmotic pumps (Alzet) containing 200 μl of 100 μg/ml goat IgG or anti-TTR antibody (C-20; Santa Cruz Biotechnology) were inserted subcutaneously in the midscapular region (flow rate, 0.5 μl/hr). Treatments were diluted in artificial CSF containing 150 mm NaCl, 1.8 mm CaCl2, 1.2 mm MgSO4, 2.0 mm K2HPO4, and 10.0 mm glucose, pH 7.4. The scalps were sutured, and mice were returned to their home cages. After 2 weeks, the goat IgG and anti-TTR-infused mice were killed and immediately perfused through the heart with ice-cold PBS followed by 4% PFA.
Stereology. Four goat IgG and four anti-TTR antibody-infused mice were sectioned at a width of 50 μm through the entire hippocampus. Every sixth section was stained with cresyl violet, and neurons with healthy and intact nuclei were counted within the cornu ammonis 1 (CA1) pyramidal neuronal field using the optical fractionator technique (West and Gundersen, 1990). Cells within the CA1 pyramidal neuronal field that possessed condensed chromatin were counted separately. Counts were made at 1000× magnification on a Zeiss Axioplan 2 microscope. Each optical dissector consisted of a 30 × 30 μm counting frame with extended exclusion lines, a height of 19 μm, and top and bottom guard zones of 3 μm. Total neuron and pyknotic cell counts were obtained from a systematic random sampling of the entire CA1 using the MicroBrightField (Colchester, VT) Stereo Investigator software.
Statistical analysis. All experimental data shown were repeated at least three times. Results are expressed as mean ± SEM. Unless otherwise stated, statistical significance was determined using a two-tailed, un-paired Student's t test, and p < 0.05 was considered significant.
Results
Aβ induces neuronal death in organotypic hippocampal cultures
Organotypic hippocampal cultures maintain the architecture of the hippocampus (Fig. 1A) and contain synaptic connections with mature synaptic properties, including long-term potentiation (Muller et al., 1993; Bahr, 1995). Compared with dissociated cultures, which are typically from embryonic brain, slices represent a more relevant model of the intact adult brain. Therefore, we used this model to study Aβ-induced neuronal toxicity and protection by sAPPα. After 2 weeks in culture, the CA neuronal fields are preserved and stain positively for the postmitotic neuronal marker NeuN (Fig. 1A,C). EthD-1 is a membrane-impermeable DNA-binding dye that is excluded from live cells with an intact plasma membrane. Calcein AM is a cell-permeant dye that fluoresces in live cells with a functional intracellular esterase. When live slices treated with reverse Aβ are incubated with EthD-1 and calcein AM, many neurons in the hippocampal neuronal fields stained positively for calcein AM, whereas only a few stained positively for EthD-1 (Fig. 1B). However, treatment with either 25 μm (Fig. 1B) or 50 μm (Fig. 1C) Aβ resulted in a dramatic increase in the number of neurons that have lost membrane integrity (EthD-1 positive).
Figure 1.

Aβ-induced cytotoxicity in organotypic hippocampal cultures. A, A hematoxylin stain of a hippocampal slice after 2 weeks in vitro (25× magnification). The inset shows a CA neuronal field at 1000× magnification. B, Laser scanning confocal analysis was performed on live hippocampal slices stained with calcein AM (green) or EthD-1 (red). Treatment with 25 μm Aβ leads to many EthD-1-positive nuclei in a CA neuronal field. Scale bar, 50 μm. C, Confocal visualization reveals costaining of NeuN (green) and EthD-1 (red) in a CA neuronal field of a slice treated with 50 μm Aβ. Few EthD-1-positive cells are found in slices treated with 50 μm reverse Aβ. Scale bar, 10 μm. D, Using confocal microscopy, no TUNEL-positive cells were observed in 50 μm reverse Aβ-treated slices. However, many cells within the hippocampal neuronal fields of 50 μm Aβ-treated slices are TUNEL positive. This TUNEL staining is prevented by pretreatment of 1 nm sAPPα. Scale bar, 10 μm. E, Counts of Nissl-stained neurons within the neuronal fields of hippocampal slices reveal a dramatic increase in the percentage of cells with condensed chromatin in Aβ-treated slices. The increase in cells with chromatin condensation is prevented by pretreatment with 1 nm sAPPα. Data are presented as mean ± SEM of three slices per treatment. *p < 0.05 compared with 50 μm reverse Aβ treatment; #p < 0.05 compared with 25 μm Aβ treatment.
Consistent with neuronal death, treatment with either 25 μm Aβ (data not shown) or 50 μm Aβ (Fig. 1D), but not reverse Aβ, resulted in DNA strand breaks, as indicated by TUNEL. The transmission image from the laser scanning confocal system demonstrated that the TUNEL-positive cells were located within the neuronal fields (Fig. 1D). Finally, the percentage of apoptosis was measured within the CA neuronal fields of Nissl-stained slices (Fig. 1E). In healthy neurons, chromatin is dispersed and does not stain intensely with cresyl violet. However, intense staining is observed in dying cells that have undergone chromatin condensation, a key feature of apoptosis. Treatment with 25 or 50 μm Aβ resulted in a significant increase in the percentage of neurons with chromatin condensation. Pretreatment with 1 nm sAPPα for 48 hr prevented the Aβ-induced TUNEL staining (Fig. 1D) and nuclear pyknosis (Fig. 1E).
Aβ induces tau phosphorylation
The monoclonal antibody AT8 recognizes phosphorylated tau that is sometimes aggregated as paired helical filaments (PHFs). The epitope includes phosphorylated serine at amino acid 202. Many neurons within AD patients contain PHFs recognized by the AT8 antibody (Fig. 2C). Vehicle and reverse Aβ-treated hippocampal slices demonstrate AT8 staining of some cells around the hilus and within the stratum radiatum. Some of these cells also stained positively when mouse IgG was used as the primary antibody, suggesting that this staining may be attributable to the presence of a nonspecific antigen or to the expression of a mouse IgG-like protein. However, no staining within the hippocampal neuronal fields occurred with a control mouse IgG (data not shown). Staining with the AT8 antibody resulted in few positively stained neurons in the CA or dentate gyrus hippocampal neuronal fields of vehicle or reverse Aβ-treated slices (Fig. 2A). In contrast, when treated with 25 μm Aβ (data not shown) or 50 μm Aβ (Fig. 2A,C), several neurons within the hippocampal neuronal fields were AT8 positive. These neurons also stained positively for EthD-1, indicating Aβ-induced cellular damage. AT8 staining revealed a neuronal cell body and dendritic redistribution of tau resembling the tau pathology that occurs in AD patients (Fig. 2C). Furthermore, many AT8-positive neurons demonstrated beaded processes consistent with neuronal degeneration (Fig. 2C). Pretreatment with sAPPα prevented the Aβ-induced AT8 staining within the hippocampal neuronal fields (Fig. 2A).
Figure 2.
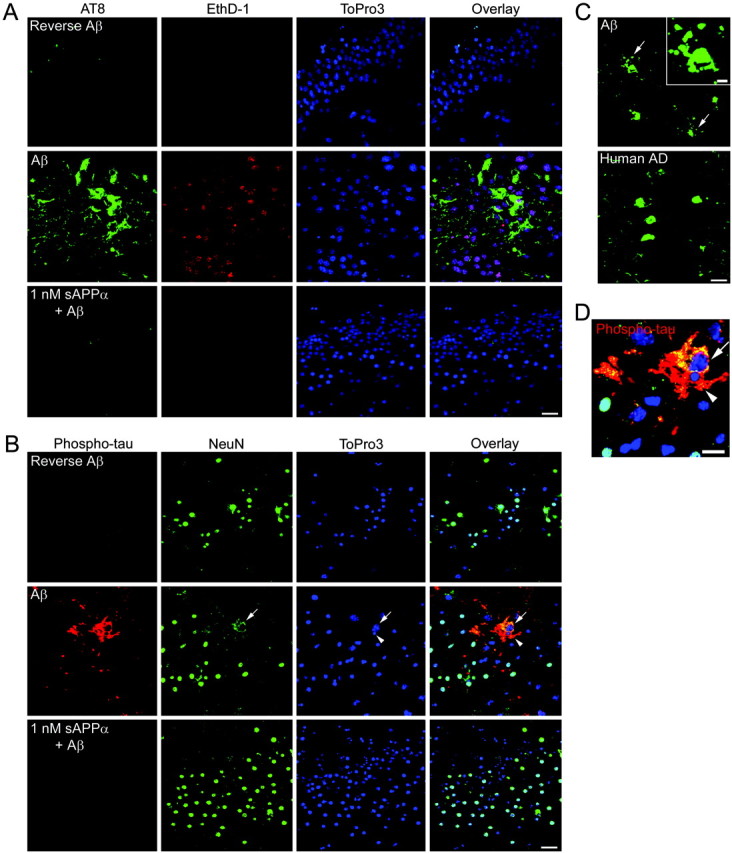
Aβ treatment induces phosphorylation of endogenous tau in mouse hippocampal neurons. A, Cells within the hippocampal neuronal fields stain positively with the AT8 antibody (green) and ethidium homodimer (red) in 50 μm Aβ-treated slices. The tau phosphorylation and plasma membrane permeability occurs infrequently in reverse Aβ-treated slices and is prevented by pretreatment with 1 nm sAPPα. The DNA-binding dye ToPro3 (blue) reveals the neuronal field architecture. Scale bar, 20 μm. B, Several neurons in 50 μm Aβ-treated slices stain positively with an antibody recognizing tau phosphorylated at Thr231 (red). The arrow points to one such neuron with NeuN staining (green) that has become diffuse and less intense. Another phospho-tau-positive neuron demonstrates diffuse NeuN staining and nuclear pyknosis (arrowhead). No neurons stain positively for phospho-tau in 50 μm reverse Aβ-treated slices, and tau phosphorylation is prevented by pretreatment with 1 nm sAPPα. Scale bar, 20 μm. C, Many neurons in Aβ-treated slices demonstrate AT8 staining in their cell bodies and in beaded processes (arrows and inset). The inset shows a higher-magnification view of one AT8 cell with beaded processes. Similar AT8 staining is observed in the CA hippocampal neurons of human patients with AD. Scale bar, 20 μm; inset, 5 μm. D, A higher magnification of the neurons containing Aβ-induced phospho-tau shown in B. Similarly, phospho-tau(Thr231) is shown in red, NeuN is shown in green, and the nuclei are shown in blue. Yellow indicates regions of phospho-tau and NeuN costaining. Scale bar, 10 μm.
No vehicle or reverse Aβ-treated slices contained neurons positive for tau phosphorylated at threonine 231. However, in slices treated with 25 μm Aβ (data not shown) or 50 μm Aβ (Fig. 2B,D), several neurons stained positively for phospho-tau(Thr231) in their cell bodies and processes. These neurons also possessed a diffuse NeuN staining (Fig. 2B,D, arrow) and pyknotic nuclei (arrowhead), indicative of neuronal degeneration. Pretreatment with 1 nm sAPPα prevented the Aβ-induced accumulation of phospho-tau(Thr231) (Fig. 2 B).
Therefore, high levels of Aβ can induce tau phosphorylation and neuronal death in mouse hippocampal slices. However, mice overexpressing mutant APP and containing micromolar levels of Aβ do not develop these pathologies. To determine whether this may be attributable to the expression of neuroprotective proteins, we examined a mouse line that overexpresses APPSw.
Neuroprotective genes and proteins are increased in aged APPSw mice but not in AD patients
In contrast to gene expression levels in AD (for review, see Stein and Johnson, 2003), the mRNA levels of several growth factors and the amyloid sequestration protein TTR are upregulated in 12-month-old APPSw mice (supplemental material, available at www.jneurosci.org). Insulin-like growth factor 2 is upregulated at both 6 months (preplaque) (Stein and Johnson, 2002) and 12 months (postplaque) (supplemental material, available at www.jneurosci.org) in APPSw mice. Furthermore, at 12 months of age, insulin is upregulated 11-fold within the hippocampus of APPSw mice (supplemental material, available at www.jneurosci.org). Both IGF-2 and insulin can bind to IGF-1R and activate a survival pathway that culminates in BAD phosphorylation.
At the protein level, 12-month-old APPSw mice possess dramatically increased levels of TTR (Fig. 3A) and IGF-2 (Fig. 3B) within the extracellular space of the hippocampus and particularly around the neuronal fields. In addition, IGF-2 is localized around some neurons and neuronal processes (Fig. 3B, arrows). BAD is a proapoptotic protein in its nonphosphorylated state. However, when phosphorylated at serine 112 or 136, BAD binds to 14-3-3 proteins and releases the anti-apoptotic BCL-2 family members BCL-2 and BCL-XL. Immunohistochemistry for phospho-BAD(112) demonstrates an increase within the NeuN-positive neurons of the hippocampal neuronal fields when compared with nontransgenic controls (Fig. 3C). In contrast, the levels of total BAD within the hippocampal neurons are unchanged between nontransgenic and APPSw mice (Fig. 3D).
Figure 3.

Increased levels of sAPPα and the neuroprotective proteins TTR, IGF-2, and phospho-BAD in 12-month-old APPSw mice. A, Immunohistochemistry with laser scanning confocal analysis demonstrates little to no TTR (red) in nontransgenic control hippocampus. However, APPSw mice possess dramatically increased levels of TTR in and around the NeuN (green)-positive hippocampal neurons of CA1. B, Little to no IGF-2 (red) is present in nontransgenic control hippocampus. APPSw mice have increased levels of IGF-2 in the extracellular space of the hippocampus and around the NeuN (green)-positive hippocampal neurons of CA1, including several neuronal processes (arrows). C, A small amount of phosphorylated BAD (red) is found within the CA1 neurons (green) of control mice. However, levels of phospho-BAD are dramatically increased within the neurons of the APPSw mice. D, Total BAD levels (red) are unchanged between nontransgenic and APPSw mice. Scale bar: (in D) A-D, 20 μm. E, TTR (green) costains with an Aβ plaque (arrowhead; red) and with intracellular Aβ in a neighboring neuron (arrow) in hippocampal sections from postmortem AD patients. Yellow indicates regions of costaining. F, A hippocampal neuron from an AD patient has accumulated intracellular Aβ that costains with TTR (arrow). Vertical sections through the plaque (E) and hippocampal neuron (F) are shown in the top panels. Scale bar: (in F) E, F, 10 μm.
In AD, many of these changes do not occur and, in fact, may be reversed. For instance, the level of total BAD is increased in the AD temporal cortex (Kitamura et al., 1998). In contrast, immunohistochemistry revealed unchanged levels of total BAD between control (n = 5) and AD (n = 6) patients within the pyramidal neurons of the hippocampus (supplemental material, available at www.jneurosci.org). IGF-2 levels were detectable around the pyramidal neurons of the hippocampal neuronal fields and were not consistently different between control and AD patients (supplemental material, available at www.jneurosci.org). Furthermore, the level of TTR has been demonstrated to be reduced in the CSF of AD patients (Serot et al., 1997). Here, little to no TTR was observed in or around the hippocampal neuronal fields of control or AD patients, as detected by immunohistochemistry of postmortem brains (supplemental material, available at www.jneurosci.org). Previous reports on the presence of TTR within plaques were conflicting (Shirahama et al., 1982; Eikelenboom and Stam, 1984). Despite this, we found that TTR deposits colocalized with many Aβ plaques within the hippocampus of all AD patients (Fig. 3E, arrowhead). Neither IGF-2 nor a control goat IgG were observed to stain Aβ plaques in the AD patients. In addition, occasional neurons within the hippocampus of AD patients contained high levels of intracellular Aβ that colocalized with TTR (Fig. 3E,F, arrow). Vertical sections demonstrate definite colocalization of TTR with Aβ (Fig. 3E,F).
sAPPα-driven gene and protein expression
One major difference between the mouse models and human AD is the overexpression of full-length APP, which may be fivefold or more in the mouse (Hsiao et al., 1996). Thus, unlike in AD, all of the cleavage products of APP likely are upregulated in the mouse models overexpressing mutant APP. Studies have demonstrated that sAPPα protects neurons against glutamate toxicity (Mattson et al., 1993; Goodman and Mattson, 1994), and we have shown protection against Aβ-induced tau phosphorylation and neuronal death (Figs. 1, 2). Thus, we reasoned that high levels of sAPPα in the APPSw mice might be responsible for the increased levels of the neuroprotective TTR, IGF-2, and phospho-BAD. In fact, nontransgenic mice have virtually undetectable levels of sAPPα in the hippocampus, whereas APPSw mice possess significantly higher amounts (Fig. 4A). The 4G8 antibody binds to full-length APP and Aβ, but not sAPPα, and does not recognize the band shown in Figure 4A. However, a weak band at ∼130 kDa that represents full-length APP is recognized by 6E10 and 4G8 in the APPSw mice (data not shown). This band is weak compared with the band for sAPPα, indicating that the majority of APP is cleaved by α- or β-secretase. Using immunoblotting, the average concentration of sAPPα in the APPSw mouse hippocampus is 1.2 ± 0.7 μm (supplemental material, available at www.jneurosci.org).
Figure 4.

sAPPα induces the expression of TTR and IGF-2 ex vivo and likely in vivo. A, Immunoblotting with the 6E10 antibody demonstrates dramatically increased sAPPα in 12-month-old APPSw mice. Below is the densitometric analysis showing the relative intensity levels of sAPPα. Values are presented as mean ± SEM (n = 4). *p < 0.05 compared with nontransgenic mice. B, SOM clustering suggests sAPPα-driven genes. The expression levels of genes and ESTs that were significantly increased (rank, ≥9) by 1 nm sAPPα in hippocampal slice cultures were clustered to identify patterns of expression. Expression patterns were examined in vehicle-treated (n = 3) and sAPPα-treated (n = 3) hippocampal slices, 6-month-old nontransgenic control (n = 3) and APPSw (n = 3) mice, and 12-month-old nontransgenic control (n = 2) and APPSw (n = 2) mice. Plotted on the ordinate is the average expression level for each cluster of genes (circular data points). The outer lines indicate the SD for each cluster. Cluster 4 (highlighted in gray) lists genes upregulated by sAPPα treatment as well as in 6- and 12-month-old APPSw mice.
Next, organotypic hippocampal slices were treated with vehicle or 1 nm sAPPα for 24 hr, the RNA was isolated, and oligonucleotide microarray analysis was performed. Samples consisted of slices treated with vehicle (n = 3) or sAPPα (n = 3), and each sample pooled 8-12 slices from four animals. The average fold change was calculated from a 3 × 3 comparison. Treatment with sAPPα resulted in a significant increase (rank, ≥9) in the expression levels of 45 genes and expressed sequence tags (ESTs) (Table 1). No genes or ESTs were significantly decreased by sAPPα treatment. Similar to the adult APPSw mice, ttr was one of the genes with the greatest fold change (8.9-fold). In addition, the mRNA levels of igf-2 and insulin-like growth factor binding protein 2 (igfbp-2) were increased by sAPPα. Several other genes involved in protective pathways such as apoptosis inhibition, detoxification, and retinol transport were upregulated by sAPPα (Table 1). For the complete microarray data, see the GEO database (available at http://www.ncbi.nlm.nih.gov/geo; accession number GSE1555).
Table 1.
Differentially expressed genes in sAPP α-treated organotypic hippocampal slice cultures
|
Classification |
Gene name |
FC |
Rank |
|---|---|---|---|
| Amyloid sequestration | Transthyretin | 8.94 ± 4.49 | 12 |
| Apoptosis | Apoptosis inhibitor 6 | 2.09 ± 1.12 | 10 |
| Calcium binding | Calmodulin-like 4 | 1.75 ± 0.42 | 12 |
| Cell cycle | Cyclin D2 | 4.22 ± 0.88 | 11 |
| Cyclin-dependent kinase 4 | 1.26 ± 0.05 | 10 | |
| Detoxification | Glutathione S-transferase, α4 | 3.52 ± 1.11 | 14 |
| Glutathione S-transferase, μ5 | 1.38 ± 0.40 | 9 | |
| Peroxiredoxin 2 | 1.19 ± 0.31 | 10 | |
| Extracellular matrix and tissue remodeling | Elastin | 1.66 ± 1.22 | 9 |
| Chondroitin sulfate proteoglycan 2 | 1.62 ± 0.21 | 11 | |
| Glycolysis | Phosphofructokinase, platelet | 1.32 ± 0.06 | 12 |
| Galactokinase 1 | 1.28 ± 0.30 | 10 | |
| Growth | Insulin-like growth factor 2 | 2.58 ± 0.42 | 14 |
| Insulin-like growth factor binding protein 2 | 1.53 ± 0.08 | 14 | |
| Immune related | Lymphocyte antigen 6 complex, locus A | 1.90 ± 0.97 | 12 |
| Intercellular adhesion molecule 2 | 1.71 ± 0.73 | 9 | |
| Ion channel | FXYD domain-containing ion transport regulator 1 | 1.20 ± 0.30 | 10 |
| Peroxisome (fatty acid transport) | ATP-binding cassette, subfamily D, member 3 | 1.35 ± 0.12 | 9 |
| Prostaglandin synthesis | Prostaglandin D2 synthase (brain) | 1.97 ± 0.53 | 12 |
| Protein catabolism | Proteasome (prosome, macropain) 28 subunit, α | 2.67 ± 0.73 | 10 |
| Praja 1, RING-H2 motif containing | 1.20 ± 0.03 | 9 | |
| Receptor | Cytokine receptor-like factor 1 | 1.54 ± 0.45 | 9 |
| Receptor (calcitonin) activity modifying protein 1 | 1.27 ± 0.06 | 9 | |
| Retionic acid binding | Cellular retinoic acid binding protein II | 2.37 ± 0.29 | 16 |
| Retinol binding protein 1, cellular | 1.69 ± 0.15 | 16 | |
| Miscellaneous | Eyes absent 4 homolog (Drosophila) | 19.26 ± 8.88 | 11 |
| H19 fetal liver mRNA | 1.95 ± 0.23 | 17 | |
| Ectonucleotide pyrophosphatase/phosphodiesterase 2 | 1.88 ± 0.82 | 11 | |
| Phosphatidic acid phosphatase 2a | 1.83 ± 0.78 | 10 | |
| Heterogeneous nuclear ribonucleoproteins methyltransferase-like 2 (Saccharomyces cerevisiae) | 1.34 ± 0.35 | 10 | |
| Topoisomerase (DNA) II α | 1.34 ± 0.96 | 10 | |
| Neuronal protein 15.6 | 1.33 ± 0.05 | 10 | |
| Ribosomal protein L8 | 1.32 ± 0.08 | 11 | |
| Ena-vasodilator stimulated phosphoprotein | 1.28 ± 0.35 | 10 | |
| Thymus cell antigen 1, θ | 1.27 ± 0.32 | 11 | |
| ADP-ribosylation-like factor 6 interacting protein 5 | 1.25 ± 0.04 | 14 | |
| STIP1 homology and U-box containing protein 1 | 1.19 ± 0.05 | 9 | |
| ESTs | AW060684 | 4.54 ± 1.78 | 9 |
| AW121164 | 2.54 ± 0.86 | 9 | |
| AW121336 | 2.50 ± 0.34 | 9 | |
| AW060956 | 1.35 ± 0.11 | 9 | |
| AW120814 | 1.32 ± 0.11 | 9 | |
| AW124069 | 1.26 ± 0.34 | 9 | |
| AW048976 | 1.24 ± 0.08 | 10 | |
|
|
AI226264 |
1.21 ± 0.03 |
10 |
Rank is based on the p value for each comparison (3 × 3). Rank values ranging from 9 to 18 indicate significantly increased gene expression. FC is expressed as mean ± SEM.
Immunohistochemistry reveals a dramatic increase in both TTR and IGF-2 within the extracellular space of 1 nm sAPPα-treated hippocampal slices. Protein levels appear highest around the hippocampal neuronal fields (supplemental material, available at www.jneurosci.org). Consistent with the increase in IGF-2 and similar to adult APPSw mice, levels of phospho-BAD(112) are increased within the NeuN-positive neurons of sAPPα-treated hippocampal slices (supplemental material, available at www.jneurosci.org).
The relative expression levels of the genes increased by sAPPα treatment in the hippocampal slice cultures were clustered together with the expression profiles of 6- and 12-month-old nontransgenic and APPSw mice. SOM clustering generated four clusters. Cluster 4 (Fig. 4B, gray) includes those genes and ESTs with low expression levels in vehicle-treated slices, 6-month-old control mice, and 12-month-old control mice and high expression levels in sAPPα-treated slices, 6-month-old APPSw mice, and 12-month-old APPSw mice. Therefore, this is a cluster of genes that is increased by sAPPα ex vivo and, likely, in vivo. Genes in this cluster include ttr, igf-2, and igfbp-2 (Fig. 4B).
TTR and IGF-2 are necessary for sAPPα-induced protection against Aβ toxicity
Organotypic hippocampal cultures were treated and incubated with EthD-1 and calcein AM. The Aβ-induced EthD-1 staining occurs within the NeuN-positive neurons of the hippocampal neuronal fields (Fig. 1C). Here, the numbers of cells stained with each fluorescent probe were counted and expressed as the percentage of cells that are EthD-1 positive and thus have lost membrane integrity (percentage of death) (Fig. 5). Treatment with either 25 or 50 μm Aβ resulted in a dramatic increase in the percentage of death compared with 50 μm reverse Aβ treatment. Pretreatment with 1 nm sAPPα for 48 hr completely protected against Aβ-induced death. When added together with Aβ, 3 μm TTR also completely protected against Aβ, whereas exogenous addition of 500 nm IGF-2 partially protected against Aβ (Fig. 5A). Furthermore, addition of an antibody that recognizes the COOH-terminal 17 aa of sAPPα (antibody 6E10) prevents the protective effect of sAPPα. This region contains part of the Aβ sequence and is unique to APP cleaved by α-secretase versus β-secretase. In fact, treatment with a 10 aa fragment that corresponds to part of this sequence mimics the protective effect of sAPPα (Fig. 5A).
Figure 5.
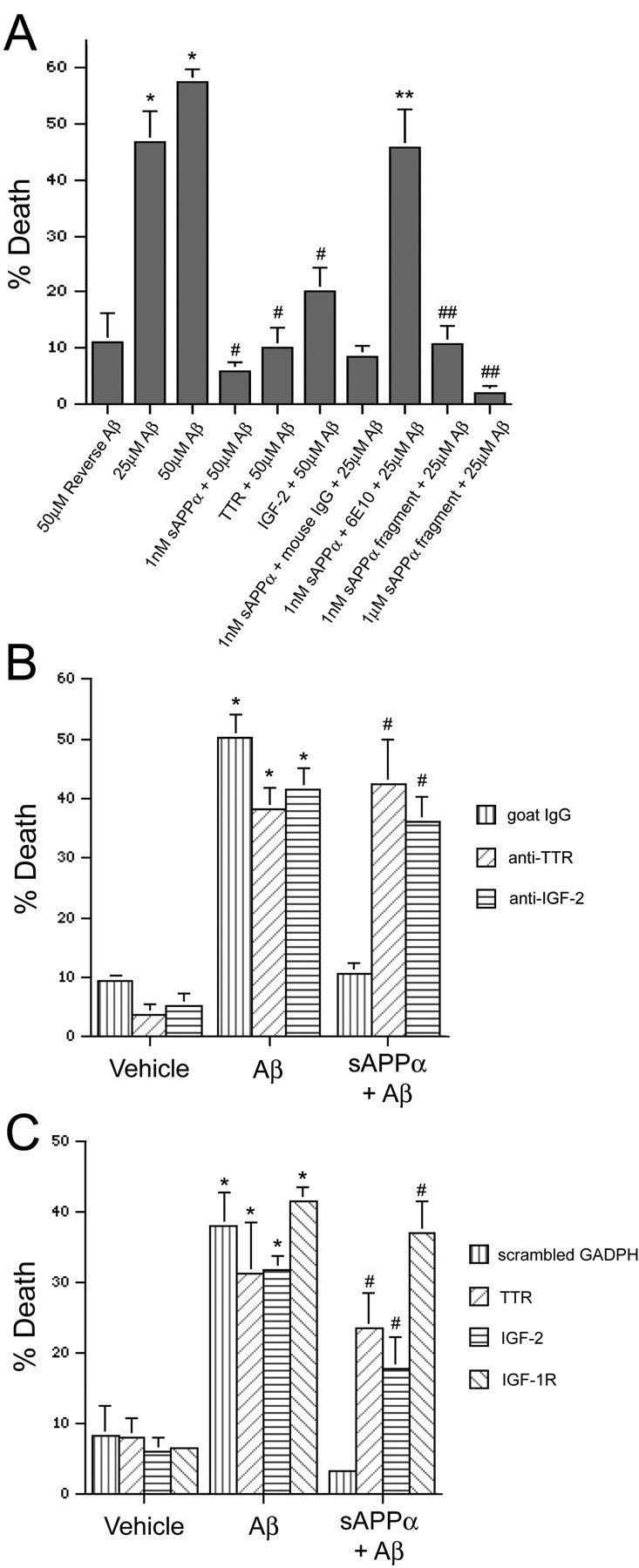
TTR and IGF-2 are necessary for sAPPα-induced protection against Aβ. The percentage of death for each treatment was quantified in neuronal fields of live hippocampal slices by counting the number of membrane-permeable, EthD-1-positive cells as well as the number of live cells that stained positively with calcein AM. Data are expressed as mean ± SEM (n = 3-5 slices per treatment). A, Aβ results in a significant increase in the percentage of death, whereas sAPPα, TTR, or IGF-2 protect against the Aβ-induced toxicity. An antibody directed against the C-terminal region of sAPPα (6E10) prevents protection by sAPPα. Finally, a fragment in the C terminus of sAPPα (SEVKMDAEFR) mimics the protective effects of sAPPα. *p < 0.05 compared with 50 μm reverse Aβ; #p < 0.01 compared with 50 μm Aβ; **p < 0.05 compared with 1 nm sAPPα plus mouse IgG plus 25 μm Aβ; ##p < 0.05 compared with 25 μm Aβ. B, Antibodies against TTR (anti-TTR) and IGF-2 (anti-IGF-2) prevent the protective effect of 1 nm sAPPα against 25 μm Aβ-induced cell death. *p < 0.01 compared with the corresponding vehicle-treated slices; #p < 0.01 compared with 1 nm sAPPα plus goat IgG plus 25 μm Aβ. C, siRNA knock-downs of TTR, IGF-2, and IGF-1R block the protective effect of 1 nm sAPPα against 25 μm Aβ-induced cell death. A scrambled glyceraldehyde-3-phosphate dehydrogenase (GAPDH) siRNA was transfected as a control. *p < 0.05 compared with the corresponding vehicle-treated slices; #p < 0.05 compared with 1 nm sAPPα plus scrambled GAPDH plus 25 μm Aβ.
Because both TTR and IGF-2 are secreted proteins, it is possible to interfere with their function by adding antibodies raised against them. Addition of a control goat IgG together with sAPPα did not interfere with sAPPα-induced protection against Aβ. However, addition of antibodies directed against TTR or IGF-2 prevented the protection by sAPPα (Fig. 5B). Supporting this, siRNA knock-down of TTR or IGF-2 partially prevented the sAPPα-induced protection against Aβ (Fig. 5C). IGF-2 may protect cells by activating the IGF-1 receptor and causing the phosphorylation of BAD. In fact, siRNA knock-down of IGF-1R also prevented protection by sAPPα (Fig. 5C). All knockdowns were confirmed by immunohistochemistry, and siRNA of either IGF-2 or IGF-1R prevented the BAD phosphorylation induced by sAPPα (data not shown). Finally, the fact that the inhibition or knock-down of either TTR or IGF-2 significantly blocked protection suggests that induction of both proteins is necessary for maximum protection against Aβ.
TTR protects APPSw mice from neurodegeneration
Mice overexpressing APPSw possess high levels of TTR within the hippocampus (Fig. 3A). Because TTR can bind Aβ and its presence is necessary for sAPPα-induced protection, we attempted to prevent the sequestration of Aβ by directly infusing an antibody against TTR into the CA1 region of the APPSw mouse hippocampus. Osmotic pumps were used to perform a continuous infusion of the anti-TTR antibody over a 2 week period. This resulted in a dramatic deposition of the anti-TTR antibody within the extracellular space of the infused hippocampus but not the noninfused hippocampus (Fig. 6A). Infusion of a control goat IgG did not result in goat IgG deposition. Moreover, APPSw mice infused with the anti-TTR antibody demonstrate markedly increased levels of Aβ in and around the hippocampal neuronal fields when compared with goat IgG or the noninfused hippocampus (Fig. 6A). This suggests that the anti-TTR antibody disrupted the TTR binding of Aβ and resulted in a locally increased Aβ load. Many plaques within the anti-TTR-infused hippocampus costained with Aβ and the anti-TTR antibody (Fig. 6B), suggesting that TTR binds to the Aβ plaques in vivo in the APPSw mice. Wild-type mice do not produce or possess any detectable TTR within their hippocampi (Fig. 3A) (Schreiber, 2002). In fact, infusion of the anti-TTR antibody into wild-type mice did not result in accumulation of the antibody (data not shown).
Figure 6.
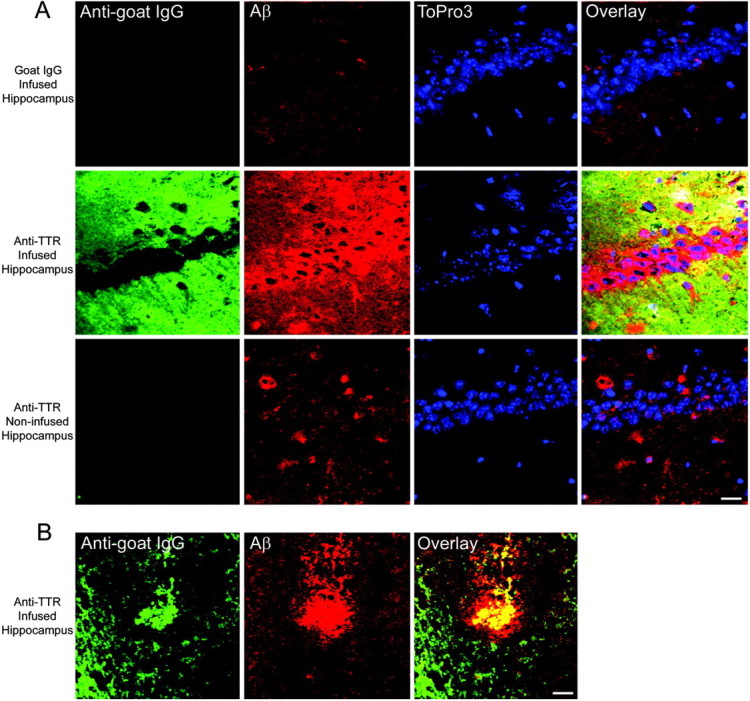
Chronic infusion of an anti-TTR antibody into the hippocampus of APPSw mice results in antibody deposition and Aβ accumulation within the infused hippocampus. Laser scanning confocal analysis was performed on hippocampal sections double immunolabeled for goat IgG or TTR (anti-goat IgG; green) and Aβ (4G8; red). Yellow indicates regions of costaining. The DNA-binding dye ToPro3 (blue) was used to stain nuclei. A, An anti-goat IgG antibody reveals that control goat IgG infused into the hippocampus is cleared after the 2 week infusion. Some Aβ staining occurs within the extracellular space and around the CA1 neurons in goat IgG-infused hippocampi and in the noninfused hippocampi of the anti-TTR-infused mice. However, mice infused with the anti-TTR antibody demonstrated a dramatic deposition of the anti-TTR antibody within the extracellular space of the infused, but not the noninfused, hippocampi. In addition, infusion of the anti-TTR antibody increased the amount of Aβ around and within the CA1 neurons near the infusion site. Scale bar, 20 μm. B, The anti-TTR antibody colabeled Aβ plaques in the infused hippocampus. Scale bar, 20 μm.
In addition to causing Aβ accumulation around the CA1 neuronal field, infusion of the anti-TTR antibody led to tau phosphorylation within many CA1 neurons (Fig. 7A,B). Several neurons immunopositive with the AT8 antibody were present within the CA1 field of the anti-TTR-infused hippocampus, whereas no cells stained positively in goat IgG-infused or noninfused hippocampi (Fig. 7A). The AT8-positive neurons often contained pyknotic nuclei (Fig. 7A, arrows and arrowhead) indicative of degeneration. An antibody that recognizes tau phosphorylated at Thr231 also stained CA1 neurons in the anti-TTR infused hippocampus. These phosphorylated tau-positive neurons costained with the neuronal marker NeuN (Fig. 7B). The number of phosphotau(Thr231)-positive cells and the intensity of staining were not altered in goat IgG and noninfused hippocampi (Fig. 7A).
Figure 7.
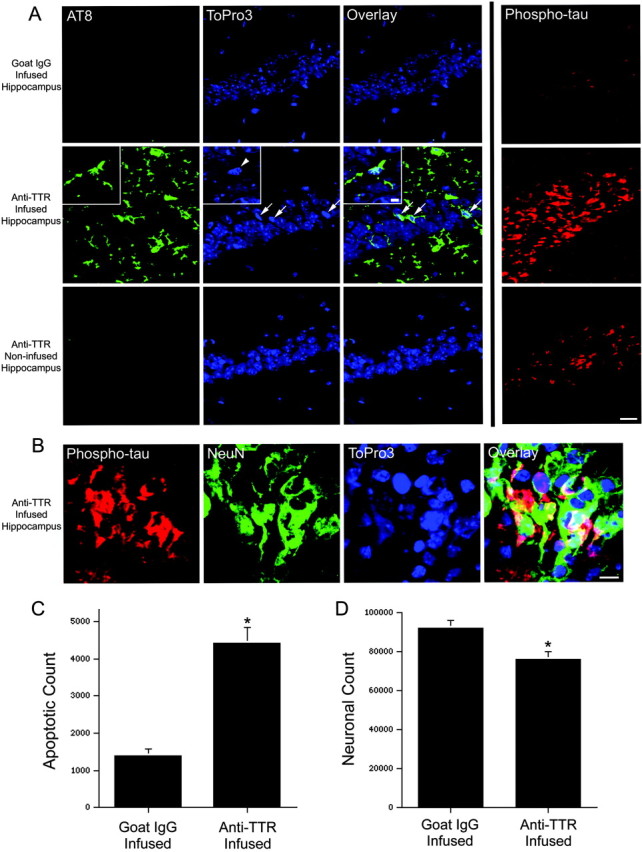
Chronic infusion of an anti-TTR antibody into the hippocampus of APPSw mice results in tau phosphorylation, apoptosis, and neuronal loss within the infused hippocampus. A, After a 2 week infusion of goat IgG, no phosphorylated tau [AT8, green; or phospho-tau(Thr231), red] is observed within the hippocampal neuronal fields. The CA1 region is shown, and the DNA-binding dye ToPro3 (blue) was used to stain nuclei. In contrast, a 2 week infusion of an antibody to TTR resulted in AT8 staining within occasional CA1 neurons. Many of these neurons possess pyknotic nuclei (ToPro3, blue; arrows). The inset shows a higher magnification within the CA1 neuronal field of a phosphorylated tau-stained neuron with a condensed nucleus (arrowhead) and beaded processes indicative of degeneration. Many neurons within the CA1 field also stain intensely for tau phosphorylated at Thr231 (phospho-tau, red). No AT8 staining and little phospho-tau(Thr231) is observed in the noninfused hippocampus. Scale bar, 20 μm; inset, 5 μm. B, CA1 neurons within the anti-TTR-infused hippocampus stain intensely for phospho-tau(Thr231) (red) and the neuronal marker NeuN (green). Scale bar, 10 μm. C, Unbiased stereology was used to determine the total number of cells with pyknotic nuclei within the infused CA1 pyramidal neuronal fields of goat IgG- (n = 4) and anti-TTR- (n = 4) infused mice. *p < 0.01, two-tailed Wilcoxon signed rank test. D, The total number of CA1 neurons was significantly reduced in anti-TTR-(n = 4) compared with goat IgG-(n = 4) infused mice. *p < 0.05, two-tailed Wilcoxon signed rank test.
To determine whether the number of apoptotic cells was increased and the total number of healthy neurons decreased in CA1 after the anti-TTR antibody infusion, unbiased stereology using the optical fractionator was performed. Apoptotic neurons are cleared within 72 hr in the in vivo nervous system (Hu et al., 1997). Despite this, after 2 weeks of continuous infusion with goat IgG, some cells with pyknotic nuclei were counted within the CA1 pyramidal neuronal field. Infusion of the anti-TTR antibody significantly increased the total number of cells with pyknotic nuclei in CA1 (Fig. 7C). As a control, no pyknotic cells could be observed in a control mouse without an infusion (data not shown). Consistent with increased neuronal apoptosis, the total number of CA1 pyramidal neurons was significantly decreased by 17.1 ± 5.3% in mice infused with the anti-TTR antibody compared with mice infused with goat IgG (Fig. 7D).
Discussion
One of the major arguments against the Aβ hypothesis in AD has been the consistent failure of mice overexpressing mutant APP to develop tau phosphorylation and neuronal loss (Masliah et al., 1996; Irizarry et al., 1997b; Takeuchi et al., 2000). However, these findings are more likely attributable to the complexity of APP. For instance, we show that the α-secretase cleaved APP can protect against the β-secretase and γ-secretase cleaved Aβ. In fact, neuroprotective pathways directed against Aβ are upregulated in the APPSw mice (Stein and Johnson, 2002). We have shown that these mice possess high levels of sAPPα and that sAPPα drives the up-regulation of the Aβ-binding protein TTR as well as the neuroprotective IGF-2 in mouse hippocampal slices. Therefore, the failure of the APP-overexpressing mouse models is likely attributable to the high levels of sAPPα, a phenomenon that does not occur in AD (Van Nostrand et al., 1992; Lannfelt et al., 1995; Olsson et al., 2003).
A recent study demonstrated that secreted APP is necessary for epidermal growth factor-induced proliferation of progenitor cells in the adult subventricular zone (Caille et al., 2004). This suggests that an endogenous function of secreted APP influences adult neurogenesis. Here, we show that sAPPα upregulates two cell cycle genes, cyclin D2 and cyclin-dependent kinase 4 (Table 1), which may contribute to progenitor proliferation in the hippocampus. Therefore, disruption of both sAPP-induced progenitor proliferation and sAPPα-induced protection against Aβ toxicity may contribute to the development of AD.
Previous studies have demonstrated various degrees of Aβ-induced tau phosphorylation in vitro (Busciglio et al., 1995; Ferreira et al., 1997) and with overexpression of human tau (Ferrari et al., 2003). Endogenous mouse tau is necessary for Aβ toxicity in dissociated hippocampal cultures (Rapoport et al., 2002). Furthermore, Aβ has been shown to increase tau phosphorylation and the number of neurofibrillary tangles (NFTs) in mice expressing a mutant tau that causes frontotemporal dementia in humans (Gotz et al., 2001; Lewis et al., 2001). However, this tau mutation alone leads to tau phosphorylation and NFTs (Lewis et al., 2000), and these studies may not address what happens in AD where tau is not mutated. We used a model system that more closely represents the architecture and chemistry of the intact brain and have shown that aggregated Aβ can induce some of the major pathological features of AD. Indeed, these slice cultures provide a means of exploring the precise mechanism by which Aβ leads to the phosphorylation of endogenous tau and neuronal death.
Transthyretin has been demonstrated to bind Aβ in vitro (Schwarzman et al., 1994; Askanas et al., 2003) and in vivo (Tsuzuki et al., 2000; Carro et al., 2002). In the hippocampus of AD patients, TTR is not markedly upregulated, as occurs in APPSw mice. However, we observed some TTR deposited within Aβ plaques and within neurons containing intracellular Aβ, demonstrating the colocalization of these two molecules in the human brain. We attempted to prevent the binding of TTR to Aβ in APPSw mice by chronic infusion into the hippocampus of an antibody that recognizes TTR. The anti-TTR antibody increased local levels of Aβ and led to tau phosphorylation, apoptosis, and neuronal loss in APPSw mice. This suggests that one endogenous function of TTR is to bind Aβ and prevent its accumulation and toxicity. Thus, TTR delivery to or induction in the brains of AD patients may act in a manner similar to Aβ immunization without the immunological sequela.
The concentration of TTR in the CSF is significantly lower in AD patients than in age-matched controls (Serot et al., 1997). This may be attributable to decreased sAPPα and may augment the Aβ accumulation that occurs in AD. Alternatively, individuals harboring polymorphisms in TTR, IGF-2, or IGF-1R may be more susceptible to the toxicity induced by Aβ and to developing sporadic AD. An analysis of a small population of AD patients did not reveal a correlation with TTR variants (Palha et al., 1996), but additional study is warranted. IGFBP-3 antagonizes the protective effects of the IGFs and may reduce IGF-1 binding to its receptor. High levels of IGFBP-3 have been demonstrated within phospho-tau-positive neurons and around some Aβ plaques of AD patients (Rensink et al., 2002). Thus, the elevated levels of IGFBP-3 in AD may shut down a normally protective pathway.
Within the cell, BAD plays a complex role in survival signaling, apoptosis, and glucose metabolism (Datta et al., 1997; Danial et al., 2003). BAD is a proapoptotic molecule that contains a BCL-2 homology 3 domain and sequesters the anti-apoptotic BCL-2 and BCL-XL. When phosphorylated at serine 112, 136, or 155 in response to survival signals, BAD releases the prosurvival BCL-2 proteins. Thus, phosphorylation of BAD couples survival signaling to the apoptotic machinery. In fact, protection by IGF-1 and brain-derived neurotrophic factor is partially dependent on BAD phosphorylation in cultured cerebellar granule neurons (Bonni et al., 1999; Datta et al., 2002). In these cells, IGF-1 acts via Akt to phosphorylate BAD at Ser136. Here, we show altered levels of BAD phosphorylated at Ser112, which can occur via protein kinase A or the MAPK (mitogen-activated protein kinase)-activated 90 kDa ribosomal S6 kinases (Bonni et al., 1999). In fact, APPSw mice have increased levels of IGF-2 and phosphorylated BAD within the hippocampus (Fig. 3) (Stein and Johnson, 2002), a phenomenon that is replicated by treatment with sAPPα in hippocampal slices. Both IGF-2 and insulin can activate IGF-1R, and knock-down of IGF-1R with siRNA prevents increased BAD phosphorylation and the protective effect of sAPPα (Fig. 5). Therefore, part of the protective effect of sAPPα likely involves IGF-2 activation of IGF-1R and phosphorylation of BAD, thus raising the threshold for induction of apoptosis by Aβ.
In conclusion, Aβ is capable of inducing tau phosphorylation and apoptosis in an ex vivo model of the mouse hippocampus. These pathologies also are observed in vivo when an antibody directed against the Aβ-binding protein TTR is infused in APPSw mice. It remains to be shown whether Aβ can induce tau phosphorylation and neuronal death in adult human tissue as well and whether the pathologies in AD can be prevented by sAPPα. If this is the case, sAPPα replacement or activation of sAPPα-induced pathways may help prevent the toxicity of Aβ and the development of AD.
Footnotes
This work was supported by National Institute of Environmental Health Sciences Grants ES08089 (J.A.J.), ES10042 (J.A.J.), and P30 AG10129 (C.D.) and by the Wisconsin Distinguished Rath Graduate Fellowship in Medicine (T.D.S.). We thank Karen Hsiao Ashe for providing Tg2576 mice, the W. M. Keck Laboratory for Biological Imaging (University of Wisconsin) for use of the laser scanning confocal system, and Matthew Slattery and the Molecular Biology Core Facility (University of Wisconsin Environmental Health Science Center) for conducting the gene array hybridizations.
Correspondence should be addressed to Jeffrey A. Johnson, University of Wisconsin-Madison, School of Pharmacy, 6125 Rennebohm Hall, 777 Highland Avenue, Madison, WI 53705-2222. E-mail: jajohnson@pharmacy.wisc.edu.
Copyright © 2004 Society for Neuroscience 0270-6474/04/247707-11$15.00/0
References
- Askanas V, Engel WK, McFerrin J, Vattemi G (2003) Transthyretin Val122Ile, accumulated Abeta, and inclusion-body myositis aspects in cultured muscle. Neurology 61: 257-260. [DOI] [PubMed] [Google Scholar]
- Bahr BA (1995) Long-term hippocampal slices: a model system for investigating synaptic mechanisms and pathologic processes. J Neurosci Res 42: 294-305. [DOI] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286: 1358-1362. [DOI] [PubMed] [Google Scholar]
- Busciglio J, Lorenzo A, Yeh J, Yankner BA (1995) Beta-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14: 879-888. [DOI] [PubMed] [Google Scholar]
- Caille I, Allinquant B, Dupont E, Bouillot C, Langer A, Muller U, Prochiantz A (2004) Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 131: 2173-2181. [DOI] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I (2002) Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med 8: 1390-1397. [DOI] [PubMed] [Google Scholar]
- Danial NN, Gramm CF, Scorrano L, Zhang CY, Krauss S, Ranger AM, Datta SR, Greenberg ME, Licklider LJ, Lowell BB, Gygi SP, Korsmeyer SJ (2003) BAD and glucokinase reside in a mitochondrial complex that integrates glycolysis and apoptosis. Nature 424: 952-956. [DOI] [PubMed] [Google Scholar]
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME (1997) Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 91: 231-241. [DOI] [PubMed] [Google Scholar]
- Datta SR, Ranger AM, Lin MZ, Sturgill JF, Ma YC, Cowan CW, Dikkes P, Korsmeyer SJ, Greenberg ME (2002) Survival factor-mediated BAD phosphorylation raises the mitochondrial threshold for apoptosis. Dev Cell 3: 631-643. [DOI] [PubMed] [Google Scholar]
- Dore S, Kar S, Quirion R (1997) Insulin-like growth factor I protects and rescues hippocampal neurons against beta-amyloid- and human amylin-induced toxicity. Proc Natl Acad Sci USA 94: 4772-4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eikelenboom P, Stam FC (1984) An immunohistochemical study on cerebral vascular and senile plaque amyloid in Alzheimer's dementia. Virchows Arch B Cell Pathol Incl Mol Pathol 47: 17-25. [DOI] [PubMed] [Google Scholar]
- Ferrari A, Hoerndli F, Baechi T, Nitsch RM, Gotz J (2003) Beta-amyloid induces PHF-like tau filaments in tissue culture. J Biol Chem 278: 40162-40168. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Lu Q, Orecchio L, Kosik KS (1997) Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar A beta. Mol Cell Neurosci 9: 220-234. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP (1996) Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem 67: 1882-1896. [DOI] [PubMed] [Google Scholar]
- Goodman Y, Mattson MP (1994) Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp Neurol 128: 1-12. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM (2001) Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293: 1491-1495. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ (2002) The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353-356. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 274: 99-102. [DOI] [PubMed] [Google Scholar]
- Hu Z, Yuri K, Ozawa H, Lu H, Kawata M (1997) The in vivo time course for elimination of adrenalectomy-induced apoptotic profiles from the granule cell layer of the rat hippocampus. J Neurosci 17: 3981-3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, McNamara M, Fedorchak K, Hsiao K, Hyman BT (1997a) APPSw transgenic mice develop age-related Abeta deposits and neuropil abnormalities, but no neuronal loss in CA1. J Neuropathol Exp Neurol 56: 965-973. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, Soriano F, McNamara M, Page KJ, Schenk D, Games D, Hyman BT (1997b) Aβ deposition is associated with neuropil changes, but not with overt neuronal loss in the human amyloid precursor protein V717F (PDAPP) transgenic mouse. J Neurosci 17: 7053-7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura Y, Shimohama S, Kamoshima W, Ota T, Matsuoka Y, Nomura Y, Smith MA, Perry G, Whitehouse PJ, Taniguchi T (1998) Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer's disease. Brain Res 780: 260-269. [DOI] [PubMed] [Google Scholar]
- Lannfelt L, Basun H, Wahlund LO, Rowe BA, Wagner SL (1995) Decreased alpha-secretase-cleaved amyloid precursor protein as a diagnostic marker for Alzheimer's disease. Nat Med 1: 829-832. [DOI] [PubMed] [Google Scholar]
- Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, Van Slegtenhorst M, Gwinn-Hardy K, Paul Murphy M, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M (2000) Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet 25: 402-405. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW, Lin WL, Chisholm L, Corral A, Jones G, Yen SH, Sahara N, Skipper L, Yager D, Eckman C, Hardy J, Hutton M, McGowan E (2001) Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science 293: 1487-1491. [DOI] [PubMed] [Google Scholar]
- Li J, Johnson JA (2002) Time-dependent changes in ARE-driven gene expression by use of a noise-filtering process for microarray data. Physiol Genomics 9: 137-144. [DOI] [PubMed] [Google Scholar]
- Masliah E, Sisk A, Mallory M, Mucke L, Schenk D, Games D (1996) Comparison of neurodegenerative pathology in transgenic mice overexpressing V717F β-amyloid precursor protein and Alzheimer's disease. J Neurosci 16: 5795-5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE (1993) Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron 10: 243-254. [DOI] [PubMed] [Google Scholar]
- Muller D, Buchs PA, Stoppini L (1993) Time course of synaptic development in hippocampal organotypic cultures. Brain Res Dev Brain Res 71: 93-100. [DOI] [PubMed] [Google Scholar]
- Olsson A, Hoglund K, Sjogren M, Andreasen N, Minthon L, Lannfelt L, Buerger K, Moller HJ, Hampel H, Davidsson P, Blennow K (2003) Measurement of alpha- and beta-secretase cleaved amyloid precursor protein in cerebrospinal fluid from Alzheimer patients. Exp Neurol 183: 74-80. [DOI] [PubMed] [Google Scholar]
- Palha JA, Moreira P, Wisniewski T, Frangione B, Saraiva MJ (1996) Transthyretin gene in Alzheimer's disease patients. Neurosci Lett 204: 212-214. [DOI] [PubMed] [Google Scholar]
- Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, Van Leuven F, Fahrenholz F (2004) A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest 113: 1456-1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapoport M, Dawson HN, Binder LI, Vitek MP, Ferreira A (2002) Tau is essential to beta-amyloid-induced neurotoxicity. Proc Natl Acad Sci USA 99: 6364-6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rensink AA, Gellekink H, Otte-Holler I, ten Donkelaar HJ, de Waal RM, Verbeek MM, Kremer B (2002) Expression of the cytokine leukemia inhibitory factor and pro-apoptotic insulin-like growth factor binding protein-3 in Alzheimer's disease. Acta Neuropathol (Berl) 104: 525-533. [DOI] [PubMed] [Google Scholar]
- Schreiber G (2002) The evolution of transthyretin synthesis in the choroid plexus. Clin Chem Lab Med 40: 1200-1210. [DOI] [PubMed] [Google Scholar]
- Schwarzman AL, Gregori L, Vitek MP, Lyubski S, Strittmatter WJ, Enghilde JJ, Bhasin R, Silverman J, Weisgraber KH, Coyle PK, Zagorski MG, Talafous J, Eisenberg M, Saunders AM, Roses AD, Goldgaber D (1994) Transthyretin sequesters amyloid beta protein and prevents amyloid formation. Proc Natl Acad Sci USA 91: 8368-8372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serot JM, Christmann D, Dubost T, Couturier M (1997) Cerebrospinal fluid transthyretin: aging and late onset Alzheimer's disease. J Neurol Neurosurg Psychiatry 63: 506-508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahama T, Skinner M, Westermark P, Rubinow A, Cohen AS, Brun A, Kemper TL (1982) Senile cerebral amyloid. Prealbumin as a common constituent in the neuritic plaque, in the neurofibrillary tangle, and in the microangiopathic lesion. Am J Pathol 107: 41-50. [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Johnson JA (2002) Lack of neurodegeneration in transgenic mice overexpressing mutant amyloid precursor protein is associated with increased levels of transthyretin and the activation of cell survival pathways. J Neurosci 22: 7380-7388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein TD, Johnson JA (2003) Genetic programming by the proteolytic fragments of the amyloid precursor protein: somewhere between confusion and clarity. Rev Neurosci 14: 317-341. [DOI] [PubMed] [Google Scholar]
- Takeuchi A, Irizarry MC, Duff K, Saido TC, Hsiao Ashe K, Hasegawa M, Mann DM, Hyman BT, Iwatsubo T (2000) Age-related amyloid beta deposition in transgenic mice overexpressing both Alzheimer mutant presenilin 1 and amyloid beta precursor protein Swedish mutant is not associated with global neuronal loss. Am J Pathol 157: 331-339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuzuki K, Fukatsu R, Yamaguchi H, Tateno M, Imai K, Fujii N, Yamauchi T (2000) Transthyretin binds amyloid beta peptides, Abeta1-42 and Abeta1-40 to form complex in the autopsied human kidney-possible role of transthyretin for abeta sequestration. Neurosci Lett 281: 171-174. [DOI] [PubMed] [Google Scholar]
- Van Nostrand WE, Wagner SL, Shankle WR, Farrow JS, Dick M, Rozemuller JM, Kuiper MA, Wolters EC, Zimmerman J, Cotman CW, Cunningham DD (1992) Decreased levels of soluble amyloid beta-protein precursor in cerebrospinal fluid of live Alzheimer disease patients. Proc Natl Acad Sci USA 89: 2551-2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Gundersen HJ (1990) Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 296: 1-22. [DOI] [PubMed] [Google Scholar]